

Abstract
Understanding viral pathogenesis is challenging because of confounding factors, including nonabrasive access to infected tissues and high abundance of inflammatory mediators that may mask mechanistic details. In diseases such as influenza and smallpox where the primary cause of mortality results from complications in the lung, the characterization of lung fluid offers a unique opportunity to study host-pathogen interactions with minimal effect on infected animals. This investigation characterizes the global proteome response in the pulmonary fluid, bronchoalveolar lavage fluid, of macaques during upper respiratory infection by monkeypox virus (MPXV), a close relative of the causative agent of smallpox, variola virus. These results are compared and contrasted against infections by vaccinia virus (VV), a low pathogenic relative of MPXV, and with extracellular fluid from MPXV-infected HeLa cells. To identify changes in the pulmonary protein compartment, macaque lung fluid was sampled twice prior to infection, serving as base line, and up to six times following intrabronchial infection with either MPXV or VV. Increased expression of inflammatory proteins was observed in response to both viruses. Although the increased expression resolved for a subset of proteins, such as C-reactive protein, S100A8, and S100A9, high expression levels persisted for other proteins, including vitamin D-binding protein and fibrinogen γ. Structural and metabolic proteins were substantially decreased in lung fluid exclusively during MPXV and not VV infection. Decreases in structural and metabolic proteins were similarly observed in the extracellular fluid of MPXV-infected HeLa cells. Results from this study suggest that the host inflammatory response may not be the only facilitator of viral pathogenesis, but rather maintaining pulmonary structural integrity could be a key factor influencing disease progression and mortality.
Monkeypox virus (MPXV)1 is a member of the Orthopoxviridae family and is closely related to variola virus, the causative agent of smallpox (1). Endemic to the rain forests of Central and West Africa, MPXV can cause potentially fatal smallpox-like disease in both human and non-human primates (NHPs) (2). Human infection typically stems from zoonotic sources such as rodents and NHPs, although occasional human-to-human transmission occurs (3). Awareness of the potential impact of this pathogen on global populations increased following the first incidence of human MPXV infection in the United States in 2003 (4, 5).
An attenuated strain of vaccinia virus (VV), a closely related orthopoxvirus that serves as an effective vaccine for smallpox, can prevent disease caused by MPXV and variola. However, worldwide eradication of smallpox led to the discontinuation of routine vaccination for variola in the 1970s. As a result, there is growing concern regarding the reemergence of pathogenic orthopoxviruses such as MPXV introduced from either a zoonotic source or from bioterrorism (2).
The replication cycle of all poxviruses occurs exclusively within the cytoplasm of an infected cell in which the virus hijacks cellular transcriptional and translational machinery to assemble viral factories, referred to as viroplasm (7), resulting in a general shift away from cellular protein expression toward viral mRNA and protein synthesis (8, 9). Importantly, the ability of poxviruses to replicate within a particular cell type is believed to typically be governed through postentry events, such as the proper recruitment of host cell machinery required for viral processes and attenuation of the innate immune response (10). Furthermore, the ability to regulate both the innate and adaptive immune responses is an extremely important aspect of poxvirus biology that governs viral replication and disease development in vivo. Some mechanisms utilized by poxviruses to inhibit the host immune response to infection include preventing major histocompatibility complex (MHC) class I expression on the cell surface, blocking inflammatory cytokine signaling, inhibition of apoptosis, and regulation of the complement system (2, 11). In humans, the primary route of monkeypox infection is through direct cutaneous or mucosal exposure, including the respiratory pathway, to infected animals (12–16). Aerosol dispersion is the most likely route of exposure if MPXV were to be used as a biological weapon (17, 18). Macaques have served as the prominent NHP model for studying MPXV infection for many years, and the clinical course of disease in this model has been well described (16, 19–22). Studies performed in NHPs utilizing an aerosol route of MPXV infection have demonstrated that the virus can spread to numerous organs within the infected animal and cause systemic tissue damage (e.g. to skin, oral mucosa, lymphoid tissue, gastrointestinal tract, liver, and reproductive system). The lungs are highly affected, and the MPXV antigen has been observed in multiple pulmonary cell types during infection (19).
In this study, we integrated observed changes in dynamic protein expression profiles of HeLa cells and macaque lung fluid during MPXV infection to gain insights into virus-host interactions and host response pathways. HeLa cell culture studies provided important information about virus-host cell interactions (8, 23), whereas the in vivo macaque studies provided new information about the highly complex nature of orthopoxvirus infection (10).
EXPERIMENTAL PROCEDURES
Viruses
Human MPXV (Zaire strain V79-I-005) was obtained from the Centers for Disease Control and Prevention (Atlanta, GA). This viral strain was originally isolated from a human victim who died of MPXV. A plaque-purified isolate of VV-WR (vaccinia virus, Western Reserve strain) was acquired from the American Type Culture Collection (catalogue number VR-1354, ATCC, Manassas, VA). Stocks of MPXV and VV were grown in BSC40 monkey kidney cells and purified by sucrose gradient ultracentrifugation using a protocol similar to one reported previously (24). To determine titers of viral stocks, standard plaque assays were performed utilizing BSC40 cells.
Cell Cultures and in Vitro Infections
HeLa cells (ATCC) were grown in DMEM + 10% FBS and adapted to growth in serum-free media (HyClone HEK293, etc., HyClone, Logan, UT). BSC40 monkey kidney cells (ATCC) were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum. For HeLa infection studies, cells were infected at a multiplicity of infection of 0.1, and supernatants and cells were collected at the indicated times postinfection.
Experimental Infection and Evaluation of Animals
All aspects of the animal studies were performed in a dedicated animal biosafety level 3 facility at the Oregon National Primate Research Center according to institutional guidelines for biosafety and animal care and use. Age- and sex-matched rhesus macaques, Macaca mulatta, were selected for the studies. Animals 23218 and 23358 were 4-year-old males infected with MPXV, and animals 22193 and 22588 were 7-year-old ovariectomized females infected with VV. Animals were infected with 2 × 105 plaque-forming units of the appropriate virus intrabronchially in 1 ml of phosphate-buffered saline (PBS), and a sample of all viral inocula was back-titered to confirm the delivery of the correct dosage during infection. Bronchoalveolar lavage fluid (BALF) samples were obtained by flushing lungs of sedated animals with PBS and collecting the resulting fluid.
