

Abstract
Coupling of neurons by electrical synapses (gap junctions) transiently increases in the mammalian CNS during development. We report here that the developmental increase in neuronal gap junction coupling and expression of connexin 36 (Cx36; neuronal gap junction protein) are regulated by an interplay between the activity of group II metabotropic glutamate receptors (mGluRs) and GABAA receptors. Specifically, using dye coupling, electrotonic coupling, Western blots and small interfering RNA in the rat and mouse hypothalamus and cortex in vivo and in vitro, we demonstrate that activation of group II mGluRs augments, and inactivation prevents, the developmental increase in neuronal gap junction coupling and Cx36 expression. However, changes in GABAA receptor activity have the opposite effects. The regulation by group II mGluRs is via cAMP/PKA-dependent signaling, and regulation by GABAA receptors is via Ca2+/PKC-dependent signaling. Furthermore, the receptor-mediated upregulation of Cx36 requires a neuron-restrictive silencer element in the Cx36 gene promoter, and the downregulation involves the 3′-untranslated region of the Cx36 mRNA, as shown using reverse-transcription quantitative real-time PCR and luciferase reporter activity analysis. In addition, the methyl thiazolyl tetrazolium analysis indicates that mechanisms for the developmental increase in neuronal gap junction coupling directly control the death/survival mechanisms in developing neurons. Together, the results suggest a multitiered strategy for chemical synapses in developmental regulation of electrical synapses.
Introduction
Transient coupling of large groups of neurons by electrical synapses (gap junctions) is a general phenomenon in the developing mammalian CNS: it has been documented in different regions of the CNS (neocortex, hippocampus, hypothalamus, striatum, locus ceruleus, spinal cord, etc.) and in different species (rat, mouse, ferret, opossum, etc.) (Bennett and Zukin, 2004). Neuronal gap junction coupling increases during embryonic and/or early postnatal development and plays an important role in a number of developmental events, including neuronal differentiation, cell death, cell migration, synaptogenesis, and neural circuit formation (Allen and Warner, 1991; Walton and Navarrete, 1991; Peinado et al., 1993; Lin et al., 1998; Bani-Yaghoub et al., 1999; Personius et al., 2001; de Rivero Vaccari et al., 2007). It is believed that the contributions of gap junctions are via the passage of Ca2+, IP3, cAMP, and small molecules between the cells and coordination of metabolic and transcriptional activities in developing neurons (Kandler and Katz, 1998). In addition, gap junctions contribute to the generation of the highly synchronized excitatory electrical activity that is a hallmark of the developing brain (Feller et al., 1996; Ben-Ari, 2001). During later stages of development, neuronal gap junction coupling decreases (Arumugam et al., 2005). However, it increases in the mature CNS during neuronal injuries, such as ischemia, traumatic brain injury, inflammation, and epilepsy (Chang et al., 2000; Frantseva et al., 2002; de Pina-Benabou et al., 2005; Nemani and Binder, 2005; Thalakoti et al., 2007).
The developmental uncoupling of neuronal gap junctions occurs in response to increasing chemical synaptic (glutamatergic) transmission and activation of NMDA receptors (NMDARs) (Arumugam et al., 2005). In addition, acute modulation of neuronal gap junction coupling by a number of neurotransmitter receptors in the developing and mature CNS has been reported (Hatton, 1998). However, whether chemical neurotransmitter receptors also regulate the increases in neuronal gap junction coupling that occur during development and injuries is not known.
Here we studied the cellular and molecular mechanisms for the developmental increase in neuronal gap junction coupling. The model system for the present study is the hypothalamus, which expresses gap junctions and is critical for homeostatic regulation and coordination of cardiovascular, nervous, and endocrine functions (Saper, 2004). We demonstrate that the developmental increase in neuronal gap junction coupling and expression of connexin 36 (Cx36; neuronal gap junction protein) are regulated by an interplay between the activity of group II metabotropic glutamate receptors (mGluRs) and GABAA receptors (GABAARs). We also show that this regulation is via the neuron-restrictive silencer element (NRSE) in the Cx36 gene promoter and posttranscriptional control of Cx36 mRNA. Finally, our data suggest that the mechanisms for the developmental increase in neuronal gap junction coupling contribute to the regulation of neuronal death/survival mechanisms in developing neurons.
Materials and Methods
Animal care.
The use of animal subjects in these experiments was approved by the University of Kansas Medical Center Animal Care and Use Committee. The experiments were conducted in accordance with National Institutes of Health guidelines. Sprague Dawley rats, Cx36 knock-out mice (C57BL/6 background strain), and wild-type mice (C57BL/6) of either sex were used. The Cx36 knock-out was originally created by Dr. David Paul (Harvard Medical School, Boston, MA). Mice were genotyped as described previously (de Rivero Vaccari et al., 2007).
Pharmacological treatments of animals.
Rat pups of either sex received daily subcutaneous injections of drugs in three increasing sets of concentrations, administered, respectively, on postnatal day 1 (P1)–P5 (dissolved in 20 μl of sterile saline), P6–P10 (40 μl), and P11–P15 (60 μl): (1R,4R,5S,6S)-4-amino-2-oxabicyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY379268), 3, 5, and 7 mg/kg; (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid (LY341495), 1.5, 2, and 2.5 mg/kg; muscimol, 0.25, 0.5, and 0.75 mg/kg; and bicuculline, 1, 1.25, and 1.5 mg/kg. Control animals received the corresponding volumes of sterile saline. The forebrains were dissected and weighted on P15, and only animals that received muscimol administrations demonstrated slightly reduced forebrain weight (data not shown). However, because the loading of protein in all Western blots was normalized (i.e., 50 μg of protein per lane) and the expression of tubulin and glycer-aldehyde-3-phosphate dehydrogenase (GAPDH) in the hypothalamus was not different between the control and muscimol-treated animals (data not shown), we concluded that a decrease in Cx36 expression in muscimol-treated rats was rather specific, i.e., it reflected that the GABAAR-dependent downregulation of Cx36 was not attributable to a nonspecific decrease in the amount of protein (e.g., because of decrease in the size of the forebrain).
Brain slice and culture preparations and culture treatments.
To prepare brain slices, rats were anesthetized (Nembutal; 70 mg/kg, i.p.), killed, the brains were removed, and 400-μm-thick coronal hypothalamic slices were prepared (at 2–4°C) and kept (at 20–22°C) in artificial CSF as described previously (Belousov and van den Pol, 1997). Cultures were prepared as reported previously (Belousov et al., 2001) from the medial hypothalamus or somatosensory cortex obtained from day 18–19 (rat) or day 16–17 (mouse) embryos. Pregnant animals were anesthetized (Nembutal; 70 mg/kg, i.p.) before embryos were removed. After disaggregation using papain, neurons were plated on glass coverslips and raised in Neurobasal medium (Invitrogen, catalog #21103), in which the percentage of neurons reaches ∼95% (Wang et al., 2008). The medium was supplemented with B-27 (Invitrogen, catalog #17504) and 0.5 mm l-glutamine. The culture medium was changed twice a week. Drug and small interfering RNA (siRNA) treatments and luciferase construct transfections were performed using sister cultures. Cell survival was estimated by analyzing the number of live neurons as described previously (Belousov et al., 2001), and none of the chronic treatments reduced neuronal survival compared with the control (only luciferase construct transfections and siRNA treatments induced neurodegeneration in cultures after, respectively, 5 and 7 d; therefore, in those experiments cells were exposed to the agents for not more than 4 and 6 d, respectively). Dendritic processes were analyzed using calcein AM staining and were only affected by activation of GABAARs (slight increase) and inactivation of GABAARs (slight decrease). However, because these changes were opposite to those in Cx36 expression and dye coupling, we concluded that the changes in dendrites were not responsible for alterations in the expression of gap junctions. For chronic GABAAR blockade in vitro, we followed a previously proposed protocol (Muller et al., 1993), where for maximal effect the use of both bicuculline and picrotoxin was proposed.
Dye coupling.
Dye coupling in slices and cultures was performed as described in detail previously (Arumugam et al., 2005). Briefly, the pipette solution contained the following (in mm): 145 KMeSO4, 10 HEPES, 2 MgCl2, 0.1 CaCl2, 1.1 EGTA, 2 Na-ATP, and 0.3 Na-GTP, with 0.2% neurobiotin (Vector Laboratories, catalog #SP-1120, MW323, gap-junction-permeable dye) and 0.04% dextran Alexa Fluor 594 (Invitrogen, catalog #D22913, MW10,000, gap-junction-impermeable dye), pH 7.2, with 3–7 MΩ electrode resistance. Cells were patched using Multiclamp 700-B amplifier and pCLAMP10 software (Molecular Devices). On the day of preparation, slices were randomly numbered and the number and condition (a drug treatment) were documented in a database (Access) for later identification. Magnocellular neurons in the hypothalamic paraventricular nucleus (PVN) and supraoptic nucleus (SON) were labeled after initial identification based on their location, size, and electrophysiological properties (Arumugam et al., 2005). Only one neuron per slice was labeled as reported (Arumugam et al., 2005). After labeling, the slices were fixed, processed, and stained with fluorescein avidin D (FITC; 1:200; Vector Laboratories). Alexa Fluor 594 fluorescence and neurobiotin staining were visualized using, respectively, Texas Red and FITC filters in a Nikon 80i fluorescent microscope, a Photometrics ES2 camera, and OpenLab software (Improvision). The incidence of gap junction coupling was analyzed blindly for experimental groups as described previously (Arumugam et al., 2005). For cultures, because different cell types are morphologically indistinguishable, and electrophysiological characterization of cell types is not elaborated, neurons in cultures were chosen randomly. Neurons were labeled, stained, and analyzed as described above for slices.
