

Abstract
BACKGROUND AND PURPOSE
Vascular endothelial growth factor (VEGF) is an angiogenic factor known to be elevated in the sputum of asymptomatic smokers as well as smokers with bronchitis type of chronic obstructive pulmonary disease. The aim of this study was to investigate whether acute exposure to cigarette smoke extract altered VEGF production in lung parenchymal cells.
EXPERIMENTAL APPROACH
We exposed human airway smooth muscle cells (ASMC), normal human lung fibroblasts (NHLF) and small airways epithelial cells (SAEC) to aqueous cigarette smoke extract (CSE) in order to investigate the effect of cigarette smoke on VEGF expression and release.
KEY RESULTS
Vascular endothelial growth factor release was elevated by sub-toxic concentrations of CSE in both ASMC and NHLF, but not in SAEC. CSE-evoked VEGF release was mimicked by its component acrolein at concentrations (10–100 µM) found in CSE, and prevented by the antioxidant and α,β-unsaturated aldehyde scavenger, N-acetylcysteine (NAC). Both CSE and acrolein (30 µM) induced VEGF mRNA expression in ASMC cultures, suggesting an effect at transcriptional level. Crotonaldehyde and 4-hydroxy-2-nonenal, an endogenous α,β-unsaturated aldehyde, stimulated VEGF release, as did H2O2. CSE-evoked VEGF release was accompanied by rapid and lasting phosphorylation of p38 MAPK (mitogen-activated protein kinase), which was abolished by NAC and mimicked by acrolein. Both CSE- and acrolein-evoked VEGF release were blocked by selective inhibition of p38 MAPK signalling.
CONCLUSIONS AND IMPLICATIONS
α,β-Unsaturated aldehydes and possibly reactive oxygen species contained in cigarette smoke stimulate VEGF expression and release from pulmonary cells through p38 MAPK signalling.
Keywords: COPD, airway smooth muscle cells, lung fibroblasts, acrolein, 4-hydroxy-2-nonenal, VEGF, p38 MAPK
Introduction
Cigarette smoke is a complex mixture of more than 4700 chemical compounds (Stedman, 1968), including free radicals and highly toxic compounds. It is generally accepted that cigarette smoke is the most important risk factor for the development and progression of chronic obstructive pulmonary disease (COPD), a syndrome characterized by progressive airflow limitation. The term COPD includes at least two main lung diseases: chronic bronchitis and emphysema, both accompanied by airways remodelling and abnormal inflammatory responses of the lungs. Increased numbers of neutrophils, macrophages and inflammatory mediators are observed in the sputum and bronchoalveolar lavage fluid of COPD patients as well as in the lung of experimental animal models exposed to cigarette smoke (D'hulst et al., 2005; Yao et al., 2008; Braber et al., 2010). Cigarette smoke (either as gas phase or aqueous extract) acutely stimulates the release of interleukin-8 (IL-8) and tumour necrosis factor-α in macrophages (e.g. Culpitt et al., 2003; Walters et al., 2005; Demirjian et al., 2006; Yang et al., 2006) as well as from lung fibroblasts (Sato et al., 1999; Numanami et al., 2003; Li et al., 2007) and airway epithelial cells (Mio et al., 1997; Masubuchi et al., 1998; Kode et al., 2006). Recently we provided evidence that the α,β-unsaturated aldehydes acrolein and crotonaldehyde are the main components of smoke responsible for IL-8 release from macrophages (Facchinetti et al., 2007) as well as from airway epithelial cells and fibroblasts through the stimulation of both extracellular signal-regulated kinase type 1 and 2 (ERK1/2) and mitogen-activated protein kinase (p38 MAPK) intracellular pathways (Moretto et al., 2009).
Vascular endothelial growth factor (VEGF) is a potent pro-angiogenic factor (Roy et al., 2006), which is widely expressed in highly vascularized organs and tissues, particularly in the lung, where it plays a key role in the maintenance of the homeostasis of the alveolar compartment (Ferrara and Bunting, 1996; Birk et al., 2008). In addition, several non-endothelial cell types also express VEGF receptors (Voelkel et al., 2006) and soluble VEGF is released by several pulmonary cells in close proximity to airways microvasculature, including smooth muscle cells (Alagappan et al., 2005), fibroblasts (Kamio et al., 2008) and epithelial cells (Thaikoottathil et al., 2009). Exposure of rodents to tobacco smoke elicits inflammatory lung responses accompanied by elevation of VEGF in the bronchoalveolar lavage fluid (Braber et al., 2010). This is consistent with the observation that, in induced sputum, VEGF levels are higher in healthy smokers as well as in patients with chronic bronchitis when compared with non-smokers (Kanazawa et al., 2003; Kanazawa, 2007; Rovina et al., 2007). Notably, elevation of VEGF levels in the sputum of asymptomatic smokers and smokers with bronchitis type of COPD correlates with smoking pack years and with IL-8 levels (Rovina et al., 2007), a chemokine up-regulated by direct cigarette smoke exposure (Facchinetti et al., 2007). Moreover, VEGF expression is increased in pulmonary muscular arteries of smokers with normal lung function when compared with non-smokers (Santos et al., 2003; Kranenburg et al., 2005). On the other hand, cigarette smoke is known to disrupt VEGF-mediated survival signalling, and decreased VEGF as well as VEGF receptor expression in COPD emphysematous lungs has been linked to increased endothelial cell death and vascular regression (Kasahara et al., 2001; Marwick et al., 2006). Therefore, it has been inferred that differences in VEGF expression may play a role in the pathophysiology of the two major phenotypes of COPD: emphysema and chronic bronchitis. Nevertheless, the mechanisms through which cigarette smoke may affect VEGF levels remain to be clarified, considering also that VEGF could be modulated by events secondary to smoking such as hypoxic conditions and/or inflammation (Rovina et al., 2007).
In order to investigate the acute effects of direct exposure to cigarette smoke on VEGF expression, we examined the effects of aqueous cigarette smoke extract (CSE) in different pulmonary cells, which are relevant both in terms of their abundance in the lung parenchyma and proximity to vasculature. We found that CSE elicited VEGF release in both airway smooth muscle cells (ASMC) and cultured normal human lung fibroblasts (NHLF) but not in small airways epithelial cells (SAEC). This effect was mimicked by the two volatile cigarette smoke components, acrolein and crotonaldehyde, and was blocked by selective p38 MAPK inhibitors.
Methods
Cell cultures
Normal human lung fibroblasts and SAEC were purchased from Lonza (Basel, CH). Normal human airway/bronchial smooth muscle cells (ASMC) were purchased from PromoCell GmbH. Both NHLF and ASMC were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, antibiotics (50 U·mL−1 penicillin and 0.05 mg·mL−1 streptomycin) and 2 mM L-glutamine in an atmosphere of 95% air and 5% CO2 at 37°C. SAEC were cultured in small airways epithelial growth factor medium (SAGM; Lonza, Basel, CH) according to supplier's instructions, as previously described (Moretto et al., 2009). NHLF, ASMC and SAEC cultures were used between passage 1 and 6.
Preparation of CSE
Cigarette smoke extract was freshly generated under standardized conditions as previously described (Sato et al., 1999; Walters et al., 2005; Facchinetti et al., 2007). Briefly, aqueous CSE was obtained by bubbling smoke generated from the combustion of four cigarettes (Marlboro Red, 12 mg tar, 0.9 mg nicotine each) through 50 mL of DMEM w/o phenol red and subsequently filtered through a 0.2 µm pore filter (Millipore). To ensure reproducibility among different CSE batches, the absorbance (optical density, OD) measured at 320 nm was used as a measure of the ‘strength’ of the extract. Subsequently, dilutions were made with culture media in order to obtain the desired absorbance. The CSE was freshly prepared on the day of the experiment and immediately used after preparation.
Cell treatments
For elisa assays, ASMC and NHLF were seeded in 48-well plates at the density of 104 cells per well in culture medium made of DMEM containing 10% fetal bovine serum, 50 U·mL−1 penicillin, 0.05 mg·mL−1 streptomycin, 2 mM L-glutamine. At ∼80–90% confluence, cells were shifted to serum-free culture medium 16–18 h before treatments. SAEC were cultured and treated in SAGM (see above). Cells were incubated with freshly prepared solutions of pharmacological compounds or vehicle 30 min before exposure to CSE, acrolein or H2O2. After 18 h, supernatants were collected and stored at −80°C for elisa determinations. For Western blot analysis, ASMC were seeded in 12-well plates at the density of 2 × 105 cells per well and cultured until confluence. After 16–18 h incubation in serum-free culture medium, media were changed and the cells were treated with either freshly prepared CSE or acrolein for increasing periods of time in serum-free medium. Whole cell extracts were prepared as described below.
