

Abstract
Follicular dendritic cells (FDCs) increase HIV replication and virus production in lymphocytes by increasing the activation of NF-κB in infected cells. Because alpha-1-antitrypsin (AAT) decreases HIV replication in PBMCs and monocytic cells and decreases NF-κB activity, we postulated that AAT might also block FDC-mediated HIV replication. Primary CD4+ T cells were infected with HIV and cultured with FDCs or their supernatant with or without AAT, and ensuing viral RNA and p24 production were monitored. NF-κB activation in the infected cells was also assessed. Virus production was increased in the presence of FDC supernatant but the addition of AAT at concentrations above 0.5 mg/ml inhibited virus replication. AAT blocked the nuclear translocation of NF-κB p50/p65 despite an unexpected elevation in associated phosphorylated and ubiquitinated IκBα (Ub-IκBα). In the presence of AAT, degradation of cytoplasmic IκBα was dramatically inhibited compared to control cultures. AAT did not inhibit the proteasome; however, it altered the pattern of ubiquitination of IκBα. AAT decreased IκBα polyubiquitination linked through ubiquitin lysine residue 48 (K48) and increased ubiquitination linked through lysine residue 63 (K63). Moreover, K63 linked Ub-IκBα degradation was substantially slower than K48 linked Ub-IκBα in the presence of AAT, correlating altered ubiquitination with a prolonged IκBα half-life. Because AAT is naturally occurring and is available clinically, examination of its use as an inhibitory agent in HIV-infected subjects may be informative and lead to the development of similar agents that inhibit HIV replication using a novel mechanism.
Introduction
HIV reservoirs pose major obstacles in the treatment and control of viral infection. The follicular dendritic cell (FDC) reservoir contains HIV that is genetically diverse, replication-competent, long-lived and contains viral quasispecies, including drug-resistant variants that are not found elsewhere in the body (1). Recently we found that FDC signaling contributes to increased HIV transcription and virus production in infected primary CD4+ T cells (2). This increased viral replication is mediated by FDC-produced TNFα, which in turn, increases the nuclear translocation of NF-κB in infected T cells. NF-κB is a prime activator of HIV transcription (3, 4) and immunohistochemical examination of secondary lymphoid tissues demonstrates the presence of TNFα in a reticular pattern consistent with FDC secretion of this cytokine. Furthermore, germinal center (GC) CD4+ T cells, that are adjacent to FDCs, show increased expression of activated NF-κB, and when infected in vitro, these cells produce more virus than other CD4+ T cells from the same tissue (2). Collectively these data suggest that FDCs and other GC cells establish a microenvironment that is highly conducive to HIV transmission and virus production.
A number of endogenous inhibitors of HIV replication have been described (5–7). Several recent reports focused on the major endogenous serine proteinase inhibitor, alpha-1-antitrypsin (AAT) (8–12). AAT has a half-life of about 4 d in the circulation and is present in the serum of healthy individuals at a concentration of 1.5 to 3.5 mg/ml, although this concentration can rapidly increase (i.e. 4-fold or more) during inflammation (13, 14). AAT is a glycoprotein consisting of 394 amino acid residues and is primarily produced in the liver. AAT is reported to inhibit HIV infection and replication in susceptible cells in vitro (8, 9). A genetic defect can result in AAT deficiency, and in one case report, pre-existing AAT deficiency was associated with accelerated HIV progression (10). While it has been shown that AAT suppresses NF-κB activation, the detailed molecular mechanism remains unknown (9).
The HIV long terminal repeat contains 2 consensus-binding sites for NF-κB, and activation of this transcription factor enhances HIV replication (3, 4). Because FDCs induce NF-κB activation in infected primary CD4+ T cells leading to increased virus replication (2), we hypothesized that AAT might interfere with this FDC-mediated effect. We report here that AAT blocks FDC-mediated HIV replication and suppresses NF-κB activation. Mechanistically, AAT inhibition appears to be mediated by blockade of IκBα degradation resulting from altered polyubiquitination linkages to this NF-κB inhibitor. These results demonstrate a unique mechanism of HIV inhibition.
Material and Methods
Alpha-1-antrypsin (AAT)
Clinical grade AAT (Aralast, Baxter Healthcare Corporation, Westlake Village, CA) purified from pooled human plasma from healthy donors was reconstituted as instructed by the manufacturer. The stock AAT concentration was approximately 20 mg/ml.
Virus preparations
HIV-1IIIB was propagated in neoplastic H9 cells and the virus from acute infection was harvested during the peak of p24 and/or reverse transcriptase (RT) production. The virus was pooled, passed through a 0.45 μm membrane, and stored in aliquots in liquid nitrogen until used. Our HIVIIIB preparations typically contained 1 μg/ml of p24 and 1×106 cpm/ml RT activity. Where noted, the primary HIV-1 isolates 91US054 (X4) or 92US714 (R5) were used after propagation in primary, PHA (5 μg/ml, Sigma) activated, IL-2 (20 U/ml, NIH AIDS Reference and Reagent Program) maintained PBMCs and harvested as described above. Primary HIV virus preparations contained 140 to 200 nanogram p24/ml and 300,000 to 450,000 cpm/ml RT activity.
HIV-1 infection and culture of neoplastic and primary CD4+ T cells
Primary CD4+ T lymphocytes were obtained from healthy, normal blood donors at BYU. Whole blood was collected aseptically and the PBMCs were isolated by density gradient separation using Ficoll-Paque Plus (GE Healthcare). After washing the PBMCs to remove any remaining Ficoll, the T cells were highly enriched via negative selection using a CD4+ T cell isolation Kit II (Miltenyi Biotech) as directed. The isolated CD4+ T cells were activated with PHA (5 μg/ml; Sigma) for 3 d in RPMI-1640 complete tissue culture medium (CM) containing HEPES (20 mM), nonessential amino acids (1x), L-glutamine (2 mM), 10% heat-inactivated, defined FBS (GIBCO/Invitrogen) and gentamicin (50 μg/ml; Life Technologies), washed to remove the mitogen and cultured overnight in IL-2 (20 U/ml; NIH AIDS Reference and Reagent Program). The neoplastic T cell lines Hut78, Jurkat and SupT1 cells were maintained in CM. Both neoplastic and primary CD4+ T cells were infected using a cell-free stock virus preparation that contained 50,000 cpm of reverse transcriptase activity per 1×106 cells. In brief, the cells were washed to remove the tissue culture medium and resuspended in the virus preparation for 2 h at 37°C after which the cells were washed to remove the unbound infecting inoculum. The infected cells were then resuspended in CM that contained IL-2 (20 U/ml) and 20% v/v FBS and cultured in the absence or presence of AAT (5 mg/ml unless otherwise stated) or PBS for the time indicated in each figure legend.
