

Abstract
Ischemic brain injury is a major problem associated with stroke. It has been increasingly recognized that acid-sensing ion channels (ASICs) contribute significantly to ischemic neuronal damage, but the underlying mechanism has remained elusive. Here, we show that extracellular spermine, one of the endogenous polyamines, exacerbates ischemic neuronal injury through sensitization of ASIC1a channels to extracellular acidosis. Pharmacological blockade of ASIC1a or deletion of the ASIC1 gene greatly reduces the enhancing effect of spermine in ischemic neuronal damage both in cultures of dissociated neurons and in a mouse model of focal ischemia. Mechanistically, spermine profoundly reduces desensitization of ASIC1a by slowing down desensitization in the open state, shifting steady-state desensitization to more acidic pH, and accelerating recovery between repeated periods of acid stimulation. Spermine-mediated potentiation of ASIC1a activity is occluded by PcTX1 (psalmotoxin 1), a specific ASIC1a inhibitor binding to its extracellular domain. Functionally, the enhanced channel activity is accompanied by increased acid-induced neuronal membrane depolarization and cytoplasmic Ca2+ overload, which may partially explain the exacerbated neuronal damage caused by spermine. More importantly, blocking endogenous spermine synthesis significantly attenuates ischemic brain injury mediated by ASIC1a but not that by NMDA receptors. Thus, extracellular spermine contributes significantly to ischemic neuronal injury through enhancing ASIC1a activity. Our data suggest new neuroprotective strategies for stroke patients via inhibition of polyamine synthesis and subsequent spermine–ASIC interaction.
Introduction
Acid-sensing ion channels (ASICs) are proton-gated members of the degenerin/epithelial Na+ channel family (Waldmann et al., 1997). Homomeric ASIC1a channels conduct Na+ and Ca2+, whereas other ASICs are mostly Ca2+ impermeable (Yermolaieva et al., 2004). In CNS neurons, ASIC1a-containing channels (hereafter referred to as ASIC1a channels) are involved in generating depolarizing currents in response to extracellular pH reduction between 6.9 and 5.0 (Waldmann et al., 1997), and both physiological and pathological acidosis activates these channels to increase neuronal firing (Vukicevic and Kellenberger, 2004). On appearance, this would seem paradoxical because the ASICs, especially ASIC1a, are known to completely desensitize within a few seconds during persistent acidosis (Krishtal, 2003). Recent studies have shown that certain ischemia-associated factors, including arachidonic acid and lactate (Immke and McCleskey, 2001; Allen and Attwell, 2002) as well as Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Gao et al., 2005), can enhance the ASIC1a current by slowing down channel desensitization, suggesting that desensitization is a critical feature that can affect the degree of acid-evoked neuronal damage through ASIC1a channels. Moreover, acidosis during ischemia is not static (Obrenovitch, 1995) (i.e., the affected brain regions are likely exposed to recurrent acidifications). Both the level of alkalinization and the rate of recovery from desensitization should strongly impact the degree of ASIC1a reactivation subject to a subsequent acidification and hence the severity of ischemic neuronal damage (DeGraba et al., 1992).
If the above idea is true, then reducing steady-state desensitization of the ASICs should facilitate channel reactivation during pH fluctuations. Given that ASICs contribute to ischemic neuronal injury (Xiong et al., 2004; Gao et al., 2005; Pignataro et al., 2007), this manipulation should enhance the ischemic damage. It was recently reported that extracellular spermine, an endogenous cationic polyamine of relatively high abundance in the mammalian brain, reduces the steady-state desensitization of ASICs, thus extending the range for their proton sensing and enhancing channel opening (Babini et al., 2002). As a result, spermine could potentially exacerbate ischemic neuronal injury through modulation of ASICs. In fact, simulated ischemia can rapidly and transiently induce the activity of ornithine decarboxylase (ODC), the rate-limiting enzyme responsible for the first step in polyamine synthesis. Pretreatment with α-difluoromethylornithine (DFMO), an irreversible and specific inhibitor of ODC, completely prevents the induction of ODC activity and the increase of polyamine levels within 24 h after ischemia, as well as ischemic injury (Kindy et al., 1994; Tantini et al., 2006).
Here, we present evidence that extracellular spermine potentiates ischemic neuronal injury in rodent models of ischemia in a manner that is primarily dependent on the activity of ASIC1a. We further show that the likely mechanism of spermine regulation of ASIC1a involves slowing down desensitization in the open state, shifting steady-state desensitization to more acidic pH, and accelerating recovery from desensitization between repeated periods of acid stimulation. Considering that acidosis may occur in a nonstatic fashion (Obrenovitch, 1995), these findings suggest a novel mechanism that underpins ASIC1a-mediated neuronal injury.
Materials and Methods
Focal ischemia.