Protein Sample Preparation and Trypsin Digestion
For infected HeLa cell samples, supernatants were first removed from the flasks after which the cells were washed with PBS and collected by scraping. Cells were pelleted by centrifugation for 5 min at 2000 rpm in a GH-3.8 rotor (Beckman Coulter, Fullerton, CA), and supernatants were clarified in a similar fashion to remove cellular debris. Samples were collected at 6 and 24 h postinfection. Cell pellets were lysed in 0.1% (w/v) RapiGest in 100 mm NH4HCO3 (pH 8.4), incubated at 100 °C for 5 min, and then cooled on ice. Clarified supernatants and BALF samples were precipitated overnight at −80 °C with 100% acetone at a ratio of 3:1, and the resulting precipitate was pelleted at 3000 rpm in a GS-6KR centrifuge (Beckman Coulter), air-dried, and resuspended in 0.1% RapiGest. Protein concentrations were then determined using BCA protein assays, and samples were digested for 1 h at 37 °C with trypsin at a ratio of 1 μg of trypsin to 50 μg of protein. Next, RapiGest was hydrolyzed by the addition of 2% TFA to achieve a pH of 2.0 and incubated for 1 h at 37 °C. Samples were flash frozen with liquid nitrogen and then thawed at room temperature to assist the precipitation of the RapiGest tail group. The samples were microcentrifuged at 16,000 × g for 10 min to pellet the RapiGest tail group, and supernatants were retained. Sample pH was restored to 7 using 30% NH4OH as the titrant. Peptide masses were determined by BCA assays.
DNA Isolation and Real-Time PCR
Parallel in vitro infections were performed in serum-free adapted HeLa cells to collect DNA for analysis of viral genome copies in infected cells. DNA was isolated from infected cells using a Puregene DNA isolation kit (Qiagen, Gaithersburg, MD), and 100 ng of each sample was subjected to real-time PCR analysis using an ABI Prism 7700 DNA sequence detector (Applied Biosystems, Foster City, CA) and primers specific for MPXV ORF F3L.
LC-MS(/MS) Proteome Analyses
Samples were blocked relative to technical replicates and randomized according to treatment to minimize the effects of systematic biases and to ensure the even distribution of known and unknown confounding factors across the entire experimental data set (25). Peptides were separated using an automated in-house-designed reverse-phase capillary nano-HPLC system as described previously (26). Eluate from the HPLC was coupled directly to an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific, San Jose, CA) using electrospray ionization (27) and an electrospray ionization interface modified in house with an electrodynamic ion funnel (28, 29).
Peptides were analyzed using the accurate mass and elution time (AMT) tag approach (30). Portions of peptide samples were pooled, separated through off-line exhaustive strong cation exchange HPLC fractionation, and analyzed by reverse-phase nanocapillary HPLC-nanoelectrospray ionization-MS/MS (31). The theoretical mass and the observed normalized elution time of each peptide were used to construct a reference database of AMT tags that served as two-dimensional markers for identifying peptides in subsequent high resolution and high mass accuracy LC-MS analyses. LC-MS features were deconvoluted by Decon2Ls (version 1.0.2; using default parameters) and aligned to the mass and time tag database using VIPER (version 3.45; using default parameters). This approach to proteomics research is enabled by a number of published and unpublished in-group-developed tools that are freely available for download (32–36) at http://omics.pnl.gov.
MS/MS spectra were searched against two dual organism (MPXV-macaque, MPXV-human, or VV-macaque) concatenated protein FASTA data files using SEQUEST (TurboSEQUEST (cluster) version 27 (revision 12), Thermo Electron Corp.). The MPXV (Zaire strain) and VV (Western Reserve strain) FASTA data files of proteins translated from genetic code contained 191 and 218 protein sequences, respectively, and were acquired from the Poxvirus Bioinformatics Resource Center (http://www.poxvirus.org/; July 4, 2005; University of Alabama, Birmingham, AL). A macaque protein list was generated by compiling Macaca sp. protein sequences from NCBI, Swiss-Prot, and TrEMBL into a single, nonredundant FASTA protein data list that contained 46,344 sequences (December 2, 2008) using Protein Digestion Simulator (http://omics.pnl.gov). The human FASTA data file of proteins translated from genetic code was obtained from the International Protein Index (75,419 protein sequences; http://www.ebi.ac.uk/IPI/; January 1, 2009; European Bioinformatics Institute, Cambridge, UK) (37). A standard parameter file without modifications to amino acid residues and without restriction to possible peptide termini (i.e. not limited to only tryptic termini) was used in the analysis. Tandem mass spectral peaks were extracted with ExtractMSn (version 5.0, Thermo Scientific) for subsequent analysis using 3.0 and 1.0 Da for parent and fragment ion tolerances, respectively. Reverse database SEQUEST search analyses were performed to estimate the error rates associated with the peptide identifications, resulting in ≥98% confidence in peptide identifications.
For each SEQUEST analysis of an MS/MS spectrum (at a given parent ion charge state), only the peptide identification with the highest XCorr (the cross-correlation score between the actual and predicted MS/MS spectrum; i.e. the “top ranked hit”) was retained. In addition, the SEQUEST peptide identifications met the following criteria (38): ΔCn (the relative difference between the correlation score for the top matching peptide and the second best matching peptide) was ≥0.1 for each parent ion charge state, and XCorr was ≥1.9 (1+), ≥2.2 (2+), and ≥3.75 (3+). An additional discriminant score was calculated from SEQUEST results based on the differences between predicted (35) and observed (39) normalized elution time values and from other factors (40).
Relative peptide abundance measurements in technical replicates were scaled and normalized to the data set with the least information using linear regression in DAnTE (41). Normalized peptide abundance values were then rolled up to proteins using RRollup; a minimum of five peptides was required for the Grubb's test with a p value cutoff of 0.05. Peptides not detected by both LC-MS and LC-MS/MS data analysis methods and proteins represented by <2 unique peptides were removed. Minimum values within data sets were imputed for missing values, and Kruskal-Wallis analyses were applied to protein abundance data sets (p value ≤0.05 and q value ≤0.01) to identify statistically significant differences in protein expression levels.
Because peptides typically map to multiple homologous proteins, i.e. protein isoforms, it is generally impossible to unambiguously assign such peptides to a specific protein (42). Signature peptides, defined as peptides unique to a single isoform, provide a differentiating identification (43) that can be used to filter ambiguous peptide assignments. However, the use of signature peptides falsely assumes that other isoforms, whose sequence may not code for identifiable signature peptides, do not exist in the sample. To address this, identified peptides were mapped to proteins and then manually grouped into protein families. Raw data and collated peptide identification information are available to the community as supplemental data at http://omics.pnl.gov/view/publication_1024.html.
Pathway Analysis
Human homologs were identified for differentially expressed macaque proteins by using InParanoid (44, 45) and ScalaBLAST (46) to compare them against the human Internation Protein Index 2009 protein collection list. For pathway analysis, human Internation Protein Index protein accession numbers were converted to NCBI Entrez gene identifiers using the Bioinformatics Resource Manager (47). Human NCBI Entrez gene identifiers corresponding to differentially expressed macaque proteins were imported into DAVID (48, 49), and the Kyoto Encyclopedia of Genes and Genomes was used to identify significantly altered functional pathways (50–52).
