

Summary
Signaling through the interleukin-2 receptor (IL-2R) contributes to T-cell tolerance by controlling three important aspects of regulatory T-cell (Treg) biology. IL-2 is essential for thymic Treg development and regulates Treg homeostasis and suppressive function. Analogous to activated conventional T lymphocytes, IL-2R signaling also plays an important part in Treg cell growth, survival, and effector differentiation. However, Treg cells somewhat distinctively assimilate IL-2R signaling. In particular, Treg cells require essentially only IL-2-dependent receptor proximal signal transducer and activator of transcription 5 (Stat5) activation, as they contain inhibitory pathways to minimize IL-2R-dependent activation of the phosphoinisitol 3-kinase/Akt pathway. Moreover, many IL-2R-dependent activities, including full induction of Foxp3 expression, in Treg cells require minimal and transient Stat5 activation. Thus, Treg cells are equipped to sense and then develop and function within biological niches containing minimal IL-2. These distinguishing features of IL-2R signaling provide a mechanistic underpinning for using IL-2 as an agent to selectively target Treg cells in immunotherapy to induce tolerance in autoimmune diseases and in allogeneic transplant recipients.
Keywords: T-regulatory cells, interleukin 2, lineage plasticity and stability, tolerance/suppression, IL-2R signaling thresholds, immunotherapy
Introduction
Interleukin-2 (IL-2) was identified nearly 30 years ago (1) based on its T-cell growth factor activity. This cytokine is produced mainly by activated T cells after engagement of the T-cell receptor (TCR) and the CD28 costimulatory molecule. Its biological functions are exhibited after binding to its high affinity receptor. The IL-2 receptor (IL-2R) contains three subunits: the α chain (IL-2Rα, also known as CD25), the β chain (IL-2Rβ, also known as CD122, shared by the IL-15R), and the common cytokine receptor γ chain (γc, also known as CD132, shared by IL-4R, IL-7R, IL-9R, IL-15R and IL-21R). All these subunits participate in high affinity binding (Kd=10−11 M) through a rapid association rate (k = 107/M/s) initiated by capture of ligand by IL-2Rα, which is then presented in cis to IL-2Rβ and γc, to form a stable quaternary complex with a slow dissociation rate (k’ = 10−4/s) (2-4).
The IL-2/IL-2R complex induces downstream signaling through IL-2Rβ and γc due to the association of the tyrosine kinases, Janus kinase 1 (JAK1) and JAK3 (5, 6), to their cytoplasmic tails, leading to phosphorylation of the JAKs as well as the three key tyrosine residues within the cytoplasmic tail of IL-2Rβ. As a consequence, three main intracellular signaling pathways are initiated (7, 8). The mitogen-activated protein kinase (MAPK) and the phosphoinositide 3-kinase (PI3K) pathways are activated primarily through the adapter Shc, which associates with the most membrane-proximal tyrosine residue (Tyr-338 in human and Tyr-341 in mouse) within the A-region and leads to Shc-dependent recruitment of the adapter proteins Grb2 and Gab2. The signal transducer and activator of transcription 5 (STAT5) pathway is predominately activated by its recruitment and association to IL-2Rβ through two other tyrosine residues (Tyr-392 and Tyr-510 in human, Tyr-395 and Tyr-498 in mouse) located within the H-region. These pathways contribute to IL-2-dependent cell cycle entry, growth, survival, and differentiation.
Both T-effector (Teff) and T-regulatory (Treg) cells utilize IL-2R signaling for important aspects of their biological response. For Teff cells, IL-2 contributes to optimal clonal expansion of antigen-activated T cells, drives terminal Teff cell differentiation, and programs memory development and survival (4). For Treg cells, IL-2 is essential during their thymic development and later for peripheral homeostasis. The common usage of IL-2R signaling by these distinct cell types represents one of the first examples of a major molecular pathway ascribed to Teff cells that is assimilated by Treg cells. This notion that Treg cells co-opt pathways of Teff cells for their unique suppressive function has been further illustrated recently, where the key transcription factors T-bet, Gata-3, Irf-4, and Stat3, essential for T-helper 1 (Th1), Th2, and Th17 development, are also utilized by Treg cells in a way that aligns their functional program for optimal suppression of these specialized Teff responses (9-12). The means by which these transcriptional regulators distinctively control Treg function are not well understood. In comparison, much more is known concerning the basis by which IL-2 induces various activities in Treg cells that are either unique or common when compared to Teff cells.
This review compares and contrasts the role of IL-2 in Treg versus Teff cells and summarizes our current understanding how Treg cells have assimilated the IL-2/IL-2R pathway for their unique functional role to suppress autoreactive T cells that escape thymic negative selection and to otherwise inhibit T-cell-dependent immune responses. First, however, the biological contribution of IL-2 to the biology of Treg cells is discussed.
IL-2R signaling is essential for Treg cells
IL-2 was the first cytokine gene to be knocked out (13). Contrary to the view at the time that IL-2 was essential for T-cell clonal expansion during immune responses, IL-2−/− mice exhibited lymphoproliferation followed by lethal autoimmunity, where activated T cells accumulated in multiple organs (14) and premature death resulted in part due to autoimmune hemolytic anemia and inflammatory bowel disease. Similar fatal autoimmunity was also found in IL-2Rα- and IL-2Rβ-deficient mice (15, 16) but not in IL-15- and IL-15Rα-deficient mice (17, 18). Although the IL-2R and IL-15R share β and γc subunits that are responsible for signal transduction, these findings indicate that autoimmunity was due to failed IL-2R signaling. Mice deficient in γc do not exhibit autoimmunity due to impaired T-cell development as the consequence of failed IL-7R signaling (19, 20). However, with time, the few T cells found in such mice exhibit an activated phenotype, which is likely accounted for by failed IL-2R signaling (21-23).
Since activated T cells that respond to IL-2 readily undergo apoptosis upon TCR signaling through re-encounter with antigen, autoimmunity was initially thought to be largely a cell-intrinsic defect due to failed IL-2-dependent signaling in maintaining the balance between the expansion and deletion of autoreactive T-lymphocyte clones (24, 25). However, the adoptive transfer of normal T cells prevented the abnormal activities seen in IL-2−/− or IL-2Rβ−/− mice (26,27), raising the possibility of a regulatory rather than an intrinsic T-cell defect. A subpopulation of CD4+ T cells, which expressed a high level of surface CD25, was identified to maintain self-tolerance by downregulating autoimmunity in adoptive transfer settings (28). These cells, which contained CD4+CD25+Foxp3+ T cells, were shown to be strikingly diminished in IL-2/IL-2R-deficient mice (27). Co-transfer of these cells with IL-2/IL-2R-deficient T cells and/or their reconstitution by providing a source of IL-2 prevented severe autoimmunity associated with IL-2/IL-2R deficiency (26, 27, 29, 30). Indeed, IL-2Rβ−/− mice lived a normal autoimmune-free life with a largely normal peripheral immune compartment after receiving a singular transfer of Treg cells during neonatal life, directly demonstrating that a defect in Treg cells was primarily responsible for the autoimmunity associated with IL-2/IL-2R deficiency (27).
Role of IL-2R signaling in Treg cells
IL-2/IL-2R signaling is essential for natural Treg cell development
Treg cells are marked by high expression of the transcriptional regulator Foxp3, which is essential for Treg lineage commitment and its suppressive program (31-33). Most Treg cells (~90%) in the mouse are thymus derived and are referred to as natural Treg (nTreg) cells. These cells suppress autoreactive T cells that escape thymic negative selection. IL-2 provides an essential role for nTreg development. The first evidence supporting this notion was that thymus-targeted transgenic expression of wildtype (WT) IL-2Rβ in IL-2Rβ−/− mice resulted in development of a normal fraction of functional Treg cells in the thymus and peripheral immune tissues that prevented autoimmunity (27, 34). As the peripheral T cells from these mice remained non-responsive to IL-2, this finding suggests that suppression of autoimmunity is independent of IL-2-mediated apoptosis of autoreactive T cells. More direct support for this notion comes from suppression of autoimmunity in IL-2Rβ−/− mice by WT Treg cells, where all other cells in the recipient are genetically non-responsive to IL-2 and IL-15. Such a finding also directly demonstrates that effective suppression is not simply due to depriving IL-2 growth-promoting signals to autoreactive T cells by Treg cell consumption of this cytokine. Furthermore, anti-IL-2 treatment of WT mice reduced the number of thymic Treg cells (35).