FDC and FDC- supernatant collection
FDCs were isolated from human tonsils as previously described (15, 16). Briefly, the tonsils were cut into small pieces and digested with an enzyme cocktail containing Blendzyme (Roche Applied Science) and DNase I (Sigma). The released cells were washed twice in RPMI 1640 and then resuspended in CM (5 ml). Goat IgG (Chrompure, Jackson ImmunoResearch) was then added and the cells were incubated for 1 h on ice to minimize non-specific binding of Ig to FcRs. Afterwards, the cells were labeled with mouse IgM, anti-human FDC mAb, HJ2 (kindly provided by Dr. M. Nahm, University of Alabama at Birmingham) and incubated overnight on ice. The labeled cells were then applied to a Percoll (GE Healthcare) gradient, centrifuged and the low-density cell fraction (1.050 – 1.060 g/ml) containing FDCs was collected, washed with RPMI 1640 and resuspended in CM. Then, the cells were labeled with a secondary, FITC-conjugated goat anti-mouse IgM (Jackson ImmunoResearch) Ab and following incubation and washing, the FDCs were obtained by FACS (BD FACS-Vantage Se equipped with the DiVa option). The resultant cells were ≥ 95% FDCs as assessed by flow cytometry. FDC-supernatant was obtained by culturing the isolated FDCs in CM for 6 d, centrifuging the cells and collecting the supernatant fluid, which was then stored at −80°C until used.
HIV RNA and p24 quantitation
Culture supernatant was obtained at the end of the stated incubation periods and used for both viral RNA and p24 quantitation. Viral RNA was isolated using the QIAamp viral RNA mini Kit (250) (Qiagen, Valencia, CA) as directed. The isolated viral RNA was reverse-transcribed into cDNA using random primers and SuperScript III RT (Invitrogen) according to the manufacturer’s instructions. Q-RT-PCR using TaqMan Universal PCR Master Mix (Applied Biosystems) used the following primers and probe: forward primer: 5’-TGGGTACCAGCACACAAAGG-3’, (nt 3696 in HXB2); reverse primer: 5’-ATCACTAGCCATTGCTCTCCAAT-3’, (nt 3850 in HXB2); probe: ATTGGAGGAAATGAAC-MBG (FAM labeled) at 900 nM (primers) and 250 nM (probe). Q-PCR conditions were as follows: 50°C × 2’ (1 cycle), 95°C × 10’ (1 cycle); followed by 60 cycles of: 95°C × 15’’, 60°C × 1’ in an ABI 7500 thermocycler. A standard curve was prepared using known concentrations (i.e. copy numbers) of ACH-2 DNA to determine the number of copies of viral RNA present in the cultures. HIV p24 concentration was measured using an ELISA kit (ZeptoMetrix Corporation, Franklin, MA) as directed.
Cell protein extraction
Cells (107 cells/sample) were collected from the cultures and washed three times with ice cold PBS. Following the last wash, the residual cell pellet was suspended in 150 μl buffer A [Tris-HCl (10 mM, pH7.9), MgCl2 (1.5 mM), KCl (10 mM), DTT (2 mM), 0.1% Triton X-100, 2.5 mM NaH2PO4, 1 mM Na3VO4, 1 mM NaF, 1 mM PMSF and 10% v/v protease inhibitor cocktail (Roche, USA)]. After gentle inversion, the mixture was incubated on ice for 15 min followed by centrifugation at 4°C (5 min at 250 X g) The supernatant was discarded and an additional 100 μl buffer A was added to the cell pellet and resuspended by drawing the mixture into a syringe having a 30-gauge needle and expelling the suspension a total of 5 times. The mixture was then centrifuged at 4°C (20 min at 8,000 X g) and the supernatant containing the cytoplasmic proteins was collected. To extract the nuclear protein, the remaining cell pellet was resuspended in 80 μl buffer B (20 mM Tris-HCl, pH7.9, 1.5 mM MgCl2, 420 mM KCl, 0.2 mM EDTA, 2 mM DTT, 1 % Igepal CA-630, 25% v/v glycerol, 2.5 mM NaH2PO4, 1 mM Na3VO4, 1 mM NaF, 1 mM PMSF and 10% v/v protease inhibitor cocktail) and the nuclei were disrupted using a syringe as described above. The resulting mixture was agitated gently at 4°C for 50 min and centrifuged for 5 min at 16,000 X g after which the supernatant containing the enriched nuclear proteins was collected. The nuclear and cytoplasmic proteins were stored at −80°C until used.
Total cellular proteins were also isolated from the cultured cells (107 cells/sample) after washing three times with ice cold PBS. The cells were solubilized using 150 μl RIPA buffer (50 mM Tris-HCl pH 8.3), 0.01% SDS, 250 mM NaCl, 0.15% Triton X-100, 0.5% w/v sodium deoxycholate, 1% v/v Igepal CA-630, 1 mM β-mercaptoethanol, 1 mM Na3VO4 , 1 mM NaF, 1 mM PMSF, 4 mM EDTA and 10% v/v protease inhibitor cocktail) and vortexed for 60 sec. The mixture was then incubated on ice for 45 min and homogenized in a syringe with a 30-gauge needle. After homogenization, the mixture was centrifuged at 4°C at 14,000 X g for 10 min after which the supernatant was collected and stored −80°C until used.
Immunoblotting
Whole cell, nuclear or cytoplasmic proteins were mixed with 4X loading buffer (250mM Tris-HCl, pH8.8, 4% SDS, 4% v/v ß-mercaptoethanol, 40 % v/v glycerol, 0.01% w/v bromophenol blue) and incubated at room temperature for 20 min. After incubation, the mixture was then heated for an additional 5 min at 100°C. The proteins were separated by 12% SDS-PAGE and transferred to Hybond-C or Hybond-P membranes (Amershan Life Science). After blocking in TBS with 5% non-fat milk for 2 h at room temperature, the membrane was incubated with the primary Ab [rabbit anti-human p50 (Chemicon international), rabbit anti-human p65 (Chemicon international), rabbit anti-human I Ba (Chemicon international), rabbit anti-human phosphorylated I Ba (Upstate), rabbit anti-human GAPDH (Abcam), mouse anti-human CD120a (TNFR1, Beckman Coulter), rabbit anti-human beta actin (Upstate), rabbit anti-human ubiquitin (Pierce), goat anti-human AAT (Bethyl) or rabbit anti-human proteasome 19S S4 or 20S alpha + beta (Abcam)] in TBS with 5% non-fat milk for 2 h at room temperature. The membranes were washed three times in TBS containing 0.1% Tween-20 and 5 % (w/v) non-fat milk. After washing, the membranes were incubated for 2 h at room temperature with an appropriate specificity of HRP-linked secondary Ab [anti-rabbit IgG-HRP (Chemicon international), anti-goat IgG-HRP (Chemicon international) or anti-mouse IgG-HRP (Chemicon international)] in TBS with non-fat milk (5% w/v). The membranes were then washed in TBS with 0.1% v/v Tween-20 and 5% w/v non-fat milk and developed using enhanced chemoluminescence. The immunoblots were quantified using a phosphorimager in chemoluminescence mode.
