

Abstract
The major objective of this paper is to address a controversial binding sequence between nucleic acid bases (NABs) and C60 by investigating adsorptions of NABs and their cations on C60 fullerene with a variety of density functional theories including two novel hybrid meta-GGA functionals, M05-2x and M06-2x, as well as a dispersion-corrected density functional, PBE-D. The M05-2x/6-311++G** provides the same binding sequence as previously reported, guanine(G) > cytosine(C) > adenine (A) > thymine (T); however, M06-2x switches the binding strengths of A and C, and PBE-D eventually results in the following sequence, G>A>T>C, which is the same as the widely accepted hierarchy for the stacking of NABs on other carbon nanomaterials such as single-walled carbon nanotube and graphite. The results indicate that the questionable relative binding strength is due to insufficient electron correlation treatment with the M05-2x or even the M06-2x method. The binding energy of G@C60 obtained with the M06-2x/6-311++G(d,p) and the PBE-D/cc-pVDZ is −7.10 and −8.07 kcal/mol, respectively, and the latter is only slightly weaker than that predicted by the MP2/6-31G(d,p) (−8.10kca/mol). Thus, the PDE-D performs better than the M06-2x for the observed NAB@C60 π-stacked complexes. To discuss whether C60 could prevent NABs from radiation-induced damage, ionization potentials of NABs and C60, and frontier molecular orbitals of the complexes NABs@C60 and (NABs@C60)+ are also extensively investigated. These results revealed that when an electron escapes from the complexes, a hole was preferentially created in C60 for T and C complexes, while for G and A the hole delocalizes over the entire complex, rather than a localization on the C60 moiety. The interesting finding might open a new strategy for protecting DNA from radiation-induced damage and offer a new idea for designing C60-based antiradiation drugs.
Keywords: radiation-induced damage, NAB, C60, dispersion-corrected DFT, binding sequence
1. Introduction
With a rapid development and wide application, the potential effect of nanoparticles on health and environment begins to be a hot topic in the nanotechnology field. A recent issue of Nano Today reported that manufacture and use of nanoparticles in medicine and other industries could pose a potential risk to human health.1 As a typical nanoparticle, since its discovery C60 has attracted considerable interest in the field of nanotechnology due to its unique properties. Because of its small size and poor solubility in polar solvents, it may penetrate through cellular membrane, resulting in adverse effects on biological systems, such as traveling through blood stream to heart and depositing on the targeted part of human body causing tissue hurt, lung disease and some toxic effects on human.2 In spite of potential unfriendly to environment, a good deal of experiments have also established prospective applications for C60 derivatives (water-soluble fullerene derivatives) in many fields such as antioxidants, neuroprotective agents, antiapoptotic activity, DNA photocleavage, enzyme inhibition, antimicrobial activity and anti-HIV activity.3 Various applications of C60 derivatives have been reviewed by Nakamura et al.4 Particularly, C60 fullerene is proposed to serve as a future antiradiation drug to protect against the side effects of cancer radiation therapy,5 which is a beneficial effect on biological systems. Understanding about the interaction mechanism between C60 or C60 derivatives and biomolecules can promote such kind of biomedical applications.
Oxidative DNA damage could cause several diseases, such as cancer6 and aging7. Radiation is one of the most general reasons for oxidative DNA damage.8 When the high-energy radiation interacts with DNA, an electron may be ejected from DNA, and a hole created in this way migrates over a long distance through a hopping mechanism,9 eventually ending on a guanine base of DNA because it has the lowest ionization potential among all DNA bases.10 The guanine radical cation (G·+) is a harbinger of many pathological conditions in living cells as a result of its involvement in many multiform reactions.11 As measured by photoionization yield, the adiabatic IP of C60 fullerene molecule is close to 7.54eV,12 which is lower than the adiabatic IP of guanine (7.77eV), the lowest one among four NABs.13 This trend is also confirmed by the PBE calculation (Table S1 in the Supporting Information). Consequently, a question is raised whether C60 could protect guanine and other DNA bases from radiation-induced damage. Upon a high-energy radiation interacting with NAB@C60 complexes, a hole will be created. If the hole localizes on the C60 moiety rather than on the NAB moiety, it may demonstrate that C60 can act as an antiradiation substance. This investigation is meaningful for the anticipative application of fullerenes in the antiradiation drug field. 5
To the best of our knowledge, there are few literatures about this issue. Shukla et al did useful pioneering research and trials in this topic, and located a stable geometry of C60-guanine.11a During the preparation of the paper, another closely relevant paper from Shukla et al was released on the website of Chem. Phys. Lett.11b In the most recent paper, a novel hybrid meta-GGA DFT, M05-2X, was used to reveal the binding strength of NABs and C60 molecules. Nevertheless, their binding energies and binding strength sequence may be questionable. The binding strength follows the order: G (−4.99kcal/mol) > C(−4.26) >A (−4.10) > T(−3.75), which is different from the well reported binding strength of DNA bases with single-walled carbon nanotube (SWNT) and graphite, G>A>T>C.14 In addition, the binding energy of G-C60 is quite far from that obtained with MP2 method (−4.99 versus −8.10 kcal/mol).11a Shukla et al 11b claimed that the binding strength order predicted by the M05-2x/6-311++G(d,p) followed a recent experimental report, G>C>A>T. 15 However, the sequence is for poly-nucleotide/SWNT hybrids, specifically, 12-base-long single stranded DNA homopolymers consisting of poly d(A)12, poly d(T)12, poly d(C)12 or poly d(G)12, rather than for the single DNA base molecules. Because of quite different self-stacking of base molecules in ssDNA, the binding between the ssDNA and SWNT may be different from that of single base and SWNT. On the other hand, even for the poly-nucleotide, a different binding strength measured with spectroscopy is G>A>T>C.16 Although the HF level together with classical force field14c and constraint optimizations with MPWB1K density functional 14a predicted the binding order of G > A > T > C in the gas phase for the interaction of NABs with (5,5) SWCNT, the binding order based upon isothermal calorimetry experiments on much larger diameter nanotube was predicted to be T > A > C.14c The solvent effects may be responsible for the diffidence.