Viability assay
Cell viability was assessed by using the 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay (Hansen et al., 1989). MTT was added to the culture medium at a final concentration of 0.5 mg·mL−1 and incubated at 37°C for 1 h. The reaction product of MTT was extracted in dimethylsulphoxide and the OD was spectrophotometrically measured at 570 nm, with dimethylsulphoxide as a blank. Viability was expressed as percentage of the values in vehicle-treated (basal) cultures, set to 100%.
Enzyme-linked immunosorbent assay
Culture media were collected following treatments and promptly used in the assay or stored at −80°C prior to analysis. Human VEGF was measured using a paired antibody quantitative elisa kit purchased by Invitrogen (detection limit: 5 pg·mL−1). The assays were performed according to the manufacturer's instructions. Data were expressed as pg·mL−1.
Real-time PCR
For real-time PCR studies, ASMC were seeded in serum-containing culture medium in 48-well plates at the density of 104 cells per well and grown to ∼80–90% confluence. Then, cultures were shifted to serum-free culture medium for 16–18 h and then exposed to CSE or acrolein. Subsequently, adherent cells were rinsed with cold phosphate buffered saline, and, by using the TaqMan Gene Expression Cells-to-Ct Kit (Applied Biosystems, Foster City, CA, USA), lysed to isolate and reversely transcribe total RNA. Briefly, cells were lysed in Cell Lysis solution containing DNAse I for 5 min, followed by 2 min incubation with the stop solution. Cell lysates were immediately used for RT reactions; 50 µL reverse transcription reactions were performed using 10 µL of each cell lysate, according to the manufacturer's instructions. Two sets of primers-probes were designed using the Primer Express Software version 3.0 (Applied Biosystems, Foster City, CA, USA). The chosen reporter fluorophores for TaqMan MGB probes were VIC for the endogenous reference β-actin gene (ACTB) and 6-carboxyfluorescein (FAM) for VEGF gene. The two sets of primers-probes were as follows: set 1, ACTB-FW (forward) 5′-GGCGGCACCACCATGTAC-3′, ACTB-RE (reverse) 5′-CAGGGCAGTGATCTCCTTCTG-3′, ACTB probe 5′-VIC-TGGCATTGCCGACAGG-3′; and set 2, VEGF-FW (forward) 5′-CCCACTGAGGAGTCCAACATC-3′, VEGF-RE (reverse) 5′-ACATTTGTTGTGCTGTAGGAAGCT and VEGF probe 5′-FAM-CCATGCAGATTATGC-3′. The chosen primers and probes were subjected to Basic Local Alignment Search Tool (blast) database searches to exclude sequence similarities with other genes. Real-time quantitative PCR was performed using StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). All samples were run in triplicate in a final volume of 25 µL containing 12.5 µL of 2× TaqMan Gene Expression PCR Master Mix, 300 nM of each primer, 250 nM of each probe and 4 µL of RT reaction, according to the manufacturer's instructions (Applied Biosystems, Foster City, CA, USA). Amplification conditions were: 50°C for 2 min and 95°C for 10 min, followed by 50 cycles of 95°C for 30 s and 60°C for 1 min. Relative expression of VEGF mRNA was calculated using the 2−Δ(ΔCT) comparative method, with VEGF gene normalized against the internal endogenous reference β-actin gene for the same sample.
Western blot analysis
Following cell treatments as described above, medium was removed and whole cell extracts were prepared by directly adding to the well the 1× loading buffer (0.06 M Tris-HCl pH 6.8, 2% SDS, 0.01% bromophenol blue, 10% glycerol and 0.4 M DTT) (Fermentas International Inc, Canada), boiled for 5 min and then electrophoresed on NuPAGE® Novex 12% Bis-Tris Gel (Invitrogen). After electrophoresis, proteins were transferred to a nitrocellulose membrane. The blot was blocked with 5% non-fat dry milk in TBS-T (TBS containing 0.1% Tween 20) for 1 h at room temperature. Blots were then incubated with primary antibodies to p38 MAPK (rabbit anti p38 MAPK, Cell Signalling Technology), phospho-p38 MAPK (mouse anti-phospho-p38 MAPK, Thr180/Tyr182, Cell Signalling Technology), ERK1 (rabbit anti-ERK1, Santa Cruz Biotechnology), phospho-ERK (mouse anti-phospho-ERK1/2, Thr202/Tyr204, Cell Signalling Technology), at room temperature for 1 h. Primary antibodies were visualized with IrDye 800CW goat anti-mouse or IrDye 680CW goat anti-rabbit from LI-COR Biosciences (Lincoln, Nebraska). Fluorescence signal was detected with Odyssey scanner (LI-COR Biosciences, Lincoln, Nebraska). Quantification was carried out by measuring the average intensity of the regions of interest using Odyssey Software. Ratios of phosphorylated/total amount of the protein were expressed as fold induction over the basal.
Statistical analysis
All values are expressed as means ± standard deviation (SD). Statistical analysis was performed using one-way analysis of variance (anova) followed by Dunnett's or Bonferroni's post hoc test for multigroup comparisons. Differences were considered statistically significant when P < 0.05.
Materials
U0126, Bis[amino[(2-aminophenyl)thio]methylene]butanedinitrile, was purchased from Upstate (Charlottesville, VA, USA). ERK inhibitor FR180204, 5-(2-phenyl-pyrazolo[1,5-a]pyridin-3-yl)-1H-pyrazolo[3,4-c]pyridazin-3-ylamine, p38 MAPK inhibitors SB202190, 4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazole and SB203580, 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole and phosphatidyl inositol 3-kinase (PI3K)-γ inhibitor II 5-(2,2-difluoro-benzo[1,3]dioxol-5-ylmethylene)-thiazolidine-2,4-dione, were purchased from Calbiochem (La Jolla, CA, USA), gefitinib (4-[3-chloro-4-fluoroanilino]-7-methoxy-6-[3-morpholinopropoxy] quinazoline), was purchased from Biaffin Gmbh & Co KG (Kassel, Germany), AP-18 (4-[4-chlorophenyl]-3-methyl-3-buten-2-one oxime) was purchased from Tocris Biosciences (Ellisville, MS, USA). Unless otherwise stated, all the other chemicals used in this study were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Results
Cigarette smoke elicits VEGF release in ASMC and NHLF but not in SAEC cultures
ASMC, NHLF and SAEC cell cultures were incubated with vehicle (basal) or increasing concentrations of CSE and, after 18 h, VEGF levels in the culture medium were measured. CSE elicited a concentration-dependent increase of VEGF release from both ASMC (maximal effect 588 ± 22% at CSE of OD = 0.1 over basal release) and NHLF (maximal effect 206 ± 37% at CSE of OD = 0.1 over basal release) cultures (Figure 1A, B). MTT viability test showed that CSE concentrations up to OD = 0.1 was not toxic to either ASMC or NHLF cultures (Figure 1C, D). In ASMC cultures, CSE at OD = 0.2 slightly but significantly decreased cell viability, and failed to enhance VEGF production over basal. Similarly, CSE (OD = 0.2) decreased cell viability also in NHLF cultures (Figure 1D), a phenomenon that was associated with a decreased VEGF release to below detectable levels (Figure 1B). In SAEC cultures, both CSE and acrolein, at concentrations capable of eliciting VEGF release in ASMC and NHLF cells, did not stimulate VEGF release (Figure 2A, B). Moreover, SAEC cultures appeared to be more sensitive to the cytotoxic effects of both acrolein and CSE than ASMC or NHLF cultures (Figure 2C, D).
Figure 1.
