

Abstract
In Shaker K+ channels, formation of an electrostatic interaction between two charged residues, D316 and K374 in transmembrane segments S3 and S4, respectively, is a key step in voltage sensor biogenesis. Mutations D316K and K374E disrupt formation of the voltage sensor and lead to endoplasmic reticulum retention. We have now investigated the fates of these misfolded proteins. Both are significantly less stable than the wild-type protein. D316K is degraded by cytoplasmic proteasomes, whereas K374E is degraded by a lactacystin-insensitive, non-proteasomal pathway. Our results suggest that the D316K and K374E proteins are misfolded in recognizably different ways, an observation with implications for voltage sensor biogenesis.
Keywords: Shaker channel, Quality control, Degradation, Biogenesis
1. Introduction
Voltage-dependent K+ channels control the excitability of nerve and muscle. Normally closed at the hyperpolarized membrane potentials characteristic of resting cells, they open upon depolarization, resulting in a K+ current across the plasma membrane [1]. K+ channels contain four subunits that surround the pore for ion conduction [2-4]. Each subunit has six transmembrane segments, S1–S6, and a reentrant P loop. Segments S1–S4 contain the voltage sensor [5,6]. Charged residues in this domain, located primarily in S4, interact electrostatically with the membrane potential [7,8]. In response to depolarization, these residues initiate conformational changes that increase the probability of pore opening. S5, S6, and the intervening P loop form the central pore domain [9]. The P loop lines the narrowest region of the pore, conferring selectivity for K+ over other cations.
Although much has been learned in recent years about the structure and function of the voltage sensor and pore in K+ channels [5,6,9-11], little is known about how these domains form during channel biogenesis [12]. We have investigated formation of the voltage sensor in Shaker K+ channels using a second site suppressor strategy [6,13,14]. The results indicate that two conserved charged residues, D316 in S3 and K374 in S4, form a short-range electrostatic interaction that is essential for generation of the voltage sensor [6,14]. Interestingly, ion pairs between residues in different transmembrane segments are seen in the high resolution X-ray structure of the voltage sensor from KvAP, a prokaryotic voltage-dependent K+ channel [15]. In Shaker, charge reversal mutations D316K and K374E disrupt folding of the protein, trapping it in the endoplasmic reticulum (ER) in an immature, core glycosylated form [14]. Combination of the two mutations in a single subunit, however, restores maturation and the ability of the subunits to incorporate into functional channels [14]. Consistent with these results, the D316K and K374E single mutant proteins lack a hallmark of the native structure, the ability to form an intersubunit disulfide bond between two cytoplasmic cysteine residues (C96 and C505) upon exposure of intact cells to mild oxidizing conditions [3,16]. In the double mutant, D316K + K374E, the ability to form this disulfide bond is recovered, consistent with restoration of the native structure [16].
Previous studies indicate that many ER-retained proteins are dislocated from the ER and degraded by cytoplasmic proteasomes [ 17-25]. Whether this pathway is used to dispose of misfolded channel subunits has been little explored [26]. In this study, we investigated the fates of the misfolded D316K and K374E single mutant proteins in mammalian cells. We find that D316K and K374E are significantly less stable than the wild-type Shaker protein. Degradation of D316K is inhibited by lactacystin, implicating cytoplasmic proteasomes in the turnover of this mutant. In contrast, K374E is degraded by a lactacystin-insensitive pathway. Our data suggest that both proteasomal and non-proteasomal pathways dispose of misfolded channel subunits.
2. Materials and methods
2.1. Cell culture, transfection, antibodies, and reagents
Human embryonic kidney cells (HEK293T) were cultured as described [27] and transfected in 35-mm wells with 1 μg of plasmid DNA encoding wild-type, D316K, D316F, D316R, K374E, or D316K + K374E Shaker using Cytofectene (BioRad, Hercules, CA). Antibodies directed against a Shaker-β-galactosidase fusion protein were the generous gift of Dr. L.Y. Jan (UCSF) [28]. An antibody against the ER marker calnexin was purchased from Affinity Biore-agents Inc. (Golden, CO). Alexa-conjugated goat anti-rabbit and goat anti-mouse secondary antibodies were obtained from Molecular Probes (Eugene, OR). Clasto-lactacystin-β-lactone (Lac), which is the active intermediate in lactacystin-mediated covalent inactivation of the 20S catalytic core of the proteasome, was from Calbiochem (La Jolla, CA) [29]. Deoxynojirimycin (dNJ) and deoxymannojirimycin (dMJ) were purchased from Toronto Research Chemicals (Toronto, Ont., Canada).
2.2. Metabolic labeling, immunoprecipitation, electrophoresis, and fluorography
Metabolic labeling, immunoprecipitation, electrophoresis, and fluorography were performed as described previously [27]. Forty-eight hours post-transfection, cells were incubated for 30 min in methionine-and cysteine-free Dulbecco’s modified Eagle’s medium (Mediatech, Herndon, VA), pulsed with 0.2–0.5 mCi/ml [35S]methionine and [35S]cysteine (Tran35S-Label, ICN, Irvine, CA) for 30 min, and chased for various times in complete, non-radioactive medium. Cells subjected to lactacystin (10 μM) treatment were pre-incubated for 12–16 h with the drug, which was also present throughout the pulse and chase periods. Cells were incubated with other drugs for 2–6 h prior to the start of metabolic labeling and during the pulse and chase periods.