The above deficiencies motivated us to revisit the system with more accurate methods dealing with “long range” electron correlation effects, such as, M06-2x and DFT-D methods. The results will allow us to get insights into whether the above strange strength order originates from the M05-2x method or it really is. As reported by Hobza et al,17 empirical dispersion-corrected density method functional (DFT-D, specifically TPSS-D) provides more accurate results for the π-stacked adenine dimers than M06-2x does. However, Sherrill et al’s assessment indicated that M06-2x performs somewhat better than PBE-D for the π-stacked NAB dimers.18 Thus, in addition to providing a benchmark binding strength for stacked complexes NABs@C60, another objective of the paper is to assess performance of M05-2x, M06-2x and DFD-D on the π-stacked NABs@C60. Furthermore, cationic systems (NABs@C60)+ were also extensively investigated in order to explore whether fullerenes could protect NABs from radiation-induced damage.
2. Computational Methods
Full geometry optimizations and property calculations for DNA bases and C60 were firstly performed with generalized gradient approximation (GGA) type of DFT, PW91 with a double numerical plus polarization function (DNP) basis sets, implemented in DMol3 package from Accelrys, Inc.19 The spin polarization scheme was utilized to deal with open-shell systems. The convergence threshold of energy was set as 10−7 a.u. Vibrational frequency analysis was also carried out at the level of geometry optimization. The optimized complexes were all minimum energy species along the potential energy surface. The less extensive method aims to extensively search for the π-stacked conformations for complexes NABs@C60.
It is known that, in spite of great improvement over LDA, a typical GGA type of DFT like the above PW91 method still underestimates the binding for the systems mainly arising from van der Waals (vdW) interaction. Hybrid meta-GGA functionals, M05-2X and M06-2x, developed by Truhlar et al 20 and implemented in Gaussian 09, 21 better describing the non-bonding interaction than PW91, were therefore also employed to fully optimize a few favorable structures for each NAB.
For an empirical dispersion-corrected density functional theory (DFT-D), Grimme’s scheme was adopted,22 where the vdw interaction term is well described by a damped interatomic potential, accounting for long-range dispersion effects in noncovalent systems. The total density functional energy can be written as follows:
| (1) |
where EDFT is the normal self-consistent density functional energy, and Evdw is the empirical dispersion correction term and is given by
| (2) |
Here, s6 is a global scaling factor only depending on the applied density functional method. As usual, the combined dispersion coefficient for atom pair i and j, , was estimated with a geometric mean of individual C6 coefficient, while Rij, the interatomic distance between atoms i and j, is an arithmetic mean value of individual vdw radius. N is the number of atoms, and fdmp (Rij), a damping function, is expressed by the following equation
| (3) |
Where α is taken to be 20 in the exponent. Perdew-Burke-Ernzerhof (PBE) 23 exchange-correlation functional in the DFT part was employed in the present work, and the DFT-D method was therefore referred to as PBE-D. The PBE-D calculations were conducted with the ORCA suite of programs.24 Dunning’s correlation-consistent polarized valence basis sets of double-zeta (cc-pVDZ) and/or triple-zeta (cc-pVTZ) were used together with the PBE-D method, as usually applied to the other π-π stacking systems.18 All of the relevant systems were also fully optimized at the PBE-D levels.
3. Results and Discussion
3.1. Neutral Complexes (NABs@C60) in the Gas Phase
With the PW91/DNP method, a variety of positions of NABs over C60 were investigated. In C60 fullerene, the hh C-C bond (the C-C bond shared by two hexagonal rings) has more π-bonding character than the hp C-C bond (the C-C bond shared by a hexagonal ring and a pentagonal ring).25 There are three distinct symmetry sites in C60 with respect to the hexagonal ring in NAB molecules: bridge, top and hollow. The adsorption on hollow site is less stable than on the other two sites. The hollow conformation therefore will not be discussed below. Various geometries and the relative energies at the PW91/DNP level are shown in Figure S1 and Table S2 in the Supporting Information.
For thymine (T), guanine (G), and cytosine (C), adsorption on the bridge site is the most stable, while for adenine the most stable one is on the top site. The fullerene molecule in these complexes does not undergo significant deformation. The binding energy Eb for the neutral complexes NAB@C60 is defined as:
| (4) |
where E(NAB-C60), E(C60), and E(NAB) are the total energies for the complex, isolated C60, and NAB, respectively. It was widely accepted that the BSSE effect is nearly negligible when the numerical basis sets are employed.19 The basis set superposition error (BSSE) was therefore not included for the PW91/DNP level with Dmol3 code, and however, it was taken into account with counterpoise scheme at the M05-2x, M06-2x and PBE-D levels.26
As shown in Table 1, the binding energies from the PW91/DNP for NAB@C60 are quite small. Obviously, all NABs undergo physisorption processes when they interact with C60. G@C60 complex has the strongest binding (−2.63 kcal/mol), followed by C-C60 (−2.30 kcal/mol) and A-C60 (−2.12 kcal/mol), and T-C60 complex has the lowest binding energy (−1.78 kcal/mol). This binding trend, G > C > A > T, is the same as that predicted by Shukla et al with M05-2X/6-311++G(d, p), although the binding is dramatically weaker than that obtained by M05-2X/6-311++G(d, p). The present full optimization with the M05-2x method generates the same binding energies for G@C60 and C@C60 complexes as reported by Shukla et al,11b and those for T@C60 and A@C60 only slightly differ (−3.70 and −4.16 kcal/mol versus −3.75 and −4.10 kcal/mol). Although their detailed geometries for the NBA@C60 complexes are not available, because of rather similar binding energies the geometries from the present and Shukla’s work should be very similar as well. Except for the similar one for G@C60 (3.031Å in the present work versus 3.041Å) the nearest distances between heavy (non-hydrogen) atom of NABs and carbon atoms of C60 differ by 0.1–0.2Å different (3.001, 2.993, and 3.131Å versus 3.249, 3.245, and 3.220Å), which may arise from different definitions.
Table 1.