The experimental protocols were approved by the Animal Care and Use Committee of Institute of Neuroscience, Shanghai, or by the Institutional Animal Care and Use Committee of Legacy Clinical Research and Technology Center (Portland, OR). Transient focal ischemia was induced by suture occlusion of the middle cerebral artery (MCAO) in male wild-type (WT) and ASIC1−/− mice (∼25 g) with congenic C57BL/6 background. Animals were anesthetized using 1.5% isoflurane, 70% N2O, and 28.5% O2 with intubation and ventilation. Rectal and temporalis muscle temperature was maintained at 37 ± 0.5°C with a thermostatically controlled heating pad and lamp. Cerebral blood flow was monitored by transcranial laser Doppler. Animals with blood flow not reduced below 20% were excluded. Intracerebroventricular injection was performed by stereotaxic technique using a microsyringe pump as described previously (Xiong et al., 2004; Pignataro et al., 2007). For each animal, 1 μl of artificial CSF (aCSF), or 1 μl of aCSF containing 2.5 mm spermine (Sigma-Aldrich), or 1 μl of aCSF containing 2.5 mm spermine plus 1 μm psalmotoxin 1 (PcTX1) (Peptides International), or 1 μl of aCSF containing 75 μg/μl DFMO (Sigma-Aldrich) alone or with 2.5 mm spermine, or 1 μl of aCSF containing 75 μg/μl DFMO plus 1 μm PcTX1 was injected 30 min before MCAO.
Animals were killed with isoflurane overdose 24 h after ischemia. Brains were removed, sectioned coronally at 1 mm intervals, and stained with the vital dye 2,3,5-triphenyltetrazolium hydrochloride (TTC). Infarction area was determined by subtracting the normal area stained with TTC in the ischemic hemisphere from the area of the nonischemic hemisphere. Infarct volume was calculated by summing infarction areas of all sections and multiplying by slice thickness. Manipulations and analyses were performed by individuals blinded to treatment groups.
Depending on the experimental design, 40 or 60 min MCAO was performed for moderate or severe ischemic model, respectively. We noticed that if the infarct volume was already above 50% (e.g., induced by 60 min MCAO) of the entire calculated area, and then the system was less sensitive to additional increase of the infarct volume (e.g., by exogenously applied spermine). For this reason, we chose a shorter duration of MCAO (40 min), by which only a small infarct volume (e.g., ∼10%) was produced in the absence of spermine.
Electrophysiological recordings.
ASIC currents were recorded using whole-cell patch-clamp techniques at room temperature (22–25°C). To illustrate the enhancing effect of spermine, the holding pH was 7.2, a condition that normally induces partial steady-state desensitization of ASIC1a channels (Babini et al., 2002). For voltage-clamp recordings, the membrane voltage was held at −60 mV unless otherwise noted. A computer-assisted solenoid valve-controlled rapid superfusion device (ALA-VM8; ALA Scientific Instruments) was used to precisely control the time intervals of activation of ASICs. The standard external solution contained the following (in mm): 150 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, buffered to various pH values with either 10 mm HEPES, pH 6.0–7.2, or 10 mm MES, pH < 6.0. The osmolarity of all solutions was kept at 300–330 mOsm/L. The patch pipette solution contained the following (in mm): 120 KCl, 30 NaCl, 1 MgCl2, 0.5 CaCl2, 5 EGTA, 2 Mg-ATP, 10 HEPES. The internal solution was adjusted to pH 7.2 with Tris base. All drugs for electrophysiological experiments were purchased from Sigma-Aldrich.
Primary neuronal cultures.
Primary cultures of rat hippocampal neurons were prepared according to previously described techniques (Gao et al., 2005). Hippocampal neurons from embryonic day 18 Sprague Dawley rats were isolated by a standard enzyme treatment protocol. Briefly, hippocampi were dissociated in Ca2+-free saline with sucrose (20 mm) and plated (1–5 × 105 cell/ml) on poly-d-lysine (Sigma-Aldrich)-coated coverglasses. The neurons were grown in DMEM (Invitrogen) with l-glutamine plus 10% fetal bovine serum (Invitrogen) and 10% F12. Neurons were cultured with Neurobasal medium supplemented with B27 and maintained at 37°C in a humidified 5% CO2 atmosphere incubator.
Primary cultures of mouse hippocampal or cortical neurons were prepared according to previously described techniques (Chu et al., 2003). Briefly, postnatal day 1 C57BL/6 WT, or ASIC1 or ASIC2 knock-out mice (with congenic C57BL/6 background) were anesthetized with halothane. Brains were removed rapidly and placed in ice-cold Ca2+- and Mg2+-free PBS. Tissues were dissected and incubated with 0.05% trypsin-EDTA for 10 min at 37°C, followed by trituration with fire-polished glass pipettes, and plated in poly-l-ornithine-coated 35 mm culture dishes at a density of 1 × 106 cells per dish. Neurons were cultured with Neurobasal medium supplemented with B27 and maintained at 37°C in a humidified 5% CO2 atmosphere incubator. Cultures were fed twice a week and used for electrophysiological recording 12–14 d after plating. For neuron cultures of both rat and mouse, glial growth was suppressed by addition of 5-fluoro-2-deoxyuridine (20 μg/ml; Sigma-Aldrich) and uridine (20 μg/ml; Sigma-Aldrich).