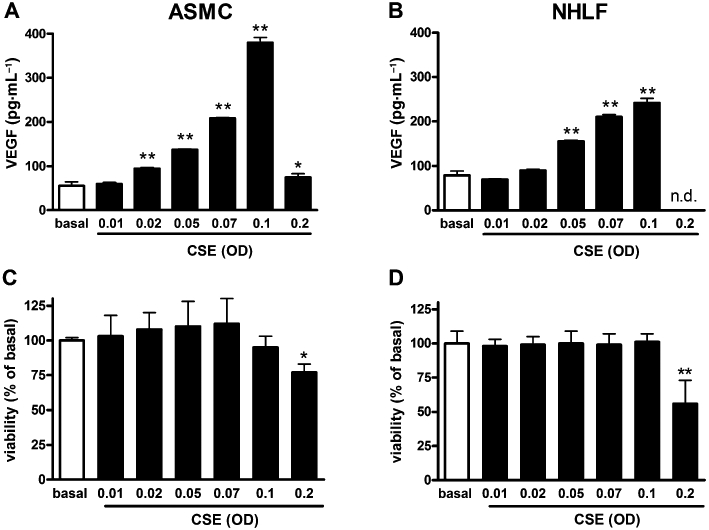
Cigarette smoke extract (CSE) enhances vascular endothelial growth factor (VEGF) release from airway smooth muscle cell (ASMC) and normal human lung fibroblast (NHLF) cells. Effects of increasing concentrations [expressed as optical density (OD) at 320 nm] of CSE on VEGF release in ASMC (A) and in NHLF (B) cultures. CSE effect on cell viability (MTT test) in ASMC (C) and NHLF (D) cultures. Each histogram is the mean ± SD of three independent experiments performed in quadruplicate. n.d., not detectable. Statistically different from basal (vehicle-treated), Dunnett's test after anova, *P < 0.05, **P < 0.01.
Figure 2.
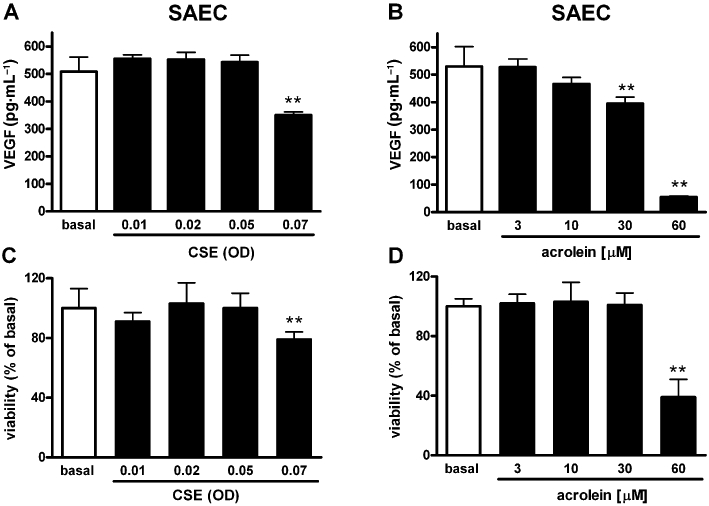
Cigarette smoke extract (CSE) does not stimulate vascular endothelial growth factor (VEGF) release from small airways epithelial cell (SAEC). Effects of increasing concentrations (expressed as optical density, OD) of CSE (A) and acrolein (B) on VEGF release in SAEC cultures. Effects on cell viability (MTT test) of CSE (C) and acrolein (D). Each histogram is the mean ± SD of three independent experiments performed in quadruplicate. Statistically different from basal (vehicle-treated), Dunnett's test after anova, **P < 0.01.
α,β-Unsaturated aldehydes mimic the effect of CSE on VEGF release
Overnight exposure to acrolein (10–100 µM) stimulated the release of VEGF from ASMC cultures in a concentration-dependent fashion. Maximal effects (1001 ± 153% over basal release) were observed at 100 µM (Figure 3A). As assessed with the MTT assay, concentrations up to 60 µM did not affect cell viability, whereas 100 µM resulted in a small but significant decrease of cell viability (Figure 3C). In NHLF cultures, acrolein significantly stimulated VEGF release (88 ± 35% over basal release) at 30 µM (Figure 3B), while higher concentrations resulted in cytotoxicity (Figure 3D). The endogenous α,β-unsaturated aldehyde 4-hydroxy-2-nonenal (4-HNE) (10–100 µM) evoked a significant release of VEGF (1051 ± 9% over basal release at 100 µM) from ASMC cultures (Figure 3E). 4-HNE up to 100 µM did not affect cell viability (Figure 3G). Crotonaldehyde (10–100 µM) elicited a concentration-dependent release of VEGF in ASMC cultures (Figure 3F) while higher concentrations resulted in significant decrement of cell viability (Figure 3H).
Figure 3.
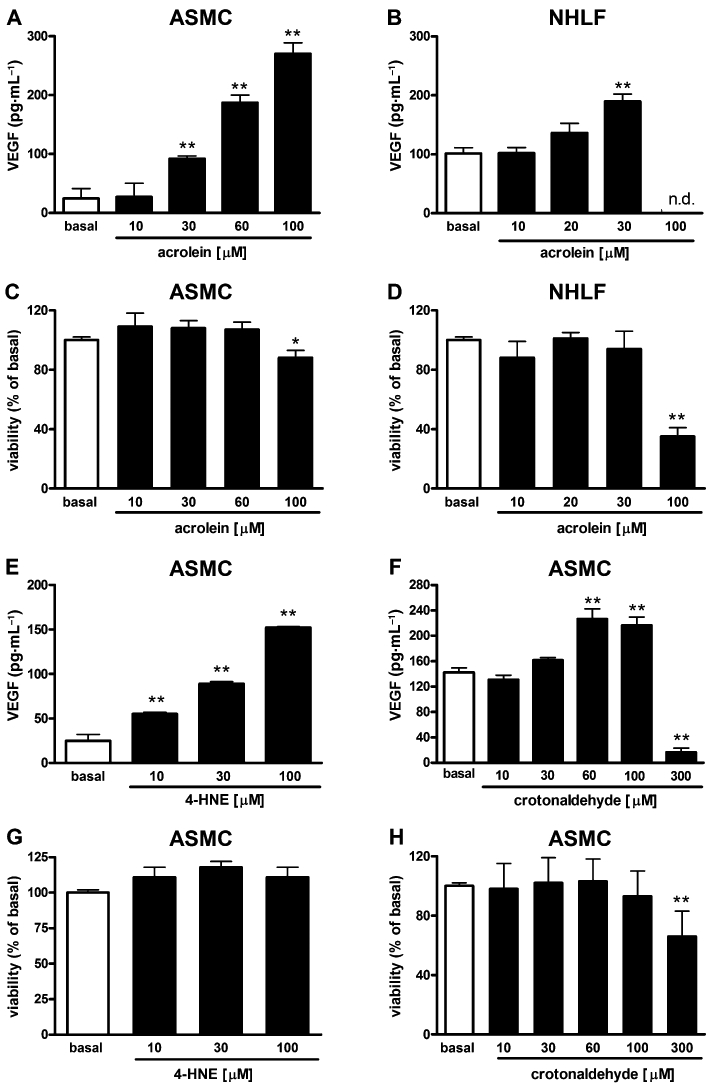
α,β-Unsaturated aldehydes stimulate vascular endothelial growth factor (VEGF) release. Effects of increasing concentrations of acrolein on VEGF release in airway smooth muscle cell (ASMC) (A) and normal human lung fibroblast (NHLF) cultures (B). Acrolein effects on cell viability (MTT test) in ASMC (C) and NHLF (D) cultures. Effects of increasing concentrations of 4-hydroxy-2-nonenal (4-HNE) (E) and crotonaldehyde (F) on the secretion of VEGF in ASMC cultures. Effects of 4-HNE (G) and crotonaldehyde (H) on cell viability (MTT test) in ASMC cultures. Each histogram is the mean ± SD of three independent experiments performed in quadruplicate. n.d., not detectable. Statistically different from basal (vehicle-treated), Dunnett's test after anova, *P < 0.05, **P < 0.01.
Both CSE and acrolein augment steady-state levels of VEGF mRNA in ASMC cultures
Airway smooth muscle cell cultures were exposed to CSE (OD = 0.075, Figure 4A) or acrolein (30 µM, Figure 4B) for 2, 5 and 24 h. Total RNAs were extracted and relative VEGF mRNA amounts were measured by TaqMan real-time PCR and normalized to β-actin mRNA. VEGF mRNA levels were found to be significantly up-regulated at 5 h in both CSE- and acrolein-treated cultures (3.2- and 4.0-fold change, respectively, over vehicle-treated basal).