Binding energies (kcal/mol) for the neutral complexes with the PW91, M05, M06 and PBE-D methods. Basis sets A, B, and C are cc-pVDZ, 6-311++G(d,p), and cc-pVTZ, respectively.
| Complex | PW91/DNP | M052x/A | M052x/B | M052x/Ba | M062x/A | M062x/Bb | PBE-D/A | PBE-D/C |
|---|---|---|---|---|---|---|---|---|
| A@C60 | −2.12 | −4.06 | −4.16 | −4.10 | −6.24 | −6.26(−6.40) | −7.36 | −7.42 |
| C@C60 | −2.30 | −3.78 | −4.26 | −4.26 | −5.64 | −6.01(−6.07) | −6.19 | −6.34 |
| G@C60 | −2.63 | −4.67 | −5.00 | −4.99 | −6.80 | −7.10(−7.55) | −8.07 | −8.45 −8.10c |
| T@C60 | −1.78 | −3.54 | −3.70 | −3.75 | −5.29 | −5.46(−5.56) | −6.53 | −6.69 |
Table 1 also shows that both the M06-2x/cc-pVDZ and M06-2x/6-311++G(d,p) significantly enhance the binding between NABs and C60 by 1–2 kcal/mol as compared with those from the M05-2x/6-311++G(d,p). It is interesting to note that the relative binding strength for C@C60 and A@C60 is opposite to that obtained by the M05-2X/6-311++G(d,p). The binding sequence for NBAs@C60 is consequently changed to G>A>C>T. Although M06-2x is considerably improved in describing the non-covalent interaction over other GGAs,20 there is still some deficiency compared with the dispersion-corrected density methods (DFT-D).17 As compared with the results from the M06-2x, the present PBE-D/cc-pVDZ does further strengthen the binding between NABs and C60 so that the BSSE-corrected binding energy for G@C60 complex (−8.07kcal/mol) is only slightly less negative than −8.10kcal/mol, obtained at the MP2/6-31G(d,p)//MP2/6-31G(d) level.11a Thus, for the examined π-stacked dimers NAB@C60, it seems that PDE-D performs better than M06 does. Since it is well known the MP2 method overestimates non-covalent interaction to some extent, the results obtained by PBE-D/cc-PVDZ method may be rather close to the real values of the binding energies. The PBE-D turns out that the binding strength between NABs@C60 follows the sequence: G > A >T >C, which is the same as the binding sequence of NABs with (5, 5) as well as (10, 0) SWNTs 14a, 14c and the hierarchy of NABs with a graphene layer as well as graphite surface.14d, 27 PBE-D/cc-PVTZ further enhances the binding of NABs with C60 by approximately 0.1kcal/mol for A, C, and T, yet by 0.35 kcal/mol for G. However, the binding strength remains the same as that from the PBE-D/cc-pVDZ. A variation of the binding strength for NABs@C60 with theoretical methods is shown in Figure 2.
Figure 2.
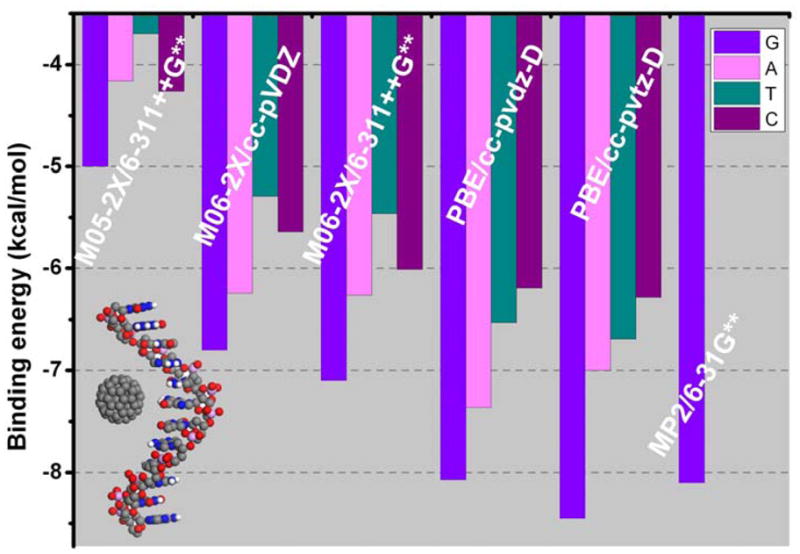
Variation of binding strength for (NAB@C60) with theoretical methods.
The displaced-stacked π-π interaction between NABs and C60 is stabilized mainly by dispersion effect depending on the surface area of buried as well as on the polarizability of the moieties.28 Since there are both five-membered imidazole ring and six-membered pyrimidine ring in guanine and adenine molecules, the π-π stacking interactions between adenine/guanine and C60 should be generally stronger than thymine/cytosine with one only six-membered ring. London’s empirical formula may help understand the above binding sequence for the NABs@C60 complexes. 29
| (5) |
where α and I are the polarizability and ionization potential for individual molecules, respectively, and R is the distance between molecules A and B. The polarizability of NABs, representing the deformability of the electronic charge distribution, is known to arise from the regions associated with the aromatic rings, lone electron pairs in nitrogen, and oxygen atoms. Guanine has the largest value of polarizability (120.8 au at the B3LYP/6-31G(d) level), followed by adenine (108.6 au), and thymine and cytosine have similar ones (94.7 and 90.5 au). This trend is the same as Gowtham’s report.27 Based on London’s equation, the ratio of the dispersion forces of the four complexes is approximately as, 100% (G@C60): 89%(A@C60): 81%(T@C60): 75%(C@C60), which also supports that A@C60 and T@C60 should be stronger than C@C60. Especially, the prediction confirms the binding sequence obtained by the PBE-D method.
To gain further insights into the electronic structures, Figure 3 illustrates the highest occupied molecular orbitals (HOMO) and the lowest unoccupied molecular orbitals (LUMO) of these complexes. LUMO of all these complexes localizes on C60. Orbital energies for the HOMO of the five individual molecules are as follows: G(−0.208 a.u.) > A(−0.218)> C60 (−0.220) > C(−0.227) > T(−0.240). As shown by the molecular orbitals in Figure 3 and the energy level diagrams in Figure 4, HOMOs from the NAB and C60 make different contributions to the HOMO of complexes. For G@C60 and A@C60, the HOMO levels of the complexes are close to the HOMO of NAB, while the HOMO levels of complexes C@C60 and T@C60 are close to the HOMO level of C60. According to Figure 3, HOMOs delocalize over both guanine/adenine moiety and C60 for G/A@C60 complexes, while they almost localize on C60 in T/C@C60 complexes. Accordingly, the presence of C60 molecule may well protect thymine and cytosine from radiation damage. However, whether fullerene could protect guanine and adenine from radiation damage needs to be further investigated.
Figure 3.