For electrophoresis, each sample loaded was derived from an aliquot of cell lysate containing an equal number of tricholoroacetic acid-precipitable counts per minute. Protein bands were quantified by densitometry (Personal Densitometer SI, Molecular Dynamics, Amersham Biosciences, Sunnyvale, CA) using Molecular Dynamics ImageQuant software (v.4.2, Molecular Dynamics).
2.3. Oxidation of C96/C505 intersubunit disulfide bonds
Intact cells expressing wild-type or mutant Shaker proteins were washed with divalent-free phosphate-buffered saline (PBS) prior to oxidation with 100 mM H2O2 for 15 min. The reaction was quenched by addition of 5 mM N-ethylmaleimide for 5 min [3]. Shaker protein was then solubilized, immunoprecipitated, and boiled for 5 min in Laemmli sample buffer containing 16 mM iodoacetamide (non-reducing conditions) [27].
2.4. Immunofluorescence microscopy
Indirect immunofluorescence microscopy was performed on fixed, permeabilized cells as described previously [30]. Cells were incubated for 2 h at room temperature with a polyclonal rabbit antiserum directed against a Shaker β-galactosidase fusion protein (1:200 dilution in 3% bovine serum albumin in PBS) and a mouse monoclonal antibody against calnexin (1:250 dilution). Cells were then washed with PBS and incubated with fluorescent-conjugated secondary antibodies (Alexa-488-conjugated goat anti-rabbit [1:1000] and Alexa-568-conjugated goat anti-mouse [1:1500]) for 1 h at room temperature. Images were acquired on an inverted laser scanning confocal microscope with a 63× quartz objective (Leica Dm IRB/E, Meyer Instruments Inc., Houston, TX).
3. Results
3.1. Different pathways dispose of the D316K and K374E proteins
To test the hypothesis that the ER-retained mutant proteins D316K and K374E are less stable than the wild-type Shaker protein, we characterized their turnover times by pulse chase analysis. HEK293T cells expressing D316K or K374E were metabolically labeled during a 30-min pulse, and were either harvested immediately or incubated in non-radioactive medium for chase periods up to 24 h (Fig. 1). After detergent extraction, the Shaker protein was immunoprecipitated and subjected to electrophoresis and fluorography. As previously reported, the D316K and K374E proteins were detected as the core-glycosylated, immature form characteristic of the ER, and did not acquire complex glycosylation [14,16,31]. The D316K protein was rapidly degraded: ~15 ± 3% of the D316K protein remained after 6 h (Fig. 1A and B). The K374E mutant was comparatively more stable: ~86 ± 14% of the protein remained after 6 h (Fig. 1C and D). However, both were significantly less stable than the wild-type protein, which shows little degradation in HEK293T cells over 24 h [30].
Fig. 1.
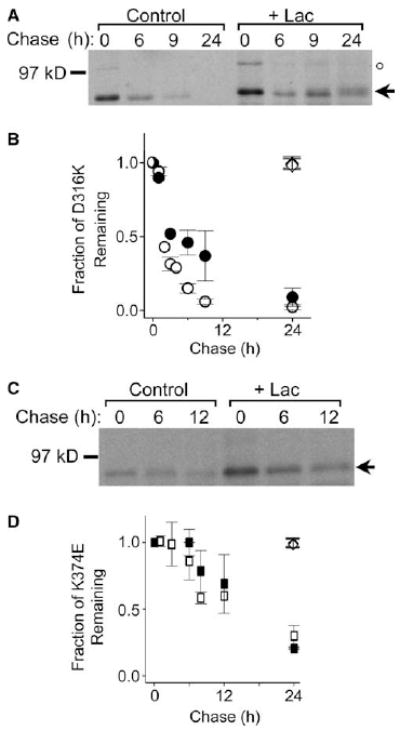
Degradation of D316K and K374E mutant proteins is differentially sensitive to lactacystin. (A,C) D316K and K374E were expressed in HEK293T cells, metabolically labeled, and chased for the indicated times in the absence or presence of the proteasomal inhibitor, lactacystin (Lac, 10 μM). Following solubilization, Shaker proteins were immunoprecipitated and subjected to electrophoresis and fluorography. Representative fluorographs for D316K (A) and K374E (C) are shown. The arrow indicates the position of the immature, core-glycosylated mutant protein. In addition to the immature band, a sharp band with an apparent molecular weight of 100 kDa is visible in (A) immediately after the pulse and then disappears rapidly thereafter (position denoted by ○). The same unstable band is seen immediately after the pulse upon expression of the wild-type protein; the band is degraded in the interval before substantial maturation of the wild-type protein occurs (see Fig. 3 in [27]). The identity of this band has not been determined. It does not correspond to the mature form of Shaker, which migrates as a broad band exhibiting high stability [27,30]. It was not included in any of the calculations described in this paper. (B,D) Summary of densitometric analysis of the turnover of the immature Shaker protein for (B) D316K (open circles, n = 8) and D316K + Lac (filled circles, n = 5), and (D) K374E (open squares, n = 4–6) and K374E + Lac (filled squares, n = 3). The amount of protein in the immature band was quantified by densitometry, normalized to the amount of Shaker protein at time 0, and plotted versus chase time. Unless otherwise stated, the data in this and subsequent figures are provided as means ± S.E.M. For the D316K mutant, the half time of degradation (t1/2) was estimated to be 2 h in the absence and 4.4 h in the presence of Lac. The t1/2 for the K374E mutant was estimated to be 15 h in the absence and 17 h in the presence of Lac. For comparison, at 24 h ~98% of the wild-type Shaker protein remained either in the absence (open diamonds, n = 14) or presence (filled diamonds, n = 4) of Lac [30].