Transfection of Chinese hamster ovary cells.
Chinese hamster ovary (CHO) cells grown in 35 mm dishes were transfected by using Lipofectamine 2000 (Invitrogen) according to standard protocols. Briefly, 1 d after plating, cells (2 ml of medium per culture) were transfected with 1–3 μg of rat cDNAs encoding ASICs (Wang et al., 2006). GFP was used for identification of the transfected cells. Electrophysiological recordings were done 1–2 d after transfection. Human ASIC1a was used for constructing all mutants in Figure 7 and supplemental Figure 5 (available at www.jneurosci.org as supplemental material).
Figure 7.
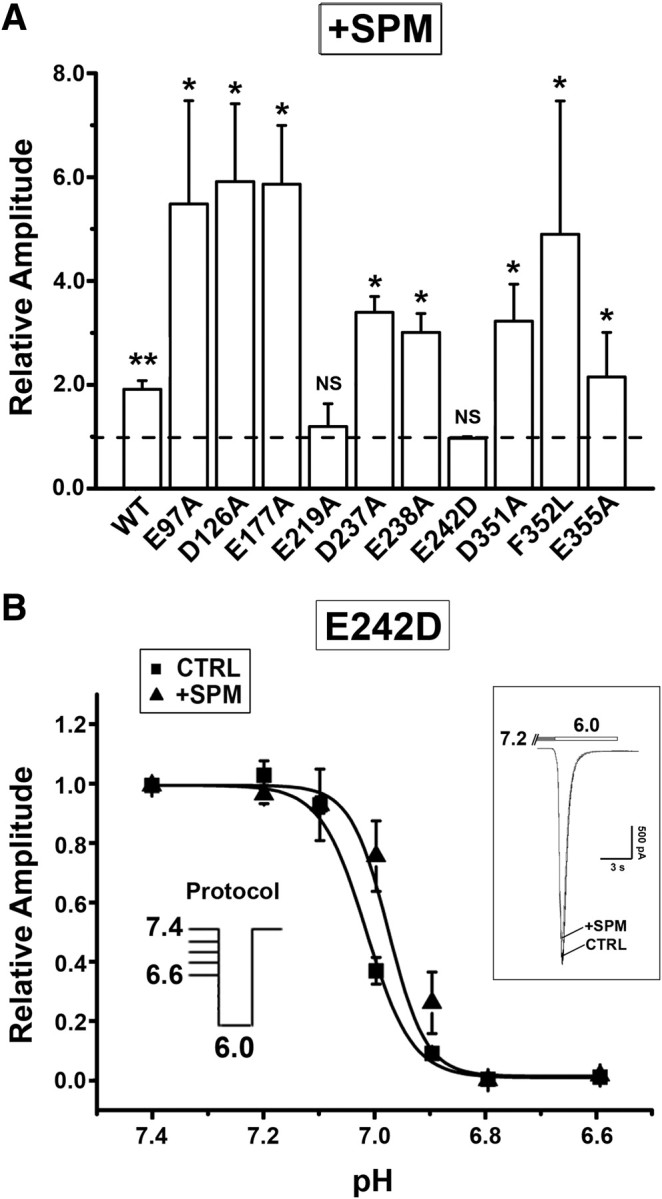
Mutations at the PcTX1-binding sites affect SPM effects. A, Summary of SPM-mediated enhancement of I6.0 in WT and mutated ASIC1a channels. Results are normalized to the peak amplitude of I6.0 in the absence of SPM, which is indicated as dashed line (the ratio of 1) (n = 3–5). *p < 0.05, **p < 0.01, compared with the ratio of 1, paired t test. B, SPM (0.25 mm) induced negligible change in the pH dependence of steady-state desensitization in ASIC1aE242D mutant (n = 3–6; unpaired t test shows no significant difference). The holding pH varied from 7.4 to 6.6. The inset shows that SPM did not affect ASIC1aE242D activation when holding pH was 7.2. Error bars indicate SEM.
Oxygen and glucose deprivation.
The cultures were transferred to an anaerobic chamber containing a 5% CO2 and 95% N2 (<0.2% O2) atmosphere. They were washed thrice with deoxygenated glucose-free bicarbonate solution and maintained anoxic for 2 h at 37°C. Oxygen and glucose deprivation (OGD) was terminated by washing the cultures with oxygenated glucose (20 mm)-containing bicarbonate solution. The cultures were maintained for an additional 24 h at 37°C in a humidified 5% CO2 atmosphere.
Cell injury assay—lactate dehydrogenase measurement.