Figure 4.
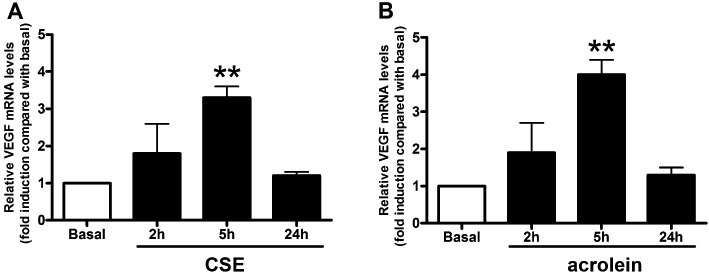
Cigarette smoke extract (CSE) and acrolein increase vascular endothelial growth factor (VEGF) mRNA expression. Airway smooth muscle cell cultures were treated with CSE (optical density = 0.075) (A) or acrolein (30 µM) (B) for the indicated periods. Total RNAs were extracted and relative VEGF mRNA amounts were measured by TaqMan real-time PCR. mRNA VEGF levels are expressed as fold change over the basal after normalizing to β-actin. Each bar is the mean ± SD of three independent experiments. Statistically different from basal (vehicle-treated), Dunnett's test after anova, **P < 0.01.
Effects of nicotinic and transient receptor potential type A1 receptor antagonists and of various kinase inhibitors against CSE-evoked VEGF release
To determine the contribution of nicotine to VEGF release induced by CSE, we exposed NHLF to CSE in the presence of mecamylamine, a selective antagonist for acetylcholine nicotinic receptors (Bacher et al., 2009). Mecamylamine (10 µM) did not affect CSE-induced VEGF release (Figure 5A). Consistent with this observation, nicotine (10 µM) failed to elevate VEGF release in NHLF cultures (Figure 5A) or in ASMC, up to 500 µM (data not shown). Acrolein is an agonist of transient receptor potential (TRP) type A1 channels (TRPA1) (Bautista et al., 2006) and can stimulate also epidermal growth factor (EGF) receptors (Takeuchi et al., 2001). However, both the TRPA1 antagonist AP-18 or the EGF receptor antagonist gefitinib (Ranson, 2002) failed to affect CSE-evoked VEGF release (Figure 5B) in NHLF cultures, ruling out the possibility that any of these receptors might account for CSE-induced VEGF release. To explore the contribution of signalling pathways known to modulate VEGF expression, we exposed ASMC cultures to the following selective inhibitors: the ERK1/2 inhibitor FR180204 (ERKi, 10 µM), the hypoxia-inducible factor type 1 (HIF-1) inhibitor 2-methoxy-estradiol (2-ME, 10 µM) (Mabjeesh et al., 2003) and 10 µM of the PI3K inhibitor II (Camps et al., 2005) as well as wortmannin (data not shown). All the above inhibitors slightly affected constitutive VEGF release (Figure 5C) and this small inhibition was also observed in cells exposed to CSE upon FR180204 and 2-ME treatments.
Figure 5.
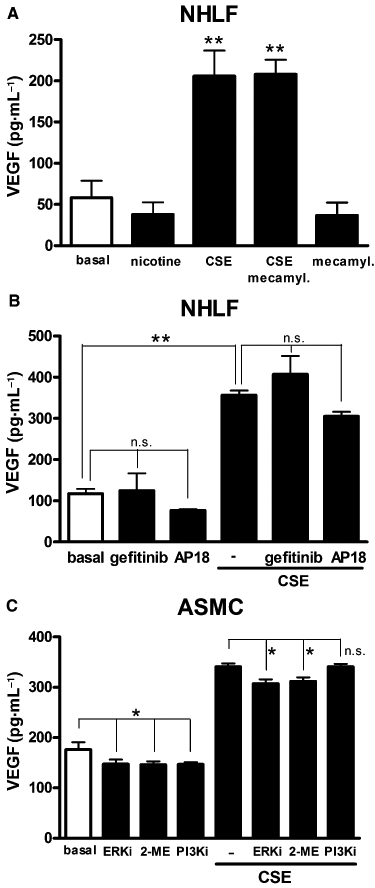
Effects of nicotinic and transient receptor potential type A1 (TRPA1) receptor antagonists and of various kinase inhibitors against cigarette smoke extract (CSE)-evoked vascular endothelial growth factor (VEGF) release. The nicotinic receptor antagonist mecamylamine (mecamyl; 10 µM) does not alter VEGF release evoked by CSE (optical density, OD = 0.075) in normal human lung fibroblast (NHLF) cultures (A). Nicotine (10 µM) did not affect constitutive VEGF release in NHLF cultures (A). Lack of effect of the EGF receptor inhibitor gefitinib (1 µM) and the TRPA1 antagonist AP-18 (10 µM) on VEGF release stimulated by CSE (OD = 0.075) in NHLF cultures (B). Effect on CSE (OD = 0.075)-evoked VEGF release of 10 µM of the following compounds: the ERK1/2 inhibitor FR180204 (ERKi), the HIF-1α inhibitor 2-methoxy-estradiol (2-ME) and the PI3K-γ inhibitor II (PI3Ki) in airway smooth muscle cell (ASMC) cultures (C). Each histogram is the mean ± SD of three independent experiments performed in quadruplicate. Dunnett's test (A) and Bonferroni's test after anova, *P < 0.05, **P < 0.01. n.s. not significant.
Effects of N-acetylcysteine and pharmacological inhibition of p38 MAPK on CSE-evoked VEGF release
Pretreatment of cells with the α,β-unsaturated aldehyde scavenger N-acetylcysteine (NAC) (0.3 mM) greatly reduced the stimulatory effect of CSE or acrolein on VEGF release in ASMC cultures (Figure 6A) and of CSE in NHLF (Figure 6B) cultures. As we have previously shown that CSE and acrolein are capable of inducing p38 MAPK phosphorylation (Moretto et al., 2009), we examined the effect of p38 MAPK inhibitors on VEGF release. For this, we used two p38 MAPK selective inhibitors: SB203580 (0.1–10 µM, Cuenda et al., 1995) and SB202190 (0.01–10 µM, Nemoto et al., 1998). Both compounds significantly inhibited CSE-induced VEGF secretion, in a concentration-dependent fashion (Figure 6C, D). Either SB203580 or SB202190 (1 µM) also completely blocked VEGF secretion induced by acrolein (Figure 6E, F). Noteworthy, both p38 MAPK inhibitors per se inhibited constitutive release of VEGF (Figure 6C–F). Because the antioxidant and aldehyde scavenger NAC reduced the effect of CSE, we tested the possibility that reactive oxygen species (ROS) contained in CSE may contribute to VEGF release (Cho et al., 2001). Indeed, ASMC cultures exposed to H2O2 (1 mM) released significantly higher concentrations of VEGF, compared with basal, suggesting that ROS may directly stimulate VEGF release (Figure 6G). Both SB203580 and SB202190 significantly inhibited H2O2-induced VEGF secretion in a concentration-dependent fashion (Figure 6G).
Figure 6.