IL-2- and IL-2R-deficient mice are not devoid of Foxp3+ T cells. Rather, the thymus contains approximately a twofold and peripheral immune tissues a 10-fold reduction in the fraction of Foxp3+ T cells relative to conventional CD4+ T cells (36, 37). This reduction was even greater when WT and IL-2Rα−/− thymocytes were in competition in vivo (36). Moreover, along with a decreased fraction of Treg cells, the expression of Foxp3 is also lower in the absence of IL-2R signaling (36, 37). These Foxp3low cells might be considered as immature non-functional Treg cells. In human, the Foxp3low phenotype is associated with activated conventional T cells rather than suppressive Treg cells (38, 39). Moreover, mice engineered to express only Foxp3low T cells were not suppressive, leading to severe lethal autoimmunity (40).
More recent studies are consistent with a two step model for IL-2-dependent Treg development (41, 42). First, TCR signaling through encounter with self-antigens results in induction of CD25 predominately by maturing CD4+ single positive thymocytes, leading to expression of the high affinity IL-2R. Developing Treg cells sensing autocrine or paracrine IL-2, the latter from neighboring thymocytes activated by TCR-dependent selection events, receive an instructive IL-2R-dependent signal. This activity induces a high level of Foxp3 and upregulates CD25, resulting in production of mature functional Treg cells competent to migrate into the periphery. Although IL-2R signaling has been associated with activation of multiple signaling pathways, IL-2-dependent activation of STAT5 is sufficient for full thymic Treg maturation (43). The precise molecular events controlled by IL-2 during Treg maturation remain unknown.
In this two step model, in the absence of IL-2R signaling, self-antigen selection leads to production of immature Treg cells characterized by the CD4+Foxp3lowCD25low phenotype. Notably, for the few T cells that develop in γc−/− mice, there is a complete absence of CD4+Foxp3+ cells in the thymus and periphery (36). Thus, another γc-dependent cytokine contributes to the expression of Foxp3. This other cytokine is IL-7, because mice double deficient in IL-7Rα and IL-2Rβ or IL-2 and IL-7 recapitulate the phenotype associated with γc-deficient mice, i.e. no Foxp3+ thymocytes (44, 45). The basis by which IL-7 regulates expression of Foxp3 for production of immature Foxp3+ T cells is unknown. However, IL-2 and IL-7 act on distinct development steps during thymic development (46). In addition, IL-2R signaling is sufficient and dominantly promotes full Treg maturation in the absence of IL-7R signaling (44).
IL-2/IL-2R signaling for induced Treg cell development and lineage plasticity Conventional T cells are also induced by environmental signals in the periphery, predominately transforming growth factor β (TGFβ), retinoic acid, and IL-2, to express Foxp3 and adopt suppressive function (47, 48). These cells are called induced Treg (iTreg) cells and are readily found in mucosal sites (48) and chronically inflamed tissues (49). Within the gut, iTreg cells have been implicated in suppressing immune responses to commensal microorganisms and food antigens (50).
Several lines of evidence support an important role for IL-2 in the production of iTreg cells in vitro. First, the generation of iTreg cells from naive T cells after the TCR stimulation readily occurs by co-culture with TGFβ and IL-2 (51-53). Importantly, neutralization of IL-2 resulted in a failure to induce Foxp3 transcription and suppressive activity, even in the presence of TGFβ (52). Second, IL-2 substitutes for the CD28 costimulatory induction of Foxp3. Lastly, iTreg cells do not develop when using conventional T cells that do not produce IL-2 or fail to exhibit normal IL-2R signaling (52, 54).
Much recent evidence indicates that the development of activated T cells into polarized functional states, e.g. Th1, Th2, Th17, and iTreg, is not fixed (55). This is particularly evident for iTreg cells, which have the potential to re-differentiate into Th17 effector cells (56). Cytokine signals not only have an important impact in driving Th and iTreg development but also in conversion between alternative cell fates. Thus, the balance between IL-2 and inflammatory signals appears to influence not only the Th17 vs. the iTreg cell fates but also the extent by which iTregs convert to Th17 cells.
This plasticity is because the expression of master transcriptional regulators driving the differentiation of these different cell types is not completely fixed but rather more flexible. For example, when T cells are activated through the TCR in the presence of TGFβ, both RORγt and Foxp3 are transcribed and interact with each other. However, in the presence of IL-2 or retinoic acid, Foxp3 transcription dominates, and this excess Foxp3 represses transcription of RORγt, promoting iTreg development (57). In contrast, under inflammatory conditions, with increased IL-6 but low or absent IL-2, the transcription of the RORγt is favored, which in turn represses Foxp3 transcription, leading to an increase of RORγt to drive Th17 development (58).
This sensing of cytokine cues normally results in a balance between these two populations that favorably maintain immune tolerance. Moreover, the re-differentiation of iTreg cells to Th17 cells likely reflects a response to increased inflammatory signaling and perhaps lower Foxp3 inductive signals, e.g. TGFβ and IL-2, that alter and repress what was dominant for Foxp3 transcription to a setting favoring RORγt, resulting in ex-iTreg cells to readily produce IL-17. Such re-differentiation represents a favorable immune outcome to enhance immune responses to a local infection. However, Th17 cells have been broadly linked to the pathogenesis of multiple autoimmune disorders. Thus, disruption of the balance between Th17 and iTreg development represents a risk for autoimmunity. Although both iTreg and Th17 cells arise from the naive CD4+ T cells, seemingly to share an intermediate stage in their development (59), the amount of IL-2 in the microenvironment may represent one of the key factors to determine which lineage these T cells adopt. However, given the abundance of retinoic acid in gut mucosa and its ability to support iTreg development in conjunction with TGFβ (48), an important unanswered question is whether IL-2 delivers a dominant signaling for iTreg development and stability in vivo.
IL-2R signaling for peripheral Treg cells
A feature of IL-2-, IL-2Rα-, and IL-2Rβ-deficient mice is their strikingly low proportion of CD4+Foxp3+ cells (36, 37). However, at the peak of the lymphoproliferative disease associated with such mice, there was only a modest reduction in absolute Treg numbers. Two factors likely contribute to this result. First, the periphery of IL-2/IL-2R-deficient mice is populated by immature CD4+CD25lowFoxp3low T cells that readily seed the periphery. Thus, their failed maturation renders them poorly functional due to lower Foxp3, which is essential to induce and maintain the Treg suppressive program. Second, IL-2 directly promotes the transcription of Foxp3 through activation of STAT5, which binds to regulatory sites within the Foxp3 gene (43, 60). The near lack of CD25 expression by these immature Treg cells renders them unable to receive normal IL-2 signals that not only interfere with Foxp3 expression but also IL-2-dependent growth and survival signals that directly maintain their peripheral numbers. Lastly, in direct competitive conditions, where autoimmunity does not occur and does not potentially confound interpretation of the results, peripheral Treg cells from WT mice greatly outnumbered those CD4+Foxp3+ T cells with impaired IL-2/IL-2R signaling (36, 37). In addition, after autoimmunity was corrected in IL-2Rβ-deficient mice by the adoptive transfer of WT Treg cells, the fraction of host Foxp3low cells remained as only a small fraction of the total Treg pool (27). Thus, even in this latter situation, where the host continually produces immature Treg cells, these cells remain at a substantial competitive disadvantage with WT Treg cells, consistent with poor homeostasis and functional activity.
However, poor Treg homeostasis is not the sole or perhaps even the major contributing reason for autoimmunity associated with impaired IL-2R. As discussed above, Treg cells with substantially impaired IL-2R signaling readily suppress autoimmunity and are found at normal numbers in non-competitive setting, raising the possibility of an effective but non-dominant IL-2-independent mechanism to maintain Treg numbers (54). Furthermore, when Bim-mediated apoptosis was abrogated in IL-2−/− Treg cells, normal numbers of Treg cells were restored (61), confirming a role for IL-2R signaling in Treg homeostasis. However, such mice still exhibited severe lethal autoimmunity. This work demonstrated that IL-2-dependent signaling for survival can be uncoupled from their functional activity. Importantly, IL-2 appears to play a central role for the suppressive function for Treg cells. Whether such a role represents a link between IL-2R signaling and maintenance of Foxp3 expression is not yet definitively established. However, this likely represents one explanation for this effect. Indeed, autoimmunity associated with IL-2Rβ-deficient mice did not occur after transgenic expression of Foxp3 under the control of a bacterial artificial chromosome (62). One caveat with this result was high Foxp3 expression was found in the most T cells in the periphery, and this effect may have also served to lower the prevalence of autoreactive T cells. In addition, it also remains to be determined whether IL-2R signaling controls Treg function primarily as a consequence of IL-2-dependent thymic Treg maturation or as a consequence of peripheral Treg cells that continually sense IL-2. These possibilities are not mutually exclusive.