Cells were washed thrice with an external solution and randomly divided into treatment groups. After acidic solution incubation or OGD, cells were washed and incubated in the normal culture medium at 37°C for 24 h. Lactate dehydrogenase (LDH) release was measured in the culture medium using the LDH assay kit (Roche Molecular Biochemicals). An aliquot of the medium (100 μl) was transferred from culture wells to wells of 96-well plates and mixed with 100 μl of reaction solution provided by the kit. Optical density was measured at 492 nm 45 min later, using a microplate reader (Spectra Max Plus; Molecular Devices). Background absorbance at 620 nm was subtracted. The maximal releasable LDH in each well was obtained by a 15 min incubation with 1% Triton X-100 at the end of each experiment. Nimodipine (5 μm), (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK-801) (20 μm), and CNQX (20 μm) were applied in all groups in acidotoxicity experiments (see Fig. 9A,B) to prevent Ca2+ entry mediated by voltage-gated Ca2+ channels and ionotropic glutamate receptors.
Figure 9.
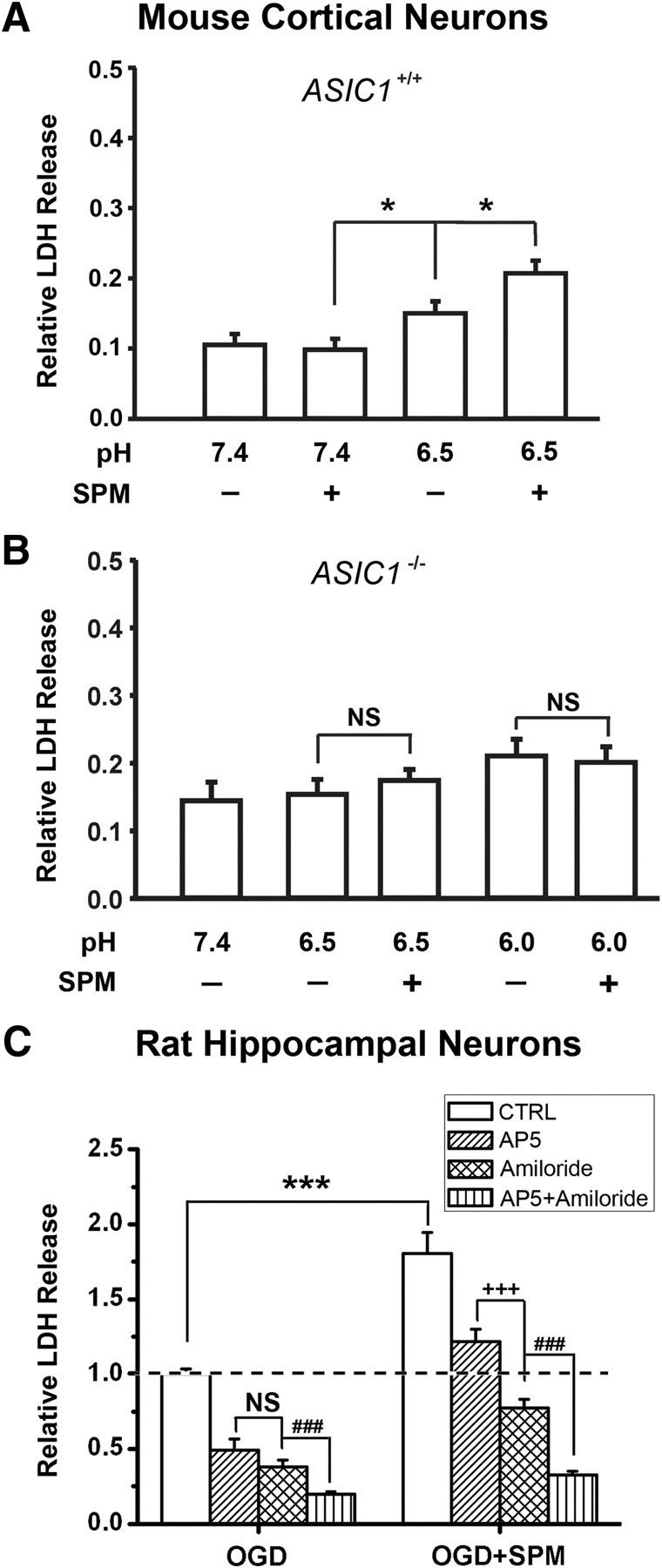
SPM exacerbated ASIC1a-mediated ischemic neuronal damage. A, SPM (0.25 mm) enhanced acid-induced LDH increase in cultured cortical neurons from WT mice (n = 12). *p < 0.05, unpaired t test. B, ASIC1 deletion attenuated the enhancing effect of SPM (n = 8; NS, no statistical significance, unpaired t test). For the experiments in A and B, MCN (MK-801, CNQX, and nimodipine) was present in the extracellular solution to block secondary activation of glutamate receptors and voltage-gated Ca2+ channels. C, SPM (0.25 mm) exacerbated OGD-induced neuronal damage in cultured rat hippocampal neurons (n = 9). ***p < 0.001 versus SPM-untreated group, unpaired t test. Amiloride (100 μm) produced significantly more neuroprotection than AP5 (20 μm) in SPM-treated group (n = 9). +++p < 0.001; NS, no statistical significance, unpaired t test. In addition, cotreatment with AP5 and amiloride produced more protection than amiloride alone in both control and SPM-treated groups (n = 9). ###p < 0.001 versus amiloride-treated group, unpaired t test. Results in A and B are normalized to the relative maximum LDH value of cell lysates. Results in C are normalized to the averaged LDH value after OGD treatment in the absence of SPM (“CTRL”; dashed line). Error bars indicate SEM.