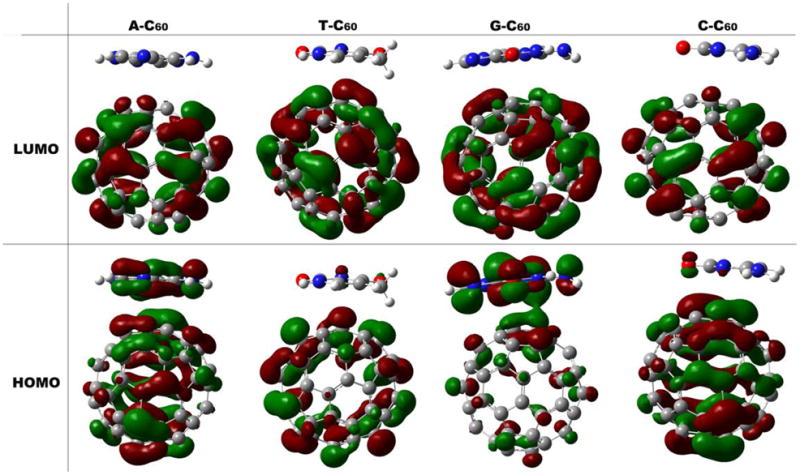
Plots of the frontier orbitals of the NAB@C60 complexes. The isovalue is 0.02 a.u.
Figure 4.
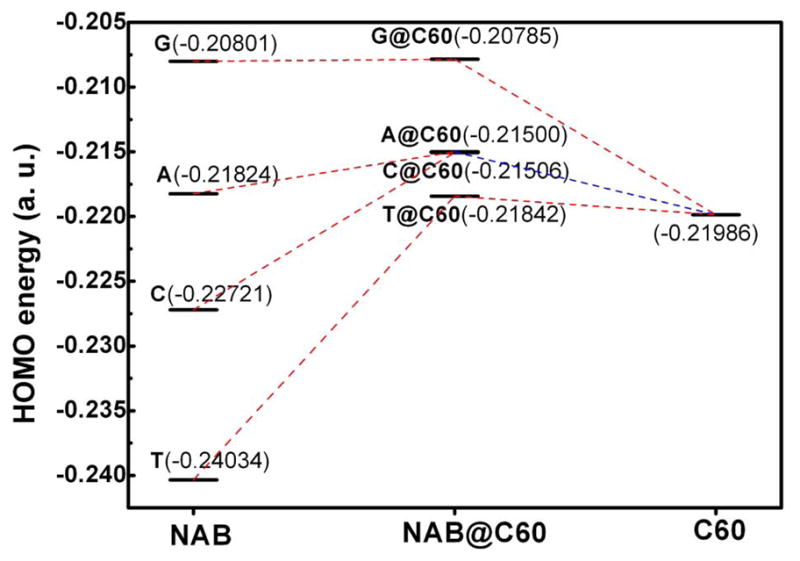
Molecular orbital energy level diagrams (au). The LUMO levels of NABs are significant higher than C60. LUMO of NABs therefore has no contribution to the LUMO of complexes. For clarity NABs’ LUMO level are not shown in this picture. HOMO energies of A@C60 and C@C60 are too close to distinguish.
3.2. Cationic Complexes (NABs@C60)+ in the Gas Phase
For the four (NABs@C60)+ complexes (NAB=A, C, G and T), both the π-π stacked and the T-shaped σ-π conformations were optimized with the PW91/DNP, M05-2x/cc-pVDZ, M06-2x/cc-pVDZ, M06-2x/6-311++G**, and PBE-D/cc-pVDZ methods. The first ionization potentials of NAB and C60 from the theoretical methods were summarized in Table 3 together with the experimental data. The M06-2x, PBE-D, and MPWB1K predicted that C60 has lower adiabatic IP than T, C and A, yet higher aIP than G. At the M06-2x/cc-pVDZ level the aIPs for the NABs are lower by 0.1–0.2eV than the experimental data, while the M06-2x/6-311++G** yields higher ones by 0.1–2eV. Thus, because of higher aIP than C60 for the ionic complexes of A, C, and T with C60 the binding energy was defined for (NABs@C60)+ as:
| (6a) |
Table 3.
The first adiabatic and vertical ionization potentials (aIP and vIP/eV) for nucleic acid bases and C60 at various theoretical levels.
| Thymine | Cytosine | Adenine | Guanine | C60 | |
|---|---|---|---|---|---|
| aIP, M052x/cc-pVDZ | 8.73 | 8.59 | 8.02 | 7.56 | 8.04 |
| aIP, M062x/cc-pVDZ | 8.71 | 8.58 | 8.02 | 7.56 | 7.96 |
| aIP, M062x/6-311++G** | 8.97 | 8.90 | 8.31 | 7.87 | 8.03 |
| aIP, PBE-D/cc-pVDZ | 8.34 | 8.14 | 7.68 | 7.23 | 7.25 |
| vIP, PBE-D/cc-pVDZ | 8.47 | 8.29 | 7.89 | 7.54 | 7.31 |
| aIP, MPWB1K/cc-pVDZ | 8.62 | 8.38 | 7.88 | 7.38 | 7.68 |
| aIP, Exp.a/b | 8.87/8.80 | 8.68/8.45 | 8.26/7.80 | 7.77/7.85 | |
| IP, Experimental rangec | 7.54–8.1 |
Because of lower aIP than C60 for the ionic complexes of G with C60,
| (6b) |
The superscript “+” represents the cationic system, the other notations are the same as them in equation (4). In the cationic complexes, because of the predictable electrostatic force, the binding energies listed in Table 2 are higher negative than those of the neutral complexes NAB@C60. Figure 5 shows the π-stacked as well as the T-shaped σ-π conformations for the complexes (NAB@C60)+, together with the nearest distances. For the π-stacked conformations, consistent with stronger binding the nearest distances for the cationic complexes are shorter than those for the neutral counterparts, e.g., at the PBE-D/cc-pVDZ level, 2.805, 2.351, 2.812 and 3.034 Å versus 2.974, 3.129, 2.913 and 3.125 Å for adenine, cytosine, guanine, and thymine, respectively.
Table 2.