Genome-wide transcriptional analyses of peripheral Foxp3+ T cells from WT and IL-2−/− mice or from mice with impaired but not absent IL-2R signaling, which effectively supports thymic Treg maturation, provide additional molecular support for IL-2-dependent signaling promoting peripheral Treg homeostasis. Both studies revealed enrichment in differentially expressed genes associated with cell cycle and survival, which were reduced without proper IL-2 signaling (36, 54). Furthermore, overnight administration of IL-2 to the IL-2−/− Treg cells in vivo increased the expression of most of these genes.
Direct evidence indicating a role of IL-2R signaling for peripheral Treg homeostasis and survival of normal WT mice was the capacity of anti-IL-2 to reduce the number of peripheral Treg cells (35, 63). This reduction was noted for both neonatal and adult peripheral Treg cells. This inhibition was particularly striking during the neonatal period, when there is substantial peripheral expansion of Treg cells due to their delayed development (64) to rapidly set a normal proportion of Treg to T conventional cells in the periphery to establish immune tolerance. Treatment of adult non-obese diabetic (NOD) mice, which spontaneously develop autoimmune type 1 diabetes (T1D), with antagonist anti-IL-2 lowered Treg numbers, compromised Treg immune-regulation, and accelerated T1D. Thus, impaired peripheral Treg homeostasis is associated with a risk for developing autoimmune disease.
Polymorphisms linked to Il2, Il2ra, and Il2rb are susceptibility factors for autoimmune disease in mouse and human (65). For the NOD mouse, the Idd3 region on mouse chromosome 3, which contains the genes for IL-2 and IL-21, represents a genetic interval contributing to spontaneous T1D associated in this mouse strain. Lower production of IL-2 is a consequence of this allele and associates with a decreased proportion of Treg cells and increased susceptibility to T1D (66). Thus, for this genetic polymorphism, low IL-2 production and poor peripheral Treg homeostasis represent a genetic risk for T1D.
In human, single nucleotide polymorphisms (SNPs) mapping to Il2, Il2ra, and Il2rb have been associated with increased risk for T1D, Grave’s disease, multiple sclerosis, rheumatoid arthritis, and celiac disease (67-70). SNPs at Il2ra represent one of the stronger non-MHC-linked genetic risks for T1D (71). For these diseases, sometimes distinct SNPs mapping to an individual gene were identified as the genetic risk factors, raising the possibility that distinct mechanisms may be responsible for an IL-2/IL-2R contribution to autoimmunity. None of the SNPs were predicted to control variants within the coding portions of these genes to yield proteins with potentially varied functional activities. Thus, it is likely that these SNPs function to regulate gene transcription. One mechanism is due to SNPs located at an extended region outside of the minimal IL-2 promoter. Alternatively, other SNPs are predicted to be associated with epigenetic changes such as modifications of histones and/or DNA methylation (72). For Il2ra the SNPs associated with T1D map to the first intron and the 5′ region of this gene and are associated with lower transcription and lower expression of CD25. In this case, lower CD25 expression was noted on the surface of memory T cells (73). However, it remains unknown for all these genetic variants what accounts for the genetic risk and whether it is related to impaired Treg cells, enhanced Teff or T memory cells, or some other cell population that expresses IL-2R.
Role of IL-2R signaling and function by Treg and Teff cells
Common and unique activities
Given the well known and dominant role for IL-2 in promoting T-cell growth, survival, and effector cell differentiation in vitro and its contribution to promoting primary and memory immune responses in vivo, it seems somewhat paradoxical that IL-2 is also such a key player in immune suppression by promoting nTreg and iTreg development and homeostasis. The obvious question, therefore, concerns how the IL-2/IL-2R pathway controls such opposing aspects within the immune system. Table 1 compares several important activities attributed to IL-2R signaling for Treg and Teff cells. On the one hand, there is an obvious overlap by which IL-2 acts on Treg and Teff populations, and this is in line with these cells being antigen-activated T lymphocytes, where Treg cells are activated by constitutive self-antigens. Thus, IL-2R signaling promotes T-cell growth and survival, upregulates CD25 expression, and importantly contributes to functional differentiation. However, more careful consideration of these and other properties, as discussed below, indicates fundamental differences by which IL-2/IL-2R system behaves in Treg and Teff cells.
Table 1.
IL-2/IL-2R system in Treg and CD4+ Teff cells
| Property | Treg | Teff |
|---|---|---|
| IL-2 production | No | Yes |
| Repression of IL-2 | Foxp3/Runx/NF-AT | T-bet, Blimp-1 |
| High affinity IL-2R expression | High, constitutive | Low, transient |
| Upregulation of IL-2Rα | Yes | Yes |
| IL-2R signaling | STAT5 | MAPK; PI3K/Akt; STAT5 |
| Activity during T cell development | Yes | No |
| Peripheral homeostasis in vivo | Yes | No |
| Growth | Yes | Yes |
| Survival | Yes | Yes |
| Regulation of function | Suppression | TH1, TH2 |
One unique activity for IL-2R signaling for nTreg cells is its role in promoting thymic nTreg maturation, as discussed above. IL-2-dependent Treg maturation depends only on STAT5 activation (43). This leads to upregulation of IL-2Rα and Foxp3, which function to induce and maintain the Treg lineage, but the essential molecular mechanisms and biological functions associated with Treg maturation remain poorly characterized. This developmental activity of IL-2 is analogous to that of IL-7 or IL-15, which are essential cytokines for the thymic T and NK and NKT cell development (74), but are also important mediators of peripheral homeostasis for naive and memory T cells rather than Treg cells (74). Some time ago, IL-2 was also thought to contribute to thymic development of conventional T lymphocytes, because developing pre-T cells are marked by very high expression of IL-2Rα (75). However, these pre-T cells do not express a functional IL-2R, as they do not express IL-2Rβ. In fact, expression of high affinity IL-2R by thymocytes is restricted to a few CD4+CD8low transitional intermediates, which are associated with a late stage of ‘double positive’ development, and Treg cells. This finding, the lack of IL-2Rα expression by all other thymocytes except pre-T cells and developing and mature Treg cells, and distinct thymic location of these pre-T and Treg cells ensures that limiting IL-2 available within the thymus is captured by developing Treg cells.
Expression of IL-2 and IL-2R
One striking difference between Treg and Teff cells partially related to IL-2R signaling is the capacity to secrete IL-2. TCR-dependent signaling results in IL-2 secretion by Teff cells whereas the inability to produce IL-2 represents one of the defining features of Treg cells. The inability of Treg cells to produce IL-2 is directly related to the repressive activity of their signature transcription factor Foxp3. This repression is due to the interaction of Foxp3/AML1/Runx1 and Foxp3/NF-AT that interact with each other and bind to IL-2 transcriptional regulatory sites (76, 77). Since IL-2-dependent STAT5 activation positively regulates Foxp3 expression, IL-2R signaling serves to reinforce this non-IL-2 producing phenotype of Treg cells. Interestingly, IL-2 transcription by Teff cells is very transient, which is in part due to repression of IL-2 transcription. However, in this case, repression of IL-2 is not due to Foxp3, which is not expressed by mouse Teff cells, but rather because TCR signaling induces T-bet (78) and IL-2-dependent activation of Blimp-1(79). Both transcriptional regulators directly bind to distinct regulatory regions within the IL-2 genes to terminate its transcription (80, 81). Thus, IL-2R signaling induces a classical positive inhibitory feedback loop unique to Teff cells to limit IL-2 production and hence IL-2-dependent responses. Moreover, Treg and Teff cells adopt distinctive strategies to repress IL-2.