Electrotonic coupling.
To determine electrotonic coupling, dual whole-cell current-clamp recordings were conducted in cultures from pairs of randomly chosen neurons. Test current steps (500 ms, −100 pA) were applied to cell 2 (injected cell), and electrotonic responses were detected in cell 1 (noninjected cell). Recordings were done at a holding potential of −65 mV. Data were monitored using an electrophysiology approach (see above) and analyzed off-line with Clampfit 10 (Molecular Devices). The coupling coefficient was calculated as the response amplitude in the noninjected cell (cell 1) divided by the amplitude in the injected cell (cell 2). Cells were considered as coupled if the coupling coefficient was above 1.6%.
Western blots.
Experiments were performed as reported in detail previously (Arumugam et al., 2005). Briefly, hypothalamic tissue or cultured cells were homogenized in a lysis buffer, and total protein was determined using the Bio-Rad DC protein assay method. Fifty micrograms of protein were loaded in each lane, transferred to a 0.45 μm polyvinylidene difluoride membrane, and processed with a blocking solution and antibodies. Rabbit anti-Cx36 (0.5 μg/ml; Zymed, catalog #51-6300), rabbit anti-connexin 43 (Cx43; 0.2 μg/ml; Zymed, catalog #71-0700), rabbit anti-mGluR2 (0.5 μg/ml; Millipore, catalog #AB9209), rabbit anti-mGluR3 (0.5 μg/ml; Sigma, catalog #G1545), mouse anti-Tubulin (1:10,000; Sigma, catalog #T6793), and rabbit anti-GAPDH (1:10,000; Cell Signaling Technology, catalog #2118) were used as the primary antibodies, and they were visualized with horseradish-peroxidase-conjugated anti-rabbit (1:10,000; Zymed, catalog #G21234) or anti-mouse (1:10,000; Zymed, catalog #G21040) antibodies. Signals were enhanced using ECL detection reagents (GE Healthcare). Band optical density was determined by using Quantity One quantification analysis software version 4.5.2 (Bio-Rad). All optical density signals were normalized relative to tubulin or GAPDH, and experimental samples were compared to controls (set at 1.0). Tubulin and GAPDH levels per unit of total protein did not vary significantly among samples used in this study.
siRNA.
The mGluR2, mGluR3, and Cx36 siRNAs were purchased from Dharmacon RNAi Technologies (catalog #M-080176-00, L-094437-01 and L-090683-00, respectively). Each siRNA consisted of four pooled 19-nucleotide duplexes and was used in a final concentration of 50 nm. mGluR2 and mGluR3 siRNA transfections were performed on day in vitro 3 (DIV3) using Lipofectamine 2000 (Invitrogen), and cells were processed on DIV7 for Western blot analysis. Cx36 siRNA transfections were conducted on DIV10, and dye coupling was analyzed on DIV15. All transfections effectively reduced protein levels. Scrambled siRNAs were used as controls and were ineffective.
Reverse-transcription quantitative real-time PCR.
Experiments were performed as reported previously (Al-Kandari et al., 2007). Total RNA was isolated from cultures using Trizol method (Invitrogen). Total RNA (1 μg) was reverse transcribed with oligo-dT primers and the SuperScript II kit (Invitrogen) according to the manufacturer's instructions. One microliter of the reverse transcription reaction material was used as template for reverse-transcription quantitative real-time PCR (RT-qPCR) using a Bio-Rad iCycler in a total volume of 20 μl with SYBR Green PCR Master Mix (Applied Biosystems) and amplified for 40 cycles for 15 s at 95°C and for 20 s at 60°C. The following primer pairs were used: rat Cx36, 5′-CTATGTGTGAAAGGGCAGGTT-3′ (sense) and 5′-AGCACTACGCAAATGAGGGCAA-3′ (antisense); rat GAPDH, 5′-CAAGGCTGTGGGCAAGGTCAT-3′ (sense) and 5′-GCAGGTTTCTCCAGGCGGCAT-3′ (antisense). RT-qPCR was performed using at least four separately prepared sets of culture samples. For each sample set, Cx36 mRNA signals were normalized to GAPDH mRNA signals, and normalized values were compared to controls (set at 1.0).
Luciferase reporter activity analysis.
Rat Cx36 gene promoter fragments were PCR amplified from −984 to +115 relative to the transcription start site of the Cx36 gene (GeneID 50564; official name, Gjd2) from a rat bacterial artificial chromosome containing the gene. Primers used were as follows: 5′-GCGAGATCTCGCTGTGCATCCGGAGGCAGC-3′ for the antisense primer, 5′-GCGGCTAGCCCCTGGTTCGCTGCTAGGCAC-3′ for the sense primer. The PCR products digested with NheI and BglII were cloned into the luciferase reporter plasmid pGL3basic (Promega). Site-directed mutagenesis was performed to produce an NRSE deleted plasmid using the Transformer Site-Directed Mutagenesis Kit (Clontech) and the following primers: 5′-TAAAATCGATAAGGGTCCGTCGACCGATGC-3′ for the selection primer and 5′-GAGACTGCGGGAGTCCGAGGTGCTGTCCAG-3′ for the mutagenic primer (the mutated nucleotide is underlined). The full-length 3′-untranslated region (3′UTR) of the rat Cx36 mRNA (GenBank accession number NM 019281) was PCR amplified using sense primer 5′-TCGAGGATCCAAAGGGCAGGTTTGGGGAAG-3′ and antisense primer 5′-GTTAGTCGACCAGGCCAAATGTCTGTCCAG-3′. The BamHI-SalI-digested products were cloned into the Cx36-promoter-containing pGL3basic vector replacing the SV40 poly A signal. All plasmid constructs were verified by sequencing. Cells were transfected on DIV3 using Lipofectamine 2000 reagent (Invitrogen) according to manufacturer's instructions. Some cultures were incubated (DIV4–DIV7) in the constant presence of receptor agonists and antagonists; cultures incubated in the absence of drugs served as a control. On DIV7, cell lysates were harvested and luciferase assay was performed with the DLR-Dual Luciferase kit (Promega) and Turner TD-20/20 Luminometer as described previously (Al-Kandari et al., 2007). All transfections for luciferase assay included an expression plasmid for Renilla luciferase. The firefly luciferase activity was normalized to Renilla luciferase activity to account for variation in transfection efficiency.
Methyl thiazolyl tetrazolium assay.
Neuronal viability in cultures was evaluated quantitatively by methyl thiazolyl tetrazolium (MTT) assay. Cultures were raised in 24-well plates. In all NMDA tests, NMDA was added to the culture medium on DIV14 (100 μm for 60 min in hypothalamic cultures or 10 μm for 30 min in cortical cultures) and then washed out. LY341495- and LY379268-treated cultures were chronically (on DIV3–DIV15) incubated in the presence of these agents. Carbenoxolone was added to the culture medium on DIV14 (alone or together with NMDA) and remained in the medium until the end of the experiment (on DIV15). On DIV15, 24 h after NMDA washout, neurons were incubated with MTT (MTT Cell Viability Assay Kit, Biotium; 40 μm; 400 μl per well) at 37°C for 4 h. Then the medium was carefully aspirated, and 400 μl of DMSO per well was added to dissolve the blue formazan product. To measure the absorbance, 200 μl of the medium from each well in the 24-well plate was transferred into an independent well in a 96-well plate. The values of absorbance at 570 nm were measured using a microplate reader (μQuant; BioTek). Furthermore, as indicated above, cultures that were raised in Neurobasal medium contained mostly neurons (up to 95%). However, to control specifically for neuronal cell death, a separate group of cultures (n = 6) was subjected to a high concentration of glutamate (500 μm), starting from DIV14 for 24 h, that killed all neurons but did not affect glial cell survival. The absorbance in these purified glial cultures was measured and averaged, and the result was subtracted from the individual absorbance data in neuronal culture groups so that the final result would represent only neuronal death/survival. Finally, the absorbance results in experimental groups were normalized to control groups.
Drugs and reagents.
All drugs were obtained from Sigma or Tocris unless specified otherwise.
Statistical analysis.
Data were analyzed using the two-tailed Student's t test (paired, when possible), ANOVA, or Fisher's exact probability test and InStat software (GraphPad Software). Data are reported as mean ± SEM for the number of samples indicated. Each date point represents the indicated day ± 1, except for siRNA and luciferase reporter activity experiments, where all dates are as indicated.
Results
Developmental increase in neuronal gap junction coupling in vivo
Developmental changes in neuronal gap junction coupling were determined in magnocellular neurons in acute slices of the PVN and SON of the rat hypothalamus. We used a dye-coupling method (Arumugam et al., 2005) that included the use of two dyes: neurobiotin, which passes through gap junctions (coupling tracer), and dextran Alexa Fluor 594, which is gap junction impermeable (Fig. 1A–C). We also performed Western blot analysis on dissected hypothalamus to confirm the dye-coupling observations by measuring developmental changes in the expression of Cx36, a gap junction protein that is neuron specific and essential for functional gap junctions in the hypothalamus (Belluardo et al., 2000; Rash et al., 2000; Long et al., 2005) (Fig. 1F,G). The incidence of dye coupling, i.e., the percentage of primary labeled neurons coupled to one or more secondary labeled neurons, and the expression of Cx36 both increased between P1 and P15 (Fig. 1). This indicates a developmental increase in neuronal gap junction coupling in the hypothalamus. Daily (on P1–P15) subcutaneous administration of the group II mGluR agonist LY379268 augmented, and group II mGluR antagonist LY341495 prevented, these developmental increases in gap junction coupling and Cx36 expression (Fig. 1D,F,H; Table 1). In contrast, daily administration of the GABAAR agonist muscimol prevented, and GABAAR antagonist bicuculline augmented, the developmental increases in gap junction coupling and expression of Cx36 (Fig. 1E,G,I; Table 1) (for drug concentrations, see Materials and Methods).