Co-Immunoprecipitation
Whole cell, nuclear or cytoplasmic proteins (1 mg at 1 mg/ml) were first mixed with protein A/G agarose beads (100 μl) and incubated for 1 h at 4°C. The beads were centrifuged and discarded to reduce nonspecific protein binding. The remaining supernatant containing the proteins of interest was incubated for 1 h at 4°C with specific Ab [anti-ubiquitin (Pierce), anti-p65 (Chemicon international), anti-p50 (Chemicon international), anti-I Ba (Chemicon international), anti-AAT (Bethyl) or anti-human proteasome 19S S4 or 20S alpha + beta (both Abcam)], after which Protein A/G beads (100 μl) were added and the mixtures incubated for 1 h at 4°C. The preparations were then centrifuged and the supernatant discarded. The beads were then washed three times with lysis buffer and 4X loading buffer was added. The beads were boiled for 10 min to separate immune complexes from the beads and the samples were centrifuged for 5 min at 3,000 X g at 4°C. The supernatant was collected, electrophoresed in 12% SDS-PAGE and the proteins identified by immunoblotting.
Pulse-Chase Assay for IκBα decay
HIV-infected CD4+ T cells (2 × 106 cells/sample) were treated with or without AAT (5 mg/ml) for 24 h after which the cells were washed 2 times with 3 ml of methionine-free RPMI 1640 medium supplemented with 10% dialyzed FBS (ΔMet-RPMI). The cells were resuspended in ΔMet-RPMI and incubated for 1 h at 37°C. The cells were then collected and suspended in 1 ml of 200 μCi/ml 35S-labeled methionine diluted in ΔMet-RPMI to initiate synthesis of labeled proteins. The cells were incubated for an additional 10 min and then washed three times with 3 ml of ΔMet-RPMI containing 4 mM unlabeled methionine and cultured in 2 ml of the same medium for the indicated times. After incubation, the cells were collected and washed three times with ice cold PBS and extracted as described earlier. The resulting mixture was incubated on ice for 45 min and homogenized with a 30-gauge needle by drawing the mixture up and ejecting 3 times. After homogenization, the mixture was treated as described above under immunoprecipitation for IκBα and the remaining radioactivity detected as described above.
Assay of 20S proteasome activity
The activity of the 20S proteasomal subunit was determined in vitro using a 20S proteasome activity assay kit (Chemicon International) according to the manufacturer’s protocol. Briefly, cell lysates containing 20S proteasomal subunits were mixed with substrate and after 2 h incubation at 37°C, the fluorescence was quantified using a fluorometer equipped with a 380/460nm filter set. The amount of fluorescence was directly proportional to the amount of 20S proteasomal activity.
AAT Biotinylation
AAT biotinylation was performed using an EZ-link® sulfo-NHS-LC-Biotinylation kit (Pierce) according to the manufacturer’s protocol. Briefly, Sulfo-NHS-LC-Biotin was added to AAT and incubated for 2 h on ice after which the excess Sulfo-NHS-LC-Biotin was separated by size exclusion chromatography. The biotinylated AAT was then eluted from the column and the level of biotin incorporation determined according to the manufacturer’s protocol.
Statistical analysis
All data were analyzed using the R statistical computing system. The nlme library was loaded so that the lme() and gls() functions could be used. These functions allow for appropriate analysis of data that have multiple sources of variability. Thus, independence of errors was not assumed and more general covariance structures were estimated. Where multiple tests were performed on a single data set, Bonferroni corrections were used to keep family alpha levels at 0.05. When all treatments were compared in a particular data set, Tukey corrections were used to keep family alpha levels at 0.05.
Studies using human cells/tissues
Studies using blood or tissue derived cells obtained from humans were reviewed and approved by an appropriate institutional review committee.
Results
We recently reported that FDCs increase HIV transcription and production in infected CD4+ T cells by producing TNFα that in turn, activates HIV-inducing NF-κB in the infected lymphocytes (2). Because AAT was reported to decrease HIV replication in infected monocytic cells and in PBMCs in a manner that decreased NF-κB activation (8, 9), we postulated that it might also inhibit FDC-mediated HIV transcription and virus production. CD4+ T cells were therefore infected with HIV in vitro and cocultured with FDC supernatant. FDC supernatant has been shown to increase HIV transcription by FDC-produced TNFα (2). AAT was added to these cultures and the resultant virus replication was monitored using quantitative PCR (Q-PCR) to determine viral RNA and ELISA to detect p24 production (Figure 1). FDC supernatant alone, as expected, increased viral RNA produced in infected T cells (panel A) and p24 production (panel B) by over three-fold. In comparison to untreated T cells, the addition of AAT at concentrations of 0.5 and 5 mg/ml significantly decreased viral RNA and p24 production in cultures of infected T cells and FDC supernatant (p<0.05). In cultures of T cells alone, addition of AAT at concentrations of 5 mg/ml reduced viral RNA and p24 production below that of untreated T cells (p<0.05). Because AAT concentrations of 5 mg/ml occur in subjects with inflammatory processes and are thus physiologically relevant and the fact that the above dose of AAT clearly suppressed HIV replication in both T cells cultured with or without FDC supernatant, we selected this concentration for the remainder of our experiments. We also compared infected T cells cultured with intact FDCs or with FDC supernatant and found both FDC preparations equally capable of augmenting HIV production (Supplemental Figure 1). Furthermore, AAT inhibited virus production under both conditions. In addition to testing the effect of AAT upon virus replication in primary CD4+ T cells, we examined its effect on HIV-infected Hut78, SupT1 and Jurkat cells that are frequently used in in vitro studies of HIV infection and replication and found a similar inhibitory effect (Supplemental Figure 2). We also tested the effect of AAT on CD4+ T cell virus replication using an R5 (92US714) or X4 (91US054) primary isolate of HIV. AAT inhibited HIV replication of the primary isolates regardless of the tropism of the virus isolate, and this effect persisted in the presence or absence of FDC supernatant (data not shown). These results established an inhibitory effect of AAT on virus replication using both primary and laboratory-adapted HIV isolates.
Figure 1.