Both Treg and Teff cells are characterized by expression of the high affinity IL-2R. The expression of IL-2Rα and IL-2Rβ depends upon TCR signaling, whereas expression of γc is constitutive. Signaling through IL-2R upregulates expression of IL-2Rα in Teff and Treg cells through STAT5 activation of IL-2Rα transcription (82), providing a positive feedback mechanism favoring continued IL-2-dependent activation. Even though this mechanism acts in parallel for Treg and Teff cells, CD25 expression in vivo for Treg cells is characterized by an apparent constitutive high level, whereas for Teff cells, it is very transient and typically low. One explanation to account for this difference is that Foxp3 acts as a direct positive activator of the IL-2Rα gene (36), and this mechanism will not be active in Teff cells. Additionally, ectodomain shedding of IL-2Rα on Teff cells in vivo readily occurs through proteolysis and represents another means to downregulate CD25 and IL-2R signals (83-85). Although it is unknown whether this mechanism targets Treg cells, the typically higher and prolonged expression of CD25 on Treg cells suggests these cells may be sequestered and protected from such degradation.
By combining mathematical modeling and direct experimental analysis, the relevance of the distinctive production of IL-2 and expression of the high affinity IL-2R by Treg cells and Teff cells has been recently considered (86). IL-2-induced upregulation of the high affinity IL-2R establishes a positive feedback loop for IL-2 signaling. This feedback not only promotes the proliferation and differentiation of Teff cells but also functions as an amplifier for IL-2 uptake by Treg cells. This IL-2/IL-2R loop enhances the capture and degradation of IL-2. Treg and Teff cells compete for IL-2 and restrict its range of action through efficient cellular uptake. Depending on the activation status and the spatial localization of the cells, IL-2 may be consumed exclusively by Treg or Teff cells or be shared between them.
We have recently proposed that such favored competition for IL-2 by Treg cells represents the basis of IL-2-dependent Treg homeostasis (87). Teff cells, or more likely T autoreactive cells, provide IL-2 to the Treg cells. Consumption of IL-2 is favored by the Treg cells due to their higher expression of IL-2Rα, promoting Treg homeostatic signals and suppressive function through activation of Foxp3. IL-2-activated Treg cells then effectively suppress the Teff or T autoreactive cells through one or more of the plethora of suppressive mechanisms at their disposal (88). This preferential consumption of IL-2 by Treg cells has also been implicated as one of the means by which Treg cells mediate suppression (89-91). Although depriving Teff cells of IL-2 only marginally affects T-cell clonal expansion in vivo, lowering IL-2 levels impairs terminal CD8+ Teff development (92) and memory programming (93) that will temper T-cell immunity and as such represents another way Treg cells might inhibit immune responses.
Epigenetic and transcriptional regulation contribute to distinct functional activity IL-2R signaling is importantly linked to functional differentiation of Treg and some Teff cells. IL-2-dependent STAT5 binding to conserved non-coding sequences (CNS) in the Foxp3 gene not only serves to reinforce repression of IL-2 but also the activation of the Treg suppressive program (94). Optimal interferon γ (IFNγ) production by Th1 cells depends on IL-2. The binding of STAT5 to a CNS associated with the IFNγ gene promotes the binding of the signature Th1 transcription factor T-bet to the IFNγ promoter (95). IL-2-dependent activation of STAT5 is essential for Th2 development in vitro in part by direct activation of Th2 targeted genes, such as IL-4, and by lowering the threshold for the binding of Th2 signature transcription factor Gata3 to Th2 genes, necessary for their activation (96). Thus, the transcription factor content and the epigenetic landscape of the activated T cells allow a common signaling intermediate induced by IL-2, i.e. STAT5, to control important and distinct cell fate outcomes for CD4+ T cells.
Genome-wide expression analysis for IL-2-dependent genes by Treg cells ex vivo and anti-CD3 activated CD4+ Teff cells identified 285 and 421 genes, respectively, that depend on IL-2, and <16% of these overlapped between these two cell populations (54). Gene enrichment analysis showed that immune system related genes were the largest functional group for both Treg and CD4+ Teff cells, but the large majority of these genes in this enrichment did not overlap. Therefore, an important aspect of IL-2R signaling in Treg and Teff cells is to promote processes known to be important for immune system function, but this common signaling resulted in regulation of distinct targets and functions. This notion is further illustrated by other major enrichment groups that were unique to Treg (cell cycle, cell death, and cytoskeleton) and CD4+ Teff (amine metabolism and differentiation) cells. The finding that IL-2 did not enrich genes related to T-cell growth and death for Teff cells likely represents the timing in which RNA was collected from the Teff cells, as initial TCR and costimulatory activation were sufficient to drive substantial proliferation. Overall, Treg and Teff cells integrate IL-2R signaling in the context of distinct transcription factors and epigenetic modifications in part as a consequence of varied environmental signals to activate a largely non-overlapping set of genes.
Distinctive IL-2R signaling by Treg cells
An important and more direct basis by which IL-2R signaling distinctively regulates Treg and Teff cells is that the IL-2R proximal pathways, MAPK, PI3K/Akt, and STAT5, are active in Teff cells, whereas STAT5 is only readily activated in Treg cells (54, 97). The poor activity of the PI3K/Akt pathway is accounted for by high expression of phosphatase and tensin homolog (PTEN) by Treg cells (98). Moreover, the level of STAT5 activation in Treg cells may be somewhat attenuated due to heightened expression of suppressor of cytokine signaling 1(SOCS1) (99, 100). Expression of SOCS1 depends on IL-2R signaling and miR155 (101). This phosphatase inhibits IL-2R signaling by interrupting the phosphorylation of STAT5 and thus provides another positive inhibitory loop to downregulate signal transduction (102, 103).
Although IL-2 is a growth factor for Treg and Teff cells, nTreg cells are much more refractory to IL-2 for T-cell growth. Indeed and in marked contrast to Teff cells, nTreg cells do not directly proliferate to exogenous IL-2 in vitro, even though they express the high affinity IL-2R. IL-2-dependent T-cell growth of nTreg cells in vitro depends on stimulation through the TCR, CD28, and the IL-2R, the latter usually with a high concentration of IL-2. CD28 signaling in Treg cells activates PI3K/Akt (104) that likely cooperates with IL-2-induced STAT5 for Treg growth. Optimal IL-2-dependent growth by Teff cells is dependent upon full integration of the MAPK, PI3K, and STAT5 pathways (8). Analogous to Treg cells, poor T-cell proliferation is associated with activation of predominately only the IL-2R-dependent STAT5 pathway. Moreover, restoring activation of PI3K in Treg cells promotes IL-2-driven T-cell growth (98). Thus, the distinctive and lower IL-2R signaling by Treg cells helps explain their more stringent requirements for T-cell growth. This lower IL-2-dependent Treg growth might represent a favorable property in vivo by rendering a consistent slower proliferative rate, characteristic of homeostatic proliferation, to maintain a proper proportion of Treg and conventional CD4+ T cells.
A low IL-2R signaling threshold supports Treg maturation and signal transduction
Mutant IL-2Rs readily support Treg cells
The effective utilization of only one component, STAT5, of the proximal signaling pathways associated with the IL-2R and the increased expression of phosphatases by Treg cells to abrogate such signaling indicate that tempered IL-2R signaling, primarily through activation of STAT5, is required for IL-2-dependent contribution to Treg development, homeostasis, and function. Recent work from our laboratory extends and better defines this notion by directly showing that many of the key activities associated with Treg cells require minimal and relatively transient IL-2R signal transduction (54). Our experimental design was to produce transgenic mice where IL-2Rβ chains are expressed in all T-lineage cells that harbor mutations in three important tyrosine residues required for activation of the MAPK, PI3K/Akt, and STAT5 pathways. These mutations, all tyrosine to phenylalanine substitutions, targeted one (Y341), two (Y395, Y498), or three (Y341, Y395, Y498) tyrosine residues. Correspondingly, the Y341 mutation prevents association to the adapter Shc and inhibits the MAPK and PI3K/Akt pathways, Y395 and Y498 mutations are important docking sites for STAT5 and inhibit the STAT5 pathway, and the Y341, Y395, and Y498 mutant was expected to abrogate these pathways (Fig. 1).
Fig. 1. Transgenic IL-2Rβ mutants and their effect on autoimmunity and Treg development after expression in IL-2Rβ−/− mice.