RESULTS
Monkeypox Infection Resulted in Distinct Effects on HeLa Proteome Partitions
The cellular response to MPXV infection was characterized in HeLa cells in part to determine whether extracellular MPXV proteins could be detected in a classical cell culture model system. Total protein content was quantified for mock- and virus-infected cellular and extracellular partitions at 6 and 24 h. After infection, MPXV genomic DNA was detectable within 6 h of infection by real-time PCR analysis and displayed a greater than 100-fold increase in genome copies between 6 and 24 h (Fig. 1) due to increased virus replication and spread. Mock-infected cultures contained no detectable viral DNA. Protein contents were similar with regard to infection state and time point. However, the cellular partition contained ∼10-fold more protein mass than the extracellular partition. Therefore, a precipitation step was necessary to concentrate proteins in the extracellular partition prior to global analysis. Both cellular and extracellular partitions were analyzed, and a substantially larger number of unique peptides were observed in the cellular partition than in the extracellular partition, i.e. ∼7600 and ∼1200 peptides, mapping to ∼3300 and ∼800 proteins, respectively (Fig. 2 and supplemental Tables S01–S04).
Fig. 1.
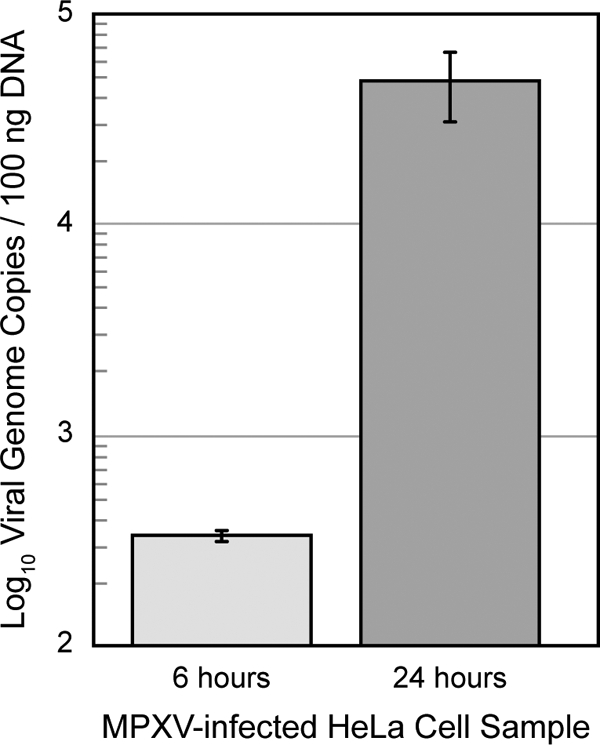
MPXV replication kinetics in HeLa cell culture over 24 h. Viral replication kinetics was assessed by using quantitative real-time PCR analysis to determine the number of viral genome copies per 100 ng of total DNA. Error bars represent standard deviations.
Fig. 2.

HeLa protein response to monkeypox infection over 24 h. Whole cell lysate proteins (A), supernatant proteins (B), viral proteins observed in the lysate (C), and viral proteins observed in the supernatant (D) are shown. Each row represents an individual protein, and each column represents an individual LC-MS analysis. Relative protein abundance is color-coded relative to the scale at the bottom.
Viral proteins were detected exclusively in MPXV-infected samples within 24 h and were found in both the intracellular and extracellular partitions. Six viral proteins were observed in the intracellular partition (E8L, M4R, F3L, H5R, I1L, and A5L), and three were identified in the extracellular partition (J1L, B9R, and B20R). Although all of these viral proteins were identified through multiple peptides, another subset included those identified by only a single peptide that passed a false discovery rate cutoff of 1%. Spectra in this subset were manually annotated to improve confidence of the peptide identifications. Four representative spectra of peptides mapping to O2L, B12R, A29L, and G4L validated the expression of these proteins exclusively during MPXV infection (Fig. 3 and supplemental Table S05).
Fig. 3.

Representative mass spectra of y and b product ions for viral peptides mapping exclusively to O2L (A), B12R (B), A29L (C), or G4L (D).
In the cellular partition, no HeLa-derived proteins displayed statistically significant differences (p ≤ 0.05, q ≤ 0.01) between mock and MPXV treatments. This is further supported by the heat map representation (Fig. 2A and supplemental Table S01) in which the samples appear to be nearly indistinguishable. In sharp contrast to the cellular partition, 614 proteins displayed statistically significant differences between mock- and MPXV-treated extracellular samples. During MPXV infection, an increase in abundance was observed for 30 unique peptides that mapped to 52 MHC-I isoforms, a large polymorphic family of related proteins that are commonly over-represented in proteome experiments because of peptide redundancy. Therefore, MHC-I proteins were manually curated to a single group and excluded from this analysis of the extracellular partition, resulting in 562 proteins (Fig. 4). Surprisingly, the greatest differences occurred at the early infection time point where the majority of proteins decreased in expression in many cases below the level of detection.
Fig. 4.
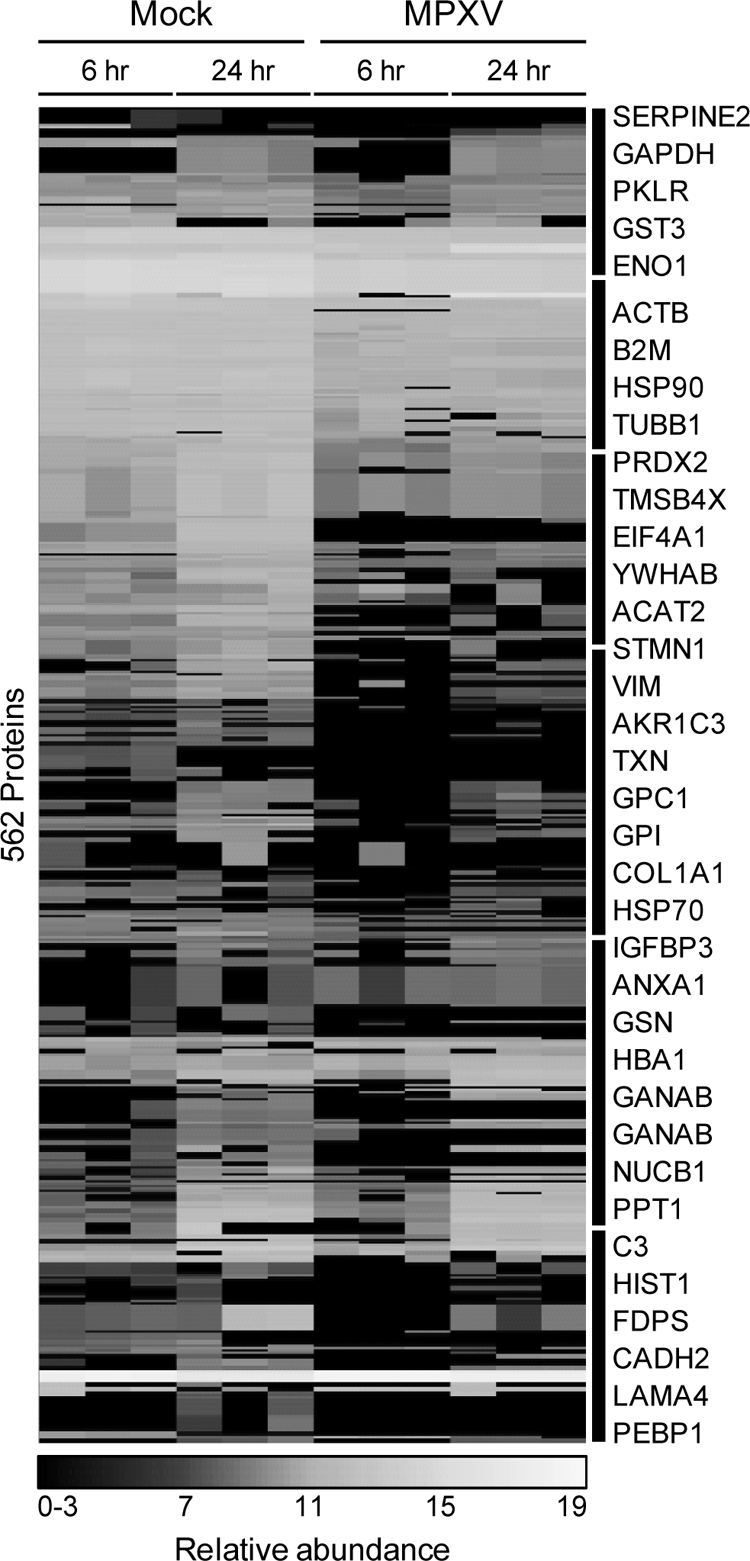
HeLa host supernatant proteins significantly altered by MPXV infection. Clusters of proteins are indicated on the right by black bars and symbols of selected proteins. PKLR, pyruvate kinase 1; GPI, phosphohexose isomerase; VIM, vimentin; FDPS, farnesyl pyrophosphate synthetase; GSN, gelsolin; GANAB, α-glucosidase II α subunit.