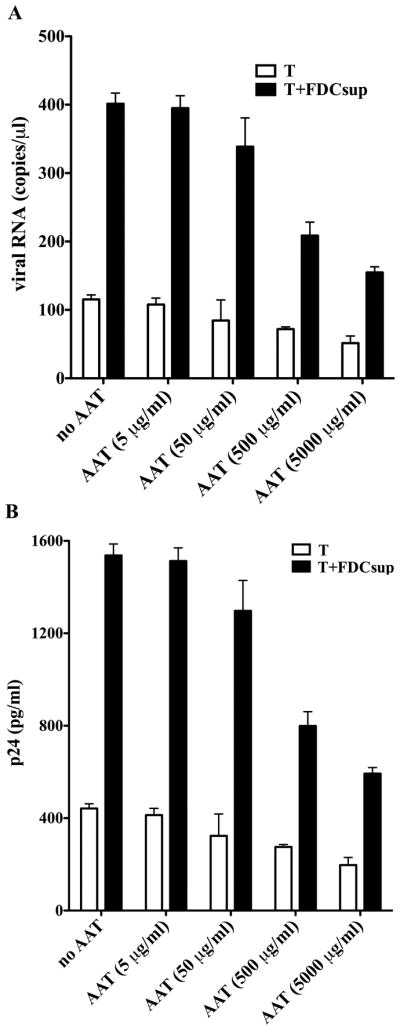
AAT inhibits HIV replication in primary CD4+ T cells cultured alone or with FDC supernatant. CD4+ T cells were isolated from the peripheral blood and infected with HIV. The infected cells were cultured for 72 h in the presence or absence of FDC supernatant (10% v/v) and with or without AAT at the concentrations indicated. The data presented are representative of 4 independent experiments and are displayed as the mean ± SD. (A) Viral RNA produced in the cultures. (B) p24 produced in the same cultures.
Because FDCs and their supernatant increase HIV transcription and virus production by increasing the activation of NF-κB, and AAT has been shown to block NF-κB activation in HIV-infected promonocytic U1 cells (17), we reasoned that AAT may inhibit FDC-mediated HIV replication by inhibiting the nuclear translocation of NF-κB induced by FDC-TNFα. CD4+ T cells infected with HIV were cultured as before with FDC supernatant in the presence or absence of AAT and immunoblotting was performed on whole cell, cytoplasmic and nuclear lysates of these cells (Figure 2, panel A). As expected, the infected T cells cultured with FDC supernatant demonstrated an increased nuclear translocation of NF-κB p50 and p65 (46% and 82% greater than infected cells without FDC supernatant, respectively, as assessed by band pixel quantitation) while simultaneously showing a decreased cytoplasmic concentration of these components (50% and 37% reduction respectively). In cultures that contained AAT, with or without FDC supernatant, decreased nuclear translocation of NF-κB p50 and p65 was observed (54% and 39% reduction for p50 and p65 without FDC supernatant; 68% and 43% reduction with FDC supernatant). However, at the same time we observed decreased nuclear NF-κB, we also detected increased amounts of cytoplasmic phosphorylated IκBα when AAT was present. This surprising finding is more consistent with increased NF-κB activation rather than the decreased activation levels we observed. To determine whether the phosphorylated IκBα remained bound to the NF-κB p50/p65 complex in the cytoplasm, the infected CD4+ T cells were cultured and lysed as before and subjected to immunoprecipitation using an Ab directed against IκBα (Figure 2B). Immunoblotting detected cytoplasmic NF-κB p50 and p65 with the immunoprecipitated IκBα indicating that NF-κB and IκBα components were associated in a complex. We also examined the ability of AAT to affect NFκB activation with a variety of stimuli that activate NF-κB in addition to FDC supernatant and with or without HIV infection and (Supplementary Figure 3). While the overall degree of activation varied somewhat between cells activated with different stimuli, AAT consistently decreased nuclear p50 and p65 in both the presence and absence of HIV infection.
Figure 2.

AAT blocks NF-κB activation despite phosphorylation and ubiquitination of IκBα. (A) Peripheral blood CD4+ T cells were isolated, stimulated and infected following the procedure in Materials and Methods. After 24 h of incubation with or without FDC supernatant (10% v/v) and in the presence or absence of AAT (5 mg/ml), the nuclear, cytoplasmic and whole cell proteins were extracted and subjected to immunoblotting for NF-κB components p50, p65 and the non- and phosphorylated forms of the NF-κB inhibitor, I Ba. As expected, β-actin was detected in both the cytoplasmic and the nuclear fractions from the CD4+ T cells (33–35). (B) Cells treated as in A were subjected to immunoprecipitation using I Ba-specific Ab and the precipitated complexes probed using the indicated Abs for IκBα, phosphorylated IκBα, and the NF-κB components p65 and p50. (C) Cells treated as in A were subjected to immunoprecipitation with ubiquitin-specific Ab and the isolated complexes subjected to immunoblotting for IκBα, phosphorylated IκBα, NF-κB p65 and p50 components and ubiquitin as indicated. The data presented in these panels are representative of 3 independent experiments.
We next determined whether IκBα was ubiquitinated by culturing cells as before and subjecting the cell lysates to immunoprecipitation using an ubiquitin-specific Ab (Figure 2C). Immunoblotting revealed the presence of phosphorylated IκBα, and the NF-κB components p65 and p50 in the ubiquitin-immunoprecipitated complexes. The amounts of these components obtained from cells incubated without AAT appeared less than those incubated with AAT (59%, 80%, 72% and 86% reduction for cytoplasmic IκBα, phospho-IκBα, p50 and p65, respectively in the absence of AAT and 61%, 67%, 76% and 88% reduction for cytoplasmic IκBα, phospho-IκBα, p65 and p50, respectively in the absence of AAT but presence of FDC supernatant). These results were consistent with the rapid degradation of phospho-ubiquitinated-IκBα in the absence of AAT. In cells exposed to FDC supernatant, the presence of AAT also increased the intensity of the IκBα, p50 and p65 bands. Comparing ubiquitin-immunoprecipitated IκBα, p50 and p65 in the supernatant from cells exposed to AAT in the presence or absence of FDC supernatant revealed a somewhat lower abundance of these molecules when FDC supernatant was present. This suggested that AAT incompletely suppressed FDC-induced TNFα-mediated NF-κB activation. Furthermore, when the ubiquitin-precipitated complexes were probed for ubiquitin (bottom panels of Figure 2C), we detected a number of proteins, further indicating that ubiquitination was ongoing and was not non-specifically blocked by AAT. Collectively these data suggested that although IκBα was both phosphorylated and ubiquitinated, it did not appear to be degraded normally. This resulted in IκBα accumulation in the cytoplasm along with the NF-κB components p50 and p65.