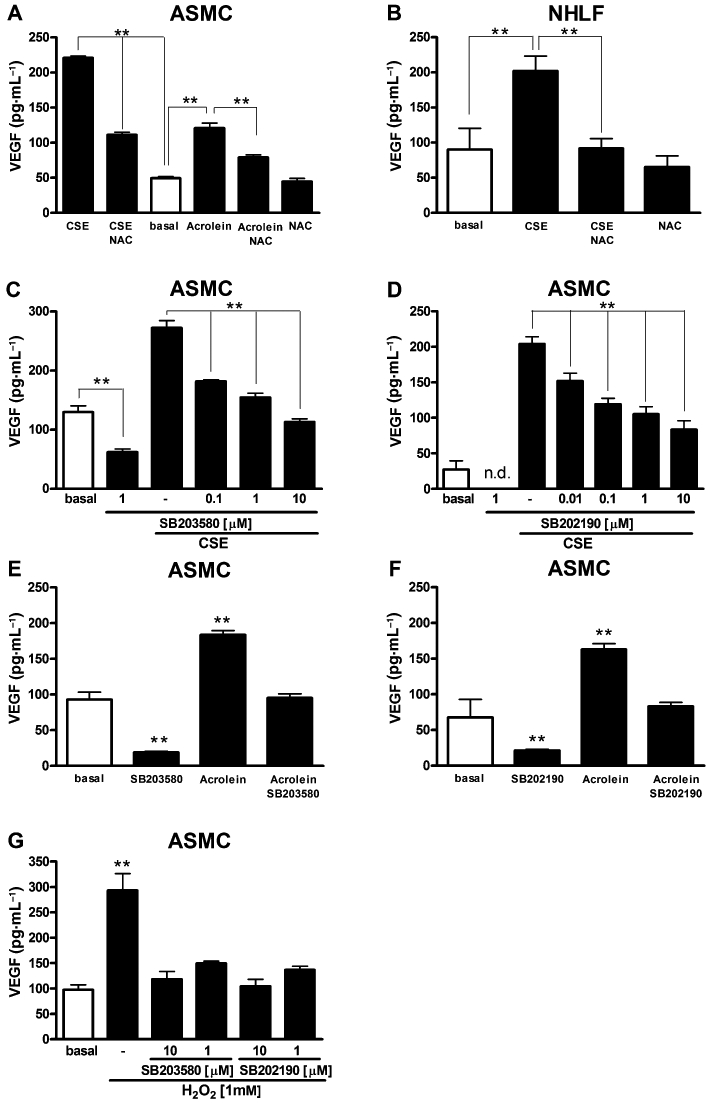
Effects of pharmacological inhibition of p38 MAPK (mitogen-activated protein kinase) and N-acetylcysteine (NAC) on vascular endothelial growth factor (VEGF) release. Effects of NAC (0.3 mM) on VEGF release elicited by cigarette smoke extract (CSE) (optical density, OD = 0.075) and acrolein (30 µM) in airway smooth muscle cell (ASMC) cultures (A). Effects of NAC (0.3 mM) on VEGF release stimulated by CSE (OD = 0.075) in normal human lung fibroblast (NHLF) cultures (B). Inhibitory effects of increasing concentrations of the p38 MAPK inhibitors SB203580 (C) and SB202190 (D) on VEGF release induced by CSE (OD = 0.075) in ASMC cultures. Effects of 1 µM of SB203580 (E) and 1 µM of SB202190 (F) on VEGF release induced by acrolein (30 µM) in ASMC cultures. Effects of H2O2 (1 mM) on VEGF release in ASMC cultures in the presence or absence of the p38 MAPK inhibitors SB203580 or SB202190 (G). Each histogram is the mean ± SD of three independent experiments performed in quadruplicate. n.d., not detectable. Bonferroni's test after anova in A–D and Dunnett's test after anova in E–G, **P < 0.01.
Effects of CSE and acrolein on p38 MAPK phosphorylation
We examined the time-dependent effect of CSE (OD = 0.1) or acrolein (60 µM) on p38 MAPK phosphorylation in ASMC cultures. As depicted in Figure 7A, CSE elicited a rapid (starting at 5 min) and marked induction of p38 MAPK phosphorylation peaking at 15 min (>16-fold increase above the basal). A decline of p38 MAPK phosphorylation to about fivefold induction with respect to the basal was observed at 2 h. A residual signal above the basal was detected up to 24 h. Acrolein elicited a rapid (starting at 5 min) and strong induction of p38 MAPK phosphorylation peaking at 60 min (>20-fold changes above the basal) (Figure 7B). At 7 h, acrolein-dependent induction of p38 MAPK phosphorylation declined to approximately sevenfold above the basal. At 24 h the signal returned to basal levels. Both CSE (Figure 7C) and acrolein (Figure 7D) elicited a rapid (starting at 5 min) induction of ERK1/2 phosphorylation peaking at 15 min (more than sixfold increase above the basal). The induction of ERK1/2 phosphorylation declined to basal levels at 4 h. Noteworthy, the p38 MAPK inhibitors SB203580 and SB202190, 1 µM each, blocked both CSE- and acrolein-evoked p38 MAPK phosphorylation (Figure 7E). p38 MAPK phosphorylation evoked by CSE or acrolein was also inhibited by 1 mM of NAC (Figure 7F).
Figure 7.
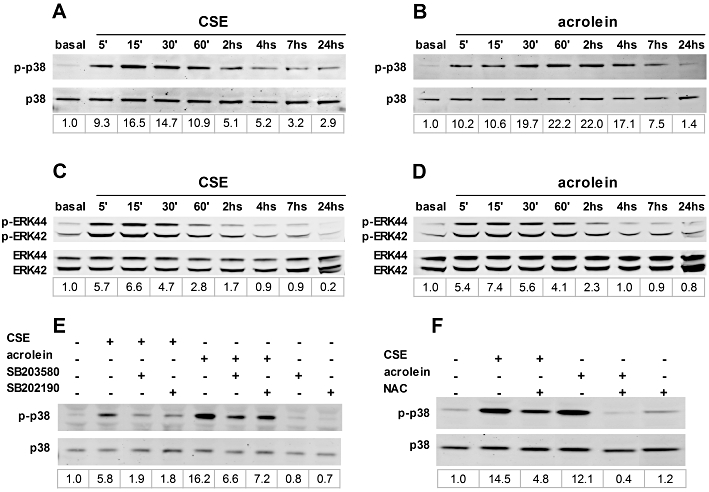
Effects of cigarette smoke extract (CSE) and acrolein on p38 MAPK (mitogen-activated protein kinase) and ERK1/2 phosphorylation. Representative blots of p38 MAPK immunoreactivity in airway smooth muscle cell cultures treated with CSE (optical density, OD = 0.1) or acrolein (60 µM) and harvested at the indicated time points (A and B, respectively). Representative blots of ERK1/2 (ERK42/44) immunoreactivity in cells treated with CSE (OD = 0.1) or acrolein (60 µM) and harvested at the indicated time points (C and D, respectively). Effects of 1 µM of the p38 MAPK inhibitors SB203580 and SB202190 (E) and of 0.3 mM of N-acetylcysteine (NAC) (F) on p38 MAPK phosphorylation induced by CSE or acrolein. Quantitative densitometry values (fold changes over basal of phospho-p38 MAPK/total p38 MAPK) are reported below each lane.
Discussion
The angiogenic factor VEGF is known to be elevated in the sputum of asymptomatic smokers and of smokers with the bronchitis type of COPD and this phenomenon correlates with smoking pack years (Rovina et al., 2007). Here, we provide for the first time evidence that CSE is capable of acutely stimulating VEGF mRNA expression and secretion in two important pulmonary cell types: ASMC and NHLF. Both NHLF and ASMC are a significant proportion of all cells present in the airways and, besides representing an important structural component of the airways wall and lung parenchyma, are also relevant sources of VEGF (Alagappan et al., 2005; Kamio et al., 2008). Considering their anatomical location with proximity to the vasculature, both ASMC and NHLF may participate in paracrine mechanisms of vascular remodelling. Interestingly, augmented VEGF release in response to cigarette smoke does not appear to be common to all pulmonary cell types as our results indicate that epithelial (SAEC) cultures exposed to CSE do not up-regulate VEGF release, a finding consistent with previous reports (St-Laurent et al., 2009; Thaikoottathil et al., 2009). Because acrolein or crotonaldehyde are two α,β-unsaturated aldehydes contained in CSE at concentrations (Facchinetti et al., 2007) capable of eliciting VEGF release, and the effect of CSE was counteracted by the α,β-unsaturated aldehyde scavenger NAC, we conclude that acrolein, crotonaldehyde and possibly other electrophilic compounds, are, at least in part, the smoke components involved in stimulation of VEGF release.
Cigarette smoking and COPD are associated with development of oxidative stress in the lung and consequent peroxidation of polyunsaturated fatty acids and generation of reactive carbonyl species, including acrolein, 4-HNE, malondialdehyde and glyoxal (Rahman and MacNee, 1996; MacNee, 2005; Aldini et al., 2006). 4-HNE in particular is markedly elevated in lungs of patients with COPD (Rahman et al., 2002). We observed that 4-HNE was capable of activating, at sub-toxic concentrations, VEGF release from ASMC cultures. Interestingly, 4-HNE also stimulated the secretion of IL-8 in macrophages (Facchinetti et al., 2007), lung fibroblasts and epithelial cells (Moretto et al., 2009) as well as eliciting neutrophil recruitment (Schaur et al., 1994). Thus, VEGF might be triggered in chronic cigarette smokers or COPD patients not only by smoke-inhaled acrolein and crotonaldehyde, but also by endogenously formed unsaturated aldehydes such as 4-HNE. In addition, by showing that H2O2 was capable of stimulating VEGF release, we strengthened the possibility that ROS and α,β-unsaturated aldehydes could cooperatively increase VEGF release.