Ca2+ imaging.
Fura-2 fluorescent Ca2+ imaging was performed as described previously (Gao et al., 2005; Duan et al., 2007). Briefly, cultured hippocampal neurons grown on 8 × 8 mm glass coverslips were washed thrice with PBS and incubated with 1 μm fura-2 acetoxymethyl ester (Sigma-Aldrich) in PBS for 20 min at 37°C. Cells were again washed thrice and then incubated in a standard extracellular solution for 30 min. The coverslip was transferred to a perfusion chamber placed on an inverted microscope (Nikon TE2000-E), which is equipped with a 40× UV fluor oil-immersion objective lens and a cooled CCD camera (Hamamatsu). The fluorescence excitation source was a 75 W xenon arc lamp. Ratio images were acquired by using alternating excitation wavelengths (340/380 nm) with a Polychrome V monochromator (TILL Photonics), and fura-2 fluorescence was detected at the emission wavelength of 510 nm. Digitized images were acquired and analyzed in a PC controlled by SimplePCI (Compix). Ratio images (340/380 nm) were analyzed by averaging pixel ratio values in circumscribed regions of cells in the field of view. The values were exported to Origin 8.0 for additional analysis.
Statistics.
Values are presented as mean ± SE. Statistical comparisons were performed using unpaired or paired Student's t tests where values of p < 0.05 are considered significant.
Results
Extracellular spermine exacerbates ASIC1-mediated ischemic brain injury in vivo
To explore the role of spermine in ASIC-mediated neuronal injury during ischemia, we first examined the effect of exogenously applied spermine in the mouse model of focal ischemia, in which ischemia was induced by MCAO (Xiong et al., 2004). We used 40 min MCAO to produce a moderate level of brain injury, which is assessed by measuring infarct volumes using vital dye TTC. In WT (ASIC1+/+) mice intracerebroventricularly injected with aCSF at 30 min before MCAO (see Materials and Methods), the infarct volume was 11.8 ± 0.2% (n = 4 mice). However, if 2.5 mm spermine was included in the aCSF, the infarct volume was dramatically increased to 44.4 ± 6.8% (n = 4 mice) (Fig. 1A,B). To test whether the spermine effect is dependent on ASIC1a, we coinjected spermine with PcTX1 (1 μm), a selective homomeric ASIC1a channel inhibitor (Escoubas et al., 2000), and observed a significant reduction of the infarct volume to 18.1 ± 4.0% (n = 4 mice) (Fig. 1A,B). As a control, heat-inactivated PcTX1 was ineffective in reducing the spermine effect (infarct volume of 46.3 ± 4.2%; n = 4 mice). These results suggest a neurotoxic effect of spermine during ischemia that depends mostly on ASIC1a channels. Consistent with this notion, when compared with the WT mice, the same concentration of spermine caused significantly less increase in the infarct volume in ASIC1−/− mice, which reached only 20.4 ± 2.5% (n = 4 mice). Notably, with the relatively short MCAO, the protective effect of ASIC1 knock-out, in the absence of exogenously applied spermine, was not obvious (Fig. 1B), indicating that ASIC1 alone is insufficient to mediate significant brain damage under moderate ischemic conditions.
Figure 1.

Neuroprotection by ASIC1a blockade and ASIC1 gene knock-out against spermine (SPM)-induced brain injury in mouse ischemia model in vivo. A, TTC-stained brain sections showing infarct area (image) in the mild ischemic brains from ASIC1+/+ (left; injected with aCSF, 2.5 mm SPM, or 1 μm PcTX1 plus SPM) and ASIC1−/− mice (right; injected with aCSF or SPM). B, Summary data for A showing infarct volumes in brains from ASIC1+/+ and ASIC1−/− under different treatments (n = 4 for each group). *,+,#p < 0.05, unpaired t test. Note SPM still caused a small but significant increase in infract volume in ASIC1−/− mice. Error bars indicate SEM.
Spermine specifically potentiates the activity of ASIC1a channels
To gain insights into the relationship between spermine and ASIC1a regulation during brain ischemia, we examined the effect of extracellular spermine on ASIC currents in cultured cortical neurons prepared from WT (ASIC+/+), ASIC1−/−, or ASIC2−/− mice. For recording acid-induced current, cells were held at −60 mV in the whole-cell configuration. As shown in Figure 2, A and B, spermine (0.25 mm) enhanced proton (pH 6.0)-induced currents (I6.0) recorded in cortical neurons from C57BL/6 mice, a WT control with the same genetic background as ASIC1−/− and ASIC2−/− mice. In cortical neurons prepared from ASIC1−/− mice, little or no current was activated at pH 6.0, but at pH 4.0, a transient inward current followed by a slow and sustained component was activated (Fig. 2C), a profile indicative of ASIC2a/2b channels (Chu et al., 2004). In these neurons, application of spermine had no effect (Fig. 2C,D), suggesting that ASIC2a/2b are not involved in the spermine-induced potentiation of ASIC currents. By contrast, spermine significantly enhanced I6.0 in cortical neurons prepared from ASIC2−/− mice (Fig. 2E,F). Moreover, the current integrals (area under the curve) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) showed a larger enhancement by spermine than the peak current amplitudes. Our results indicate that the potentiating effect of spermine is specific to ASIC1 but not ASIC2 isoforms.