We next focused on whether exogenous AAT was internalized by CD4+ lymphocytes. HIV-infected CD4+ T cells were cultured for 1 h with biotin-labeled AAT, after which the cells were harvested and examined for the presence of AAT in the nucleus, cytoplasm and intact whole cell (Figure 3A). We detected the presence of biotin-labeled AAT in each of these cellular compartments. To verify the internalization of AAT and determine if it associated with components of the NF-κB complex, we performed immunoprecipitation using Abs to AAT or IκBα followed by immunoblotting to detect these components (Figure 3B). Immunoprecipitation indicated that AAT formed a complex with phospho-IκBα and the p50/p65 components of NF-κB. Moreover, when IκBα was precipitated, we detected AAT only when it was exogenously added, indicating that there was no detectable endogenous intracellular AAT. We explored the possibility that AAT inhibited proteasomal degradation of IκBα as a mechanism of cytoplasmic retention. As shown in Figure 3B, precipitation of the 19S and 20S proteasome subunits failed to detect the presence of AAT or components of the NF-κB complex. When we performed pulse-chase experiments to detect the kinetics of AAT entry and half-life, we found it could be detected within 30 min of treatment and persisted inside the cells for at least 72 h (data not shown). Immunoprecipitation of IκBα revealed that association with AAT was detected within 3 h and remained in association for at least 72 h. These data were consistent with the entry of exogenous AAT into the cell and its association with the NF-κB-IκBα complex leading to interference with IκBα proteasomal degradation.
Figure 3.

AAT enters CD4+ T cells and associates with IκBα and NF-κB components p50 and p65 but not with the proteasome. CD4+ T cells were treated as in Figure 2. After 24 h of incubation with or without AAT (5 mg/ml), the nuclear, cytoplasmic and whole cell proteins were isolated and analyzed. (A) Isolated proteins were subjected to SDS-PAGE followed by immunoblotting for AAT. A β-actin immunoblot served as a protein loading control. As expected, β-actin was present in both nuclear and cytoplasmic fractions (33–35). AAT and AAT* represent purified AAT and biotin-labeled purified AAT, respectively. (B) Isolated proteins from the cells were immunoprecipitated using Abs specific for I Ba, AAT, or the 19S or 20S proteasomal subunits. The data presented in these panels are representative of 3 independent experiments. The Abs used in immunoprecipitation are indicated at the top of panel B while those used in immunoblotting are indicated at the left of each blot. IP, Immunoprecipitation.
Because AAT appeared to inhibit NF-κB activation downstream of phosphorylation and ubiquitination of IκBα, we determined whether AAT inhibited the enzymatic activity of the proteasome. Infected CD4+ T cells were again cultured with or without AAT, the cells lysed and the proteasome complex was immunoprecipitated using Ab specific to the 20S subunits. The enzymatic activity of the immunoprecipitated 20S subunit was then measured (Figure 4). When we immunoprecipitated the proteasome using Ab specific to the 20S subunits, we found no significant difference between cultures with or without AAT, even when we added AAT directly to the immunoprecipitated complex (Figure 4, see far right bars). Thus, AAT did not inhibit proteasomal activity.
Figure 4.

AAT does not inhibit the 20S enzymatic activity of the proteasome. CD4+ T cells were obtained and treated as in Figure 2. The cells were then lysed, the A) cytoplasmic or B) nuclear fraction obtained and the proteasomes immunoprecipitated using Ab directed against the 20S subunit. The precipitated complexes were washed and tested for 20S proteasomal activity as indicated in Materials and Methods. The data are presented as box plots where the thick horizontal lines represent the median, the box extensions (whiskers) represent the data range and the box itself covers from the 25th to the 75th percentile. The data depicted are representative of 3 independent experiments. T, lysates of untreated T cells; T+IP, immunoprecipitation of 20S subunit from lysates of untreated T cells; T+AAT+IP, immunoprecipitation of 20S subunit from lysates of AAT (5 mg/ml) treated T cells; T+AAT+IP+AAT, immunoprecipitation of 20S subunit from lysates of AAT (5 mg/ml) treated T cells with additional AAT (5 mg/ml) added after precipitation. No significant change in 20S enzymatic activity was observed (cytoplasmic protein, p<0.50; nuclear protein p<0.55).
We reasoned that if AAT inhibited the ability of IκBα to be targeted for proteasomal degradation, it should similarly affect other molecules that are routinely degraded in the proteasome while having no impact on those that are degraded elsewhere (i.e. lysosome). We also postulated that the effect of AAT-mediated blockade of IκBα proteasomal degradation might be replicated by inhibiting the proteasome itself. We tested these postulates using AAT treated cells with or without FDC supernatant and in the presence or absence of the proteasome inhibitor MG132. We first examined HIV-infected, AAT treated CD4+ T cells for the relative abundance of the TNF receptor 1 (TNFR1) that is degraded in the proteasome and GAPDH that is not (Figure 5A). In the presence of AAT, the concentration of TNFR1 clearly increased above that present in untreated cells while GAPDH levels remained unchanged. The presence of FDC supernatant had no discernable effect upon the intensity of either TNFR1 or GAPDH under the same conditions. When we examined the relative concentration of TNFR1 and GAPDH in the presence of the proteasome inhibitor MG132, we found that MG132 replicated the AAT effect, although the MG132 effect appeared to be more potent than that of AAT at the concentrations we used.
Figure 5.

AAT inhibits proteasomal degradation of TNFRI and increases IκBα half-life. (A) CD4+ T cells were isolated and infected as before and cultured for 24 h in the presence or absence of FDC supernatant, AAT (5 mg/ml) and the proteasome inhibitor, MG132 (40 μM). After treatment, the cells were washed, lysed, and subjected to immunoblotting for TNFR1 (a protein degraded in the proteasome) or GAPDH (a protein degraded in the lysosome). β-actin was immunostained as a protein loading control. (B) The time-course of I Ba detection in infected CD4+ T cells was examined with or without FDC supernatant or AAT. Cells were cultured for 24 h with or without FDC supernatant or AAT (5 mg/ml) and then washed, pulsed for 10 min with 35S-Met (30 μCi/ml), washed and cultured (“chased”) with complete medium containing unlabeled Met. Samples were collected over a 2 h period when FDC supernatant was present or over a 32 h period when AAT (without FDC supernatant) was added. After culture, IκBα was immunoprecipitated and the resulting complexes were then analyzed by SDS-PAGE. Radiolabeled IκBα was detected using a phosphorimager. The data presented are representative of 3 independent experiments.
We further reasoned that if AAT inhibited the degradation of IκBα, then its half-life should be increased. We therefore cultured infected CD4+ T cells with AAT and monitored IκBα decay in a pulse-chase experiment (Figure 5B). In cells incubated without AAT, in the presence or absence of FDC supernatant, IκBα was degraded to undetectable levels between 60-90 min. However, in the presence of AAT, IκBα decay was almost completely blocked, with detectable levels persisting for at least 32 h. Therefore, AAT appears to inhibit NF-κB activation in HIV infected target cells by interfering with IκBα degradation with resultant blockade of the nuclear translocation of the p50/p65 NF-κB heterodimer.