Figure 1.
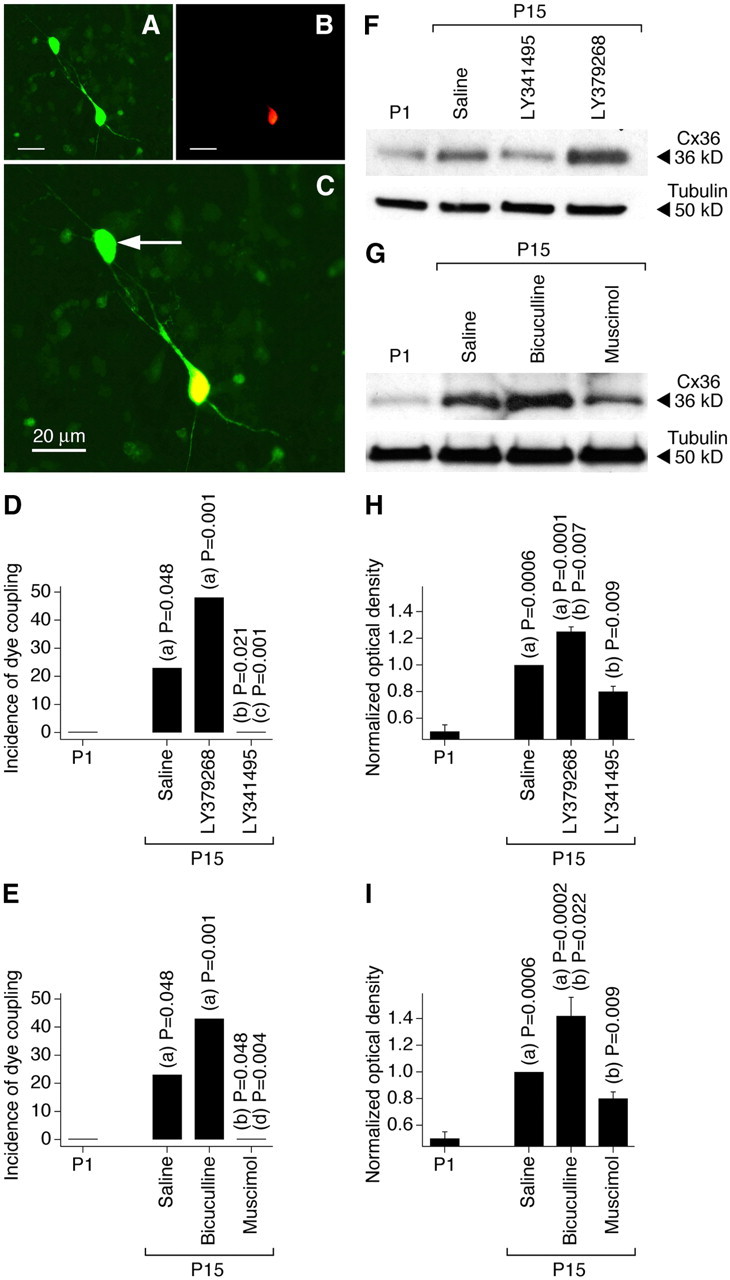
Developmental increase in neuronal gap junction coupling in the rat hypothalamus in vivo is regulated by an interplay between the activity of group II mGluRs and GABAARs. A–C, Images of neurobiotin (A, green) and dextran Alexa Fluor 594 (B, red) staining in an SON slice (P15; bicuculline-treated rat) are superimposed in C (shown at a higher magnification). Yellow indicates dye colocalization in the primary labeled neuron. The arrow indicates the secondary labeled neuron. Scale bars: A–C, 20 μm. D, E, Incidence of dye coupling. Statistical significance was calculated using the Fisher's exact probability test (19–22 primary labeled neurons per data point; data for SON and PVN are combined) (Table 1). F–I, Expression of Cx36 protein in the hypothalamus. Optical density signals are normalized relative to tubulin and P15 saline-treated controls are set at 1.0. Statistical analysis (H, I) was done with Student's t test (mean ± SEM); n = 10 in each group. In all graphs, statistical difference is shown relative to (a) P1, (b) P15 saline, (c) LY379268, and (d) bicuculline.
Table 1.
Neuronal dye coupling in the hypothalamus in vivo and in vitro
| Conditions | Total number of primary labeled neurons | Number of dye-coupled primary labeled neurons (%) | Coupling indexa |
|||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||
| Slice, ratb | ||||||
| P1 | 19 | 0 (0), p = 0.049 | ||||
| P15, saline | 21 | 5 (23.8) | 3 | 1 | 1 | |
| P15, bicuculline | 21 | 10 (47.6), p = 0.197 | 6 | 3 | 1 | |
| P15, muscimol | 19 | 0 (0), p = 0.049 | ||||
| P15, LY341495 | 22 | 0 (0), p = 0.021 | ||||
| P15, LY379268 | 21 | 9 (42.9), p = 0.326 | 6 | 3 | ||
| Culture, ratc | ||||||
| DIV3, control | 32 | 0 (0), p = 0.024 | ||||
| DIV15, control | 32 | 6 (18.8) | 5 | 1 | ||
| DIV15, Bic/PiTX+A/C | 25 | 11 (44.0), p = 0.047 | 10 | 1 | ||
| DIV15, muscimol | 35 | 1 (2.9), p = 0.048 | 1 | |||
| DIV15, LY341495 | 24 | 0 (0), p = 0.032 | ||||
| DIV15, LY379268 | 20 | 6 (33.3), p = 0.500 | 4 | 2 | ||
| DIV15, Cx36 siRNA | 25 | 0 (0), p = 0.029 | ||||
| DIV15, SCR siRNA | 21 | 4 (19.0), p = 1.0c | 2 | 1 | 1 | |
| p = 0.037d | ||||||
| Culture, wild-type mousec | ||||||
| DIV15, control | 26 | 3 (11.5) | 2 | 1 | ||
| DIV15, Bic/PiTX+A/C | 21 | 8 (38.1), p = 0.043 | 5 | 2 | 1 | |
| DIV15, LY379268 | 24 | 9 (37.5), p = 0.047 | 6 | 1 | 1 | 1 |
| Culture, Cx36 knock-out mousec | ||||||
| DIV15, control | 20 | 0 (0) | ||||
| DIV15, Bic/PiTX+A/C | 21 | 0 (0)e | ||||
| DIV15, LY379268 | 21 | 1 (4.8), p = 1.0 | 1 | |||
Bic/PiTX+A/C, Culture treated with bicuculline, picrotoxin, AP5, and CNQX. For drug concentrations, see the text.
aCoupling index is the number of secondary labeled neurons coupled to the primary labeled neuron.
bStatistical significance was calculated using the Fisher's exact probability test relative to P15 controls.
cStatistical significance was calculated using the Fisher's exact probability test relative to the corresponding DIV15 controls.
dFor SCR siRNA (scrambled siRNA) group, statistical significance is shown relative to the controlc and Cx36 siRNA.d
eThe p value cannot be calculated.
Developmental increase in neuronal gap junction coupling in vitro
A previous study in primary rat hypothalamic cultures indicated (Arumugam et al., 2005) that a developmental increase in neuronal gap junction coupling occurs during the first 2 weeks of in vitro development. This increase was prevented by blockade of action potentials (with 2 μm tetrodotoxin, a voltage-gated sodium channel blocker) and was not affected by inactivation of NMDARs (with 100 μm AP5). These results suggested that the developmental increase in neuronal gap junction coupling is regulated via action-potential-dependent (synaptic) release of neurotransmitters, but NMDARs are not involved in this regulation.
In the present study, using neurobiotin with dextran Alexa Fluor 594 and Western blot analysis, we observed increases in neuronal dye coupling and Cx36 expression in developing rat hypothalamic neuronal cultures between DIV3 and DIV15 (Fig. 2; Table 1). In cultures, as in the hypothalamus in vivo, the developmental increases in dye coupling and Cx36 expression were augmented by the chronic (on DIV3–DIV15) activation of group II mGluRs with LY379268 (2 μm) or DCG-IV [(2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine] (10 μm; another group II mGluR agonist) and were prevented by inactivation of group II mGluRs with LY341495 (2 μm) or EGLU [(2S)-α-ethylglutamic acid] (100 μm; another group II mGluR antagonist) (Fig. 2D,F,H; Table 1; see Fig. 5A). Furthermore, the developmental increases in gap junction coupling and the expression of Cx36 were prevented by activation of GABAARs with muscimol (25 μm) or GABA (100 μm) and were augmented by GABAAR inactivation with bicuculline plus picrotoxin (100 + 500 μm; in these tests, ionotropic glutamate receptor antagonists, 100 μm AP5 and 10 μm CNQX, were coadministered with the GABAAR antagonists to prevent an increase in glutamate-dependent activity) (Fig. 2E,G,I; Table 1; see Fig. 5B). Similar results were obtained in cultures using electrotonic coupling analysis (Fig. 3). However, no difference in Cx36 expression was observed between the controls and cultures that were chronically treated (on DIV3–DIV15) with AP5 plus CNQX (100 and 10 μm), DHPG [(S)-3,5-dihydroxyphenylglycine] (10 μm; group I mGluR agonist), AIDA [(RS)-1-aminoindan-1,5-dicarboxylic acid] (100 μm; group I mGluR antagonist), PPG [(RS)-α-cyclopropyl-4-phosphonophenyl glycine] (10 μm; group III mGluR agonist), MSOP [(RS)-methylserine-O-phosphate] (100 μm; group III mGluR antagonist), nicotine (100 μm; nicotinic acetylcholine receptor agonist), atropine plus mecamylamine (100 μm each; muscarinic and nicotinic acetylcholine receptor antagonists), baclofen (20 μm; GABAB receptor agonist), or phaclofen (100 μm; GABAB receptor antagonist) (Fig. 4).