Figure 2.

SPM enhanced ASIC currents in cultured cortical neurons from ASIC2−/− but not ASIC1−/− mice. Representative current traces (left) and summary data (mean ± SE; right) are shown. A, B, SPM (0.25 mm) enhanced I6.0 in neurons from WT control mice (n = 6). *p < 0.05, paired t test. C, D, SPM (0.25 mm) had no effect on I4.0 induced by pH 4.0 in neurons from ASIC1−/− mice. NS, No statistical significance, paired t test (n = 11). E, F, Knock-out of ASIC2 did not affect the enhancement of I6.0 by SPM (0.25 mm) (n = 11). **p < 0.01, paired t test.
The specificity of spermine on ASIC1 currents was further examined in CHO cells expressing various ASIC subunits. Consistent with the findings in cortical neurons, spermine (0.25 mm) markedly enhanced the peak amplitudes of proton-evoked currents mediated by homomeric ASIC1a or heteromeric 1a/2a channels, but not those mediated by homomeric ASIC1b, 2a, and 3 channels, or by heteromeric ASIC1a/1b and 1a/2b channels (Fig. 3A,B). Thus, spermine specifically affects the two major forms of ASICs (1a, 1a/2a) known to be present in CNS neurons (Baron et al., 2002; Askwith et al., 2004). This ASIC1a channel-specific regulation may explain the spermine-dependent and ASIC1a-mediated ischemic brain damage (Fig. 1A,B).
Figure 3.
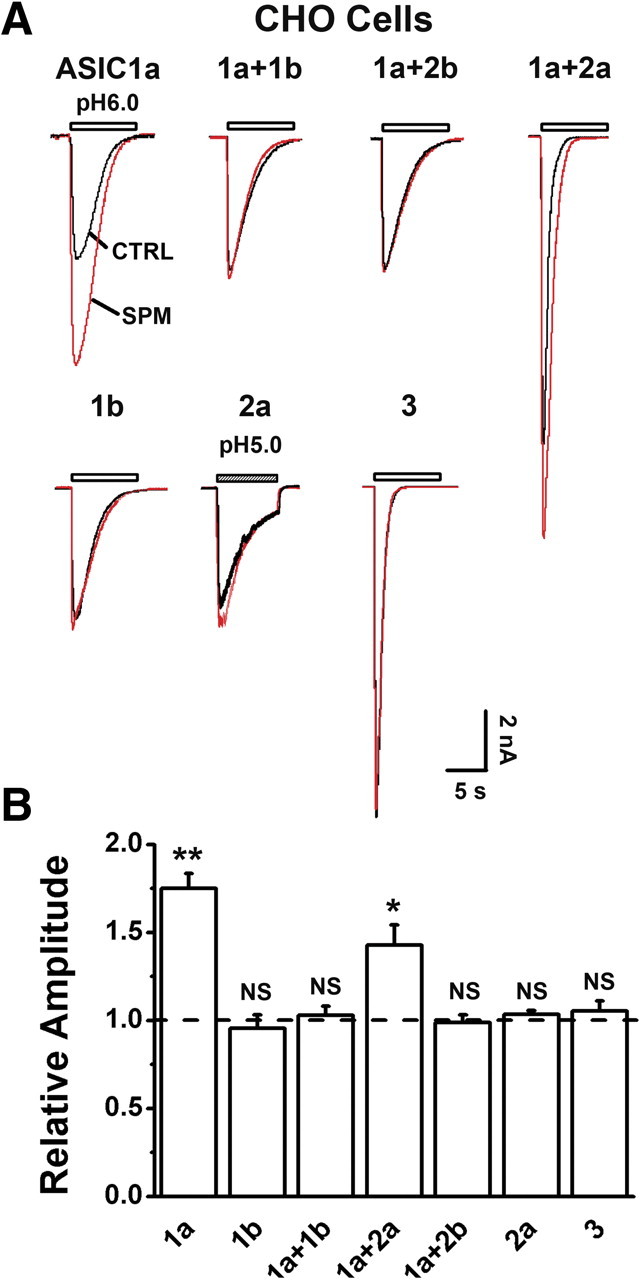
Subunit specificity of SPM enhancement of ASIC currents in CHO cells. A, Representative current traces showing that SPM (0.25 mm) enhanced ASIC currents mediated by homomeric ASIC1a and heteromeric ASIC1a/2a, but not those by homomeric ASIC1b, ASIC2a, and ASIC3 or heteromeric ASIC1a/1b, 1a/2b channels. B, Pooled data from experiments in A, normalized to the mean amplitude of I6.0 (I5.0 for ASIC2a) before SPM application (dashed line) for each cell (n = 15 for 1a; n = 4–8 for the rest of combinations). **p < 0.01; *p < 0.05; NS, no statistical significance, paired t test. Error bars indicate SEM.