Conjugation of polyubiquitin to target proteins occurs via linkage to different lysine molecules on ubiquitin and the linkages utilized can affect the fates of the targeted proteins (18-20). When the target protein is conjugated via ubiquitin lysine residue 48 (K48), the molecule is then targeted for proteasomal degradation, while ubiquitin conjugation via lysine residue 63 (K63) does not result in degradation of the protein. To determine whether IκBα polyubiquitination occurred via residue K48 or K63, we immunoprecipitated the IκBα complex in the presence or absence of AAT and performed immunoblotting using ubiquitin linkage-specific Abs (Figure 6A). In the presence of AAT, we saw a shift from conjugation of ubiquitin molecules through residue K48 to linkage with K63 and this intensified between 24 and 72 h of culture. Concomitantly, an increase in phospho-IκBα was observed, as seen previously. Thus it appears that a mechanism of AAT-mediated inhibition of NF-κB nuclear translocation is alteration of the linkages used for polyubiquitination of phospho-IκBα, which results in decreased degradation of this NF-κB activation inhibitor. We also immunoprecipitated IκBα in the presence and absence of infection to determine whether HIV affected AAT-mediated differential polyubiquitination (Supplementary Figure 3B). Similar band intensities were observed regardless of the presence of HIV and the kinetics of altered ubiquitination followed the same pattern as seen previously (i.e. in Figure 6A).
Figure 6.

AAT treatment alters polyubiquitination linkages resulting in the prolonged half-life of IκBα in CD4+ T cells. (A) CD4+ T cells were treated as before and incubated in the presence or absence of AAT (5 mg/ml) and harvested at the indicated times, lysed and IκBα was immunoprecipitated and subjected to immunoblotting with Abs specific for polyubiquitination on either ubiquitin lysine residue K48 or K63. (B) Cells were treated as in Figure 5 to radiolabel proteins (pulse-chase) and after immunoprecipitation of IκBα at the indicated times, the agarose A/G beads were boiled and the supernatant collected. The IκBα complexes were subjected to a second immunoprecipitation using Abs specific for polyubiquitination linked through ubiquitin residues K48 or K63. The precipitated complexes were then examined using a phosphorimager to detect the radiolabeled IκBα that remained over the period of culture. The top two rows of this panel present data from CD4+ T cell cultures with or without FDC supernatant while the remaining rows are from cells cultured with or without AAT but not FDC supernatant.
To determine if the shift in ubiquitination patterns resulted in the prolonged half-life of IκBα that we observed previously (Figure 5B), we performed a pulse-chase experiment with a double immunoprecipitation, first with an IκBα-specific Ab, and then after releasing IκBα from the beads, a second immunoprecipitation was performed using Abs specific for ubiquitin linked via residue K48 or K63 (Figure 6B). Consistent with our hypothesis that a prolonged half-life was associated with increased ubiquitin linkage with K63, IκBα with ubiquitin conjugated via K48 was not observed after 120 min in the presence of AAT, while in the same cultures, K63 ubiquitinated IκBα persisted for at least 32 h. Collectively, these data indicate that AAT prolongs the half-life of the NF-κB inhibitor, IκBα, by interacting with IκBα and shifting its pattern of ubiquitination from a predominant K48 linkage to a K63 linkage.
Discussion
FDCs provide a long-term reservoir of infectious HIV that can transmit infection to surrounding GC cells (1, 16). In addition, FDCs contribute a number of signals that increase both virus transmission and propagation, including generation of TNFα that increases HIV replication (2, 21–23). In this study we report the ability of AAT, a naturally occurring prototypic serine protease inhibitor, to suppress HIV replication including that induced by FDCs. AAT inhibited the activation of NF-κB with or without FDC supernatant by prolonging the half-life of IκBα, which in turn remained bound to cytoplasmic NF-κB in the infected T cells. IκBα preservation in the cytoplasm blocked NF-κB nuclear translocation and subsequent activation of HIV transcription that occurs by attachment to NF-κB binding sites in the HIV promoter.
Initially, we were surprised by the observation that AAT decreased nuclear NF-κB while at the same time increasing levels of cytoplasmic phospho-IκBα. We first reasoned that AAT might block the subsequent ubiquitination of phospho-IκBα resulting in failure of IκBα to dissociate from NF-κB thereby suppressing the activation of this transcription factor. However, in subsequent testing, we found that cytoplasmic phospho-IκBα was ubiquitinated. We then reasoned that AAT might directly inhibit the proteolytic activity of the proteasome. However, when intact immunoprecipitated proteasomes were examined, we found no AAT-induced depression of the 20S proteolytic activity, suggesting that AAT did not inhibit NF-κB activation by suppressing proteasome function. Furthermore, immunoprecipitation of both the 19S and 20S proteasomal subunits failed to detect AAT association with the proteasome. Collectively, these data suggested that AAT inhibited IκBα degradation following phosphorylation and ubiquitination, but prior to proteasomal degradation. Therefore, our attention was directed toward transportation of ubiquitin-tagged IκBα into the proteasome.
It is now understood that different patterns of polyubiquitination on proteins lead to different outcomes (19, 24). Polyubiquitination occurs via an isopeptide linkage between the carboxy-terminal glycine residue of a ubiquitin molecule bound to a target protein and one of seven lysine residues of additional ubiquitin molecules that form a polyubiquitin complex. The two most prevalent polyubiquitination patterns presently characterized are linkage at lysine residues K48 or K63. Polyubiquitination through K48 residues targets proteins for proteasomal degradation while polyubiquitination via K63 residues is associated with protein-protein interactions, cell signaling, localization within the cell and functional regulation of proteins (18, 19). We observed an AAT-induced shift in the pattern of polyubiquitination of IκBα consisting of a decrease in K48 linkages and a concomitant increase in polyubiquitination using K63 linkages. These data directly correlated with a significantly increased half-life of IκBα and the resulting inhibition of NF-κB activation.
The mechanism whereby AAT alters the pattern of IκBα polyubiquitination is unknown. FDC-produced TNFα leads to NF-κB activation (2), and TNFα induces NF-κB activation by phosphorylation of IκBα on serine residues 32 and 36. Following phosphorylation, initial IκBα ubiquitination occurs on residue K19 (24). IκBα is subsequently polyubiquitinated through the activity of the Skp1, Cullin 1 and F-box protein β transducin repeat containing protein (βTRCP) SCFIκ B E3 ligase complex. Normally the SCFIκ B E3 ligase complex leads to ubiquitination using ubiquitin residue K48, which is followed by proteasomal degradation of the ubiquitinated protein (24, 25). There are a number of possible causes for the observed change in polyubiquitination of IκBα. When the NF-κB-IκBα complex was precipitated using Abs specific for IκBα, we found AAT in the complex. AAT binding to IκBα may alter the way in which the SCFIκ B E3 ligase complex interacts with IκBα. The initially bound ubiquitin molecule may be conformationally altered in a way that its K63 attacks the thioester linkage of the incoming E2 enzyme-bound ubiquitin and favors linkage through the K63 residue. It is also possible that AAT-binding to IκBα influences other components of the SCFIκ B E3 ligase complex changing its composition or specificity. However, we detected normal components of the SCFIκ B E3 ligase complex in the presence of AAT, suggesting that no overt changes in the SCFIκ B E3 ligase components occurred (data not shown). It is also possible that AAT affects deubiquitinating enzymes, and this may alter polyubiquitin linkages. Whatever the mechanism for altered linkage of ubiquitin, it is intriguing that an abundant serum protein has such a dramatic effect on NF-κB activation and HIV replication. It is possible that the mechanism of AAT modulation of HIV replication that we describe may occur naturally in infected subjects.