MPXV infection resulted in reduced abundances of several cellular structural and metabolic proteins. Among the structural proteins that decreased during MPXV infection were β-actin (ACTB), collagen type I α1 (COL1A1), gelsolin (GSN), stathmin 1 (STMN1), thymosin β-4 (TMSB4X), tubulin β1 (TUBB1), and vimentin (VIM). Additionally, the expression levels of the following metabolic proteins decreased: acetyl-CoA acetyltransferase 2 (ACAT2), enolase 1 (ENO1), farnesyl pyrophosphate synthetase (FDPS), α-glucosidase II α subunit (GANAB), aldo-keto reductase family 1 member C1 (AKR1C1), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), phosphohexose isomerase (GPI), pyruvate kinase 1 (PKLR), peroxiredoxin 2 (PRDX2), and thioredoxin (TXN).
Macaques Display a Temporal and Dynamic Response to Orthopoxvirus
Results from the viral infections of HeLa cultures suggested that significant changes occur to the protein composition of extracellular fluid early upon MPXV infection. To determine whether similar observations also occur during in vivo infection, similar proteome measurements were extended to an NHP model system of MPXV infection. BALF samples were collected from the lungs of rhesus macaques at various time points preinfection (healthy, uninfected controls) and postinfection following intrabronchial inoculation with either MPXV or VV. Global proteome analysis of BALF samples identified ∼4200 unique peptides (∼2600 from the MPXV-infected animals and ∼3100 from the VV-infected animals) that map to ∼2100 proteins (∼1500 from the MPXV-infected animals and ∼1800 from the VV-infected animals). Approximately 1200 proteins (>80%) from the MPXV samples were observed in common with the VV samples.
Protein expression profiles derived from MPXV- and VV-infected lung fluid exhibited three major temporal expression patterns that correlate roughly to pre-, early (<16 days postinfection), and late infection (>16 days postinfection). Kruskal-Wallis analysis identified 996 proteins (555 proteins from MPXV and 747 proteins from VV) with significant changes in expression (p ≤ 0.05 and q ≤ 0.01) during pre-, early, and late stages of infection (Fig. 5 and supplemental Tables S06 and S07). Three MPXV proteins (J1L, B9R, and H5R), all of which were detected earlier in the extracellular partition of infected HeLa cells, were statistically altered in expression and observed exclusively during the early stages of MPXV infection. In contrast, no viral proteins were detected throughout the course of VV infection.
Fig. 5.

Significantly altered proteins in macaque lung fluid upon inoculation with MPXV (A) or VV (B). Time points above heat maps refer to prior to viral infection (PRE), <16 days (EARLY), or >16 days (LATE).
The subset of proteins that significantly increased in response to MPXV fell into two major categories, i.e. transient and persistent “increasers,” depending on whether the increase in abundance was resolved by the late stage infection or not, respectively (Fig. 6). Transiently increasing proteins included α1-acid glycoprotein 1 precursor (AGP1), C-reactive protein (CRP), complement factor H (CFH), serum amyloid A4 (SAA4), and S100 calcium-binding protein A8 (S100A8). Persistently increasing proteins included α1-B glycoprotein (A1BG), vitamin D-binding protein (GC), fibrinogen γ (FGG), apolipoprotein A-I (APOA1), and S100 calcium-binding protein A9 (S100A9). Similar expression dynamics were observed during VV infection (Fig. 6) with the exception of AGP1. The increased abundance level of AGP1 during viral infection persisted throughout the late phase of VV infection. Although trends were similar for both MPXV and VV infections, a significantly larger number of peptides corresponding to the immunoglobulin family were observed during VV infection (1624 peptides) compared with MPXV infection (428 peptides).
Fig. 6.

Selected proteins from macaque lung fluid observed in both MPXV (A) and VV (B) infections. Viral proteins are designated by a blue bar on the left, proteins that increased during infection are designated by a red bar, and proteins that decreased are designated by a green bar. Persistent and transient increasers are indicated to the right of the heat maps. Averaged and normalized protein abundances are graphed (C) relative to pretreatment. Solid lines with diamonds represent MPXV infection, and dotted lines with squares represent VV infection. Red represents proteins that increased during infection, green represents proteins that decreased, and blue represents the viral proteins. The gray box designates one standard deviation from the mean. Asterisks (*) indicate viral proteins that were only detected in the MPXV infection during the early phase of infection and were given an arbitrary score of +3. GC, vitamin D-binding protein; FGG, fibrinogen γ; CRP, C-reactive protein; CFH, complement factor H; AFP, α-fetoprotein.
Relative to the proteins that exhibited increases in abundance, a much larger group of proteins displayed dramatic decreases in expression upon MPXV infection, some of which fell below the level of detection. In contrast, only moderate, if any, decreases were observed in these proteins upon VV infection. Similar to the HeLa cell supernatant, structural and metabolic proteins in lung fluid decreased in abundance during the early phases of MPXV infection. The structural proteins included periostin osteoblast-specific factor (POSTN), tropomyosin 1 (TPM1), and COL1A1. Metabolic proteins that decreased included AKR1C1, ENO1, aldehyde dehydrogenase family 1 subfamily A1 (ALDH1A1), and tripartite motif-containing 5 (TRIM5). Similarly, the transport protein α-fetoprotein (AFP), which was present prior to MPXV inoculation, was not detected during the early phase of infection.