To determine whether cytoplasmic proteasomes contribute to the degradation of the D316K and K374E proteins, turnover rates were compared in the presence and absence of lactacystin, a proteasome-specific inhibitor [29]. Treatment with lactacystin significantly slowed degradation of the D316K protein: at 6 h, ~46 ± 8% of the mutant protein remained (Fig. 1A and B). In contrast, lactacystin had no significant effect on the turnover of the K374E protein (Fig. 1C and D). These results indicate that the D316K and K374E proteins have different fates. D316K is degraded, at least in part, by cytoplasmic proteasomes, whereas disposal of K374E occurs via a lactacystin-insensitive pathway.
3.2. Multiple D316 mutants are degraded by proteasomes
To investigate whether the nature of the mutation at D316 was important for targeting the Shaker protein to cytoplasmic proteasomes for degradation, we generated the D316R and D316F mutations. Pulse chase analysis revealed that the D316F and D316R mutant proteins did not mature and were rapidly degraded: at 6 h, ~10% of the D316F (Fig. 2A and C) and ~5% of the D316R proteins remained (Fig. 2B and C). Similar to the D316K mutant, the degradation of both D316F and D316R was significantly inhibited by lactacystin: at 6 h, ~45% of both proteins remained (Fig. 2A–C). These results indicate that several mutations of residue 316 destabilize the Shaker protein and target it for degradation by cytoplasmic proteasomes. Consistent with the lack of maturation and instability of D316F and D316R, immunofluorescence microscopy indicated that these proteins were retained intracellularly where they partially co-localized with the ER marker, calnexin (Fig. 2D). In contrast, the wild-type Shaker protein was detected primarily at the cell surface with little or no overlap with calnexin (Fig. 2D).
Fig. 2.
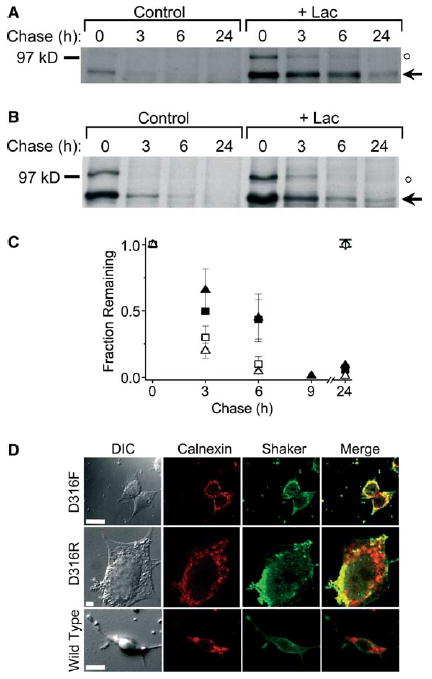
Different D316 mutations target the Shaker protein to proteasomes for degradation. (A,B) D316F and D316R mutant Shaker proteins were metabolically labeled and treated as described under Fig. 1. Representative fluorographs are shown for D316F (A) and D316R (B). The arrow indicates the position of the immature, core-glycosylated mutant protein. An unstable band of unknown identity with an apparent molecular weight of 100 kDa is also visible (denoted by ○, see Fig. 1 legend). (C) Summary of densitometric analysis of turnover for D316F (open squares, n = 3), D316F + Lac (filled squares, n = 3), D316R (open triangles, n = 3; except n = 1 for the 9 h and n = 2 for the 24 h time points) and D316R + Lac (filled triangles, n = 3; except n = 1 for the 9 h and n = 2 for the 24 h time points). The amount of protein in the bands was quantified by densitometry, normalized to the amount of immature Shaker protein at time 0, and plotted versus chase time. The t1/2 for the D316F mutant was estimated to be 1 h in the absence and 5 h in the presence of Lac. The t1/2 for the D316R mutant was estimated to be 1.5 h in the absence and 3 h in the presence of Lac. The diamond symbols have the same meaning as in Fig. 1B and D. Data are provided as mean ± S.E.M. except for the 9 and 24 h time points. (D) Representative differential image contrast and confocal images of HEK293T cells transiently transfected with D316F, D316R, or wild-type Shaker. Forty eight hours post-transfection, cells were permeabilized and labeled with a rabbit polyclonal antiserum directed against a Shaker-β-galactosidase fusion protein and a mouse monoclonal antibody directed against calnexin (an ER-resident protein) and visualized by incubation with fluorescent-conjugated Alexa-488 goat anti-rabbit (Shaker, green) and Alexa-568 goat anti-mouse (calnexin, red) secondary antibodies, respectively. For each panel, the same confocal plane was used for acquisition. Yellow regions in the merged images represent co-localization of mutant Shaker proteins and calnexin. Calibration bar, 10 μm.