Other studies have examined a relationship between AAT and HIV. AAT inhibited HIV infection or production in infected cell lines and in primary PBMCs (8, 9). In a case report, an HIV-infected individual with genetic AAT-deficiency experienced unusually rapid decline in CD4+ T cell concentrations (10). It has also been demonstrated that the addition of AAT to cell cultures inhibits the replication of HIV in stimulated U1 monocytic cells and in infected primary PBMC (9). AAT inhibition of HIV replication in this previous study was linked to decreased NF-κB activation suggesting that this transcription factor is regulated by AAT, although the specific mechanism of this regulation was not addressed. In the present report, we extend these observations to show AAT inhibition of HIV replication in stimulated, HIV-infected primary CD4+ T cells cultured alone or with FDCs or their supernatant. We also addressed the mechanism whereby AAT inhibits NF-κB activation and implicated altered IκBα ubiquitination.
Throughout the natural course of HIV infection, HIV replication persists in secondary lymphoid tissues surrounding sites where FDCs and CD4+ GC T cells reside in close proximity (26–29). FDCs trap and retain large quantities of infectious HIV and maintain the infectious nature of this virus for months and years (1, 16, 21). Moreover, the FDC reservoir of HIV contains virus having a high level of genetic diversity and archived virus that includes antiretroviral drug-resistant mutants. FDCs also interact with surrounding GC CD4+ T cells and increase expression of CXCR4 that increases susceptibility to X4 HIV variants (15). FDCs produce CXCL13 that attracts GC T cells into the lymphoid follicles where they can interact with FDC-trapped virus. Furthermore, FDC interactions with CD4+ T cells induces the expression of two regulators of G- protein coupled signaling, RGS13 and RGS16, that can impede GC T cell migration to CXCL12 signals that may help them exit the GCs (30). Finally, FDCs produce TNFα that increases HIV replication (2). Taken together, these FDC-mediated signals induce a microenvironment that is highly conducive to transmission, propagation and persistence of HIV. During immune responses, FDCs are stimulated resulting in the activation of NF-κB (31, 32). It is intriguing to conjecture that AAT, in addition to decreasing HIV replication, may also have inhibitory effects on FDCs that could potentially decrease some of their contributions to HIV pathogenesis. The clinical availability of AAT and its in vitro effect on HIV replication suggest that further studies of this molecule in the HIV/AIDS setting may be highly informative.
Supplementary Material
Acknowledgments
We gratefully acknowledge the assistance of Drs. Kipp M. Robins and Randal B. Gibb, the Utah Valley Regional Medical Center, the Health South Provo Surgical Center, and the Central Utah Surgical Center for providing tissues from which FDCs and other lymphoid cells were obtained. We also acknowledge the NIH AIDS Reference and Reagent Program and Dr. Moon Nahm, University of Alabama at Birmingham, for providing reagents and Drs. Gregory B. Pott and Nana Sono-Koree for helpful discussions and technical advice.
Abbreviations used in this paper
- AAT
alpha-1-antitrypsin
- FDC
follicular dendritic cell
- RT
reverse transcriptase
- GC
germinal center
Footnotes
This work was supported by Public Health Service grants AI39963 and AI91517 from the National Institute of Allergy and Infectious Disease (GFB), GM078550, from the National Institute of General Medical Sciences (BMW), the Campbell Foundation (LS) and by a Brigham Young University Grant from the College of Physical and Mathematical Sciences (GFB).
References
- 1.Keele BF, Tazi L, Gartner S, Liu Y, Burgon TB, Estes JD, Thacker TC, Crandall KA, McArthur JC, Burton GF. Characterization of the follicular dendritic cell reservoir of Human Immunodeficiency Virus Type 1. J Virol. 2008;82:5548–5561. doi: 10.1128/JVI.00124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thacker TC, Zhou X, Estes JD, Jiang Y, Keele BF, Elton TS, Burton GF. Follicular dendritic cells and human immunodeficiency virus type 1 transcription in CD4+ T cells. J Virol. 2009;83:150–158. doi: 10.1128/JVI.01652-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Osborn L, Kunkel S, Nabel GJ. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc Natl Acad Sci U S A. 1989;86:2336–2340. doi: 10.1073/pnas.86.7.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duh EJ, Maury WJ, Folks TM, Fauci AS, Rabson AB. Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-kappa B sites in the long terminal repeat. Proc Natl Acad Sci USA. 1989;86:5974–5978. doi: 10.1073/pnas.86.15.5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Venkataraman N, Cole AL, Ruchala P, Waring AJ, Lehrer RI, Stuchlik O, Pohl J, Cole AM. Reawakening Retrocyclins: Ancestral Human Defensins Active Against HIV-1. PLoS Biol. 2009;7:e95. doi: 10.1371/journal.pbio.1000095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bishop KN, Verma M, Kim EY, Wolinsky SM, Malim MH. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008;4:e1000231. doi: 10.1371/journal.ppat.1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiu YL, Greene WC. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu Rev Immunol. 2008;26:317–353. doi: 10.1146/annurev.immunol.26.021607.090350. [DOI] [PubMed] [Google Scholar]
- 8.Munch J, Standker L, Adermann K, Schulz A, Schindler M, Chinnadurai R, Pohlmann S, Chaipan C, Biet T, Peters T, Meyer B, Wilhelm D, Lu H, Jing W, Jiang S, Forssmann WG, Kirchhoff F. Discovery and optimization of a natural HIV-1 entry inhibitor targeting the gp41 fusion peptide. Cell. 2007;129:263–275. doi: 10.1016/j.cell.2007.02.042. [DOI] [PubMed] [Google Scholar]