Figure 2.
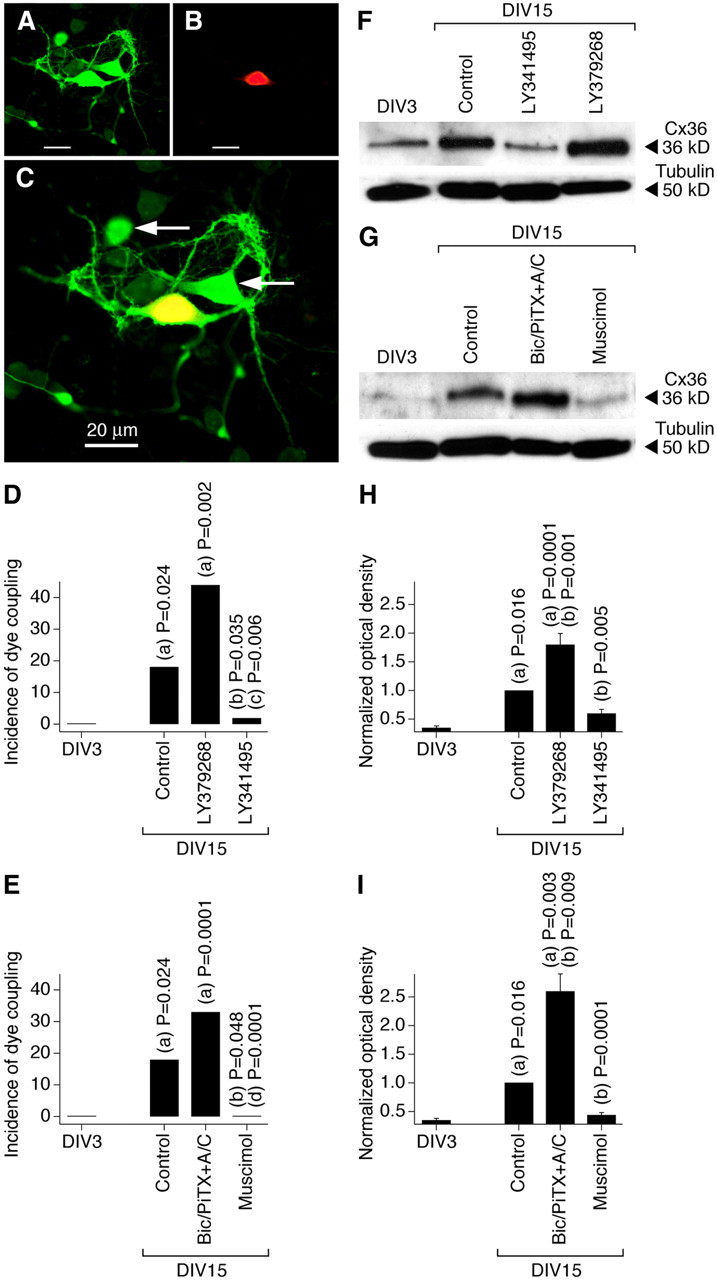
Developmental increase in neuronal gap junction coupling in the rat hypothalamus in vitro is regulated by an interplay between the activity of group II mGluRs and GABAARs. A–C, Images of neurobiotin (A) and dextran Alexa Fluor 594 (B) staining and their overlap (C) are shown [DIV15; culture treated with bicuculline, picrotoxin, AP5, and CNQX (Bic/PiTX+A/C)]. Yellow indicates dye colocalization in the primary labeled neuron. Arrows indicate the secondary labeled neurons. Scale bars: A–C, 20 μm. D, E, Incidence of dye coupling. Statistical significance was calculated using the Fisher's exact probability test (20–35 primary labeled neurons per data point) (Table 1). F–I, Expression of Cx36 protein. Optical density signals are normalized relative to tubulin, and DIV15 controls are set at 1.0. Statistical analysis (H, I) was done using Student's t test (mean ± SEM); n = 6 in each group. In all graphs, statistical difference is shown relative to (a) DIV3, (b) DIV15 control, (c) LY379268, and (d) Bic/PiTX+A/C.
Figure 5.

Signal transduction pathways. Pharmacological treatments were performed in rat hypothalamic cultures on DIV3-DIV15 followed by assessment of Cx36 protein expression on DIV15 (see text for details on the used drug treatments and concentrations). A, The regulation of Cx36 by group II mGluRs is through cAMP/PKA-dependent signaling. B, The regulation of Cx36 by GABAARs is through Ca2+/L-type VGCC/PKC-dependent signaling. CaMKII is not involved in the developmental regulation of Cx36. In both panels, the top shows statistical data; the bottom shows representative blots for cAMP/PKA-dependent (A) and Ca2+/PKC-dependent (B) signaling. Statistical analysis was done with paired Student's t test (mean ± SEM); n = 6 in each group. Optical density values are normalized to tubulin and compared to controls (set at 1.0). Bic/PiTX+A/C, Bicuculline, picrotoxin, AP5, and CNQX; cAMP, 8-Br-cAMP plus IBMX; GFX, GF 109203X; NS, not significant.
Figure 3.

Developmental increase in neuronal gap junction coupling is regulated by an interplay between the activity of group II mGluRs and GABAARs. Data from electrotonic coupling experiments in rat hypothalamic cultures are presented. The coupling was determined as described in Materials and Methods. The number of coupled pairs was as follows: DIV3, 0 of 21; DIV15, control, 6 of 28; LY379268, 8 of 15; LY341495, 1 of 32; muscimol, 0 of 22; Bic/PiTX+A/C (bicuculline, picrotoxin, AP5, and CNQX), 10 of 19. A, B, Incidence of electrotonic coupling represents the percentage of neuronal pairs that demonstrated the coupling. Statistical significance was calculated using Fisher's exact probability test relative to (a) DIV3 and (b) the DIV15 control. C, Representative traces of electrotonic responses are shown (each trace is the average voltage response from 5 sequential steps). D, Statistical analysis of the coupling coefficient (see Materials and Methods) was done using Student's t test relative to the DIV15 control; mean ± SEM (responses from all of the tested pairs are included in the analysis). Note that on DIV15 the incidence of electrotonic coupling and the coupling coefficient both are higher (relative to the control) in cultures subjected to chronic activation of group II mGluRs and inactivation of GABAARs and lower in cultures subjected to inactivation of group II mGluRs and activation of GABAARs.
Figure 4.

Ionotropic glutamate receptors, group I mGluRs, group III mGluRs, acetylcholine receptors, and GABAB receptors are not involved in the regulation of developmental increase in neuronal gap junction coupling. Pharmacological treatments were performed in rat hypothalamic cultures on DIV3–15 followed by assessment of Cx36 protein expression on DIV15 (see text for details on the used drug treatments and concentrations). Optical density signals are normalized relative to tubulin, and normalized values are compared to the control (set at 1.0). Statistical data are presented: paired Student's t test relative to control; mean ± SEM; n = 6 in each group. No significant difference between the control and indicated treatments was found. A/C, AP5 plus CNQX; Atr/Mec, atropine plus mecamylamine.
Together, our in vivo and in vitro data suggest that the developmental increase in gap junction coupling in hypothalamic neurons is regulated by an interplay between the activity of group II mGluRs and GABAARs. In addition, group I mGluR, group III mGluR, NMDA, non-NMDA, acetylcholine, and GABAB receptors are not involved in these regulatory mechanisms.
Cellular mechanisms for developmental increase in gap junction coupling
Signaling pathways for group II mGluRs and GABAARs are well characterized. Group II mGluRs (that include mGluR2 and mGluR3) negatively regulate cAMP/PKA-dependent signaling (De Blasi et al., 2001; Conn et al., 2005). GABAAR is a Cl−-permeable ion channel. In mature neurons, an activation of GABAARs causes Cl− influx and cell hyperpolarization; in developing neurons, an activation of GABAARs causes an efflux of Cl− ions and cell depolarization that results in a Ca2+ influx through voltage-gated Ca2+ channels (VGCCs) and activation of Ca2+-dependent protein kinases (Stein and Nicoll, 2003). In primary rat hypothalamic cultures, GABAAR-mediated responses are excitatory during the first 1–2 weeks of development, and then the excitation is replaced by inhibitory responses (Obrietan and van den Pol, 1995).
To determine whether group II mGluRs and GABAARs regulate the developmental increase in neuronal gap junction coupling via, respectively, cAMP/PKA- and Ca2+-dependent signaling pathways, we performed additional pharmacological manipulations in rat hypothalamic cultures on DIV3–DIV15 and detected Cx36 expression by Western blot analysis (Fig. 5). An increase in cytoplasmic levels of cAMP [by 8-bromo-cAMP (8-Br-cAMP) plus 3-isobutyl-1-methylxanthine (IBMX); 100 μm + 50 μm; a cell-permeable analog of cAMP that activates PKA and a nonspecific phosphodiesterase inhibitor that increases intracellular cAMP levels] reduced, and the blockade of PKA [by N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide dihydrochloride (H89); 1 μm; PKA antagonist] augmented, the developmental increase in Cx36 expression (Fig. 5A). Furthermore, the effects of group II mGluR agonist and antagonist in the developmental regulation of Cx36 (Fig. 2) were prevented, respectively, by the increase in cAMP levels and inactivation of PKA (Fig. 5A).