3.3. Differential stability of mature and immature forms of the D316K + K374E double mutant
We have previously shown that combining the D316K and K374E mutations in the same subunit restores maturation to a complex glycosylated form [14]. Upon expression in Xenopus oocytes, approximately 85% of the D316K + K374E double mutant protein matures [14]. Two lines of evidence indicate that this mature protein is properly folded. First, the double mutant subunit incorporates efficiently into functional channels in which the voltage dependence of activation is shifted in the depolarized direction [14]. Second, the D316K + K374E double mutant protein has the ability to form an intersubunit disulfide bond between two cytoplasmic cysteine residues, C96 and C505 upon exposure of intact cells to mild oxidizing conditions [16]. This disulfide bond can be efficiently oxidized in the wild-type Shaker protein, whether it is located at the cell surface or in the ER, and in mutant proteins that fold and assemble properly as evidenced by maturation and functional activity [16,32]. However, the C96/C505 disulfide bond cannot be detected in a variety of ER-retained mutant proteins [16]. Thus, the C96/C505 disulfide bond serves as a hallmark for the native Shaker structure, providing an indirect measure of the ability of a mutant protein to fold and assemble properly [3,16,30,32,33].
In HEK293T cells, maturation of the D316K + K374E protein was somewhat reduced compared to oocytes, but still substantial, with about 50% of the protein being converted to the complex glycosylated form (Fig. 3A). To compare the ability of the wild-type, D316K, K374E, and D316K + K374E proteins to form the C96/C505 disulfide bond in mammalian cells, intact cells expressing these proteins were metabolically labeled and incubated with 100 mM H2O2. Remaining free sulfhydryl groups were protected with N-ethylmaleimide prior to solubilization and immunoprecipitation. Electrophoresis under non-reducing conditions revealed that the Shaker wild-type protein and the D316K + K374E double mutant were converted to higher molecular weight adducts corresponding to dimer, trimer, linear tetramer, and circular tetramer (Fig. 3B) [3,30]. In contrast, disulfide-bonded adducts could not be detected in the D316K or K374E single mutant proteins (Fig. 3B). These results are consistent with the idea that the D316K and K374E proteins fail to fold properly, whereas some of the D316K + K374E protein adopts the native structure in mammalian cells [14,16].
Fig. 3.
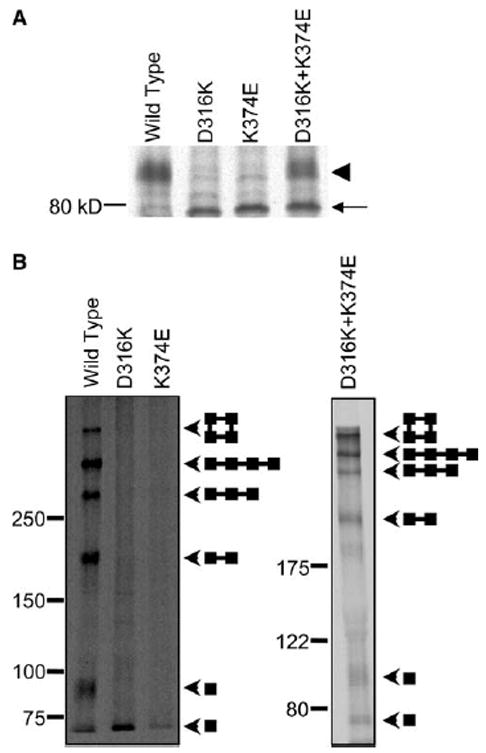
D316K + K374E protein forms C96/C505 disulfide bond in mammalian cells. (A) The wild-type, D316K, K374E, and D316K + K374E proteins were expressed in HEK293T cells, metabolically labeled for 30 min and chased for 3 h in non-radioactive medium. Shaker protein was immunoprecipitated, and electrophoresis was performed under reducing conditions. A representative fluorograph is shown (n = 3). The arrowhead indicates the position of the mature, complex glycosylated protein. The arrow indicates the position of the immature, core-glycosylated protein. (B) Intact cells were metabolically labeled for 30 min, chased in non-radioactive medium for 3 h, and oxidized with 100 mM hydrogen peroxide for 15 min. Remaining free sulfhydryl groups were protected with N-ethylmaleimide. Shaker protein was immunoprecipitated, and electrophoresis was performed under non-reducing conditions on 5–20% gradient (left panel) or 7.5% (right panel) SDS–PAGE gels. Arrowheads indicate the positions of the bands corresponding to monomer, dimer, trimer, linear tetramer, and circular tetramer. Diagrams show schematically the predicted structures of each band [3].
We investigated the stability of the mature and immature forms of the double mutant protein, D316K + K374E, by pulse chase analysis. The mature form of the D316K + K374E protein was extremely stable with little or no degradation for up to 36 h (Fig. 4A). Lactacystin had no effect on the mature D316K + K374E protein (Fig. 4A). Thus, the stability of the mature form of D316K + K374E is comparable to that of the wild-type protein, consistent with the conclusion that it folds and assembles properly [14,16].
Fig. 4.