Binding energies (kcal/mol) for (NAB@C60)+ at various calculation levels.
| Complexes | PW91/DNP | M05-2x/cc-pVDZ | M06-2x/cc-pVDZa | M06-2x/6-311++G** | PBE-D/cc-pVDZa | |
|---|---|---|---|---|---|---|
| (A@C60)+ | stacking | −11.1 | −16.4 | −17.6 (−14.3) | −15.7 | −22.2(−19.0) |
| (A@C60)+ | σ-π | — | −9.7 | −10.7 | −7.1 | — |
| (C@C60)+ | stacking | — | — | −15.4 | −14.5 | −22.0(−17.6) |
| (C@C60)+ | σ-π | −14.4 | −31.5 | −29.9 (−24.1) | −25.3 | −26.8(−20.3) |
| (G@C60)+ | stacking | — | −12.8 | −14.5 | −16.5 | −26.1(−22.70) |
| (G@C60)+ | σ-π | −18.7 | −6.8 | −10.7 | −14.2 | — |
| (T@C60)+ | stacking | — | −8.3 | −11.0 | −11.2 | −15.9(−13.5) |
| (T@C60)+ | σ-π | −9.4 | −9.1 | −9.7 | −9.7 | −13.2(−10.6) |
The data in parentheses include BSSE correction.
Figure 5. π-stacked and T-shaped [NAB@C60]+ complexes.

The dark, blue and red data are the nearest distance between non-hydrogen atom of NBA and C60 with M05-2x/cc-PVDZ, M06-2x/cc-PVDZ, and PBE-D/cc-pVDZ methods, respectively.
At the PW91/DNP level, except for the π-stacked geometry of (A@C60)+ that still maintains the favorable conformation similar to its neutral complex, for the other three complexes C60+ binds with NABs through π and carbonyl oxygen, referred to as σ-π conformation. When electron correlation effects are well taken into account by the M05-2x, M06-2x and PBE-D methods, the located conformations are significant different from those obtained with the PW91/DNP method. At the M05-2x/cc-pVDZ level, for (C@C60)+ the initial π-stacked conformations always converge to the σ-π conformation, while for other three cationic complexes (NAB@C60)+ both the π-π stacked and T-shaped σ-π conformations were located. The stacked (T@C60)+ is only 0.8 kcal/mol less stable than the T-shaped one. According to Table 2, however, the π-stacked conformation is more stable than the σ-π one by 6.7 and 6.0 kcal/mol for the (A@C60)+ and (G@C60)+ complexes, respectively.
As shown in Figure 5, as electron correlation is better included by the M06-2x/cc-pVDZ and M06-2x/6-311++G** methods, a rather tilted stacked conformation for (C@C60)+ was also predicted, but it is considerably less stable than the T-shaped σ-π conformation by 14.5 and 10.8 kcal/mol. At the PBE-D/cc-pVDZ level, although the stacked conformation for (C@C60)+ is still less stable than the T-shaped one, their binding energy difference is just 4.8 kcal/mol. The M06-2x/cc-pVDZ, M06-2x/6-311++G**, and PBE-D/cc-pVDZ also switch the relative binding for the (T@C60)+ predicted by the M05-2x, e.g., the stacking is stronger than the σ-π interaction by approximately 1–2 kcal/mol. At the PBE/cc-pVDZ level, the binding strength including BSSE corrections decrease in the sequence, G (−22.7kcal/mol)> C(−20.3) >A(−19.0) >T (−13.6).
Obviously, favorable geometries of the cationic complexes are very sensitive to the applied theoretical methods. For the (NBAs@C60)+ complexes, two principal forces, dispersion and electrostatic force, may determine the conformations of (NBAs@C60)+. Because of possessing two aromatic rings, guanine and adenine interact more strongly with C60+ via dispersion force than one-ring cytosine and thymine. When the high level methods are applied, such as M06-2x and PBE-D that can accurately deal with electron correlation inducing dispersion force, for adenine and guanine the favorable conformations are therefore the π-stacked ones. On the other hand, because of the positively charged surface in C60+, electrostatic force in (NBAs@C60)+ plays more important role on determining the conformation than in the neutral complexes. The unique oxygen atom in cytosine carries more negative charge (−0.362e) than carbonyl oxygen atoms (−0.338 and −0.354e) in thymine. The stronger electrostatic interaction between the oxygen of cytosine and C60+ induces a favorable T-shaped σ-π conformation for (C@C60)+ complex. Actually, as shown in Table 2 and Figure 5 only M06-2x and PBE-D predicts a rather tilted partially stacked conformation that is less stable than the T-shaped (C@C60)+. For the (T@C60)+ complex, the two major driving forces are well balanced, and both the stacked and T-shaped conformations are brought about at the M05-2x, M06-2x and PBE-D levels. Since the PW91 considerably underestimates dispersion force, at this level electrostatic force may determine the optimal complexes. Thus, for (C@C60)+, (G@C60)+, and (T@C60)+ complexes the conformations obtained by PW91/DNP are T-shaped, which also supports the claim that the T-shaped configuration has more stabilizing electrostatic interaction than the displaced π-π stacking configuration due to favorable quadrupole-quadrupole interaction.30
3.3 Hole Distribution and Its Biological Implication
To address the other objective whether fullerene can protect DNA bases from radiation damage, the spin densities for (NBAs@C60)+ were illustrated in Figure 6 together with the atomic charge fitted from electrostatic potential and the total spin density on the C60 moiety. The total atomic charges on the C60 are +0.632e, +0.731e, +0.850e and +0.863e for the π-stacked complexes (G@C60)+, (A@C60)+, (T@C60)+, and (C@C60)+, respectively, while for the T-shaped (C@C60)+ and (T@C60)+ the charges are even larger (+0.888e and +0.975e). This shows that an electron can be dominantly ejected from the C60 moiety for the (C@C60) and (T@C60) complexes upon photoionization or radiation. As a result, the trapped hole also mainly distributes over the C60 portion in (C@C60)+ and (T@C60)+, which is further supported by the high spin density in the C60 moiety ranging from 0.764 for the π-stacked (T@C60)+ to 0.924 for the T-shaped (T@C60)+. The above significant spin density localization over the C60 moiety in the (C@C60)+ and (T@C60)+ complexes demonstrates that the presence of C60 could well prevent C and T from radiation or photoionization. The spin density on the C60 of (A@C60)+ is lower than those of the π-stacked (C@C60)+ and (T@C60)+ by approximately 0.1, (0.670 versus 0.849 and 0.760). For (G@C60)+, the spin density almost equivalently delocalizes over guanine and C60 (0.461 versus 0.539). Thus, C60 could only partially protect A and G from radiation damage by sharing the injected hole.