The developmental increase in Cx36 expression was also prevented by KCl (1–5 mm, which causes cell depolarization and Ca2+ influx through L-type VGCCs) (Bessho et al., 1994) and was augmented by the blockade of PKC [with 2-[1-(3-dimethylaminopropyl)indol-3-yl]-3-(indol-3-yl) maleimide (GF 109203X); 0.5–1 μm] (Fig. 5B). Furthermore, the effect of the GABAAR agonist muscimol (25 μm) was prevented by blockade of L-type VGCCs with nifedipine (10 μm) or PKC inactivation (Fig. 5B), whereas the effect of GABAAR antagonists (Fig. 2) was prevented by activation of PKC [with PMA (phorbol 12-myristate 13-acetate); 1 μm] (Fig. 5B). However, the developmental increase in Cx36 expression was not affected by inactivation of another calcium-regulated protein kinase, Ca2+/calmodulin-dependent protein kinase II [(CaMKII); with 4-[(2S)-2-[(5-isoquinolinylsulfonyl)methylamino]-3-oxo-3-(4-phenyl-1-piperazinyl)propyl] phenyl isoquinolinesulfonic acid ester (KN-62); 2.5 μm] (Fig. 5B).
These data suggest that the developmental regulation of Cx36 by group II mGluRs is via cAMP/PKA-dependent signaling, that regulation by GABAARs is via Ca2+/L-type VGCC/PKC-dependent signaling, and that CaMKII is not involved in control of the developmental increase in Cx36 expression.
The roles of mGluR2 vs mGluR3 and group II mGluRs vs GABAARs
We used an siRNA approach in rat hypothalamic cultures to determine the importance of mGluR2 versus mGluR3 in the developmental increase in expression of Cx36 (Fig. 6A,B). The genetic suppression of both mGluR2 and mGluR3 (on DIV3–DIV7) decreased Cx36 protein levels, suggesting that both receptors are important for the developmental increase in neuronal gap junction coupling.
Figure 6.

Characterization of the mechanisms for developmental increase in neuronal gap junction coupling. A–D, Western blot (A–C) and dye coupling (D) tests were performed in neuronal cultures prepared from the rat (A–C) and mouse (D) hypothalamus. A, B, siRNA suppression (on DIV3–DIV7) of mGluR2 (A) and mGluR3 (B) decreases both the receptor and Cx36 protein levels. Representative images (top) and statistical data (bottom; paired Student's t test relative to control; mean ± SEM) are shown. Stainings were done sequentially on one membrane. SCR, scrambled siRNA. C, A combined activation (on DIV3–DIV15) of group II mGluRs and GABAARs (with LY379268 plus muscimol) does not affect significantly the developmental upregulation of Cx36 expression. Statistical analysis was done using ANOVA with Tukey's post hoc test relative to (a) DIV15 control and (b) LY379268 plus muscimol (mean ± SEM). D, In wild-type cultures, dye coupling increases between DIV3 (data not shown) and DIV15 (control), and this increase is augmented by inactivation of GABAARs [Bic/PiTX+A/C (bicuculline, picrotoxin, AP5, and CNQX)] and by activation of group II mGluRs (LY379268). In Cx36 knock-out cultures, neither the developmental nor the treatment-mediated increases occur. The number of dye-coupled primary labeled neurons of the total number of primary labeled neurons and statistical significance (Fisher's exact probability test; relative to the corresponding control) on DIV15 are shown. Pi, p value cannot be calculated.
We also tested the importance of group II mGluRs versus GABAARs (Fig. 6C). Rat hypothalamic cultures were chronically treated (on DIV3–DIV15) with LY379268 (2 μm) and muscimol (25 μm). A combined activation of group II mGluRs and GABAARs did not affect significantly the developmental upregulation of Cx36. However, the effect of activating GABAARs dominated the effect of activating group II mGluRs: on DIV15, the expression of Cx36 was not different between muscimol-treated and LY379268 plus muscimol-treated cultures, but it was significantly different between LY379268-treated and LY379268 plus muscimol-treated cells (Fig. 6C). These data are in agreement with the observation that, in cell cultures on DIV3–DIV15, blockade of GABAARs increases Cx36 expression to a higher level than activation of group II mGluRs (2.64 ± 0.35 vs 1.81 ± 0.20 normalized optical density; p = 0.045; unpaired Student's t test; n = 6 in each group) (Fig. 2H,I).
Specificity of the mechanisms for increase in gap junction coupling
We tested whether the regulation by group II mGluRs and GABAARs is specific for neuronal Cx36-containing gap junctions. First, we found that the developmental increase in neuronal dye coupling in rat hypothalamic cultures is prevented by Cx36 siRNA (transfected on DIV10 and tested on DIV15; Table 1). Second, neuronal dye coupling was measured in hypothalamic cultures prepared from wild-type and Cx36 knock-out mice. As expected, in wild-type cultures, the coupling was observed on DIV15 and was increased by both LY379268 and bicuculline plus picrotoxin (in the presence of AP5 and CNQX, in concentrations as in rat cultures) (Fig. 6D; Table 1). However, the coupling was not observed in Cx36 knock-out cultures, whether untreated or treated (Fig. 6D; Table 1). Moreover, in the rat hypothalamus in vivo and in vitro, the expression of a presumptive astrocytic connexin, Cx43 (Rash et al., 2000), was not different between the control and the treatment conditions, including the treatments with group II mGluR and GABAAR agonists and antagonists (Fig. 7). The data suggest that the group II mGluR/GABAAR-dependent regulation is specific for neuronal Cx36-containing, but not glial Cx43-containing gap junctions. Moreover, this regulation is exclusive for Cx36 because no increase in gap junction coupling occurs in Cx36-deficient neurons.
Figure 7.

Group II mGluRs and GABAARs are not involved in the developmental regulation of hypothalamic expression of glial Cx43. A–F, Western blots for Cx43 (A, B, D, E) and statistical data (C, F) in the developing rat hypothalamus (A–C) and hypothalamic cultures (D–F) are presented. In C and F, statistical analysis is done using the paired Student's t test relative to P15 saline or DIV15 control (mean ± SEM); n = 6 in each group. Data are normalized and analyzed as described in Figure 1 and 2 for Cx36. Cx43 expression increases during development both in vivo and in vitro; however, the increase is not affected by group II mGluR and GABAAR agonists or antagonists (i.e., no statistical significance between the saline/control and experimental groups on P15 or DIV15 is detected). Bic/PiTX+A/C, Bicuculline, picrotoxin, AP5, and CNQX.
We also examined whether these developmental mechanisms operate in other CNS regions and species, and whether they are active in mature neurons. The activity of group II mGluRs and GABAARs was pharmacologically modulated (on DIV3–DIV15) in developing neuronal cultures prepared from the mouse somatosensory cortex, and Western blot analysis of Cx36 expression revealed changes that were similar to those in rat hypothalamic cultures (Fig. 8). This suggests that the same regulatory mechanisms are employed in the developing mouse cortex and rat and mouse hypothalamus. Furthermore, we tested mature cultures prepared from the rat hypothalamus. In these cultures, chronic (DIV30–DIV36) activation of group II mGluRs (with LY379268), but not inactivation of GABAARs (with bicuculline plus picrotoxin in the presence of AP5 and CNQX, in concentrations as above), increased the expression of Cx36 (Fig. 9). Similar results were obtained in mature cultures prepared from the mouse cortex (data not shown). These data suggest that the group II mGluRs may contribute to the upregulation of neuronal gap junction coupling in mature neurons, though GABAARs, which are inhibitory in the mature CNS (Obrietan and van den Pol, 1995; Stein and Nicoll, 2003), presumably do not regulate the coupling after neuronal maturation. [The receptor-mediated inhibition of Cx36 expression was not tested as the expression of Cx36 is low in mature neuronal cultures (Arumugam et al., 2005).]
Figure 8.

Developmental increase in neuronal gap junction coupling in the mouse cortex in vitro is regulated by an interplay between the activity of group II mGluRs and GABAARs. A, B, Expression of Cx36 protein. Data are normalized and analyzed as described in Figure 2. Statistical analysis was done using Student's t test relative to (a) DIV3 and (b) the DIV15 control (mean ± SEM); n = 6 in each group. Drug concentrations are as in experiments in rat hypothalamic cultures shown in Figure 2. Bic/PiTX+A/C, Bicuculline, picrotoxin, AP5, and CNQX.
Figure 9.

Regulation of Cx36 expression in mature neurons. Pharmacological treatments were conducted in rat hypothalamic neuronal cultures on DIV30–DIV36 followed by assessment of Cx36 protein expression on DIV36. Statistical analysis was done using paired Student's t test relative to control (mean ± SEM); LY3792688, n = 7; Bic/PiTX+A/C (bicuculline, picrotoxin, AP5, and CNQX), n = 5. Data are normalized and analyzed as described in Figure 2.
Molecular mechanisms for developmental increase in gap junction coupling
In response to either group II mGluR activation or GABAAR inactivation, Cx36 protein levels increase during development, and we tested whether this is associated with increased Cx36 mRNA. The expression of Cx36 mRNA was evaluated in rat hypothalamic cultures by RT-qPCR (Fig. 10A). We observed an increase in Cx36 mRNA levels between DIV3 and DIV15. Furthermore, this increase was augmented by both activation of group II mGluRs (with LY379268) and inactivation of GABAARs (with bicuculline plus picrotoxin in the presence of AP5 and CNQX, in concentrations as above). However, the increase was not affected by either inactivation of group II mGluRs (with LY341495) and activation of GABAARs (with muscimol). The data suggest that the receptor-regulated increase, but not the decrease, in Cx36 expression during development may be controlled by transcriptional mechanisms.
Figure 10.