Each of these transgenic lines was bred to IL-2Rβ-deficient mice to yield T cells that only expressed the mutant IL-2Rβ molecules. There are two important considerations for this particular experimental design. First, this approach allows examining the outcome of these mutations on both Treg and Teff cells. Second, this design takes advantage of the fact that mouse IL-2 does not readily bind to IL-2Rβ and γc. This property coupled to the known expression of IL-2Rα, which is restricted essentially to pre-T cells and Treg cells in a normal naive mouse, predicts that constitutive expression of the mutant or even WT IL-2Rβ will not cause generalized aberrant IL-2R signaling in non-relevant T cells. Indeed, control IL-2Rβ−/− mice that expressed transgenic WT IL-2Rβ in all T-lineage cells closely paralleled normal C57BL/6 mice in T-cell composition as well as their activation status and functions in the thymus and peripheral immune tissues (54). Another consideration is that IL-15 through trans-presentation to IL-2Rβ and γc by IL-15Rα/IL-15 on another cell might alter the T cells in these transgenic models. However, we did not find any abnormal accumulation of CD8+ T cells in mice expressing the WT transgene, where IL-15 is known to direct homeostatic signaling. Furthermore, unpublished genome-wide expression profiles of activated CD4+ and CD8+ T cells from normal control and IL-2Rβ-deficient mice expressing transgenic WT IL-2Rβ were strikingly comparable. These data further indicate that constitutive transgenic IL-2Rβ expression in all T-lineage cells does not impose intrinsic alterations in these cells and provides additional support for the validity of this approach for tissue-specific targeting of IL-2Rβ mutations. The only alteration in the T-cell compartment observed thus far not related to activated T cells or Treg cells is a lower number of peripheral CD8+ T cells bearing mutant IL-2Rβ, which is anticipated based on the role for IL-15 in CD8+ T-cell homeostasis.
For these mutant IL-2Rβs, the striking finding was a normal compartment of functional mature Treg cells. Each transgenic IL-2Rβ−/− line lacked lethal autoimmune disease associated with the non-transgenic parental mice, whereas the Teff compartment as assessed in vitro was severely impaired (Fig. 1). We were quite surprised that the triple Y341, Y395, Y498 mutation supported Treg maturation and function, as IL-2R signaling was expected to be abrogated by this severe mutation. However, direct analysis of this mutation showed that it still supported weak transient STAT5 activation in both Treg and Teff cells that was similar to that found for the Y395, Y498 mutation, which interferes with STAT5 association to IL-2Rβ. In this latter mutation, weak STAT5 activation was anticipated as this has already noted by others as a consequence of the recruitment of the Shc adapter (105).
Based on the apparent IL-2Rβ-tyrosine independent activation of STAT5, we considered more recently whether this represents a novel mode of STAT5 activation, potentially independent of IL-2Rβ, i.e. dependent only on γc, or independent of the H- and A-regions of the IL-2Rβ cytoplasmic domain, which contain these three tyrosine residues. Therefore, we prepared additional transgenic mice where the cytoplasmic H- or both the H- and A-regions were deleted and expressed in T cells of IL-2Rβ−/− mice (Fig. 1). Treg maturation and function were also largely normal in mice with the H-region deletion, which also deletes Y395 and Y498. However, mice with the deletion of both the H- and A-regions behaved as if this IL-2Rβ was completely non-functional, as they exhibited lethal autoimmunity comparable to non-transgenic IL-2Rβ-deficient mice. Moreover, the immature Treg cells from the transgenic A- and H-region deleted IL-2Rβ-deficient mice failed to activate STAT5 in a manner identical to immature Treg cells from IL-2Rβ−/− mice (Fig. 2). Thus, these experiments map the weak STAT5 activation associated with the triple tyrosine mutations to the A-region. What accounts for this activation, however, is not known as both direct recruitment and interaction of Shc and STAT5 are not expected to occur.
Fig. 2. Treg cells that express IL-2Rβ lacking both the A- and H-regions (ΔAH) do not activate STAT5.

These results show that even without optimal IL-2R signaling, thymic Treg development and peripheral homeostasis effectively occur. Weak IL-2R signaling is linked to upregulation of Foxp3, which likely represents one of the determining reasons for effective Treg maturation and suppression of autoimmunity. However, those mice with the lowest STAT5 activation with time eventually exhibit an activated T-cell phenotype and inflammatory infiltrates, most commonly in the lungs and salivary glands but also sometimes in the gut, pancreas, and liver. Thus, impaired IL-2R signaling represents a risk for developing autoimmune disease. This notion is reminiscent of NOD mice, where lower IL-2 production has been associated with impaired Treg cells and one of the genetic risk factors for T1D in this strain (66). Based on these mouse studies, we believe that a careful assessment of Treg cell impairment is warranted as the main genetic risk posed by SNPs associated with the human Il2, Il2rα, and Il2rb genes.
Minimal expression of IL-2R supports Treg cells
Another well-characterized model developed in our laboratory is expression of transgenic WT IL-2Rβ predominately in the thymus of the IL-2Rβ−/− (IL-2RβWT/Thymus) mice by using the proximal lck promoter to control transgenic IL-2Rβ expression. This model was useful to first show that IL-2R signaling plays an important role in the thymus, as both the thymic and peripheral Treg compartments were normalized in these largely autoimmune-free mice (27, 34). The normal peripheral T-cell compartment in these mice was interesting, because the Treg and Teff cells expressed barely detectable levels of IL-2Rβ such that their conventional activated T cells were virtually non-responsive to IL-2.
These findings led us to propose that IL-2R signaling within the thymus was the only important IL-2-dependent step for Treg cells. This idea seems to contradict the notion that IL-2 is also required for peripheral Treg homeostasis. We considered that perhaps constant thymic Treg output by IL-2RβWT/Thymus compensated for poor IL-2R signaling by Treg cells in the periphery. However, after adult thymectomy, IL-2RβWT/Thymus mice retained a normal number and proportion of Treg cells in peripheral immune tissues and did not exhibit early symptoms of autoimmune disorders associated with IL-2Rβ−/− mice (37). Nevertheless, the peripheral Treg compartment was not entirely normal in IL-2RβWT/Thymus mice. First, BrdU incorporation studies showed that proliferation of peripheral IL-2RβWT/Thymus Treg cells was lower than WT Treg cells. Peripheral IL-2RβWT/Thymus Treg cells also showed increased survival and lower turnover, which represent one means by which these Treg cells compensate for impaired IL-2R expression to maintain peripheral Treg numbers (37). Second, in mixed chimeras using bone marrow from WT and IL-2RβWT/Thymus mice, near equivalent competition between WT and IL-2RβWT/Thymus Treg cells was noted in the thymus, but WT Treg cells greatly outnumbered IL-2RβWT/Thymus Treg cells in the spleen (37).
More recent work indicates that the very low expression of IL-2Rβ by IL-2RβWT/Thymus peripheral Treg cells supports weak and transient STAT5 that was essentially comparable to that found for Treg cells bearing the triple Y341, Y395, Y498 IL-2Rβ mutation (54). Thus, the behavior of peripheral Treg cells from IL-2RβWT/Thymus mice is entirely in line with the notion that low IL-2Rβ signaling supports peripheral Treg cells. Lower IL-2R signaling by peripheral Treg cells provides an explanation for the altered proliferative activity and poor competitive behavior with WT Treg cells. Furthermore, genome-wide transcriptional profiling between peripheral control WT and IL-2RβWT/Thymus Treg cells showed differential expression of genes related to immune system, cell cycle, cell death, and the cytoskeleton (54). This pattern of gene expression is consistent with an important role for IL-2 in peripheral Treg homeostasis.
This same gene expression analysis also revealed that a substantial fraction (approximately 20%) of the Treg gene signature (106) was IL-2-dependent. This finding places relatively high IL-2R-signaling in a pivotal role to fully specifying the Treg cell phenotype. However, other key targets such as Foxp3, TGFβ, and CTLA4 were similarly expressed by WT and IL-2RβWT/Thymus peripheral Treg cells. Thus, the low IL-2R signaling, as exemplified by mutant IL-2Rβs or low expression of WT IL-2Rβ, as in IL-2RβWT/Thymus Treg cells, readily supports Foxp3 expression, and this activity induces in the thymus and maintains in the periphery many of the fundamental activities of these suppressive T cells.