Figure 6.
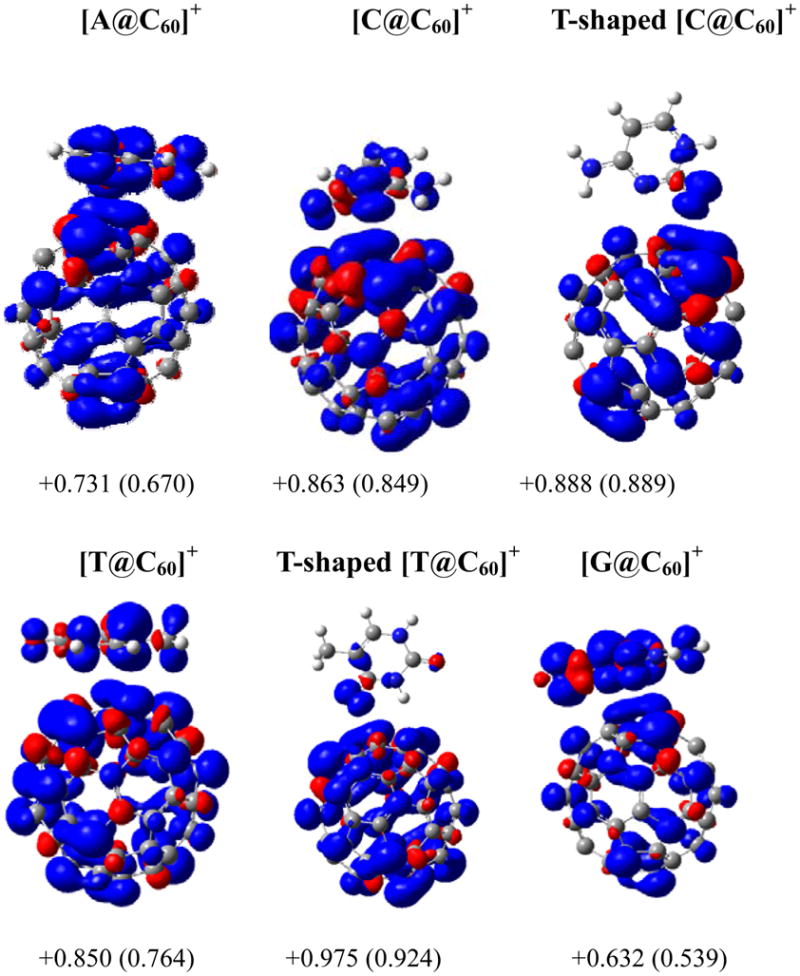
The spin density surface of cationic complexes (NABs@C60)+ with an isovalue of 0.004au. The numbers below each panel stand for the charge from ESP fitting and total spin density (in parentheses) in C60 moiety.
The above spin density distribution scenario somehow agrees well with the IP difference between C60 and NABs. The lager the IP difference, the higher the degree of the spin density delocalization over C60. To obtain a more remarkable performance for NABs protection, it is therefore suggested to synthesize and utilize new fullerene derivatives that have a significant IP gap with guanine.
IP values for the complexes were also calculated, as defined below,
| (7) |
Combination of equations (4), (6) and (7), results in,
| (8) |
Binding energies of the cationic systems (Eb+) are much more negative than their corresponding neutral systems (Eb). Thus, IP of the complex is smaller than C60 as well as NABs. As shown in Figure 7, the discrepancies between IPs of four NAB@C60 complexes are smaller than those of four NABs. These observations indicate that the binding of NABs with C60 can reduce the IP of whole system by sharing the hole as possible, leading NAB@C60 to promising hole carriers or relays for the long-range charge migration. Further, the trend of IPs for NAB@C60 is also different from that for the isolated NAB molecules. For the NAB@C60 complexes, A@C60 has larger IP than C@C60, which is opposite to the isolated NAB (IP(C) > IP(A)). G@C60 still has the lowest IP in the four complexes just as guanine in the four NABs. This implies that the NAB@C60 carriers or relays of a hole are property-tunable via varying the base partners of C60.
Figure 7.
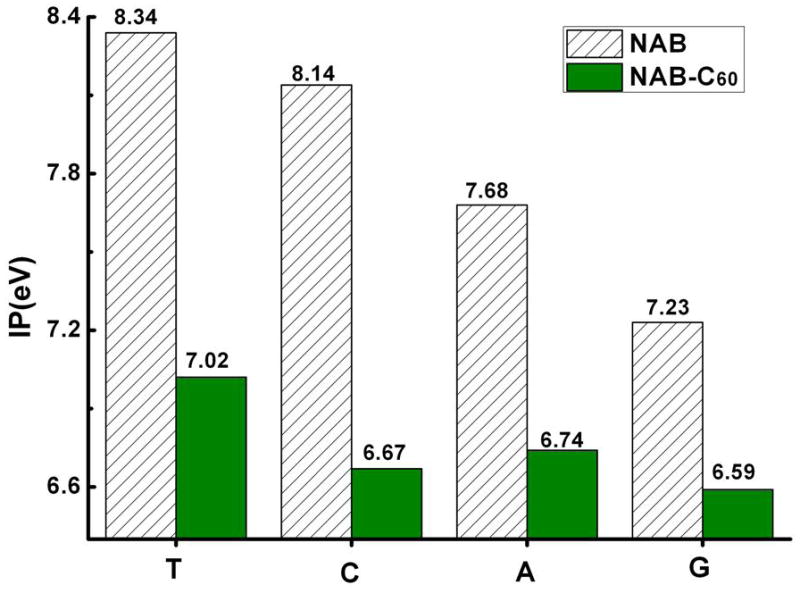
Adiabatic IP values of the individual NABs and NABs@C60 complexes in the gas phase at the PBE-D/cc-pVDZ level, while that of C60 is 7.25eV.
Clearly, all above predictions are derived from the steady-state analysis on the properties of the NAB@C60 complexes. However, some dynamics information are still unknown which hopefully provide the dynamic knowledge for a deep understanding of the interaction of the shortly-separated NABs and C60 and their charge transfers. In addition, the protein surroundings and solvent molecules may yield considerable effects on the interaction modes between NABs and C60, the charge transfer mechanism among them, and tunability of the carrier/relay properties of the NAB@C60 complexes. Further investigations are certainly needed for a clarification of all these details.