Effect of spermine on ASIC1a channels is unique among polyamines
During polyamine biosynthesis, the primary rate-limiting enzyme ODC catalyzes the synthesis of diamine putrescine, which is then converted to triamine spermidine by spermidine synthase and to tetraamine spermine by spermine synthase (Bernstein and Müller, 1999). To determine how common among polyamines the regulation of ASICs is, we compared the effect of putrescine, spermidine, and spermine on proton-evoked currents in cultured rat hippocampal neurons, a region known to be vulnerable to brain ischemia (Gao et al., 2005). In these neurons, I6.0 exhibits properties indistinguishable from those of the mouse cortical neurons but of smaller magnitude. Similar to its effect on ASIC1a currents in mouse cortical neurons (Fig. 2A,B), spermine (0.25 mm) markedly increased I6.0 in rat hippocampal neurons in a reversible and dose-dependent manner (Fig. 4A,C), suggesting that spermine exerts a similar potentiation effect on ASIC1a currents in both mice and rats and in both cortical and hippocampal neurons. The extent of potentiation in rat (∼60%) (Fig. 4B) is larger than in mouse neurons (∼20%) (Fig. 2B). This may be attributed to the difference in ASIC subunit composition (i.e., ratio of 1a to 1a/2a) in CNS neurons between the two species (Baron et al., 2008) and in different brain regions, an idea supported by our observation that spermine had larger enhancement effect on homomeric ASIC1a than heteromeric 1a/2a channels (Fig. 3B). Dose–response analysis revealed a half-maximum concentration (EC50) of 495 ± 39 μm and a Hill coefficient of 1.07 ± 0.02 for spermine in enhancing I6.0 in rat hippocampal neurons (Fig. 4C). In contrast to spermine, putrescine had no effect on I6.0 (Fig. 4A,B) when applied at higher concentrations (up to 10 mm), and slightly decreased I6.0 at low concentrations (0.1 and 0.25 mm). The structurally closer polyamine, spermidine, mildly enhanced I6.0 only at concentrations >1 mm (Fig. 4C). Thus, spermine is unique among polyamines in its modulation of ASIC1a channels.
Figure 4.

SPM, but not putrescine and spermidine, enhanced ASIC currents in cultured rat hippocampal neurons. A, B, Representative current traces (A) and summary data (B) (n = 6) showing the effects of putrescine (PUT), spermidine (SPD), and SPM at 0.25 mm on I6.0. **p < 0.01; NS, no statistical significance, compared with control I6.0 (dashed line), paired t test. C, Dose–response curves for polyamine enhancement of ASIC currents. Data (n = 6–8 for each group) are normalized to the mean amplitude of I6.0 before polyamine application (CTRL) (data not shown) for each neuron before averaging. The inset shows representative current traces at CTRL and different concentrations of SPM. D, SPM (0.25 mm) has no effect on pH50 (6.18 ± 0.04 without SPM; 6.20 ± 0.04 with SPM) or Hill coefficient (1.16 ± 0.05 without SPM; 1.14 ± 0.07 with SPM) of proton-induced activation of ASIC currents. For each point, n = 6–8. The holding pH was 7.2. Error bars indicate SEM.
Spermine alters the pH sensitivity of ASIC1a desensitization and recovery
We next performed a series of experiments to investigate the underlying mechanisms by which spermine exerts its modulation. First, we examined the effect of membrane potential because spermine is positively charged at or below physiological pH, pH 7.2, and thus could exert its effect by screening surface charges. However, when ASIC currents were activated at different holding potentials (Vh, from −60 to +60 mV), spermine enhanced the I6.0 to a similar extent (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This observation, together with the notion that spermine is unique among multiple polyamines in potentiating I6.0, indicates that spermine modulates ASIC1a channels via specific mechanisms rather than nonspecific charge screening. Second, the ion selectivity of ASICs does not appear to be altered by spermine since the reversal potential of I6.0 remained unchanged (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Third, the pH sensitivity of ASICs was unaffected by spermine because the half-maximum effective pH value (pH50) and the Hill coefficient remained unchanged (Fig. 4D). Fourth, because extracellular spermine can be transported into the cytoplasm via plasma membrane transporters (Seiler et al., 1996; Masuko et al., 2003), we examined the possibility that spermine may act from the cytoplasmic side. However, intracellular loading of spermine did not influence I6.0; subsequent bath application of spermine still enhanced the ASIC currents (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). In addition, bath application of spermine effectively potentiated homomeric ASIC1a channels in outside-out patches (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material), which are devoid of intracellular cytosol, indicating that the action of spermine does not involve soluble cytoplasmic components.