Integrating Interspecies Response Profiles to Poxvirus Infection
To highlight common virus-host response profiles characteristic of orthopoxvirus infections in general (i.e. independent of host species or cell type), statistically altered homologs across macaques and humans were selected based on the most dramatic changes in expression during infection and assembled relative to virus and host (Fig. 7 and supplemental Tables S08–S10). Several structural proteins (e.g. tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, beta polypeptide (YWHAB), TPM3, TUBB4, ACTB, and TMSB4X) and metabolic proteins (e.g. TXN, PKM2, and AKR1C3) displayed decreased expression during viral infection. Although expression of HSP90 in both macaque lung fluid and HeLa cell supernatant decreased during MPXV infection, the suppressive effect was less apparent during VV infection. In contrast, increased expression of host serine protease inhibitors (SERPINs) is a common response between species and is a general response to orthopoxvirus infection. MPXV also encodes multiple SERPINs, including B19R, C2L, and B12R. SERPINs are used by several viruses to subvert the host immune response and inflammation (53, 54). The observed interspecies increase from both host- and virus-encoded SERPINs highlights the important role this class of proteins plays in orthopoxvirus infection.
Fig. 7.
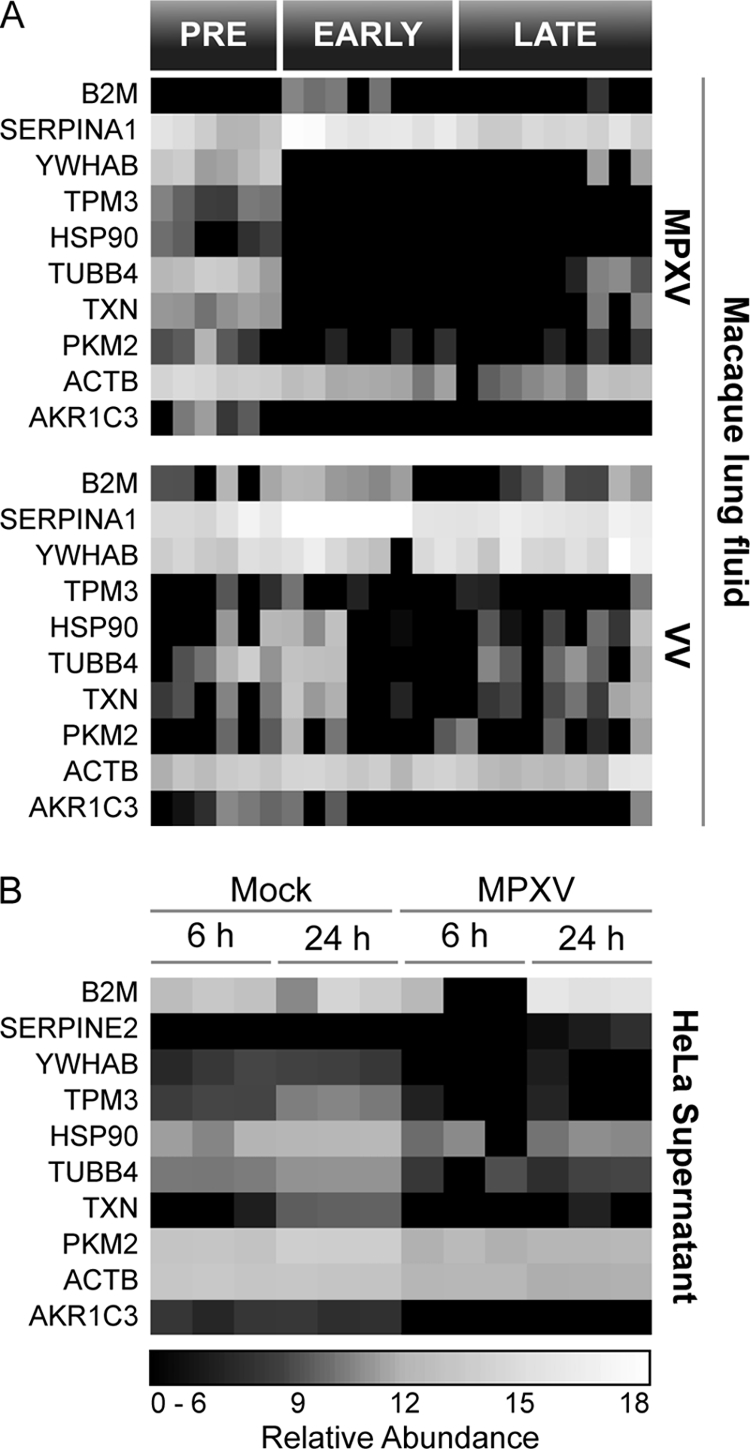
Common interspecies response profile in MPXV- and VV-infected macaque lung fluid (A) and supernatant from MPXV-infected HeLa cells (B).
Inflammatory Response to Viral Infection
Macaque proteins classified as statistically altered during MPXV and VV infections were mapped to human homologs for pathway analysis. Human Entrez gene identifiers for altered macaque proteins were uploaded to DAVID, and Kyoto Encyclopedia of Genes and Genomes pathways were selected for analyses based on global coverage. The complement cascade pathway was identified by DAVID as the most highly affected by MPXV (p = 1.3 × 10−22 and Benjamini = 2.6 × 10−20) and VV (p = 4.4 × 10−30 and Benjamini = 8.9 × 10−28) (Fig. 8). Among this group of 25 proteins, the expression of 19 (76%) increased early during the course of both MPXV and VV infection, which suggests that both viruses initiated qualitatively similar inflammatory responses in lung fluid. Activation of the complement cascade is a common innate immune response against invading pathogens, including viruses. Complement activation results in inflammation, immune cell recruitment, viral clearance, and death of infected cells while also having effects on the development of the adaptive immune response against invading pathogens (55, 56). Importantly, a virus-encoded complement inhibitor has been suggested to be a major virulence factor for some strains of MPXV (21), further suggesting an important role for the complement system in MPXV pathogenesis.
Fig. 8.

MPXV and VV infection impacted the complement cascade in macaque lung fluid. The complement cascade was identified by DAVID as significantly affected (MPXV: p = 1.3 × 10−22 and Benjamini = 2.6 × 10−20; VV: p = 4.4 × 10−30 and Benjamini = 8.9 × 10−28) (A). Proteins increased exclusively by MPXV are colored orange, proteins increased exclusive by VV are green, and proteins increased during both infections are red. Expression profiles for proteins involved in the complement pathway are displayed in B. FG, fibrinogen; CF, complement factor H/B, VWF, von Willebrand factor; BDKR, bradykinin receptor; THBD, thrombomodulin; MBL, mannose-binding lectin; PLG, plasminogen; KNG, kininogen; BF, factor B; DF, factor D; IF, factor I; MCP, membrane cofactor protein; DAF, decay-accelerating factor; PLAU, plasminogen activator, urokinase; PLAT, plasminogen activator, tissue; PLAUR, plasminogen activator, urokinase receptor; PROC, protein C; CRRY, CR1-related y.