4. Conclusions
In summary, geometric and electronic properties of NAB@C60 and (NAB@C60)+ were extensively investigated by the M05-2x, M06-2x and PBE-D methods. The binding strength sequence for the neutral complex NAB@C60 varies with the applied theoretical methods. The M05-2x/6-311++G** provides the same binding sequence as previously reported by Shukla et al.,11b guanine (G) > cytosine (C) > adenine (A) > thymine (T); however, the binding strengths of A and C are switched at the M06-2x level, resulting in the binding sequence, G > A > C > T. When dispersion force is explicitly included by the PBE-D method, the above sequence is further revised to, G>A>T>C, which is the same as the binding sequence of NABs with SWNTs, a graphene layer as well as a graphite surface,14a, 14c–d, 27 and also follows the sequence predicted by the London’s dispersion equation. The results indicate that the previous questionable relative binding strength is due to insufficient electron correlation treatment with the M05-2x. A comparison among the predicted binding energy with different methods for G@C60 shows that PDE-D performs better than M06-2x. At both the M06-2x and PBE-D levels, the favorable conformation for (A@C60)+, (G@C60)+, and (T@C60)+ is the π-stacked one; while for (C@C60)+ the T-shaped σ-π conformation is more favorable. The frontier molecular orbital analysis reveals that when an electron escapes from the complexes, the hole dominantly localizes on C60 for (C@C60)+ and (T@C60)+, while for (A@C60)+ and (G@C60)+ delocalizes over the entire complex. Thus, the presence of C60 may well prevent C and T from radiation damage, while only partially protect A and G. It should be noted that protein surrounding and solvent effect could modify the binding modes and even the charge distributions, thus providing a possibility in regulating the interactions and charge exchange mechanism between the NABs and C60 and competition among four NAB bases. This work is being expanded with great efforts in our co-laboratory.
Supplementary Material
Figure 1. Side- and top views of π-stacked NAB@C60 complexes.

The dark, blue and red data are the nearest distance between non-hydrogen atom of NBA and C60 with the M05-2x/6-311++G(d,p), M06-2x/6-311++G(d,p) and PBE-D/cc-pVDZ methods, respectively.
Acknowledgments
This work at Shandong University (Sun and Bu) is supported by NSFC (20633060, 20973101), NCET, and the Independent Innovation Foundation (2009JC020) of Shandong University. The project described here was also supported in part by the National Institute of General Medical of the National Institute of Health (SC3GM082324), the American Recovery and Reinvestment Act (3SC3GM082324-02S1) and ACS PRF (47286-GB5) in terms of scholarly development (Wang at Albany State University). We are also grateful to Dr. Zhengfeng Xu for optimizing the complexes (NAB@C60)+ at M06-2x/6-311++G(d,p) level.
Footnotes
Supporting Information Available: Figure S1 displays the geometries of various NAB@C60 systems obtained at the PW91/DNP level in the gas phase. Table S1 shows the adiabatic and vertical IP calculated with B3LYP/6-31G(d) for the isolated molecules optimized at the PBE-D/cc-pVDZ level. Table S2 summarizes the relative energies (in kcal/mol) of various configurations at the PW91/DNP level. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Wenming Sun, The Center for Modeling & Simulation Chemistry, Institute of Theoretical Chemistry, Shandong University, Jinan, 250100, People’s Republic of China.
Yuxiang Bu, The Center for Modeling & Simulation Chemistry, Institute of Theoretical Chemistry, Shandong University, Jinan, 250100, People’s Republic of China.
Yixuan Wang, Department of Natural Science, Albany State University, Albany, Georgia 31705.
References
- 1.Sealy C. Nanoparticles cause DNA damage, according to new reports. Nano Today. 2010;5(1):1. [Google Scholar]
- 2.Nel A, Xia T, Madler L, Li N. Toxic Potential of Materials at the Nanolevel. Science. 2006;311(5761):622–627. doi: 10.1126/science.1114397. [DOI] [PubMed] [Google Scholar]
- 3.Bosi S, Da Ros T, Spalluto G, Prato M. Fullerene derivatives: an attractive tool for biological applications. Euro J Med Chem. 2003;38(11–12):913–923. doi: 10.1016/j.ejmech.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Nakamura E, Isobe H. Functionalized Fullerenes in Water. The First 10 Years of Their Chemistry, Biology, and Nanoscience. Acc Chem Res. 2003;36(11):807–815. doi: 10.1021/ar030027y. [DOI] [PubMed] [Google Scholar]
- 5.Bhatia A. Nuclear Medicine In 21st Millennium: An Approach Via Nanotechnology. Asian J Exp Sci. 2008;22(2):12. [Google Scholar]
- 6.Loft S, Poulsen HE. Cancer risk and oxidative DNA damage in man. J Mol Med. 1996;74(6):297–312. doi: 10.1007/BF00207507. [DOI] [PubMed] [Google Scholar]
- 7.Mecocci P, Fanó G, Fulle S, MacGarvey U, Shinobu L, Polidori MC, Cherubini A, Vecchiet J, Senin U, Beal MF. Age-dependent increases in oxidative damage to DNA, lipids, and proteins in human skeletal muscle. Free Rad Bio Med. 1999;26(3–4):303–308. doi: 10.1016/s0891-5849(98)00208-1. [DOI] [PubMed] [Google Scholar]
- 8.Burrows CJ, Muller JG. Oxidative Nucleobase Modifications Leading to Strand Scission. Chem Rev. 1998;98(3):1109–1152. doi: 10.1021/cr960421s. [DOI] [PubMed] [Google Scholar]
- 9.Giese B, Amaudrut J, Kohler AK, Spormann M, Wessely S. Direct observation of hole transfer through DNA by hopping between adenine bases and by tunnelling. Nature. 2001;412(6844):318–320. doi: 10.1038/35085542. [DOI] [PubMed] [Google Scholar]
- 10.Steenken S. Purine bases, nucleosides, and nucleotides: aqueous solution redox chemistry and transformation reactions of their radical cations and e- and OH adducts. Chem Rev. 1989;89(3):503–520. [Google Scholar]