IL-2 as a selective agent for Treg immunotherapy
Although IL-2 targets both Treg and Teff cells, two important features of the IL-2/IL-2R system provide a mechanistic underpinning for using IL-2 as an agent to selectively promote Treg cells in immunotherapy to promote T-cell tolerance. First, the STAT5 pathway is predominantly utilized by Treg cells activated through the IL-2R. Therefore, inhibition of the MAPK and PI3K pathways are predicted to preferentially affect Teff cells. Second, weak transient IL-2R-dependent STAT5 activation effectively induces many critical IL-2-dependent activities in Treg cells. Thus, controlling the available levels of IL-2 has the potential to target therapy to Treg versus Teff cells.
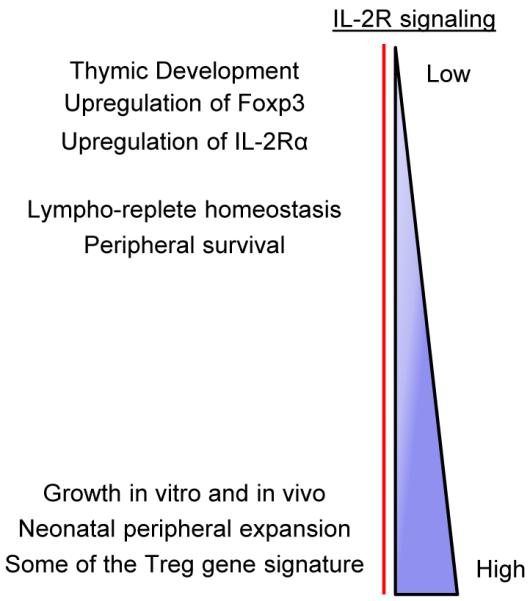
When extrapolating data from our mouse models concerning IL-2R signaling in Treg cells (Fig. 3), induction of Foxp3 is associated with the lowest level of signaling and is associated with substantial benefit, as it readily supports Treg maturation, homeostasis, and suppression of autoimmunity. Somewhat more extensive signaling is required for upregulation of IL-2Rα, and even more extensive signaling is associated with a host of other targets linked in part to the Treg signature and to Treg growth and death. IL-2-dependent Treg growth appears to require the most extensive IL-2R signaling, probably to accommodate the reduced activity of the PI3K/Akt pathway in Treg cells. Importantly, IL-2-dependent activities in Teff cells have not yet been shown to occur with the type of low transient IL-2R signaling that effectively functions for Treg cells. T-cell growth, optimal induction of IL-2-dependent effector molecules, such as perforin and granzyme B, and terminal differentiation of CD8+ Teff cells all depend on high IL-2R signaling (54, 92, 93). Lower IL-2R signaling may promote CD8+ memory T-cell development and/or programming. However, whether this potentially lower signaling is analogous to the low signaling for full expression of Foxp3 remains to be determined.
Fig.3. Varied levels of IL-2R signaling strength support distinct functional activities of nTreg cells.
Thymus and lymph node cells from mice of the indicated IL-2Rβ genotype (2Rβ) were rested for 30 min in medium and then stimulated with IL-2 (10 ng/ml) for the indicated time. Activation of tyrosine-phosphorylated STAT5 (pSTAT5) was determined by flow cytometry after gating on CD4+Foxp3+ T cells. Data were normalized to the maximal response by control 2Rβ+/− Treg cells. Data represent 2-3 mice/group.
This signaling threshold model predicts that applying the proper relatively low dose of IL-2 in vivo should preferentially augment Treg activity and favor immune tolerance. Several studies support this notion. In the first such study, the agonistic activity of an IL-2/anti-IL-2 complex was exploited as a therapeutic agent for T1D in NOD mice (107). This complex displays IL-2 for binding by IL-2Rα, IL-2Rβ, and γc in the context of the high affinity IL-2R (108) and acts to greatly extend the effective half-life of IL-2 from approximate 30 min for free IL-2 to 48 to 72 h for the IL-2/anti-IL-2 complex (109). Importantly, a low dose of the complex caused increased expression of IL-2Rα and pro-survival Bcl-2 by Treg cells and prevented the onset of T1D. However, a high dose of the same complex accelerated the progression to T1D (107). This latter effect is likely due to enhancing autoreactive Teff cells that express the high affinity IL-2R. Pre-treating mice with the same IL-2/anti-IL-2 complex, which transiently increased Treg numbers, also prevented the induction of experimental autoimmune encephalomyelitis (EAE) and promoted long-lasting tolerance to tissue allografts (110). More recent studies also showed that treating mice with new onset diabetes with conventional IL-2 reversed T1D in NOD mice (111).
Treg cells are relatively resistant to rapamycin due in part to the minimal activation of PI3K/Akt and downstream mTORC1, the target of rapamycin. This property has been exploited to suppress the outgrowth of Teff cells in cultures aimed to expand Treg cells. Furthermore, agonist IL-2/anti-IL-2 complexes in conjunction with rapamycin ameliorated the symptoms associated with ongoing EAE (110). These preclinical studies provide initial validation that under the proper conditions, IL-2 mono-therapy offers the potential to selectively augment Treg function. Using IL-2 in conjunction with drugs that block mTORC1 provides a parallel means promoting Treg cells while inhibiting Teff responses. Either approach represents modalities to treat autoimmune diseases or to elicit transplantation tolerance. A particular advantageous feature of these approaches is that they represent a direct drug-based biotherapy. This therapy does not require the personalized approach inherent with adoptive Treg cell therapy and avoids the current technical and regulatory drawbacks associated with the production of sufficient cells for this type of immunotherapy.
Further definition, therefore, of the basis by which IL-2 uniquely controls the Treg and the Teff arms of the immune system should help to further refine the potential for selectively targeting the IL-2R to enhance tolerance versus immunity. In particular, better understanding of these differences in the human immune system will likely be required to develop effective immunotherapy that targets IL-2/IL-2R or to identify relevant biomarkers to predict the IL-2/IL-2R genetic risk for developing several common autoimmune diseases.
Acknowledgments
Our work is supported by the NIH (R01AI055815, R01CA045957, P01CA109094). The authors declare no conflicts of interest.