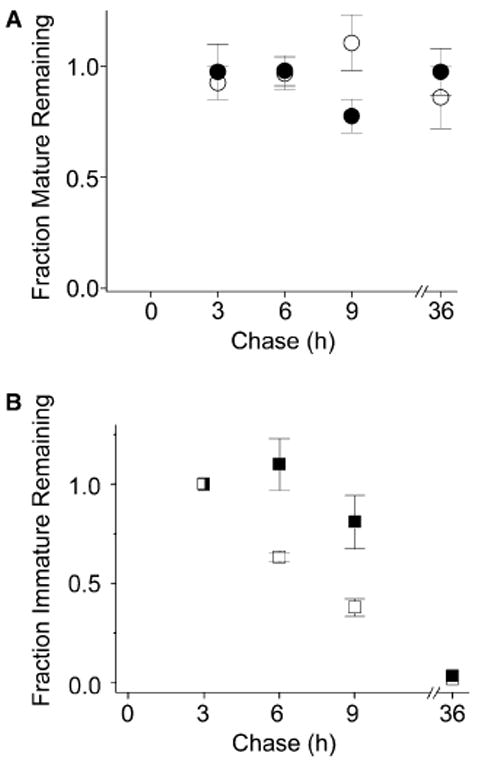
The mature and immature forms of the D316K + K374E protein are differentially stable. The D316K + K374E double mutant was metabolically labeled and treated as described under Fig. 1. (A) Summary of densitometric analysis of protein turnover for the mature band of D316K + K374E (open circles, n = 3) and D316K + K374E + Lac (filled circles, n = 3). The amount of protein in the mature band was quantified by densitometry, normalized to the amount of mature Shaker protein present at time 3 h, and plotted versus chase time. (B) Summary of densitometric analysis of protein turnover for the immature band of D316K + K374E (open squares, n = 3) and D316K + K374E + Lac (filled squares, n = 3). The amount of protein in the immature band was quantified by densitometry, normalized to the amount of immature Shaker protein present at time 3 h, and plotted versus chase time. Note that at time 0, the total D316K + K374E protein is in the form of an immature band that represents a mixed population. About 50% of this protein will mature, a process that should be complete by approximately 2 h of chase [27]. After 3 h of chase, maturation is expected to be complete, so further change in the amounts of the mature and immature bands primarily reflects turnover. Therefore, the turnover of the mature and immature bands was assessed relative to the 3 h time point. The half time of degradation (t1/2) for the immature form of D316K + K374E was estimated to be 4.5 h in the absence of Lac, a value that is intermediate between estimated t1/2 values for the D316K and K374E single mutants, which were 2 and 15 h, respectively (see Fig. 1 legend).
The behavior of the immature form of D316K + K374E differed from that of the wild-type protein. We have previously shown that the immature form of the wild-type Shaker protein matures in HEK293T cells with a half time of approximately 45 min [31]. Maturation is virtually complete in 2 h [27]. In contrast, as noted above, the maturation of D316K + K374E is incomplete in mammalian cells. A fraction of the immature form of D316K + K374E failed to mature, persisting in cells past 2 h of chase. This fraction of persistent immature protein was subsequently degraded with rapid kinetics intermediate to those of the single D316K and K374E mutants (Fig. 4B). Degradation of the immature D316K + K374E protein was inhibited by lactacystin, implicating proteasomes in its disposal (Fig. 4B). Thus, the immature and mature forms of the double mutant protein are differentially stable. The results suggest that the immature form that persists past 2 h of chase represents a fraction of D316K + K374E protein that fails to fold properly in mammalian cells.
3.4. Role of glycan processing in the differential turnover of D316K and K374E
Several recent reports indicate that interaction with the lectin chaperone calnexin and trimming of the core glycan play key roles in targeting glycoproteins for ER-associated degradation (ERAD) [ 17-25,34]. For some proteins, interaction with calnexin has been shown to inhibit ERAD, whereas processing of the core glycan by ER mannosidase I promotes degradation [19-21,35]. We have previously shown that calnexin interacts transiently with the Shaker protein soon after its synthesis [31]. Interestingly, the time course of association with calnexin is similar for the Shaker wild-type, D316K, and K374E proteins, indicating that the misfolded mutant proteins escape the folding sensor of the calnexin chaperone system [31]. We have also presented evidence that transient interaction with calnexin confers long-term protection from ERAD on properly folded Shaker protein that is localized in the ER [36]. We therefore investigated the role of calnexin interaction and glycan trimming in the quality control pathways that dispose of D316K and K374E.
To prevent interaction with calnexin, cells expressing D316K or K374E were incubated co- and post-translationally with dNJ, which inhibits ER glucosidases I and II and thereby prevents formation of the monoglucosylated core glycan that is recognized by calnexin. We have previously shown that treatment with dNJ abolishes association of the Shaker protein with calnexin [36]. In parallel experiments, cells were incubated with dMJ, a general inhibitor of mannosidase activity. Neither drug altered the turnover of the D316K and K374E proteins (Fig. 5). We conclude that processing of the core glycan is not required for degradation of D316K or K374E.
Fig. 5.
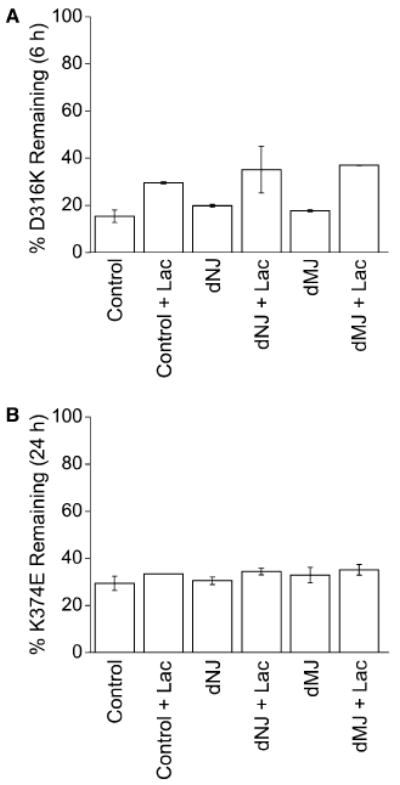
Processing of core glycan is not required for turnover of D316K or K374E. (A) Cells expressing D316K were metabolically labeled for 30 min and either harvested immediately or chased for 6 h. Cells were either untreated (control) or treated with Lac alone (control + Lac), or treated with dNJ or dMJ in the absence or presence of Lac. The percent protein remaining at 6 h of chase is shown for each condition, n = 2. Values shown represent means ± S.D. (B) Cells expressing K374E were labeled for 30 min and either harvested immediately or chased for 24 h. Cells were treated as described in (A). The percent protein remaining at 24 h of chase is shown for each condition, n = 2, except for “control + Lac”, n = 1. Values shown represent means ± S.D., except for “control + Lac”.