DISCUSSION
Analysis of longitudinal lung fluid samples obtained from MPXV-infected macaques revealed a biphasic response to infection with distinct proteome patterns corresponding to pre-, early, and late stages of viral disease. Although both pathogenic MPXV and nonpathogenic VV infections produced increased levels of proteins indicative of inflammation, only MPXV infection produced a dramatic decrease of structural and metabolic proteins. It is generally believed that inflammation is the primary cause of mortality, contributing to fibrinonecrotic bronchopneumonia (19, 57). In this study, the increased levels of inflammatory proteins occurred simultaneously with specific loss of structural and metabolic proteins, which suggests that inflammation may not be the direct or sole contributor to disease progression. Instead, these structural and metabolic proteins may represent essential factors for controlling and resolving pulmonary inflammation during viral infection.
The extracellular partition of HeLa cell cultures also displayed substantial decreases in the abundance of several proteins within 6 h of MPXV application, which may reflect virus-specific targeted inhibition of cellular transcription and/or translation. However, in this study, we observed no significant differences in protein expression in the intracellular partition in contrast to previous findings (8, 23, 58, 59). Several scenarios could explain this apparent discordance with existing literature, including the conservative statistical tests used, differences between transcriptional and translational studies, or the combination of these specific biological organisms. For example, a subset of proteins was observed to be differentially expressed in the intracellular partition between the uninfected control and MPXV-infected samples, including the viral proteins observed exclusively by 24 h of virus infection. However, the large number of observations and the conservative statistical model (Kruskal-Wallis) made it difficult to identify differences that exceed the statistical threshold for the results from the lysate (Fig. 2A). In fact, the results illustrate the high reproducibility and specificity of the AMT tag approach for analyzing these samples. Another possible explanation for the lack of substantial differences in the intracellular proteins is that MPXV may have evolved evasion strategies that minimize intracellular proteome responses to ensure viral replication within the host (60).
Interpreting changes in the abundance of proteins in extracellular fluid is especially challenging as this could reflect a decrease in protein synthesis, increased protein turnover, or post-translational modification affecting transport or function of the protein. The important aspect to focus on in this study is the changing trends in protein expression profiles during the course of viral infection. Overall, the trend in extracellular protein abundance observed during VV infection was very similar to that of MPXV infection, which is expected as these are closely related viruses and likely reflects the general response to orthopoxvirus infection. The major differences observed between MPXV and VV were that responses to VV were more delayed, short lived, and reduced in magnitude as compared with those responses observed for MPXV, which appears to correspond to the difference in virulence between the two viral strains. In contrast, the response to MPXV was observed in the lung fluid at the earliest time point, suggesting that the most important impact of the virus occurs early during infection through perturbation of cell-cell communications and/or disruption of extracellular structures.
A critical observation was that although viral proteins were observed exclusively during infection as expected clearly distinct viral proteins were identified between HeLa cell lysate and supernatant samples. Six MPXV proteins were identified from the complex intracellular samples: M4R, F3L, E8L, A5L, I1L, and H5R. Three of the intracellular viral proteins are known structural components of the virion: M4R and A5L are viral core proteins, and E8L is a surface membrane protein of cell-associated viral particles, all of which would be expected to be relatively abundant. H5R and I1L are virosomal proteins that are essential for viral replication. F3L inhibits intracellular innate immune pattern recognition molecules, such as double-stranded RNA protein kinase and oligoadenylate synthetase. Three viral proteins were detected in the supernatant of infected HeLa cells within 24 h: B20R, J1L, and B9R. These proteins were also observed in the lung fluid of infected macaques, demonstrating similarity between the in vitro HeLa cell and the in vivo rhesus macaque infection models. J1L and B9R are known secreted receptor mimics for CC chemokines and IFN-γ, respectively, which promote viral replication in the host by blocking immune response. In contrast, B20R is derived from a predicted protein-coding gene within the MPXV genome and is functionally uncharacterized. All observed viral proteins have previously been identified at the RNA level by microarray analysis during MPXV infection of HeLa cells, primary human monocytes, and fibroblasts (6).
Overall, the insights gained by applying the AMT tag proteomics approach to studying both the HeLa cell and macaque lung fluid improve our understanding of disease by human MPXV. In particular, the reduced abundance of a large number of proteins in the extracellular fluid and lung fluid suggests the possibility that increased mortality as a result of MPXV infection may largely be due to reduction in expression of proteins important to host integrity.
Supplementary Material
Acknowledgments
We gratefully acknowledge the contributions and assistance of Penny Colton. Human MPXV (Zaire strain V79-I-005) was graciously provided by Dr. Inger Damon (Centers for Disease Control and Prevention, Atlanta, GA). Portions of the work were performed in the Environmental Molecular Science Laboratory, a United States Department of Energy (DOE) Office of Biological and Environmental Research national scientific user facility at Pacific Northwest National Laboratory (PNNL) in Richland, WA. PNNL is a multiprogram national laboratory operated by Battelle Memorial Institute for the DOE under Contract DE-AC05-76RLO-1830.
Footnotes
* This work was supported, in whole or in part, by National Institutes of Health Grants RR00163 and RR18522 from the National Center for Research Resources and Interagency Agreements Y1-AI-4894–01 and Y1-A1–8401 from the NIAID/United States Department of Health and Human Services. This work was also supported by United States Department of Defense Grant W81XWH-05-1-0046) and Battelle Internal Research and Development.
 This article contains supplemental Tables S01–S10.
This article contains supplemental Tables S01–S10.
1 The abbreviations used are:
- MPXV
- monkeypox virus
- VV
- vaccinia virus
- NHP
- nonhuman primate
- BALF
- bronchoalveolar lavage fluid
- AMT
- accurate mass and elution time
- NCBI
- National Center for Biotechnology Information
- DAVID
- Database for Annotation, Visualization, and Integrated Discovery
- ACTB
- β-actin
- COL1A1
- collagen type I α1
- TMSB4X
- thymosin β-4
- ENO1
- enolase 1
- AKR1C1
- aldo-keto reductase family 1 member C1
- TXN
- thioredoxin
- AGP1
- α1-acid glycoprotein 1 precursor
- TUBB
- tubulin β
- SERPIN
- serine protease inhibitor.