Molecular mechanisms for the developmental regulation of Cx36. A, RT-qPCR analysis in rat hypothalamic cultures demonstrates that Cx36 mRNA expression increases during development and this increase is augmented by activation of group II mGluRs (LY379268) and inactivation of GABAARs (Bic/PiTX+A/C). However, it is not affected by inactivation of group II mGluRs (LY341495) and activation of GABAARs (muscimol). Relative Cx36 transcript levels are normalized to GAPDH, and normalized values are compared to DIV15 controls (set to 1.0). Statistical analysis was done using paired Student's t test relative to control (mean ± SEM). B, Dual-luciferase reporter assay. Cells were transfected on DIV3 with the luciferase (LUC) reporter plasmids driven by the rat Cx36 promoter (PCx36), incubated in the absence or in the presence of indicated agents and then assayed on DIV7. The NRSE deleted plasmid was produced using site-directed mutagenesis. The plasmid containing the Cx36 3′UTR was constructed by replacing the original SV40 poly A signal (SV40 pA) in the plasmid containing PCx36 with the full-length rat Cx36 3′UTR. Firefly luciferase values are normalized relative to Renilla luciferase values to control for transfection efficiency, and the results are presented as relative activity of the promoter constructs compared to the pGL3basic vector (set to 1.0). Statistical analysis was done using paired Student's t test relative to the corresponding nontreated control. Bic/PiTX+A/C, Bicuculline, picrotoxin, AP5, and CNQX.
Based on RT-qPCR experiments, we set out to determine whether an element within the Cx36 proximal promoter is responsible for the developmental increase in Cx36 mRNA levels (Fig. 10B). Cultures were transfected (on DIV3) with a plasmid containing the rat Cx36 promoter linked to the firefly luciferase gene and treated (on DIV4–DIV7) with the group II mGluR and GABAAR agonists and antagonists. On DIV7, cells were harvested, and luciferase assay was performed. The Cx36 proximal promoter (−984/+115) supported robust transcription. As in RT-qPCR experiments, the Cx36 promoter activity was augmented by activation of group II mGluRs and inactivation of GABAARs, and was not affected by LY341495 and muscimol. The increases in Cx36 promoter activity were completely abolished by deletion of an NRSE located at nucleotide position −164/−144 (Martin et al., 2003). In addition, NRSE deletion increased the basal expression of Cx36 promoter (Fig. 10B). The data suggest that NRSE has a repressive effect on Cx36 promoter activity, and this repression is removed by activation of group II mGluRs or inactivation of GABAARs.
Next, we conducted experiments to determine whether the mechanism for the receptor-mediated decrease in Cx36 expression during development involves posttranscriptional regulation (Fig. 10B). We constructed the plasmid containing the full-length Cx36 3′UTR (∼1.5 kb) and the luciferase reporter driven by the Cx36 promoter (−984/+115). Whereas this construct demonstrated reduced luciferase activity (compared to the plasmid containing the Cx36 promoter alone), it showed not only significant increases but also decreases in response to the treatments that, respectively, increase (group II mGluR agonists and GABAAR antagonists) or decrease (group II mGluR antagonists and GABAAR agonists) Cx36 protein expression (Fig. 10B). Together, the results indicate that the mechanisms that orchestrate the developmental changes in Cx36 expression involve both the Cx36 promoter-mediated transcriptional regulation through NRSE and regulatory events mediated via the 3′UTR, perhaps involving post-transcriptional mechanisms.
Functional implications
de Rivero Vaccari et al. (2007) demonstrated that neuronal gap junctions play a critical role in the mechanisms of neuronal death/survival during development. Specifically, in rat and wild-type mouse hypothalamic neuronal cultures, we found that hyperactivation and inactivation of NMDARs both induced neuronal death exclusively during the peak of developmental gap junction coupling (i.e., on DIV14–DIV17). Furthermore, both types of NMDAR-dependent neuronal death were completely prevented by pharmacological inactivation of gap junctions (with carbenoxolone and 18α-glycyrrhetinic acid) or genetic knock-out of Cx36. In the present study, we set out to determine whether manipulation of the mechanisms regulating the developmental increase in neuronal gap junction coupling affects the death/survival mechanisms in developing neurons. We used a model of NMDAR-dependent excitotoxicity. Experiments were conducted in rat hypothalamic cultures, nontreated or chronically treated (on DIV3–DIV15) with the group II mGluR agonist (LY379268) or antagonist (LY341495, in concentrations as above). Administration of NMDA (100 μm; on DIV14 for 60 min) induced significant neuronal death in nontreated cultures that was detected 24 h later (on DIV15) using MTT assay (Fig. 11A). NMDA-mediated neuronal death was more pronounced in LY379268-treated cultures, i.e., in the cultures with high levels of neuronal gap junction coupling and Cx36 expression. Furthermore, NMDA-mediated neuronal death was not observed in LY341495-treated cultures, i.e., in the cultures that lack neuronal gap junction coupling because of low levels of Cx36. NMDA-mediated neuronal death also was prevented by the gap junction blocker carbenoxolone (25 μm) (Fig. 11B), supporting the notion that gap junctions participate in NMDAR-dependent death in developing neurons. Similar results were obtained in wild-type mouse cortical neuronal cultures (data not shown). The results indicate that the mechanisms for the developmental increase in neuronal gap junction coupling play a role in regulation of neuronal death/survival during development.
Figure 11.

Mechanisms for the developmental increase in neuronal gap junction coupling play a role in regulation of neuronal death/survival during development. Experiments were conducted in rat hypothalamic neuronal cultures. NMDA (100 μm) was added to the culture medium on DIV14 for 60 min and then washed out. The MTT analysis of neuronal death was conducted 24 h later (on DIV15). A, NMDA administration induces neuronal death in nontreated cultures. NMDA-mediated neuronal death is augmented in cultures chronically treated (on DIV3–DIV15) with the group II mGluR agonist (LY379268) and does not occur in cultures chronically treated with the group II mGluR antagonist (LY341495). The mGluR agents by themselves do not affect cell survival. B, NMDA-mediated neuronal death also is prevented by coadministration of the gap junctional blocker carbenoxolone (CBX). In both graphs, statistical data are shown. Statistical analysis was done using ANOVA with Tukey's post hoc test relative to (a) control and (b) nontreated plus NMDA conditions (mean ± SEM); n = 6 in each group.
Discussion
We demonstrated here that the developmental increase in neuronal gap junction coupling in the rat and mouse hypothalamus and cortex is regulated by an interplay between the activity of group II mGluRs and GABAARs. The regulation by group II mGluRs is via cAMP/PKA-dependent signaling. The regulation by GABAARs, which cause neuronal excitation during development (Obrietan and van den Pol, 1995; Stein and Nicoll, 2003), is via Ca2+ influx through VGCCs and activation of PKC. We also showed that other glutamate receptors, acetylcholine receptors, GABAB receptors, and CaMKII are not involved in these regulatory mechanisms. A previous study in the rat hypothalamus indicated (Arumugam et al., 2005) that blockade of action potentials prevents the developmental increase in neuronal gap junction coupling. Together with the results described here, this implicates a role for action-potential-dependent synaptic release of glutamate and GABA in regulation of the developmental increase in neuronal gap junction coupling.
Group II mGluRs include mGluR2 and mGluR3, and our experiments suggested that both of them are important for the developmental increase in neuronal gap junction coupling. Data also indicated that the effects of GABAAR agents in modulation of gap junctions are stronger than those of group II mGluR agents. This supports a more important role for GABAARs than group II mGluRs in these regulatory mechanisms, which may be explained by the fact that formation of GABAergic synapses in the CNS precedes the formation of glutamatergic synapses (Ben-Ari, 2002). It is possible, however, that the pattern of expression of particular neurotransmitter receptors and the timing of the switch of GABAARs from excitation to inhibition during development determine when the developmental increase in neuronal gap junction coupling occurs. Future experiments are needed to evaluate this prediction.
Cx43 is a presumptive glial connexin (Rash et al., 2000) that is also involved in a number of developmental events, including neuronal migration (Elias et al., 2007). Our study showed that the expression of Cx43 also increases during development; however, the increase is not affected by the group II mGluR and GABAAR agents. This suggests that the regulation by group II mGluRs and GABAARs is specific for Cx36-containing, but not Cx43-containing gap junctions. Moreover, this regulation is exclusive for Cx36, because no increase in gap junction coupling (either developmental or treatment mediated) occurs in Cx36-deficient neurons (Fig. 6D).
NRSE is a DNA sequence element in a promoter region of a number of neuronal genes that binds RE1-silencing transcription factor (REST) and regulates the transcriptional activity of these genes. REST activity is influenced by both Ca2+- and cAMP-dependent signaling (Nadeau and Lester, 2002; Somekawa et al., 2009). Rat and mouse Cx36 genes contain NRSEs (−164/−144 and +201/+221, respectively) (Cicirata et al., 2000; Martin et al., 2003), and occupancy of the Cx36 NRSE by REST is observed in mouse neuronal stem cells (Johnson et al., 2008). Furthermore, REST/NRSE-dependent signaling negatively regulates Cx36 mRNA expression in insulin-producing cells (Martin et al., 2003). Our study in developing hypothalamic neurons agrees with those findings and demonstrates that deletion of the NRSE in the Cx36 promoter region results in both higher basal transcription and loss of responsiveness to the group II mGluR agonist and GABAAR antagonists. Thus, the data suggest that the receptor-mediated increase in Cx36 expression during development is regulated by group II mGluRs and GABAARs via removal of the NRSE-dependent repression of Cx36 promoter activity.