4. Discussion
4.1. Evidence for proteasomal and non-proteasomal disposal pathways involved in quality control of misfolded channel mutants
In Shaker channels, mutations D316K in S3 and K374E in S4 disrupt an electrostatic structural interaction that is essential for biogenesis of the voltage sensor [14,16]. In this study, we have investigated pathways used by mammalian cells to dispose of the misfolded D316K and K374E proteins. We find that these mutations differentially target the Shaker protein to lactacystin-sensitive and -insensitive disposal pathways. D316K is degraded, at least in part, by the lactacystin-sensitive proteasome pathway. In contrast, disposal of K374E is unaffected by lactacystin, suggesting that a non-proteasomal pathway degrades the K374E protein.
Although a number of studies have suggested that non-proteasomal pathways are involved in ERAD, the molecular components involved have not been identified [37,38]. As a result, it has been suggested that proteasomes are the main pathway responsible for ERAD [39]. Although we have been unable to identify the pathway responsible for disposal of the K374E protein (see below), our results appear to be incompatible with the idea that proteasomes are solely responsible for ERAD of misfolded Shaker proteins. The D316K and K374E proteins differ from each other by only one amino acid residue, yet their degradation is differentially sensitive to lactacystin. Although proteasomes contain multiple proteolytic activities [40-42], the overall sequence identity of the D316K and K374E proteins suggests that the same protease components would degrade both of them, and that these proteases would therefore be equally sensitive to inhibition by lactacystin. For these reasons, we conclude that both proteasomal and non-proteasomal pathways are likely to participate in the disposal of misfolded, ER-retained Shaker proteins in mammalian cells.
Despite earnest attempts to characterize the lactacystin-insensitive pathway, its identity remains elusive. Degradation of D316K and K374E was insensitive to drugs that inhibit lysosomal proteases or a cytoplasmic “giant” protease that under some circumstances substitutes for the proteasome (data not shown) [43,44]. Furthermore, no evidence was found that D316K and K374E form aggresomes, an alternative disposal pathway in which misfolded protein forms an inclusion body located at the microtubule organizing center (data not shown) [45]. One study reported a phenylarsine oxide-sensitive pathway in ERAD, implicating a tyrosine phosphatase activity in a non-proteasomal ER disposal pathway [37]. We have been unable to evaluate this pathway in our work due to the extreme toxicity of the drug during long pulse chase experiments (data not shown) [ 25]. Further work will be required to characterize the proposed non-proteasomal ERAD pathway.
4.2. Possible implications for voltage sensor biogenesis
Our results indicate that the cellular quality control machinery is able to distinguish between D316K and K374E, suggesting that these proteins are misfolded in recognizably different ways. If so, our results have implications for the mechanism of voltage sensor biogenesis. Data from in vitro models suggests that the folding of polytopic membrane proteins involves at least two stages [46,47]. In the first stage, the protein establishes its secondary structure and membrane topology. In the second stage, tertiary interhelical interactions are formed, leading to the condensed, properly folded structure. Our previous results suggest that the D316K and K374E mutations affect the formation of a tertiary interaction between transmembrane segments S3 and S4 [14]. One possibility suggested by the two-stage model is that the two mutant proteins might be able to establish the correct membrane topology but fail to form the proper condensed structure. However, this predicts that the mutant proteins would be misfolded in similar ways, which appears to be inconsistent with our findings. An alternative possibility is that the mutant proteins differ in their ability to establish the proper membrane topology. For instance, insertion of S3 and S4 into the membrane may not be independent events during biogenesis of the voltage sensor. Interestingly, Deutsch and colleagues [48] have presented evidence that S3 and S4 interact during biogenesis, whereas Sato et al. [49] report that membrane insertion of an S4 segment derived from a plant K+ channel depends on the presence of S3. If so, it may be necessary to form an interaction between positions 316 and 374 before the S4 segment can be inserted into the membrane.
Although the differences between the misfolded structures adopted by D316K and K374E are unknown, at least one possibility can be suggested. In the presence of the D316K mutation, the S4 segment may fail to insert into the membrane, leading to an aberrant topology. In the presence of the K374E mutation, D316 in S3 may mispair with one of the other positively charged residues in S4, leading to insertion of both the S3 and S4 segments but with an incorrect set of tertiary contacts. In this case the proper condensed structure would not be formed despite the presence of the appropriate topology.