REFERENCES
- 1. Di Giulio D. B., Eckburg P. B. (2004) Human monkeypox: an emerging zoonosis. Lancet Infect. Dis. 4, 15–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weaver J. R., Isaacs S. N. (2008) Monkeypox virus and insights into its immunomodulatory proteins. Immunol. Rev. 225, 96–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Henderson D. A., Inglesby T. V., Bartlett J. G., Ascher M. S., Eitzen E., Jahrling P. B., Hauer J., Layton M., McDade J., Osterholm M. T., O'Toole T., Parker G., Perl T., Russell P. K., Tonat K. (1999) Smallpox as a biological weapon: medical and public health management. Working Group on Civilian Biodefense. JAMA 281, 2127–2137 [DOI] [PubMed] [Google Scholar]
- 4. Reed K. D., Melski J. W., Graham M. B., Regnery R. L., Sotir M. J., Wegner M. V., Kazmierczak J. J., Stratman E. J., Li Y., Fairley J. A., Swain G. R., Olson V. A., Sargent E. K., Kehl S. C., Frace M. A., Kline R., Foldy S. L., Davis J. P., Damon I. K. (2004) The detection of monkeypox in humans in the Western Hemisphere. N. Engl. J. Med. 350, 342–350 [DOI] [PubMed] [Google Scholar]
- 5. Centers for Disease Control and Prevention (CDC) (2003) Multistate outbreak of monkeypox—Illinois, Indiana, and Wisconsin, 2003. MMWR Morb. Mortal. Wkly. Rep. 52, 537–540 [PubMed] [Google Scholar]
- 6. Rubins K. H., Hensley L. E., Bell G. W., Wang C., Lefkowitz E. J., Brown P. O., Relman D. A. (2008) Comparative analysis of viral gene expression programs during poxvirus infection: a transcriptional map of the vaccinia and monkeypox genomes. PLoS One 3, e2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Katsafanas G. C., Moss B. (2007) Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2, 221–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brum L. M., Lopez M. C., Varela J. C., Baker H. V., Moyer R. W. (2003) Microarray analysis of A549 cells infected with rabbitpox virus (RPV): a comparison of wild-type RPV and RPV deleted for the host range gene, SPI-1. Virology 315, 322–334 [DOI] [PubMed] [Google Scholar]
- 9. Guerra S., López-Fernández L. A., Pascual-Montano A., Muñoz M., Harshman K., Esteban M. (2003) Cellular gene expression survey of vaccinia virus infection of human HeLa cells. J. Virol. 77, 6493–6506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McFadden G. (2005) Poxvirus tropism. Nat. Rev. Microbiol. 3, 201–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Seet B. T., Johnston J. B., Brunetti C. R., Barrett J. W., Everett H., Cameron C., Sypula J., Nazarian S. H., Lucas A., McFadden G. (2003) Poxviruses and immune evasion. Annu Rev. Immunol. 21, 377–423 [DOI] [PubMed] [Google Scholar]
- 12. Arita I., Jezek Z., Khodakevich L., Ruti K. (1985) Human monkeypox: a newly emerged orthopoxvirus zoonosis in the tropical rain forests of Africa. Am. J. Trop. Med. Hyg. 34, 781–789 [DOI] [PubMed] [Google Scholar]
- 13. Breman J. G., Kalisa-Ruti Steniowski M. V., Zanotto E., Gromyko A. I., Arita I. (1980) Human monkeypox, 1970–79. Bull. World Health Organ. 58, 165–182 [PMC free article] [PubMed] [Google Scholar]
- 14. Jezek Z., Grab B., Szczeniowski M., Paluku K. M., Mutombo M. (1988) Clinico-epidemiological features of monkeypox patients with an animal or human source of infection. Bull. World Health Organ. 66, 459–464 [PMC free article] [PubMed] [Google Scholar]
- 15. Lourie B., Bingham P. G., Evans H. H., Foster S. O., Nakano J. H., Herrmann K. L. (1972) Human infection with monkeypox virus: laboratory investigation of six cases in West Africa. Bull. World Health Organ. 46, 633–639 [PMC free article] [PubMed] [Google Scholar]
- 16. Cho C. T., Wenner H. A. (1973) Monkeypox virus. Bacteriol. Rev. 37, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ryan C. P. (2008) Zoonoses likely to be used in bioterrorism. Public Health Rep. 123, 276–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garnier L., Gaudin J. C., Bensadoun P., Rebillat I., Morel Y. (2009) Real-time PCR assay for detection of a new simulant for poxvirus biothreat agents. Appl. Environ. Microbiol. 75, 1614–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zaucha G. M., Jahrling P. B., Geisbert T. W., Swearengen J. R., Hensley L. (2001) The pathology of experimental aerosolized monkeypox virus infection in cynomolgus monkeys (Macaca fascicularis). Lab. Invest. 81, 1581–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Saijo M., Ami Y., Suzaki Y., Nagata N., Iwata N., Hasegawa H., Iizuka I., Shiota T., Sakai K., Ogata M., Fukushi S., Mizutani T., Sata T., Kurata T., Kurane I., Morikawa S. (2009) Virulence and pathophysiology of the Congo Basin and West African strains of monkeypox virus in non-human primates. J. Gen. Virol. 90, 2266–2271 [DOI] [PubMed] [Google Scholar]
- 21. Chen N., Li G., Liszewski M. K., Atkinson J. P., Jahrling P. B., Feng Z., Schriewer J., Buck C., Wang C., Lefkowitz E. J., Esposito J. J., Harms T., Damon I. K., Roper R. L., Upton C., Buller R. M. (2005) Virulence differences between monkeypox virus isolates from West Africa and the Congo basin. Virology 340, 46–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rubins K. H., Hensley L. E., Jahrling P. B., Whitney A. R., Geisbert T. W., Huggins J. W., Owen A., Leduc J. W., Brown P. O., Relman D. A. (2004) The host response to smallpox: analysis of the gene expression program in peripheral blood cells in a nonhuman primate model. Proc. Natl. Acad. Sci. U.S.A. 101, 15190–15195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moss B. (1968) Inhibition of HeLa cell protein synthesis by the vaccinia virion. J. Virol. 2, 1028–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Doms R. W., Blumenthal R., Moss B. (1990) Fusion of intra- and extracellular forms of vaccinia virus with the cell membrane. J. Virol. 64, 4884–4892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oberg A. L., Vitek O. (2009) Statistical design of quantitative mass spectrometry-based proteomic experiments. J. Proteome Res. 8, 2144–2156 [DOI] [PubMed] [Google Scholar]
- 26. Livesay E. A., Tang K., Taylor B. K., Buschbach M. A., Hopkins D. F., LaMarche B. L., Zhao R., Shen Y., Orton D. J., Moore R. J., Kelly R. T., Udseth H. R., Smith R. D. (2008) Fully automated four-column capillary LC-MS system for maximizing throughput in proteomic analyses. Anal. Chem. 80, 294–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kelly R. T., Page J. S., Tang K., Smith R. D. (2007) Array of chemically etched fused-silica emitters for improving the sensitivity and quantitation of electrospray ionization mass spectrometry. Anal. Chem. 79, 4192–4198 [DOI] [PubMed] [Google Scholar]