References
- 1.Taniguchi T, et al. Structure and expression of a cloned cDNA for human interleukin-2. Nature. 1983;302:305–310. doi: 10.1038/302305a0. [DOI] [PubMed] [Google Scholar]
- 2.Wang HM, Smith KA. The interleukin 2 receptor. Functional consequences of its bimolecular structure. J Exp Med. 1987;166:1055–1069. doi: 10.1084/jem.166.4.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson K, et al. Soluble IL-2 receptor β and γ subunits: ligand binding and cooperativity. Eur Cytokine Netw. 1994;5:23–34. [PubMed] [Google Scholar]
- 4.Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26:453–479. doi: 10.1146/annurev.immunol.26.021607.090357. [DOI] [PubMed] [Google Scholar]
- 5.Nelson BH, Lord JD, Greenberg PD. Cytoplasmic domains of the interleukin-2 receptor β and γ chains mediate the signal for T-cell proliferation. Nature. 1994;369:333–336. doi: 10.1038/369333a0. [DOI] [PubMed] [Google Scholar]
- 6.Russell SM, et al. Interaction of IL-2R β and γc chains with Jak1 and Jak3: implications for XSCID and XCID. Science. 1994;266:1042–1045. doi: 10.1126/science.7973658. [DOI] [PubMed] [Google Scholar]
- 7.Nelson BH, Willerford DM. Biology of the interleukin-2 receptor. Adv Immunol. 1998;70:1–81. doi: 10.1016/s0065-2776(08)60386-7. [DOI] [PubMed] [Google Scholar]
- 8.Gaffen SL. Signaling domains of the interleukin 2 receptor. Cytokine. 2001;14:63–77. doi: 10.1006/cyto.2001.0862. [DOI] [PubMed] [Google Scholar]
- 9.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaudhry A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. 2009;326:986–991. doi: 10.1126/science.1172702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng Y, et al. Regulatory T cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. 2009;458:351–356. doi: 10.1038/nature07674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim BS, et al. Conversion of Th2 memory cells into Foxp3+ regulatory T cells suppressing Th2-mediated allergic asthma. Proc Natl Acad Sci USA. 2010;107:8742–8747. doi: 10.1073/pnas.0911756107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schorle H, Holtschke T, Hunig T, Schimpl A, Horak I. Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature. 1991;352:621–624. doi: 10.1038/352621a0. [DOI] [PubMed] [Google Scholar]
- 14.Sadlack B, et al. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur J Immunol. 1995;25:3053–3059. doi: 10.1002/eji.1830251111. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki H, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor β. Science. 1995;268:1472–1476. doi: 10.1126/science.7770771. [DOI] [PubMed] [Google Scholar]
- 16.Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor α chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3:521–530. doi: 10.1016/1074-7613(95)90180-9. [DOI] [PubMed] [Google Scholar]
- 17.Lodolce JP, et al. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. 1998;9:669–676. doi: 10.1016/s1074-7613(00)80664-0. [DOI] [PubMed] [Google Scholar]
- 18.Kennedy MK, et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med. 2000;191:771–780. doi: 10.1084/jem.191.5.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Freeden-Jeffry U, Vieira P, Lucian LA, McNeil T, Burdach SE, Murray R. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J Exp Med. 1995;181:1519–1526. doi: 10.1084/jem.181.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peschon JJ, et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J Exp Med. 1994;180:1955–1960. doi: 10.1084/jem.180.5.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao X, et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor γ chain. Immunity. 1995;2:223–238. doi: 10.1016/1074-7613(95)90047-0. [DOI] [PubMed] [Google Scholar]
- 22.DiSanto JP, Muller W, Guy-Grand D, Fischer A, Rajewsky K. Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor γ chain. Proc Natl Acad Sci USA. 1995;92:377–381. doi: 10.1073/pnas.92.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by γc family cytokines. Nat Rev Immunol. 2009;9:480–490. doi: 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lenardo MJ. Interleukin-2 programs mouse αβ T lymphocytes for apoptosis. Nature. 1991;353:858–861. doi: 10.1038/353858a0. [DOI] [PubMed] [Google Scholar]
- 25.Critchfield JM, et al. T cell deletion in high antigen dose therapy of autoimmune encephalomyelitis. Science. 1994;263:1139–1143. doi: 10.1126/science.7509084. [DOI] [PubMed] [Google Scholar]
- 26.Wolf M, Schimpl A, Hunig T. Control of T cell hyperactivation in IL-2-deficient mice by CD4+CD25− and CD4+CD25− T cells: evidence for two distinct regulatory mechanisms. Eur J Immunol. 2001;31:1637–1645. doi: 10.1002/1521-4141(200106)31:6<1637::aid-immu1637>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 27.Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rβ-deficient mice. Implications for the nonredundant function of IL-2. Immunity. 2002;17:167–178. doi: 10.1016/s1074-7613(02)00367-9. [DOI] [PubMed] [Google Scholar]
- 28.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 29.Almeida AR, Legrand N, Papiernik M, Freitas AA. Homeostasis of peripheral CD4+ T cells: IL-2Rα and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J Immunol. 2002;169:4850–4860. doi: 10.4049/jimmunol.169.9.4850. [DOI] [PubMed] [Google Scholar]
- 30.Furtado GC, Curotto de Lafaille MA, Kutchukhidze N, Lafaille JJ. Interleukin 2 signaling is required for CD4+ regulatory T cell function. J Exp Med. 2002;196:851–857. doi: 10.1084/jem.20020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 32.Kim JM, Rudensky A. The role of the transcription factor Foxp3 in the development of regulatory T cells. Immunol Rev. 2006;212:86–98. doi: 10.1111/j.0105-2896.2006.00426.x. [DOI] [PubMed] [Google Scholar]
- 33.Josefowicz SZ, Rudensky A. Control of regulatory T cell lineage commitment and maintenance. Immunity. 2009;30:616–625. doi: 10.1016/j.immuni.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malek TR, Porter BO, Codias EK, Scibelli P, Yu A. Normal lymphoid homeostasis and lack of lethal autoimmunity in mice containing mature T cells with severely impaired IL-2 receptors. J Immunol. 2000;164:2905–2914. doi: 10.4049/jimmunol.164.6.2905. [DOI] [PubMed] [Google Scholar]
- 35.Bayer AL, Yu A, Adeegbe D, Malek TR. Essential role for interleukin-2 for CD4+CD25+ T regulatory cell development during the neonatal period. J Exp Med. 2005;201:769–777. doi: 10.1084/jem.20041179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 37.Bayer AL, Yu A, Malek TR. Function of the IL-2R for thymic and peripheral CD4+CD25+ Foxp3+ T regulatory cells. J Immunol. 2007;178:4062–4071. doi: 10.4049/jimmunol.178.7.4062. [DOI] [PubMed] [Google Scholar]
- 38.Wang J, Ioan-Facsinay A, van der Voort EI, Huizinga TW, Toes RE. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur J Immunol. 2007;37:129–138. doi: 10.1002/eji.200636435. [DOI] [PubMed] [Google Scholar]
- 39.Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+ FOXP3 T cells by T cell receptor stimulation is transforming growth factor β dependent but does not confer a regulatory phenotype. Blood. 2007;110:2983–2990. doi: 10.1182/blood-2007-06-094656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–770. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- 41.Lio CW, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity. 2008;28:100–111. doi: 10.1016/j.immuni.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burchill MA, et al. Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity. 2008;28:112–121. doi: 10.1016/j.immuni.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor β dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178:280–290. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- 44.Bayer AL, Lee JY, de la Barrera A, Surh CD, Malek TR. A function for IL-7R for CD4+CD25+Foxp3+ T regulatory cells. J Immunol. 2008;181:225–234. doi: 10.4049/jimmunol.181.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vang KB, Yang J, Mahmud SA, Burchill MA, Vegoe AL, Farrar MA. IL-2, -7, and -15, but not thymic stromal lymphopoeitin, redundantly govern CD4+Foxp3+ regulatory T cell development. J Immunol. 2008;181:3285–3290. doi: 10.4049/jimmunol.181.5.3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu A, Malek TR. Selective availability of IL-2 is a major determinant controlling the production of CD4+CD25+Foxp3+ T regulatory cells. J Immunol. 2006;177:5115–5121. doi: 10.4049/jimmunol.177.8.5115. [DOI] [PubMed] [Google Scholar]
- 47.Chen W, et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGFβ induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun CM, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Curotto de Lafaille MA, Kutchukhidze N, Shen S, Ding Y, Yee H, Lafaille JJ. Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity. 2008;29:114–126. doi: 10.1016/j.immuni.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 50.Izcue A, Powrie F. Special regulatory T-cell review: Regulatory T cells and the intestinal tract--patrolling the frontier. Immunology. 2008;123:6–10. doi: 10.1111/j.1365-2567.2007.02778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGFβ induces a regulatory phenotype in CD4+CD25− T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172:5149–5153. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- 52.Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting Edge: IL-2 is essential for TGFβ-mediated induction of Foxp3+ T regulatory cells. J Immunol. 2007;178:4022–4026. doi: 10.4049/jimmunol.178.7.4022. [DOI] [PubMed] [Google Scholar]
- 53.Zheng SG, Wang J, Horwitz DA. Cutting edge: Foxp3+CD4+CD25+ regulatory T cells induced by IL-2 and TGFβ are resistant to Th17 conversion by IL-6. J Immunol. 2008;180:7112–7116. doi: 10.4049/jimmunol.180.11.7112. [DOI] [PubMed] [Google Scholar]