In contrast, the mechanisms for the receptor-mediated decrease in expression of Cx36 during development may not be attributable to new mRNA synthesis, but rather may involve posttranscriptional mechanisms dependent on sequences within the 3′UTR. Although we cannot totally rule out the presence of a transcriptional regulatory element in the region of 3′UTR, given that the Cx36 mRNA levels did not change in response to inactivation of group II mGluRs or activation of GABAARs, this possibility seems unlikely. Rather, we suspect that this regulation may be via microRNAs, small noncoding RNAs, which bind to complementary sites on 3′UTR of target mRNAs and reduce gene expression primarily through translational repression (Bartel, 2004). Many microRNAs are brain specific (Sempere et al., 2004) and are involved in the regulation of neuronal development, differentiation, and morphogenesis (Smirnova et al., 2005; Vo et al., 2005). The 3′UTR of Cx36 mRNA contains binding sites for a number of brain-specific microRNAs, including miR-9, miR-128a, and miR-128b (Rash et al., 2005). A focus of future studies will be to identify which, if any, of these miRNAs target Cx36 and to determine the specific signals that regulate their action.
During pharmacological manipulations, changes occur not only in the incidence of dye and electrotonic coupling, but also in the coupling coefficient. Together with data demonstrating alterations in Cx36 protein expression, these results suggest the possibility of adding/removing Cx36 molecules to/from individual neurons.
We also postulate that group II mGluR and GABAAR agonists and antagonists exert specific, physiological effects on neuronal gap junction coupling. First, the agents modulate the developmental increases in the coupling and Cx36 expression in a similar way in vivo and in vitro. Second, the increases are modulated not only by receptor agonists and antagonists, but also by manipulating the corresponding receptor-coupled intracellular signaling. Third, it is unlikely that the effects occur simply through the regulation of electrical activity because the developmental increases in coupling and/or Cx36 expression are prevented both by tetrodotoxin (that reduces the action potential activity) (Arumugam et al., 2005) and by KCl (which depolarizes neurons and increases the action potential activity) (Fig. 5B). Finally, the developmental increase in Cx36 is affected not only by pharmacological treatments, but also by genetic manipulations for the expression of mGluR2 and mGluR3.
A previous study demonstrated a role for glutamate-dependent synaptic transmission in the developmental uncoupling of neuronal gap junctions (that occurs in the hypothalamus between days 15 and 30 in vivo and in vitro) (Arumugam et al., 2005). The data indicated that developmental uncoupling is attributable to activation of NMDARs and Ca2+-, CaMKII-, PKC- and Ca2+/cAMP response element-binding protein (CREB)-dependent downregulation of Cx36. Together with the present work, our studies strongly suggest that developing chemical synapses regulate electrical synapses. We postulate that during early postnatal development, GABAAR-dependent excitation maintains the expression of Cx36 in neuronal circuits at a low level (via Ca2+/PKC signaling and the 3′UTR of the Cx36 mRNA). The subsequent transition from GABAAR excitation to inhibition, in combination with increased activity of the group II mGluRs, results in the developmental upregulation of Cx36 (via the NRSE in the Cx36 gene) and increased neuronal gap junction coupling. However, the developmental increase in the activity of NMDARs then causes downregulation of Cx36 (via Ca2+-dependent signaling, including CREB) and gap junction uncoupling. If mechanisms for the developmental regulation of gap junctions have universal character in the CNS (as we show here for the hypothalamus and cortex), the variations among different CNS regions in the timing of neuronal gap junction coupling and uncoupling (Bennett and Zukin, 2004) presumably can be explained by the interregional differences in the activity of these mechanisms (i.e., receptor and synaptic activity and timing of the excitation/inhibition switch for GABAARs). Other additional, region-specific factors probably also contribute.
In the developing and mature CNS, neuronal gap junction coupling also may be modulated acutely by changes in the activity of neurotransmitter receptors (Hatton, 1998). We believe that acute and developmental alterations in the coupling represent different functional aspects of gap junction physiology. The acute modulation of gap junctions by neurotransmitter receptors likely involves gating mechanisms and plays a role in rapid modifications in neuronal connectivity and signaling in response to changes in chemical synaptic activity (Hatton, 1998). In contrast, the developmental changes in gap junctions are likely related to specific genetic programs and/or developmental pathways during the period when chemical synapses are still being established. It is possible, however, that the acute modulation of coupling may translate into a prolonged modification if the changed level of receptor activity sustains.
In the developing CNS, programmed cell death helps to establish the final number of neurons and contributes to the distribution of various cell classes and neuronal circuit formation (Nijhawan et al., 2000). The activity of NMDARs is also the factor that plays a role in cell survival versus death decisions during neuronal development (Scheetz and Constantine-Paton, 1994; de Rivero Vaccari et al., 2006). It has been suggested that during development gap junctions are involved in the regulation of apoptosis and NMDAR-dependent neuronal death (Cusato et al., 2003; de Rivero Vaccari et al., 2007). We showed here that the NMDAR-mediated excitotoxicity is eliminated or augmented if the amount of gap junctional coupling is reduced or increased, respectively. This suggests that mechanisms for the developmental increase in neuronal gap junction coupling directly regulate death/survival mechanisms in developing neurons. This also implies a role for gap junctions in the formation of neuronal circuits via regulation of neuronal death/survival.
In conclusion, we have characterized the mechanisms for developmental increase in neuronal gap junction coupling. The coupling also increases in the mature CNS during neuronal injuries (Chang et al., 2000; Frantseva et al., 2002; de Pina-Benabou et al., 2005; Nemani and Binder, 2005; Thalakoti et al., 2007), and selective blockade of Cx36-containing gap junctions is neuroprotective (Wang et al., 2010). Given that neuronal injuries are characterized by excessive release of glutamate (Arundine and Tymianski, 2004) and that activation of group II mGluRs increases Cx36 expression in mature neurons (present study), it will be interesting to see whether mechanisms for the injury-related increases in neuronal gap junction coupling are group II mGluR-dependent and whether inactivation of these mechanisms has a neuroprotective effect.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants R01 NS064256 and R01 DA015088, NIH/National Center for Research Resources Grant P20 RR024214, the Kansas IDeA Network of Biomedical Research Excellence, and University of Kansas Medical Center funds (A.B.B.), and by the Biomedical Research Training Program award (Y.W.). Core support was provided by NIH Grant HD002528. We are grateful to Drs. D. Paul and M. Feller for providing us with the Cx36 knock-out mice.
References
- Al-Kandari W, Jambunathan S, Navalgund V, Koneni R, Freer M, Parimi N, Mudhasani R, Fontes JD. ZXDC, a novel zinc finger protein that binds CIITA and activates MHC gene transcription. Mol Immunol. 2007;44:311–321. doi: 10.1016/j.molimm.2006.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen F, Warner A. Gap junctional communication during neuromuscular junction formation. Neuron. 1991;6:101–111. doi: 10.1016/0896-6273(91)90125-j. [DOI] [PubMed] [Google Scholar]
- Arumugam H, Liu X, Colombo PJ, Corriveau RA, Belousov AB. NMDA receptors regulate developmental gap junction uncoupling via CREB signaling. Nat Neurosci. 2005;8:1720–1726. doi: 10.1038/nn1588. [DOI] [PubMed] [Google Scholar]
- Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci. 2004;61:657–668. doi: 10.1007/s00018-003-3319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bani-Yaghoub M, Underhill TM, Naus CC. Gap junction blockage interferes with neuronal and astroglial differentiation of mouse P19 embryonal carcinoma cells. Dev Genet. 1999;24:69–81. doi: 10.1002/(SICI)1520-6408(1999)24:1/2<69::AID-DVG8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Belluardo N, Mudo G, Trovato-Salinaro A, Le Gurun S, Charollais A, Serre-Beinier V, Amato G, Haefliger JA, Meda P, Condorelli DF. Expression of connexin36 in the adult and developing rat brain. Brain Res. 2000;865:121–138. doi: 10.1016/s0006-8993(00)02300-3. [DOI] [PubMed] [Google Scholar]
- Belousov AB, van den Pol AN. Local synaptic release of glutamate from neurons in the rat hypothalamic arcuate nucleus. J Physiol. 1997;499:747–761. doi: 10.1113/jphysiol.1997.sp021966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belousov AB, O'Hara BF, Denisova JV. Acetylcholine becomes the major excitatory neurotransmitter in the hypothalamus in vitro in the absence of glutamate excitation. J Neurosci. 2001;21:2015–2027. doi: 10.1523/JNEUROSCI.21-06-02015.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y. Developing networks play a similar melody. Trends Neurosci. 2001;24:353–360. doi: 10.1016/s0166-2236(00)01813-0. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Bennett MV, Zukin RS. Electrical coupling and neuronal synchronization in the mammalian brain. Neuron. 2004;41:495–511. doi: 10.1016/s0896-6273(04)00043-1. [DOI] [PubMed] [Google Scholar]