- 28. Manes N. P., Estep R. D., Mottaz H. M., Moore R. J., Clauss T. R., Monroe M. E., Du X., Adkins J. N., Wong S. W., Smith R. D. (2008) Comparative proteomics of human monkeypox and vaccinia intracellular mature and extracellular enveloped virions. J. Proteome Res. 7, 960–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Page J. S., Tolmachev A. V., Tang K., Smith R. D. (2006) Theoretical and experimental evaluation of the low m/z transmission of an electrodynamic ion funnel. J. Am. Soc. Mass Spectrom. 17, 586–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zimmer J. S., Monroe M. E., Qian W. J., Smith R. D. (2006) Advances in proteomics data analysis and display using an accurate mass and time tag approach. Mass Spectrom. Rev. 25, 450–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adkins J. N., Mottaz H. M., Norbeck A. D., Gustin J. K., Rue J., Clauss T. R., Purvine S. O., Rodland K. D., Heffron F., Smith R. D. (2006) Analysis of the Salmonella typhimurium proteome through environmental response toward infectious conditions. Mol. Cell. Proteomics 5, 1450–1461 [DOI] [PubMed] [Google Scholar]
- 32. Jaitly N., Monroe M. E., Petyuk V. A., Clauss T. R., Adkins J. N., Smith R. D. (2006) Robust algorithm for alignment of liquid chromatography-mass spectrometry analyses in an accurate mass and time tag data analysis pipeline. Anal. Chem. 78, 7397–7409 [DOI] [PubMed] [Google Scholar]
- 33. Kiebel G. R., Auberry K. J., Jaitly N., Clark D. A., Monroe M. E., Peterson E. S., Toliæ N., Anderson G. A., Smith R. D. (2006) PRISM: a data management system for high-throughput proteomics. Proteomics 6, 1783–1790 [DOI] [PubMed] [Google Scholar]
- 34. Monroe M. E., Toliæ N., Jaitly N., Shaw J. L., Adkins J. N., Smith R. D. (2007) VIPER: an advanced software package to support high-throughput LC-MS peptide identification. Bioinformatics 23, 2021–2023 [DOI] [PubMed] [Google Scholar]
- 35. Petritis K., Kangas L. J., Yan B., Monroe M. E., Strittmatter E. F., Qian W. J., Adkins J. N., Moore R. J., Xu Y., Lipton M. S., Camp D. G., 2nd, Smith R. D. (2006) Improved peptide elution time prediction for reversed-phase liquid chromatography-MS by incorporating peptide sequence information. Anal. Chem. 78, 5026–5039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Monroe M. E., Shaw J. L., Daly D. S., Adkins J. N., Smith R. D. (2008) MASIC: a software program for fast quantitation and flexible visualization of chromatographic profiles from detected LC-MS(/MS) features. Comput. Biol. Chem. 32, 215–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kersey P. J., Duarte J., Williams A., Karavidopoulou Y., Birney E., Apweiler R. (2004) The International Protein Index: an integrated database for proteomics experiments. Proteomics 4, 1985–1988 [DOI] [PubMed] [Google Scholar]
- 38. Washburn M. P., Wolters D., Yates J. R., 3rd (2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 [DOI] [PubMed] [Google Scholar]
- 39. Liu T., Belov M. E., Jaitly N., Qian W. J., Smith R. D. (2007) Accurate mass measurements in proteomics. Chem. Rev. 107, 3621–3653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Strittmatter E. F., Kangas L. J., Petritis K., Mottaz H. M., Anderson G. A., Shen Y., Jacobs J. M., Camp D. G., 2nd, Smith R. D. (2004) Application of peptide LC retention time information in a discriminant function for peptide identification by tandem mass spectrometry. J. Proteome Res. 3, 760–769 [DOI] [PubMed] [Google Scholar]
- 41. Polpitiya A. D., Qian W. J., Jaitly N., Petyuk V. A., Adkins J. N., Camp D. G., 2nd, Anderson G. A., Smith R. D. (2008) DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinformatics 24, 1556–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Adkins J. N., Monroe M. E., Auberry K. J., Shen Y., Jacobs J. M., Camp D. G., 2nd, Vitzthum F., Rodland K. D., Zangar R. C., Smith R. D., Pounds J. G. (2005) A proteomic study of the HUPO Plasma Proteome Project's pilot samples using an accurate mass and time tag strategy. Proteomics 5, 3454–3466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Keshishian H., Addona T., Burgess M., Kuhn E., Carr S. A. (2007) Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 6, 2212–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Berglund A. C., Sjölund E., Ostlund G., Sonnhammer E. L. (2008) InParanoid 6: eukaryotic ortholog clusters with inparalogs. Nucleic Acids Res. 36, D263–D266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O'Brien K. P., Remm M., Sonnhammer E. L. (2005) Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 33, D476–D480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oehmen C., Nieplocha J. (2006) ScalaBLAST: a scalable implementation of BLAST for high-performance data-intensive bioinformatics analysis. IEEE Trans. Parallel Distrib. Syst. 17, 740–749 [Google Scholar]
- 47. Shah A. R., Singhal M., Klicker K. R., Stephan E. G., Wiley H. S., Waters K. M. (2007) Enabling high-throughput data management for systems biology: the Bioinformatics Resource Manager. Bioinformatics 23, 906–909 [DOI] [PubMed] [Google Scholar]
- 48. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 49. Dennis G., Jr., Sherman B. T., Hosack D. A., Yang J., Gao W., Lane H. C., Lempicki R. A. (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, P3. [PubMed] [Google Scholar]
- 50. Kanehisa M., Araki M., Goto S., Hattori M., Hirakawa M., Itoh M., Katayama T., Kawashima S., Okuda S., Tokimatsu T., Yamanishi Y. (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kanehisa M., Goto S. (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kanehisa M., Goto S., Hattori M., Aoki-Kinoshita K. F., Itoh M., Kawashima S., Katayama T., Araki M., Hirakawa M. (2006) From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34, D354–D357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lucas A., Liu L., Dai E., Bot I., Viswanathan K., Munuswamy-Ramunujam G., Davids J. A., Bartee M. Y., Richardson J., Christov A., Wang H., Macaulay C., Poznansky M., Zhong R., Miller L., Biessen E., Richardson M., Sullivan C., Moyer R., Hatton M., Lomas D. A., McFadden G. (2009) The serpin saga; development of a new class of virus derived anti-inflammatory protein immunotherapeutics. Adv. Exp. Med. Biol. 666, 132–156 [DOI] [PubMed] [Google Scholar]
- 54. Amati L., Passeri M. E., Lippolis A., Lio D., Caruso C., Jirillo E., Covelli V. (2006) Taking advantage of viral immune evasion: virus-derived proteins represent novel biopharmaceuticals. Curr. Med. Chem. 13, 325–333 [DOI] [PubMed] [Google Scholar]
- 55. Lambris J. D., Ricklin D., Geisbrecht B. V. (2008) Complement evasion by human pathogens. Nat. Rev. Microbiol. 6, 132–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Carroll M. C. (2004) The complement system in regulation of adaptive immunity. Nat. Immunol. 5, 981–986 [DOI] [PubMed] [Google Scholar]
- 57. Stanford M. M., McFadden G., Karupiah G., Chaudhri G. (2007) Immunopathogenesis of poxvirus infections: forecasting the impending storm. Immunol. Cell Biol. 85, 93–102 [DOI] [PubMed] [Google Scholar]
- 58. Moss B., Salzman N. P. (1968) Sequential protein synthesis following vaccinia virus infection. J. Virol. 2, 1016–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rice A. P., Roberts B. E. (1983) Vaccinia virus induces cellular mRNA degradation. J. Virol. 47, 529–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Alcami A. (2007) New insights into the subversion of the chemokine system by poxviruses. Eur. J. Immunol. 37, 880–883 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.