We found that several D316 mutations resulted in proteasomal degradation of the Shaker protein. Furthermore, the persistent immature form of the D316K + K374E double mutant was degraded by proteasomes with rapid kinetics. The structural epitope(s) involved in targeting these proteins to proteasomes for degradation have not been identified. One possibility is that D316K, D316R, D316F, and the immature form of the D316K + K374E protein have a common misfolded structure, which differs from that of the K374E single mutant. In this regard, the double mutant protein is of particular interest. When the D316K and K374E mutations are combined in a single subunit, much of the protein is able to fold properly and mature [14,16]. A fraction, however, remains misfolded.
In summary, our results provide evidence that voltage sensor mutations differentially target the Shaker protein to proteasomal and non-proteasomal disposal pathways. The results suggest that D316K and K374E are misfolded in recognizably different ways. One possibility is that the mutations have differential effects on topogenesis, consistent with previous reports suggesting that the insertion of an S4 segment is dependent on S3 [48,49]. If so, this would suggest that the interaction between D316 in S3 and K374 in S4 helps to generate the correct membrane topology of the Shaker protein, as well as the condensed tertiary structure of the voltage sensor.
Acknowledgments
We thank Drs. Lily Jan (UCSF) for antibodies to Shaker, Michael Haykinson (Biological Chemistry Imaging Facility, UCLA) for help with densitometric analyses, Matthew J. Schibler (Carol Moss Spivak Cell Imaging Facility, UCLA) for assistance with confocal microscopy, and Allan Mock for excellent technical assistance. We are grateful to Drs. John Bannister, Ji-jun Wan, and Joanna Jen for helpful comments on the manuscript. This work was supported by grants to D.M. Papazian from the National Institutes of Health (GM66686) and the American Heart Association, Western States Affiliate. M.P. Myers was partially supported by an NIH post-doctoral training grant (NS07101-20). R. Khanna was supported by post-doctoral fellowships from the Natural Sciences and Engineering Research Council of Canada (PDF231011) and the American Heart Association, Western States Affiliate (0120119Y).
Abbreviations
- dMJ
deoxymannojirimycin
- dNJ
deoxynojirimycin
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- HEK293T
human embryonic kidney 293T cells
- PBS
phosphate-buffered saline
References
- 1.Hille B. Ionic Channels of Excitable Membranes. 4. Sinauer Associates; Sunderland, MA: 2003. [Google Scholar]
- 2.MacKinnon R. Nature. 1991;350:232–235. doi: 10.1038/350232a0. [DOI] [PubMed] [Google Scholar]
- 3.Schulteis CT, Nagaya N, Papazian DM. Biochemistry. 1996;35:12133–12140. doi: 10.1021/bi961083s. [DOI] [PubMed] [Google Scholar]
- 4.Sokolova O, Kolmakova-Partensky L, Grigorieff N. Structure. 2001;9:215–220. doi: 10.1016/s0969-2126(01)00578-0. [DOI] [PubMed] [Google Scholar]
- 5.Bezanilla F. Physiol Rev. 2000;80:555–592. doi: 10.1152/physrev.2000.80.2.555. [DOI] [PubMed] [Google Scholar]
- 6.Papazian DM, Silverman WR, Lin MA, Tiwari-Woodruff SK, Tang CY. Novartis Found Symp. 2002;245:178–190. [PubMed] [Google Scholar]
- 7.Seoh SA, Sigg D, Papazian DM, Bezanilla F. Neuron. 1996;16:1159–1167. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- 8.Aggarwal SK, MacKinnon R. Neuron. 1996;16:1169–1177. doi: 10.1016/s0896-6273(00)80143-9. [DOI] [PubMed] [Google Scholar]
- 9.Doyle DA, Morais-Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 10.Morais-Cabral JH, Zhou Y, MacKinnon R. Nature. 2001;414:37–42. doi: 10.1038/35102000. [DOI] [PubMed] [Google Scholar]
- 11.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 12.Deutsch C. Annu Rev Physiol. 2002;64:19–46. doi: 10.1146/annurev.physiol.64.081501.155934. [DOI] [PubMed] [Google Scholar]
- 13.Papazian DM, Shao XM, Seoh SA, Mock AF, Huang Y, Wainstock DH. Neuron. 1995;14:1293–1301. doi: 10.1016/0896-6273(95)90276-7. [DOI] [PubMed] [Google Scholar]
- 14.Tiwari-Woodruff SK, Schulteis CT, Mock AF, Papazian DM. Biophys J. 1997;72:1489–1500. doi: 10.1016/S0006-3495(97)78797-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 16.Schulteis CT, Nagaya N, Papazian DM. J Biol Chem. 1998;273:26210–26217. doi: 10.1074/jbc.273.40.26210. [DOI] [PubMed] [Google Scholar]