Nicotine, one of the important constituents of cigarette smoke, has been shown to augment VEGF release in rat vascular smooth muscle cells (Kanda and Watanabe, 2007). To determine the contribution, if any, of nicotine in eliciting VEGF release we exposed NHLF to CSE in the presence of mecamylamine, a selective antagonist for nicotinic receptors. However, mecamylamine did not affect CSE-induced VEGF release. Moreover, nicotine per se failed to increase VEGF release either in NHLF or in ASMC cultures. Thus, we conclude that, in both ASMC and NHLF, nicotine does not appear to contribute to VEGF release elicited by CSE.
Intracellular signalling pathways such as p38 MAPK and ERK1/2 have been shown to modulate VEGF expression in various cell types including human vascular smooth muscle cells (Pagès et al., 2000a,b;). In particular, p38 MAPK signalling seems to play a role in the development and/or maintenance of a number of pulmonary inflammatory conditions, including asthma, cystic fibrosis, idiopathic pulmonary fibrosis and COPD (Chopra et al., 2008; Chung and Marwick, 2010). In addition, VEGF is a key target of the intracellular PI3K/Akt pathway in various cells acting mainly through the activation of HIF-1 (Gao et al., 2002; Hellwig-Bürgel et al., 2005; Riazy et al., 2009). We have previously shown that both CSE and acrolein are capable of eliciting the activation of the p38 MAPK and ERK1/2 pathways in NHLF cultures and that both these pathways are required for IL-8 production (Moretto et al., 2009). We have extended those previous findings by showing a detailed time-course of p38 MAPK and ERK1/2 phosphorylation induced by CSE or acrolein in ASMC cultures. Induction of both p38 MAPK and ERK1/2 by either CSE or acrolein in ASMC cultures is rapid. Up-regulation of p38 MAPK phosphorylation is larger and longer lasting (up to 24 h) than ERK1/2 phosphorylation, which is back to basal levels after 4 h. Our observations with Western blot analysis in ASMC cultures can be summarized as follows: (i) acrolein induced over the time a pattern of induction of p38 MAPK or ERK1/2 phosphorylation very similar to that observed with CSE; (ii) NAC blocked both CSE- and acrolein-induced p38 MAPK activation; and (iii) p38 MAPK phosphorylation elicited by acrolein and CSE was inhibited by pharmacological blockade of p38 MAPK. In addition, we observed that pharmacological blockade of p38 MAPK phosphorylation inhibits also constitutive VEGF release; a finding consistent with a previous report showing that p38 MAPK plays a key role in the stabilization of VEGF mRNA (Pagès et al., 2000a). Taken together, our data provide substantial evidence that p38 MAPK signalling was required for both CSE and acrolein to elicit VEGF release. Although also ERK1/2 signalling is activated by CSE, as well as by acrolein, it does not significantly contribute to VEGF release. In addition neither HIF-1 nor PI3K pathways appear to play a role in VEGF release in ASMC as either the anti-angiogenic agent 2-ME known to inhibit HIF-1-induced transcriptional activation of VEGF expression (Mabjeesh et al., 2003; Mooberry, 2003), or the PI3K-γ inhibitor II, as well as wortmannin (data not shown), did not affect VEGF release evoked by CSE.
A potential mechanism underlying CSE-evoked p38 MAPK phosphorylation, and possibly VEGF release, is through acrolein-mediated activation of EGF receptors (Takeuchi et al., 2001). However, gefitinib, a selective EGF receptor inhibitor (Ranson, 2002), did not modify VEGF release in NHLF cultures exposed to CSE, thus excluding the involvement of EGF receptor. We also investigated the possibility that TRPA1 cation channels, known to be activated by acrolein (Bautista et al., 2006), could mediate CSE-induced VEGF release. However, the selective TRPA1 antagonist AP-18 (Petrus et al., 2007) did not modify CSE-induced VEGF release, thus ruling out TRPA1 contribution.
Overall, our data indicate that cigarette smoke is capable of directly stimulating VEGF release from pulmonary cells, an effect that might contribute to the observed elevated levels of VEGF measured in the sputum obtained from smokers. Present findings, together with the reported stimulatory effect of CSE on IL-8 release (Moretto et al., 2009), are consistent with the positive correlation between IL-8 and VEGF levels in the sputum of healthy smokers as well as in smokers with bronchitis type of COPD (Rovina et al., 2007). VEGF, by increasing vascular permeability, may favour lung infiltration of leukocytes, including neutrophils, from the blood. On the other hand, VEGF up-regulation may also serve as a compensatory pro-survival response to the negative effects of cigarette smoke on VEGF receptor-mediated pro-survival signalling (Marwick et al., 2006; Edirisinghe et al., 2008). Indeed, COPD emphysematous patients have reduced VEGF levels in sputum, a variation that correlates with a fall in FEV1, so that reduced VEGF expression has been implicated in the destruction of alveolar wall components, including the microvasculature (Kanazawa, 2007; Siafakas et al., 2007). Therefore, it remains to be clarified in which pathological condition (e.g. bronchitis vs. emphysema), pharmacological modulation of VEGF production consequent to p38 MAPK inhibition might be beneficial.
In summary, our study provides for the first time evidence that cigarette smoke acutely stimulates release of VEGF from human pulmonary cells and that such effect is mediated chiefly by acrolein and other unsaturated aldehydes (e.g. crotonaldehyde) contained in cigarette smoke. In addition, ROS and aldehydes endogenously generated by lipid peroxidation, such as 4-HNE, can similarly elevate VEGF production from pulmonary cells. Our findings provide, at least in part, a mechanistic explanation of the observed increase of VEGF expression in the lung of chronic smokers, including COPD patients, and highlight the role played by the intracellular p38 MAPK pathway in mediating CSE-evoked VEGF release. Such mechanisms may contribute to the enhanced bronchial expression of VEGF observed in smokers and COPD patients, which may play a role in the development of vascular remodelling (Kessler et al., 2001; Santos et al., 2003). However, other studies suggest that VEGF may have a protective role against hypoxia-induced pulmonary hypertension (Partovian et al., 2000; Taraseviciene-Stewart et al., 2001). Because p38 MAPK selective inhibitors are currently under clinical development for COPD (Singh et al., 2010), a question arises as to whether VEGF inhibition resulting from prolonged treatment with this class of drugs might result in a benefit for patients or, conversely, into a worsening of their condition. On the basis of present knowledge, it may be that COPD patients characterized by the chronic bronchitis phenotype might derive more benefit from p38 MAPK inhibitors than emphysematous COPD patients.
Acknowledgments
This study was supported in part by the Italian Ministry for University and Scientific Research (MiUR), grant code: RBIP06YM29.
Glossary
Abbreviations
- ASMC
human airway smooth muscle cells
- COPD
chronic obstructive pulmonary disease
- CSE
aqueous cigarette smoke extract
- ERK1/2
extracellular signal-regulated kinase type 1 and 2
- 4-HNE
4-hydroxy-2-nonenal
- MAPK
mitogen-activated protein kinase
- 2-ME
2-methoxy-estradiol
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NAC
N-acetylcysteine
- SAEC
small airways epithelial cells
- VEGF
vascular endothelial growth factor
Conflict of interest
On behalf of all the authors, I declare that we have no conflict of interest. All the authors are employed at Chiesi Farmaceutici S.p.A.
Supporting Information
Teaching Materials; Figs 1–7 as PowerPoint slide.