- 9.Shapiro L, Pott GB, Ralston AH. Alpha-1-antitrypsin inhibits human immunodeficiency virus type 1. FASEB J. 2001;15:115–122. doi: 10.1096/fj.00-0311com. [DOI] [PubMed] [Google Scholar]
- 10.Potthoff AV, Munch J, Kirchhoff F, Brockmeyer NH. HIV infection in a patient with alpha-1 antitrypsin deficiency: a detrimental combination? Aids. 2007;21:2115–2116. doi: 10.1097/QAD.0b013e3282f08b97. [DOI] [PubMed] [Google Scholar]
- 11.Congote LF. The C-terminal 26-residue peptide of serpin A1 is an inhibitor of HIV-1. Biochem Biophys Res Commun. 2006;343:617–622. doi: 10.1016/j.bbrc.2006.02.190. [DOI] [PubMed] [Google Scholar]
- 12.Hayes VM, Gardiner-Garden M. Are polymorphic markers within the alpha-1-antitrypsin gene associated with risk of human immunodeficiency virus disease? J Infect Dis. 2003;188:1205–1208. doi: 10.1086/378641. [DOI] [PubMed] [Google Scholar]
- 13.Kushner I, Mackiewicz A. Acute Phase Proteins:Molecular Biology, Biochemistry and Clinical Applications. CRC Press; 1993. The acute phase response: an overview; pp. 3–20. [Google Scholar]
- 14.Voulgari F, Cummins P, Gardecki TI, Beeching NJ, Stone PC, Stuart J. Serum levels of acute phase and cardiac proteins after myocardial infarction, surgery, and infection. Br Heart J. 1982;48:352–356. doi: 10.1136/hrt.48.4.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Estes JD, Keele BF, Tenner-Racz K, Racz P, Redd MA, Thacker TC, Jiang Y, Lloyd MJ, Gartner S, Burton GF. Follicular Dendritic Cell-Mediated Up-Regulation of CXCR4 Expression on CD4 T Cells and HIV Pathogenesis. J Immunol. 2002;169:2313–2322. doi: 10.4049/jimmunol.169.5.2313. [DOI] [PubMed] [Google Scholar]
- 16.Smith BA, Gartner S, Liu Y, Perelson AS, Stilianakis NI, Keele BF, Kerkering TM, Ferreira-Gonzalez A, Szakal AK, Tew JG, Burton GF. Persistence of infectious HIV on follicular dendritic cells. J Immunol. 2001;166:690–696. doi: 10.4049/jimmunol.166.1.690. [DOI] [PubMed] [Google Scholar]
- 17.Pott GB, Chan ED, Dinarello CA, Shapiro L. a-1-antitrypsin is an endogenous inhibitor of proinflammatory cytokine production in whole blood. J Leukoc Biol. 2009;85:886–895. doi: 10.1189/jlb.0208145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 19.Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, Tan J, Dugger D, Gordon N, Sidhu SS, Fellouse FA, Komuves L, French DM, Ferrando RE, Lam C, Compaan D, Yu C, Bosanac I, Hymowitz SG, Kelley RF, Dixit VM. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell. 2008;134:668–678. doi: 10.1016/j.cell.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 20.Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation. Nat Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- 21.Burton GF, Keele BF, Estes JD, Thacker TC, Gartner S. Follicular dendritic cell contributions to HIV pathogenesis. Seminars in Immunology. 2002:275–284. doi: 10.1016/s1044-5323(02)00060-x. [DOI] [PubMed] [Google Scholar]
- 22.Burton GF, Masuda A, Heath SL, Smith BA, Tew JG, Szakal AK. Follicular dendritic cells (FDC) in retroviral infection: host/pathogen perspectives. Immunol Rev. 1997;156:185–197. doi: 10.1111/j.1600-065x.1997.tb00968.x. [DOI] [PubMed] [Google Scholar]
- 23.Heath SL, Tew JG, Tew JG, Szakal AK, Burton GF. Follicular dendritic cells and human immunodeficiency virus infectivity. Nature. 1995;377:740–744. doi: 10.1038/377740a0. [DOI] [PubMed] [Google Scholar]
- 24.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki H, Chiba T, Kobayashi M, Takeuchi M, Suzuki T, Ichiyama A, Ikenoue T, Omata M, Furuichi K, Tanaka K. IkappaBalpha ubiquitination is catalyzed by an SCF-like complex containing Skp1, cullin-1, and two F-box/WD40-repeat proteins, betaTrCP1 and betaTrCP2. Biochem Biophys Res Commun. 1999;256:127–132. doi: 10.1006/bbrc.1999.0289. [DOI] [PubMed] [Google Scholar]
- 26.Pantaleo G, Fauci AS. HIV-1 infection in the lymphoid organs: a model of disease development. J NIH Res. 1993;5:68–72. [Google Scholar]
- 27.Pantaleo G, Graziosi C, Butini L, Pizzo PA, Schnittman SM, Kotler DP, Fauci AS. Lymphoid organs function as major reservoirs for human immunodeficiency virus. Proc Natl Acad Sci USA. 1991;88:9838–9842. doi: 10.1073/pnas.88.21.9838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fox CH, Cottler-Fox M. The pathobiology of HIV infection. Immunol Today. 1992;13:353–356. doi: 10.1016/0167-5699(92)90171-3. [DOI] [PubMed] [Google Scholar]
- 29.Embretson J, Zupancic M, Ribas JL, Burke A, Racz P, Tenner-Racz K, Haase AT. Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature. 1993;362:359–362. doi: 10.1038/362359a0. [DOI] [PubMed] [Google Scholar]
- 30.Estes JD, Thacker TC, Hampton DL, Kell SA, Keele BF, Palenske EA, Druey KM, Burton GF. Follicular dendritic cell regulation of CXCR4-mediated germinal center CD4 T cell migration. J Immunol. 2004;173:6169–6178. doi: 10.4049/jimmunol.173.10.6169. [DOI] [PubMed] [Google Scholar]
- 31.El Shikh ME, El Sayed R, Szakal AK, Tew JG. Follicular dendritic cell (FDC)-FcgammaRIIB engagement via immune complexes induces the activated FDC phenotype associated with secondary follicle development. Eur J Immunol. 2006;36:2715–2724. doi: 10.1002/eji.200636122. [DOI] [PubMed] [Google Scholar]
- 32.El Shikh ME, El Sayed RM, Wu Y, Szakal AK, Tew JG. TLR4 on follicular dendritic cells: an activation pathway that promotes accessory activity. J Immunol. 2007;179:4444–4450. doi: 10.4049/jimmunol.179.7.4444. [DOI] [PubMed] [Google Scholar]
- 33.Bettinger BT, Gilbert DM, Amberg DC. Actin up in the nucleus. Nat Rev Mol Cell Biol. 2004;5:410–415. doi: 10.1038/nrm1370. [DOI] [PubMed] [Google Scholar]
- 34.de Lanerolle P, Johnson T, Hofmann WA. Actin and myosin I in the nucleus: what next? Nat Struct Mol Biol. 2005;12:742–746. doi: 10.1038/nsmb983. [DOI] [PubMed] [Google Scholar]
- 35.Grummt I. Actin and myosin as transcription factors. Curr Opin Genet Dev. 2006;16:191–196. doi: 10.1016/j.gde.2006.02.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.