- 17.Keller SH, Lindstrom J, Taylor P. J Biol Chem. 1998;273:17064–17072. doi: 10.1074/jbc.273.27.17064. [DOI] [PubMed] [Google Scholar]
- 18.de Virgilio M, Kitzmuller C, Schwaiger E, Klein M, Kreibich G, Ivessa NE. Mol Biol Cell. 1999;10:4059–4073. doi: 10.1091/mbc.10.12.4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung DH, Ohashi K, Watanabe M, Miyasaka N, Hirosawa S. J Biol Chem. 2000;275:4981–4987. doi: 10.1074/jbc.275.7.4981. [DOI] [PubMed] [Google Scholar]
- 20.Chillaron J, Adan C, Haas IG. Biol Chem. 2000;381:1155–1164. doi: 10.1515/BC.2000.143. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, White AL. Biochemistry. 2000;39:8993–9000. doi: 10.1021/bi000027v. [DOI] [PubMed] [Google Scholar]
- 22.Wilson CM, Farmery MR, Bulleid NJ. J Biol Chem. 2000;275:21224–21232. doi: 10.1074/jbc.M000567200. [DOI] [PubMed] [Google Scholar]
- 23.Jakob CA, Chevet E, Thomas DY, Bergeron JJ. Results Probl Cell Differ. 2001;33:1–17. doi: 10.1007/978-3-540-46410-5_1. [DOI] [PubMed] [Google Scholar]
- 24.Fagioli C, Sitia R. J Biol Chem. 2001;276:12885–12892. doi: 10.1074/jbc.M009603200. [DOI] [PubMed] [Google Scholar]
- Tokunaga F, Hara K, Koide T. Arch Biochem Biophys. 2003;411:235–242. doi: 10.1016/s0003-9861(02)00717-8. [DOI] [PubMed] [Google Scholar]
- 26.Kagan A, Yu Z, Fishman GI, McDonald TV. J Biol Chem. 2000;275:11241–11248. doi: 10.1074/jbc.275.15.11241. [DOI] [PubMed] [Google Scholar]
- 27.Schulteis CT, John SA, Huang Y, Tang CY, Papazian DM. Biochemistry. 1995;34:1725–1733. doi: 10.1021/bi00005a029. [DOI] [PubMed] [Google Scholar]
- 28.Schwarz TL, Papazian DM, Caretto RC, Jan YN, Jan LY. Neuron. 1990;4:119–127. doi: 10.1016/0896-6273(90)90448-o. [DOI] [PubMed] [Google Scholar]
- 29.Fenteany G, Schreiber SL. J Biol Chem. 1998;273:8545–8548. doi: 10.1074/jbc.273.15.8545. [DOI] [PubMed] [Google Scholar]
- 30.Khanna R, Myers MP, Lainé M, Papazian DM. J Biol Chem. 2001;276:34028–34034. doi: 10.1074/jbc.M105248200. [DOI] [PubMed] [Google Scholar]
- 31.Nagaya N, Schulteis CT, Papazian DM. Receptors Channels. 1999;6:229–239. [PubMed] [Google Scholar]
- 32.Nagaya N, Papazian DM. J Biol Chem. 1997;272:3022–3027. doi: 10.1074/jbc.272.5.3022. [DOI] [PubMed] [Google Scholar]
- 33.Papazian DM. Neuron. 1999;23:7–10. doi: 10.1016/s0896-6273(00)80746-1. [DOI] [PubMed] [Google Scholar]
- 34.Ellgaard L, Helenius A. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 35.Jakob CA, Burda P, Roth J, Aebi M. J Cell Biol. 1998;142:1223–1233. doi: 10.1083/jcb.142.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khanna R, Lee EJ, Papazian DM. J Cell Sci. 2004 doi: 10.1242/jcs.01141. in press. [DOI] [PubMed] [Google Scholar]
- 37.Cabral CM, Choudhury P, Liu Y, Sifers RN. J Biol Chem. 2000;275:25015–25022. doi: 10.1074/jbc.M910172199. [DOI] [PubMed] [Google Scholar]
- 38.Fayadat L, Siffroi-Fernandez S, Lanet J, Franc JL. J Biol Chem. 2000;275:15948–15954. doi: 10.1074/jbc.M905763199. [DOI] [PubMed] [Google Scholar]
- 39.Gelman MS, Kannegaard ES, Kopito RR. J Biol Chem. 2002;277:11709–11714. doi: 10.1074/jbc.M111958200. [DOI] [PubMed] [Google Scholar]
- 40.Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- 41.Loayza D, Michaelis S. Mol Cell Biol. 1998;18:779–789. doi: 10.1128/mcb.18.2.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Umebayashi K, Fukuda R, Hirata A, Horiuchi H, Nakano A, Ohta A, Takagi M. J Biol Chem. 2001;276:41444–41454. doi: 10.1074/jbc.M105829200. [DOI] [PubMed] [Google Scholar]
- 43.Geier E, Pfeifer G, Wilm M, Lucchiari-Hartz M, Baumeister W, Eichmann K, Niedermann G. Science. 1999;283:978–981. doi: 10.1126/science.283.5404.978. [DOI] [PubMed] [Google Scholar]
- 44.Wang EW, Kessler BM, Borodovsky A, Cravatt BF, Bogyo M, Ploegh HL, Glas R. Proc Natl Acad Sci USA. 2000;97:9990–9995. doi: 10.1073/pnas.180328897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnston JA, Ward CL, Kopito RR. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Popot JL, Engelman DM. Annu Rev Biochem. 2000;69:881–922. doi: 10.1146/annurev.biochem.69.1.881. [DOI] [PubMed] [Google Scholar]
- 47.Popot JL, Engelman DM. Biochemistry. 1990;29:4031–4037. doi: 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- 48.Tu L, Wang J, Helm A, Skach WR, Deutsch C. Biochemistry. 2000;39:824–836. doi: 10.1021/bi991740r. [DOI] [PubMed] [Google Scholar]
- 49.Sato Y, Sakaguchi M, Goshima S, Nakamura T, Uozumi N. Proc Natl Acad Sci USA. 2002;99:60–65. doi: 10.1073/pnas.012399799. [DOI] [PMC free article] [PubMed] [Google Scholar]