References
- Alagappan VK, McKay S, Widyastuti A, Garrelds IM, Bogers AJ, Hoogsteden HC, et al. Proinflammatory cytokines upregulate mRNA expression and secretion of vascular endothelial growth factor in cultured human airway smooth muscle cells. Cell Biochem Biophys. 2005;43:119–129. doi: 10.1385/CBB:43:1:119. [DOI] [PubMed] [Google Scholar]
- Aldini G, Dalle-Donne I, Colombo R, Maffei Facino R, Milzani A, Carini M. Lipoxidation-derived reactive carbonyl species as potential drug targets in preventing protein carbonylation and related cellular dysfunction. ChemMedChem. 2006;1:1045–1058. doi: 10.1002/cmdc.200600075. [DOI] [PubMed] [Google Scholar]
- Bacher I, Wu B, Shytle DR, George TP. Mecamylamine – a nicotinic acetylcholine receptor antagonist with potential for the treatment of neuropsychiatric disorders. Expert Opin Pharmacother. 2009;10:2709–2721. doi: 10.1517/14656560903329102. [DOI] [PubMed] [Google Scholar]
- Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, et al. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell. 2006;124:1269–1282. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- Birk DM, Barbato J, Mureebe L, Chaer RA. Current insights on the biology and clinical aspects of VEGF regulation. Vasc Endovascular Surg. 2008;42:517–530. doi: 10.1177/1538574408322755. [DOI] [PubMed] [Google Scholar]
- Braber S, Henricks PA, Nijkamp FP, Kraneveld AD, Folkerts G. Inflammatory changes in the airways of mice caused by cigarette smoke exposure are only partially reversed after smoking cessation. Respir Res. 2010;11:99. doi: 10.1186/1465-9921-11-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps M, Rückle T, Ji H, Ardissone V, Rintelen F, Shaw J, et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med. 2005;11:936–943. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- Cho M, Hunt TK, Hussain MZ. Hydrogen peroxide stimulates macrophage vascular endothelial growth factor release. Am J Physiol Heart Circ Physiol. 2001;280:H2357–H2363. doi: 10.1152/ajpheart.2001.280.5.H2357. [DOI] [PubMed] [Google Scholar]
- Chopra P, Kanoje V, Semwal A, Ray A. Therapeutic potential of inhaled p38 mitogen-activated protein kinase inhibitors for inflammatory pulmonary diseases. Expert Opin Investig Drugs. 2008;17:1411–1425. doi: 10.1517/13543784.17.10.1411. [DOI] [PubMed] [Google Scholar]
- Chung KF, Marwick JA. Molecular mechanisms of oxidative stress in airways and lungs with reference to asthma and chronic obstructive pulmonary disease. Ann NY Acad Sci. 2010;1203:85–91. doi: 10.1111/j.1749-6632.2010.05600.x. [DOI] [PubMed] [Google Scholar]
- Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, et al. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- Culpitt SV, Rogers DF, Fenwick PS, Shah P, De Matos C, Russell RE, et al. Inhibition by red wine extract, resveratrol, of cytokine release by alveolar macrophages in COPD. Thorax. 2003;58:942–946. doi: 10.1136/thorax.58.11.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'hulst AI, Vermaelen KY, Brusselle GG, Joos GF, Pauwels RA. Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur Respir J. 2005;26:204–213. doi: 10.1183/09031936.05.00095204. [DOI] [PubMed] [Google Scholar]
- Demirjian L, Abboud RT, Li H, Duronio V. Acute effect of cigarette smoke on TNF-alpha release by macrophages mediated through the erk1/2 pathway. Biochim Biophys Acta. 2006;1762:592–597. doi: 10.1016/j.bbadis.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Edirisinghe I, Yang SR, Yao H, Rajendrasozhan S, Caito S, Adenuga D, et al. VEGFR-2 inhibition augments cigarette smoke-induced oxidative stress and inflammatory responses leading to endothelial dysfunction. FASEB J. 2008;22:2297–2310. doi: 10.1096/fj.07-099481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facchinetti F, Amadei F, Geppetti P, Tarantini F, Di Serio C, Dragotto A, et al. Alpha,beta-unsaturated aldehydes in cigarette smoke release inflammatory mediators from human macrophages. Am J Respir Cell Mol Biol. 2007;37:617–623. doi: 10.1165/rcmb.2007-0130OC. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Bunting S. Vascular endothelial growth factor, a specific regulator of angiogenesis. Curr Opin Nephrol Hypertens. 1996;5:35–44. doi: 10.1097/00041552-199601000-00008. [DOI] [PubMed] [Google Scholar]
- Gao N, Ding M, Zheng JZ, Zhang Z, Leonard SS, Liu KJ, et al. Vanadate-induced expression of hypoxia-inducible factor 1 alpha and vascular endothelial growth factor through phosphatidylinositol 3-kinase/Akt pathway and reactive oxygen species. J Biol Chem. 2002;277:31963–31971. doi: 10.1074/jbc.M200082200. [DOI] [PubMed] [Google Scholar]
- Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- Hellwig-Bürgel T, Stiehl DP, Katschinski DM, Marxsen J, Kreft B, Jelkmann W. VEGF production by primary human renal proximal tubular cells: requirement of HIF-1, PI3-kinase and MAPKK-1 signaling. Cell Physiol Biochem. 2005;15:99–108. doi: 10.1159/000083642. [DOI] [PubMed] [Google Scholar]
- Kamio K, Sato T, Liu X, Sugiura H, Togo S, Kobayashi T, et al. Prostacyclin analogs stimulate VEGF production from human lung fibroblasts in culture. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1226–L1232. doi: 10.1152/ajplung.00129.2007. [DOI] [PubMed] [Google Scholar]
- Kanazawa H. Role of vascular endothelial growth factor in the pathogenesis of chronic obstructive pulmonary disease. Med Sci Monit. 2007;13:189–195. [PubMed] [Google Scholar]
- Kanazawa H, Asai K, Hirata K, Yoshikawa J. Possible effects of vascular endothelial growth factor in the pathogenesis of chronic obstructive pulmonary disease. Am J Med. 2003;114:413–414. doi: 10.1016/s0002-9343(02)01562-0. [DOI] [PubMed] [Google Scholar]
- Kanda Y, Watanabe Y. Nicotine-induced vascular endothelial growth factor release via the EGFR-ERK pathway in rat vascular smooth muscle cells. Life Sci. 2007;80:1409–1414. doi: 10.1016/j.lfs.2006.12.033. [DOI] [PubMed] [Google Scholar]
- Kasahara Y, Tuder RM, Cool CD, Lynch DA, Flores SC, Voelkel NF. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med. 2001;163:737–744. doi: 10.1164/ajrccm.163.3.2002117. [DOI] [PubMed] [Google Scholar]
- Kessler R, Faller M, Weitzenblum E, Chaouat A, Aykut A, Ducoloné A, et al. ‘Natural history’ of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am J Respir Crit Care Med. 2001;164:219–224. doi: 10.1164/ajrccm.164.2.2006129. [DOI] [PubMed] [Google Scholar]
- Kode A, Yang SR, Rahman I. Differential effects of cigarette smoke on oxidative stress and proinflammatory cytokine release in primary human airway epithelial cells and in a variety of transformed alveolar epithelial cells. Respir Res. 2006;7:132. doi: 10.1186/1465-9921-7-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranenburg AR, de Boer WI, Alagappan VK, Sterk PJ, Sharma HS. Enhanced bronchial expression of vascular endothelial growth factor and receptors (Flk-1 and Flt-1) in patients with chronic obstructive pulmonary disease. Thorax. 2005;60:106–113. doi: 10.1136/thx.2004.023986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CJ, Ning W, Matthay MA, Feghali-Bostwick CA, Choi AM. MAPK pathway mediates EGR-1-HSP70-dependent cigarette smoke-induced chemokine production. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1297–L1303. doi: 10.1152/ajplung.00194.2006. [DOI] [PubMed] [Google Scholar]
- Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS, et al. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003;3:363–375. doi: 10.1016/s1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- MacNee W. Oxidants and COPD. Curr Drug Targets Inflamm Allergy. 2005;4:627–641. doi: 10.2174/156801005774912815. [DOI] [PubMed] [Google Scholar]