- Bessho Y, Nawa H, Nakanishi S. Selective up-regulation of an NMDA receptor subunit mRNA in cultured cerebellar granule cells by K(+)-induced depolarization and NMDA treatment. Neuron. 1994;12:87–95. doi: 10.1016/0896-6273(94)90154-6. [DOI] [PubMed] [Google Scholar]
- Chang Q, Pereda A, Pinter MJ, Balice-Gordon RJ. Nerve injury induces gap junctional coupling among axotomized adult motor neurons. J Neurosci. 2000;20:674–684. doi: 10.1523/JNEUROSCI.20-02-00674.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicirata F, Parenti R, Spinella F, Giglio S, Tuorto F, Zuffardi O, Gulisano M. Genomic organization and chromosomal localization of the mouse Connexin36 (mCx36) gene. Gene. 2000;251:123–130. doi: 10.1016/s0378-1119(00)00202-x. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Battaglia G, Marino MJ, Nicoletti F. Metabotropic glutamate receptors in the basal ganglia motor circuit. Nat Rev Neurosci. 2005;6:787–798. doi: 10.1038/nrn1763. [DOI] [PubMed] [Google Scholar]
- Cusato K, Bosco A, Rozental R, Guimaraes CA, Reese BE, Linden R, Spray DC. Gap junctions mediate bystander cell death in developing retina. J Neurosci. 2003;23:6413–6422. doi: 10.1523/JNEUROSCI.23-16-06413.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Blasi A, Conn PJ, Pin J, Nicoletti F. Molecular determinants of metabotropic glutamate receptor signaling. Trends Pharmacol Sci. 2001;22:114–120. doi: 10.1016/s0165-6147(00)01635-7. [DOI] [PubMed] [Google Scholar]
- de Pina-Benabou MH, Szostak V, Kyrozis A, Rempe D, Uziel D, Urban-Maldonado M, Benabou S, Spray DC, Federoff HJ, Stanton PK, Rozental R. Blockade of gap junctions in vivo provides neuroprotection after perinatal global ischemia. Stroke. 2005;36:2232–2237. doi: 10.1161/01.STR.0000182239.75969.d8. [DOI] [PubMed] [Google Scholar]
- de Rivero Vaccari JC, Casey GP, Aleem S, Park WM, Corriveau RA. NMDA receptors promote survival in somatosensory relay nuclei by inhibiting Bax-dependent developmental cell death. Proc Natl Acad Sci U S A. 2006;103:16971–16976. doi: 10.1073/pnas.0608068103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rivero Vaccari JC, Corriveau RA, Belousov AB. Gap junctions are required for NMDA receptor-dependent cell death in developing neurons. J Neurophysiol. 2007;98:2878–2886. doi: 10.1152/jn.00362.2007. [DOI] [PubMed] [Google Scholar]
- Elias LA, Wang DD, Kriegstein AR. Gap junction adhesion is necessary for radial migration in the neocortex. Nature. 2007;448:901–907. doi: 10.1038/nature06063. [DOI] [PubMed] [Google Scholar]
- Feller MB, Wellis DP, Stellwagen D, Werblin FS, Shatz CJ. Requirement for cholinergic synaptic transmission in the propagation of spontaneous retinal waves. Science. 1996;272:1182–1187. doi: 10.1126/science.272.5265.1182. [DOI] [PubMed] [Google Scholar]
- Frantseva MV, Kokarovtseva L, Naus CG, Carlen PL, MacFabe D, Perez Velazquez JL. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J Neurosci. 2002;22:644–653. doi: 10.1523/JNEUROSCI.22-03-00644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton GI. Synaptic modulation of neuronal coupling. Cell Biol Int. 1998;22:765–780. doi: 10.1006/cbir.1998.0386. [DOI] [PubMed] [Google Scholar]
- Johnson R, Teh CH, Kunarso G, Wong KY, Srinivasan G, Cooper ML, Volta M, Chan SS, Lipovich L, Pollard SM, Karuturi RK, Wei CL, Buckley NJ, Stanton LW. REST regulates distinct transcriptional networks in embryonic and neural stem cells. PLoS Biol. 2008;6:e256. doi: 10.1371/journal.pbio.0060256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandler K, Katz LC. Coordination of neuronal activity in developing visual cortex by gap junction-mediated biochemical communication. J Neurosci. 1998;18:1419–1427. doi: 10.1523/JNEUROSCI.18-04-01419.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Weigel H, Cotrina ML, Liu S, Bueno E, Hansen AJ, Hansen TW, Goldman S, Nedergaard M. Gap-junction-mediated propagation and amplification of cell injury. Nat Neurosci. 1998;1:494–500. doi: 10.1038/2210. [DOI] [PubMed] [Google Scholar]
- Long MA, Jutras MJ, Connors BW, Burwell RD. Electrical synapses coordinate activity in the suprachiasmatic nucleus. Nat Neurosci. 2005;8:61–66. doi: 10.1038/nn1361. [DOI] [PubMed] [Google Scholar]
- Martin D, Tawadros T, Meylan L, Abderrahmani A, Condorelli DF, Waeber G, Haefliger JA. Critical role of the transcriptional repressor neuron-restrictive silencer factor in the specific control of connexin36 in insulin-producing cell lines. J Biol Chem. 2003;278:53082–53089. doi: 10.1074/jbc.M306861200. [DOI] [PubMed] [Google Scholar]
- Muller M, Gahwiler BH, Rietschin L, Thompson SM. Reversible loss of dendritic spines and altered excitability after chronic epilepsy in hippocampal slice cultures. Proc Natl Acad Sci U S A. 1993;90:257–261. doi: 10.1073/pnas.90.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau H, Lester HA. NRSF causes cAMP-sensitive suppression of sodium current in cultured hippocampal neurons. J Neurophysiol. 2002;88:409–421. doi: 10.1152/jn.2002.88.1.409. [DOI] [PubMed] [Google Scholar]
- Nemani VM, Binder DK. Emerging role of gap junctions in epilepsy. Histol Histopathol. 2005;20:253–259. doi: 10.14670/HH-20.253. [DOI] [PubMed] [Google Scholar]
- Nijhawan D, Honarpour N, Wang X. Apoptosis in neural development and disease. Annu Rev Neurosci. 2000;23:73–87. doi: 10.1146/annurev.neuro.23.1.73. [DOI] [PubMed] [Google Scholar]
- Obrietan K, van den Pol AN. GABA neurotransmission in the hypothalamus: developmental reversal from Ca2+ elevating to depressing. J Neurosci. 1995;15:5065–5077. doi: 10.1523/JNEUROSCI.15-07-05065.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado A, Yuste R, Katz LC. Extensive dye coupling between rat neocortical neurons during the period of circuit formation. Neuron. 1993;10:103–114. doi: 10.1016/0896-6273(93)90246-n. [DOI] [PubMed] [Google Scholar]
- Personius K, Chang Q, Bittman K, Panzer J, Balice-Gordon R. Gap junctional communication among motor and other neurons shapes patterns of neural activity and synaptic connectivity during development. Cell Commun Adhes. 2001;8:329–333. doi: 10.3109/15419060109080748. [DOI] [PubMed] [Google Scholar]
- Rash JE, Staines WA, Yasumura T, Patel D, Furman CS, Stelmack GL, Nagy JI. Immunogold evidence that neuronal gap junctions in adult rat brain and spinal cord contain connexin-36 but not connexin-32 or connexin-43. Proc Natl Acad Sci U S A. 2000;97:7573–7578. doi: 10.1073/pnas.97.13.7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rash JE, Davidson KG, Kamasawa N, Yasumura T, Kamasawa M, Zhang C, Michaels R, Restrepo D, Ottersen OP, Olson CO, Nagy JI. Ultrastructural localization of connexins (Cx36, Cx43, Cx45), glutamate receptors and aquaporin-4 in rodent olfactory mucosa, olfactory nerve and olfactory bulb. J Neurocytol. 2005;34:307–341. doi: 10.1007/s11068-005-8360-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB. Hypothalamus. In: Paxinos G, Mai JK, editors. The human nervous system. Ed 2. San Diego: Academic; 2004. pp. 513–550. [Google Scholar]
- Scheetz AJ, Constantine-Paton M. Modulation of NMDA receptor function: implications for vertebrate neural development. FASEB J. 1994;8:745–752. doi: 10.1096/fasebj.8.10.8050674. [DOI] [PubMed] [Google Scholar]
- Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004;5:R13. doi: 10.1186/gb-2004-5-3-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova L, Grafe A, Seiler A, Schumacher S, Nitsch R, Wulczyn FG. Regulation of miRNA expression during neural cell specification. Eur J Neurosci. 2005;21:1469–1477. doi: 10.1111/j.1460-9568.2005.03978.x. [DOI] [PubMed] [Google Scholar]
- Somekawa S, Imagawa K, Naya N, Takemoto Y, Onoue K, Okayama S, Takeda Y, Kawata H, Horii M, Nakajima T, Uemura S, Mochizuki N, Saito Y. Regulation of aldosterone and cortisol production by the transcriptional repressor neuron restrictive silencer factor. Endocrinology. 2009;150:3110–3117. doi: 10.1210/en.2008-1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein V, Nicoll RA. GABA generates excitement. Neuron. 2003;37:375–378. doi: 10.1016/s0896-6273(03)00056-4. [DOI] [PubMed] [Google Scholar]
- Thalakoti S, Patil VV, Damodaram S, Vause CV, Langford LE, Freeman SE, Durham PL. Neuron-glia signaling in trigeminal ganglion: implications for migraine pathology. Headache. 2007;47:1008–1023. doi: 10.1111/j.1526-4610.2007.00854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo N, Klein ME, Varlamova O, Keller DM, Yamamoto T, Goodman RH, Impey S. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc Natl Acad Sci U S A. 2005;102:16426–16431. doi: 10.1073/pnas.0508448102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton KD, Navarrete R. Postnatal changes in motoneurone electrotonic coupling studied in the in vitro rat lumbar spinal cord. J Physiol. 1991;433:283–305. doi: 10.1113/jphysiol.1991.sp018426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zheng H, Liu C, Zhu C, Wang W, Li Z. Ciliary neurotrophic factor-treated astrocyte conditioned medium regulates the L-type calcium channel activity in rat cortical neurons. Neurochem Res. 2008;33:826–832. doi: 10.1007/s11064-007-9514-7. [DOI] [PubMed] [Google Scholar]
- Wang Y, Denisova JV, Kang KS, Fontes JD, Zhu BT, Belousov AB. Neuronal gap junctions are required for NMDA receptor-mediated excitotoxicity: implications in ischemic stroke. J Neurophysiol. 2010;104:3551–3556. doi: 10.1152/jn.00656.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]