- 54.Yu A, Zhu L, Altman NH, Malek TR. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity. 2009;30:204–217. doi: 10.1016/j.immuni.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 2010;327:1098–1102. doi: 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang XO, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou L, et al. TGFβ induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORγt function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 59.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 60.Murawski MR, Litherland SA, Clare-Salzler MJ, Davoodi-Semiromi A. Upregulation of Foxp3 expression in mouse and human Treg is IL-2/STAT5 dependent: implications for the NOD STAT5B mutation in diabetes pathogenesis. Ann N Y Acad Sci. 2006;1079:198–204. doi: 10.1196/annals.1375.031. [DOI] [PubMed] [Google Scholar]
- 61.Barron L, et al. Cutting Edge: Mechanisms of IL-2-Dependent Maintenance of Functional Regulatory T Cells. J Immunol. doi: 10.4049/jimmunol.0903940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Soper DM, Kasprowicz DJ, Ziegler SF. IL-2Rβ links IL-2R signaling with Foxp3 expression. Eur J Immunol. 2007;37:1817–1826. doi: 10.1002/eji.200737101. [DOI] [PubMed] [Google Scholar]
- 63.Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3+ CD25+ CD4+ regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. 2005;201:723–735. doi: 10.1084/jem.20041982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fontenot JD, Dooley JL, Farr AG, Rudensky AY. Developmental regulation of Foxp3 expression during ontogeny. J Exp Med. 2005;202:901–906. doi: 10.1084/jem.20050784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Todd JA. Etiology of type 1 diabetes. Immunity. 2010;32:457–467. doi: 10.1016/j.immuni.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 66.Yamanouchi J, et al. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat Genet. 2007;39:329–337. doi: 10.1038/ng1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Todd JA, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhernakova A, et al. Novel association in chromosome 4q27 region with rheumatoid arthritis and confirmation of type 1 diabetes point to a general risk locus for autoimmune diseases. Am J Hum Genet. 2007;81:1284–1288. doi: 10.1086/522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matesanz F, et al. Allelic expression and interleukin-2 polymorphisms in multiple sclerosis. J Neuroimmunol. 2001;119:101–105. doi: 10.1016/s0165-5728(01)00354-x. [DOI] [PubMed] [Google Scholar]
- 70.van Heel DA, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet. 2007;39:827–829. doi: 10.1038/ng2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lowe CE, et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2Rα region in type 1 diabetes. Nat Genet. 2007;39:1074–1082. doi: 10.1038/ng2102. [DOI] [PubMed] [Google Scholar]
- 72.Wang J, Wicker LS, Santamaria P. IL-2 and its high-affinity receptor: genetic control of immunoregulation and autoimmunity. Semin Immunol. 2009;21:363–371. doi: 10.1016/j.smim.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 73.Dendrou CA, et al. Cell-specific protein phenotypes for the autoimmune locus IL2Rα using a genotype-selectable human bioresource. Nat Genet. 2009;41:1011–1015. doi: 10.1038/ng.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol. 2006;24:657–679. doi: 10.1146/annurev.immunol.24.021605.090727. [DOI] [PubMed] [Google Scholar]
- 75.Raulet DH. Expression and function of interleukin-2 receptors on immature thymocytes. Nature. 1985;314:101–103. doi: 10.1038/314101a0. [DOI] [PubMed] [Google Scholar]
- 76.Ono M, et al. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446:685–689. doi: 10.1038/nature05673. [DOI] [PubMed] [Google Scholar]
- 77.Wu Y, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 78.Placek K, et al. Integration of distinct intracellular signaling pathways at distal regulatory elements directs T-bet expression in human CD4+ T cells. J Immunol. 2009;183:7743–7751. doi: 10.4049/jimmunol.0803812. [DOI] [PubMed] [Google Scholar]
- 79.Gong D, Malek TR. Cytokine-dependent Blimp-1 expression in activated T cells inhibits IL-2 production. J Immunol. 2007;178:242–252. doi: 10.4049/jimmunol.178.1.242. [DOI] [PubMed] [Google Scholar]
- 80.Hwang ES, Hong JH, Glimcher LH. IL-2 production in developing Th1 cells is regulated by heterodimerization of RelA and T-bet and requires T-bet serine residue 508. J Exp Med. 2005;202:1289–1300. doi: 10.1084/jem.20051044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martins GA, et al. Transcriptional repressor Blimp-1 regulates T cell homeostasis and function. Nat Immunol. 2006;7:457–465. doi: 10.1038/ni1320. [DOI] [PubMed] [Google Scholar]
- 82.Kim HP, Kelly J, Leonard WJ. The basis for IL-2-induced IL-2 receptor α chain gene regulation: importance of two widely separated IL-2 response elements. Immunity. 2001;15:159–172. doi: 10.1016/s1074-7613(01)00167-4. [DOI] [PubMed] [Google Scholar]
- 83.Fernandez-Botran R, Chilton PM, Ma Y. Soluble cytokine receptors: their roles in immunoregulation, disease, and therapy. Adv Immunol. 1996;63:269–336. doi: 10.1016/s0065-2776(08)60858-5. [DOI] [PubMed] [Google Scholar]
- 84.Arribas J, Borroto A. Protein ectodomain shedding. Chem Rev. 2002;102:4627–4638. doi: 10.1021/cr010202t. [DOI] [PubMed] [Google Scholar]
- 85.Kheradmand F, Werb Z. Shedding light on sheddases: role in growth and development. Bioessays. 2002;24:8–12. doi: 10.1002/bies.10037. [DOI] [PubMed] [Google Scholar]
- 86.Busse D, et al. Competing feedback loops shape IL-2 signaling between helper and regulatory T lymphocytes in cellular microenvironments. Proc Natl Acad Sci USA. 2010;107:3058–3063. doi: 10.1073/pnas.0812851107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity. 2010;33:153–165. doi: 10.1016/j.immuni.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 89.Scheffold A, Huhn J, Hofer T. Regulation of CD4+CD25+ regulatory T cell activity: it takes (IL-)two to tango. Eur J Immunol. 2005;35:1336–1341. doi: 10.1002/eji.200425887. [DOI] [PubMed] [Google Scholar]
- 90.Barthlott T, et al. CD25+ CD4+ T cells compete with naive CD4+ T cells for IL-2 and exploit it for the induction of IL-10 production. Int Immunol. 2005;17:279–288. doi: 10.1093/intimm/dxh207. [DOI] [PubMed] [Google Scholar]
- 91.Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8:1353–1362. doi: 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- 92.Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged interleukin-2Rα expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity. 2010;32:91–103. doi: 10.1016/j.immuni.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 93.Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32:79–90. doi: 10.1016/j.immuni.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T cell fate. Nature. 2010;463:808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aune TM, Collins PL, Chang S. Epigenetics and T helper 1 differentiation. Immunology. 2009;126:299–305. doi: 10.1111/j.1365-2567.2008.03026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol. 2010;10:225–235. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bensinger SJ, et al. Distinct IL-2 receptor signaling pattern in CD4+CD25+ regulatory T cells. J Immunol. 2004;172:5287–5296. doi: 10.4049/jimmunol.172.9.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Walsh PT, et al. PTEN inhibits IL-2 receptor-mediated expansion of CD4+ CD25+ Tregs. J Clin Invest. 2006;116:2521–2531. doi: 10.1172/JCI28057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gavin MA, Clarke SR, Negrou E, Gallegos A, Rudensky A. Homeostasis and anergy of CD4+CD25+suppressor T cells in vivo. Nat Immunol. 2002;3:33–41. doi: 10.1038/ni743. [DOI] [PubMed] [Google Scholar]
- 100.McHugh RS, et al. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–323. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 101.Lu LF, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30:80–91. doi: 10.1016/j.immuni.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fujimoto M, Naka T. Regulation of cytokine signaling by SOCS family molecules. Trends Immunol. 2003;24:659–666. doi: 10.1016/j.it.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 103.Sporri B, Kovanen PE, Sasaki A, Yoshimura A, Leonard WJ. JAB/SOCS1/SSI-1 is an interleukin-2-induced inhibitor of IL-2 signaling. Blood. 2001;97:221–226. doi: 10.1182/blood.v97.1.221. [DOI] [PubMed] [Google Scholar]
- 104.Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity. 2010;33:301–311. doi: 10.1016/j.immuni.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lockyer HM, Tran E, Nelson BH. STAT5 is essential for Akt/p70S6 kinase activity during IL-2-induced lymphocyte proliferation. J Immunol. 2007;179:5301–5308. doi: 10.4049/jimmunol.179.8.5301. [DOI] [PubMed] [Google Scholar]
- 106.Hill JA, et al. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. 2007;27:786–800. doi: 10.1016/j.immuni.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 107.Tang Q, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity. 2008;28:687–697. doi: 10.1016/j.immuni.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311:1924–1927. doi: 10.1126/science.1122927. [DOI] [PubMed] [Google Scholar]
- 109.Letourneau S, et al. IL-2/anti-IL-2 antibody complexes show strong biological activity by avoiding interaction with IL-2 receptor α subunit CD25. Proc Natl Acad Sci USA. 2010;107:2171–2176. doi: 10.1073/pnas.0909384107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Webster KE, et al. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med. 2009;206:751–760. doi: 10.1084/jem.20082824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Grinberg-Bleyer Y, et al. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med. 2010;207:1871–1878. doi: 10.1084/jem.20100209. [DOI] [PMC free article] [PubMed] [Google Scholar]