- Marwick JA, Stevenson CS, Giddings J, MacNee W, Butler K, Rahman I, et al. Cigarette smoke disrupts VEGF165-VEGFR-2 receptor signaling complex in rat lungs and patients with COPD: morphological impact of VEGFR-2 inhibition. Am J Physiol Lung Cell Mol Physiol. 2006;290:L897–L908. doi: 10.1152/ajplung.00116.2005. [DOI] [PubMed] [Google Scholar]
- Masubuchi T, Koyama S, Sato E, Takamizawa A, Kubo K, Sekiguchi M, et al. Smoke extract stimulates lung epithelial cells to release neutrophil and monocyte chemotactic activity. Am J Pathol. 1998;153:1903–1912. doi: 10.1016/S0002-9440(10)65704-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mio T, Romberger DJ, Thompson AB, Robbins RA, Heires A, Rennard SI. Cigarette smoke induces interleukin-8 release from human bronchial epithelial cells. Am J Respir Crit Care Med. 1997;155:1770–1776. doi: 10.1164/ajrccm.155.5.9154890. [DOI] [PubMed] [Google Scholar]
- Mooberry SL. Mechanism of action of 2-methoxyestradiol: new developments. Drug Resist Updat. 2003;6:355–361. doi: 10.1016/j.drup.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Moretto N, Facchinetti F, Southworth T, Civelli M, Singh D, Patacchini R. alpha,beta-Unsaturated aldehydes contained in cigarette smoke elicit IL-8 release in pulmonary cells through mitogen-activated protein kinases. Am J Physiol Lung Cell Mol Physiol. 2009;296:L839–L848. doi: 10.1152/ajplung.90570.2008. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Xiang J, Huang S, Lin A. Induction of apoptosis by SB202190 through inhibition of p38beta mitogen-activated protein kinase. J Biol Chem. 1998;273:16415–16420. doi: 10.1074/jbc.273.26.16415. [DOI] [PubMed] [Google Scholar]
- Numanami H, Koyama S, Nelson DK, Hoyt JC, Freels JL, Habib MP, et al. Serine protease inhibitors modulate smoke-induced chemokine release from human lung fibroblasts. Am J Respir Cell Mol Biol. 2003;29:613–619. doi: 10.1165/rcmb.2003-0113OC. [DOI] [PubMed] [Google Scholar]
- Pagès G, Berra E, Milanini J, Levy AP, Pouysségur J. Stress-activated protein kinases (JNK and p38/HOG) are essential for vascular endothelial growth factor mRNA stability. J Biol Chem. 2000a;275:26484–26491. doi: 10.1074/jbc.M002104200. [DOI] [PubMed] [Google Scholar]
- Pagès G, Milanini J, Richard DE, Berra E, Gothié E, Viñals F, et al. Signaling angiogenesis via p42/p44 MAP kinase cascade. Ann NY Acad Sci. 2000b;902:187–200. doi: 10.1111/j.1749-6632.2000.tb06313.x. [DOI] [PubMed] [Google Scholar]
- Partovian C, Adnot S, Raffestin B, Louzier V, Levame M, Mavier IM, et al. Adenovirus-mediated lung vascular endothelial growth factor overexpression protects against hypoxic pulmonary hypertension in rats. Am J Respir Cell Mol Biol. 2000;23:62–71. doi: 10.1165/ajrcmb.23.6.4106. [DOI] [PubMed] [Google Scholar]
- Petrus M, Peier AM, Bandell M, Hwang SW, Huynh T, Olney N, et al. A role of TRPA1 in mechanical hyperalgesia is revealed by pharmacological inhibition. Mol Pain. 2007;3:40–48. doi: 10.1186/1744-8069-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman I, MacNee W. Oxidant/antioxidant imbalance in smokers and chronic obstructive pulmonary disease. Thorax. 1996;51:348–350. doi: 10.1136/thx.51.4.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman I, van Schadewijk AA, Crowther AJ, Hiemstra PS, Stolk J, MacNee W, et al. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166:490–495. doi: 10.1164/rccm.2110101. [DOI] [PubMed] [Google Scholar]
- Ranson M. ZD1839 (Iressa): for more than just non-small cell lung cancer. Oncologist. 2002;7:16–24. doi: 10.1634/theoncologist.7-suppl_4-16. [DOI] [PubMed] [Google Scholar]
- Riazy M, Chen JH, Steinbrecher UP. VEGF secretion by macrophages is stimulated by lipid and protein components of OxLDL via PI3-kinase and PKCzeta activation and is independent of OxLDL uptake. Atherosclerosis. 2009;204:47–54. doi: 10.1016/j.atherosclerosis.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Rovina N, Papapetropoulos A, Kollintza A, Michailidou M, Simoes DC, Roussos C, et al. Vascular endothelial growth factor: an angiogenic factor reflecting airway inflammation in healthy smokers and in patients with bronchitis type of chronic obstructive pulmonary disease? Respir Res. 2007;8:53. doi: 10.1186/1465-9921-8-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy H, Bhardwaj S, Ylä-Herttuala S. Biology of vascular endothelial growth factors. FEBS Lett. 2006;580:2879–2887. doi: 10.1016/j.febslet.2006.03.087. [DOI] [PubMed] [Google Scholar]
- Santos S, Peinado VI, Ramirez J, Morales-Blanhir J, Bastos R, Roca J, et al. Enhanced expression of vascular endothelial growth factor in pulmonary arteries of smokers and patients with moderate chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2003;167:1250–1256. doi: 10.1164/rccm.200210-1233OC. [DOI] [PubMed] [Google Scholar]
- Sato E, Koyama S, Takamizawa A, Masubuchi T, Kubo K, Robbins RA, et al. Smoke extract stimulates lung fibroblasts to release neutrophil and monocyte chemotactic activities. Am J Physiol. 1999;277:L1149–L1157. doi: 10.1152/ajplung.1999.277.6.L1149. [DOI] [PubMed] [Google Scholar]
- Schaur RJ, Dussing G, Kink E, Schauenstein E, Posch W, Kukovetz E, et al. The lipid peroxidation product 4-hydroxynonenal is formed by and is able to attract rat neutrophils in vivo. Free Radic Res. 1994;20:365–373. doi: 10.3109/10715769409145636. [DOI] [PubMed] [Google Scholar]
- Siafakas NM, Antoniou KM, Tzortzaki EG. Role of angiogenesis and vascular remodeling in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2007;2:453–462. [PMC free article] [PubMed] [Google Scholar]
- Singh D, Smyth L, Borrill Z, Sweeney L, Tal-Singer R. A randomized, placebo-controlled study of the effects of the p38 MAPK inhibitor SB-681323 on blood biomarkers of inflammation in COPD patients. J Clin Pharmacol. 2010;50:94–100. doi: 10.1177/0091270009347873. [DOI] [PubMed] [Google Scholar]
- Stedman RL. The chemical composition of tobacco and tobacco smoke. Chem Rev. 1968;68:153–207. doi: 10.1021/cr60252a002. [DOI] [PubMed] [Google Scholar]
- St-Laurent J, Proulx LI, Boulet LP, Bissonnette E. Comparison of two in vitro models of cigarette smoke exposure. Inhal Toxicol. 2009;21:1148–1153. doi: 10.3109/08958370902926692. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Kato M, Suzuki H, Akhand AA, Wu J, Hossain K, et al. Acrolein induces activation of the epidermal growth factor receptor of human keratinocytes for cell death. J Cell Biochem. 2001;81:679–688. doi: 10.1002/jcb.1105. [DOI] [PubMed] [Google Scholar]
- Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, et al. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15:427–438. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- Thaikoottathil JV, Martin RJ, Zdunek J, Weinberger A, Rino JG, Chu HW. Cigarette smoke extract reduces VEGF in primary human airway epithelial cells. Eur Respir J. 2009;33:835–843. doi: 10.1183/09031936.00080708. [DOI] [PubMed] [Google Scholar]
- Voelkel NF, Vandivier RW, Tuder RM. Vascular endothelial growth factor in the lung. Am J Physiol Lung Cell Mol Physiol. 2006;290:L209–L221. doi: 10.1152/ajplung.00185.2005. [DOI] [PubMed] [Google Scholar]
- Walters MJ, Paul-Clark MJ, McMaster SK, Ito K, Adcock IM, Mitchell JA. Cigarette smoke activates human monocytes by an oxidant-AP-1 signaling pathway: implications for steroid resistance. Mol Pharmacol. 2005;68:1343–1353. doi: 10.1124/mol.105.012591. [DOI] [PubMed] [Google Scholar]
- Yang SR, Chida AS, Bauter MR, Shafiq N, Seweryniak K, Maggirwar SB, et al. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol. 2006;291:L46–L57. doi: 10.1152/ajplung.00241.2005. [DOI] [PubMed] [Google Scholar]
- Yao H, Edirisinghe I, Rajendrasozhan S, Yang SR, Caito S, Adenuga D, et al. Cigarette smoke-mediated inflammatory and oxidative responses are strain-dependent in mice. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1174–L1186. doi: 10.1152/ajplung.00439.2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.