- 11.(a) Shukla MK, Leszczynski J. Fullerene (C60) forms stable complex with nucleic acid base guanine. Chem Phys Lett. 2009;469(1–3):207–209. [Google Scholar]; (b) Shukla MK, Dubey M, Zakar E, Namburu R, Leszczynski J. Interaction of nucleic acid bases and Watson-Crick base pairs with fullerene: Computational study. Chem Phys Lett. 2010;493(1–3):130–134. [Google Scholar]
- 12.Hertel IV, Steger H, de Vries J, Weisser B, Menzel C, Kamke B, Kamke W. Giant plasmon excitation in free C_{60} and C_{70} molecules studied by photoionization. Phys Rev Lett. 1992;68:784. doi: 10.1103/PhysRevLett.68.784. [DOI] [PubMed] [Google Scholar]
- 13.Orlov VM, Smirnov AN, Varshavsky YM. Ionization potentials and electron-donor ability of nucleic acid babes and their analogues. Tetra Lett. 1976;17(48):4377–4378. [Google Scholar]
- 14.(a) Wang Y. Theoretical Evidence for the Stronger Ability of Thymine to Disperse SWCNT than Cytosine and Adenine: Self-Stacking of DNA Bases vs Their Cross-Stacking with SWCNT. J Phys Chem C. 2008;112(37):14297–14305. doi: 10.1021/jp803917t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang YX, Bu YX. Noncovalent Interactions between Cytosine and SWCNT: Curvature Dependence of Complexes via π•••π Stacking and Cooperative CH•••π/NH•••π. J Phys Chem B. 2007;111:6520–6526. doi: 10.1021/jp0700433. [DOI] [PubMed] [Google Scholar]; (c) Das A, Sood AK, Maiti PK, Das M, Varadarajan R, Rao CNR. Binding of nucleobases with single-walled carbon nanotubes: Theory and experiment. Chem Phys Lett. 2008;453(4–6):266–273. [Google Scholar]; (d) Sowerby SJ, Cohn CA, Heck WM, Holm NG. Differential adsorption of nucleic acid bases: Relevance to the origin of life. Proc Natl Acad Sci. 2001;98:3. doi: 10.1073/pnas.98.3.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Albertorio F, Hughes ME, Golovchenko JA, Branton D. Base dependent DNA-carbon nanotube interactions: activation enthalpies and assembly-disassembly control. Nanotech. 2009:20. doi: 10.1088/0957-4484/20/39/395101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manohar S, Mantz AR, Bancroft KE, Hui C-Y, Jagota A, Vezenov DV. Peeling single-stranded DNA from graphite surface to determine oligonucleotide binding energy by force spectroscopy. Nano Lett. 2008;8:8. doi: 10.1021/nl8022143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgado CA, Jurecka P, Svozil D, Hobza P, Sponer J. Reference MP2/CBS and CCSD(T) quantum-chemical calculations on stacked adenine dimers. Comparison with DFT-D, MP2.5, SCS(MI)-MP2, M06-2X, CBS(SCS-D) and force field descriptions. Phys Chem Chem Phys. 2010;12:3522. doi: 10.1039/b924461a. [DOI] [PubMed] [Google Scholar]
- 18.Sherrill CDT Tait, Hohenstein Edward G. An Assessment of Theoretical Methods for Nonbonded Interactions: Comparison to Complete Basis Set Limit Coupled-Cluster Potential Energy Curves for the Benzene Dimer, the Methane Dimer, Benzene-Methane, and Benzene-H2S. J Phys Chem A. 2009;113:10146–10159. doi: 10.1021/jp9034375. [DOI] [PubMed] [Google Scholar]
- 19.(a) Delley B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J Chem Phys. 1990;92(1):508–517. [Google Scholar]; (b) Delley B. From molecules to solids with the DMol[sup 3] approach. J Chem Phys. 2000;113(18):7756–7764. [Google Scholar]
- 20.Zhao Y, Schultz NE, Truhlar DG. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J Chem Theory Comput. 2006;2(2):364–382. doi: 10.1021/ct0502763. [DOI] [PubMed] [Google Scholar]
- 21.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, JEP, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision A.02. Gaussian, Inc; Pittsburgh PA: 2009. [Google Scholar]
- 22.(a) Grimme S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J Comput Chem. 2004;25(12):1463–1473. doi: 10.1002/jcc.20078. [DOI] [PubMed] [Google Scholar]; (b) Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J Comput Chem. 2006;27(15):1787–1799. doi: 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- 23.Perdew JP, Burke K, Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys Rev Lett. 1996;77(18):3865. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 24.Neese F. ORCA-an ab initio, density functional and semiempirical program package, Ver. 2.8.0. Max-Planck-Institut fur Bioanorganische Chemie; Mulheim an der Ruhr: 2010. [Google Scholar]
- 25.Wu X, Zeng XC. First-Principles Study of a Carbon Nanobud. ACS Nano. 2008;2(7):1459–1465. doi: 10.1021/nn800256d. [DOI] [PubMed] [Google Scholar]
- 26.Boys SF, Bernardi F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys. 1970;19(4):553–566. [Google Scholar]
- 27.Gowtham S, Scheicher RH, Ahuja R, Pandey R, Karna SP. Physisorption of nucleobases on graphene: Density-functional calculations. Phys Rev B. 2007;76(3):033401. [Google Scholar]
- 28.Guckian KM, Schweitzer BA, Ren RXF, Sheils CJ, Tahmassebi DC, Kool ET. Factors Contributing to Aromatic Stacking in Water: Evaluation in the Context of DNA. J Am Chem Soc. 2000;122(10):2213–2222. doi: 10.1021/ja9934854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.London F. Zur Theorie und Systematik der Molekularkräfte. Zeitschrift für Physik A Hadrons and Nuclei. 1930;63(3):245–279. [Google Scholar]
- 30.Sinnokrot MO, Sherrill CD. Highly Accurate Coupled Cluster Potential Energy Curves for the Benzene Dimer: Sandwich, T-Shaped, and Parallel-Displaced Configurations. J Phys Chem A. 2004;108(46):10200–10207. [Google Scholar]
- 31.Lias SG, Bartmess JE, Liebman JF, Holmes JL, Levin RD, Mallard WG. Gas-phase ion and neutral thermochemistry. J Phys Chem Ref Data, supplement. 1988;17:1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.