Next, we compared the desensitization properties of ASIC currents in the absence or presence of spermine. Steady-state desensitization was examined by superfusion of cultured hippocampal neurons for 2 min in solutions with pH values ranging from 7.8 to 6.8 before application of the pH 6.0 solution. The pH50 value for steady-state desensitization displays a rightward shift from 7.39 ± 0.05 to 7.22 ± 0.03 with the presence of 0.25 mm spermine (n = 6) (Fig. 5A), indicative of a decreased apparent H+ affinity under steady-state conditions. The Hill coefficient was 0.75 ± 0.10 with spermine and 0.89 ± 0.08 without spermine. Interestingly, the shift in pH50 value was more pronounced when human ASIC1a was tested (pH50 was 7.22 ± 0.01 in the absence of spermine, and 7.00 ± 0.01 in the presence of spermine) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). The shift of the steady-state desensitization curve to lower pH suggests that, during recurrent acidifications, reactivation of the ASIC1a channel can occur with more moderate alkalinization when spermine is present. Furthermore, as indicated by the time constant (τ) of the decay of I6.0, spermine significantly slowed down the desensitization rate of the ASIC currents (Fig. 5B). These results suggest that spermine alters the proton dependence of ASIC1a desensitization and contributes significantly to repeated ASIC1a channel activation during pH fluctuations under acidosis conditions.
Figure 5.

SPM reduced desensitization of homomeric ASIC1a channels expressed in CHO cells. A, SPM (0.25 mm) induced a rightward shift of the pH dependence of steady-state desensitization. For each point, n = 6–8. The holding pH varied from 7.8 to 6.8. B, Effect of SPM (0.25 mm) on time constant (τ) of open-state desensitization of ASIC1a channels. The dashed line indicates control τ without SPM (n = 10). **p < 0.01, paired t test. The inset shows scaled and superimposed traces: gray, without SPM; black, with SPM. C, Representative current traces and statistic data showing the acceleration of ASIC1a channel reactivation by SPM (0.25 mm). Cells were held at −60 mV and currents activated by repetitive pH drops from pH 7.2 to 6.0. For each cell, several traces are superimposed, including one control (I1) and several subsequent reactivated currents (I2) recorded at 4, 8, 20, 40, and 60 s after the control pH drop. The amplitude ratios of reactivated ASIC1a current to control current (I2/I1) are plotted as a function of time intervals between the end of the control pH drop and the onset of the subsequent pH drop. Data (n = 4–7) are fitted with a single exponential function (τ = 19.1 s for control group, and 9.3 s for SPM-treated). Error bars indicate SEM.
The decrease in pH sensitivity of desensitization suggests that spermine may facilitate recovery of ASIC1a channels from desensitization. To test this possibility, we used a paired stimulation protocol with varying intervals between two consecutive applications of the pH 6.0 solution. The holding pH was 7.2, which is in the range greatly affected by spermine as shown in Figure 5A. In ASIC1a-expressing CHO cells, spermine (0.25 mm) significantly accelerated the recovery of the ASIC1a currents from desensitization, as indicated by the ratio of the peak I6.0 between the second and the first acid stimulation (τ = 19.1 s for control group, and 9.3 s for SPM-treated) (Fig. 5C). Therefore, the recovery from desensitization of ASIC1a channels is greatly accelerated by spermine.
Spermine competes with PcTX1 in binding to ASIC1a
It has been previously reported that the specific ASIC1a channel antagonist PcTX1 acts by desensitizing the channel through increasing its apparent H+ affinity (Chen et al., 2005). Since spermine reduces the H+ affinity (Fig. 5A), it might compete with PcTX1 in modulating ASIC1a currents. Indeed, as shown in Figure 6A, spermine (0.25 mm) reversed the inhibitory effect of low concentrations of PcTX1, but failed to affect the inhibition by high concentrations of PcTX1 (e.g., 1 and 5 nm) on I6.0 in ASIC1a-expressing CHO cells. However, at higher spermine concentrations (>1 mm), the inhibition by 5 nm PcTX1 was attenuated (Fig. 6B). Since spermine also shifted steady-state desensitization of ASIC1a to lower pH, we tested whether PcTX1 affects such a function. In the presence of PcTX1 (5 nm), spermine failed to shift the steady-state desensitization of ASIC1a channels (pH50 was 7.55 ± 0.04 in the absence of spermine, and 7.54 ± 0.02 in the presence of spermine; n = 6). Together, these results, showing antagonizing effects between spermine and PcTX1, suggest that the two drugs may compete for common binding site(s) and affect the ASIC1a channel function in opposite ways.
Figure 6.

SPM effect was occluded by PcTX1. A, Dose–response of PcTX1 for the inhibition of I6.0 in the absence (square) or presence (triangle) of 0.25 mm SPM in CHO cells expressing ASIC1a. All responses were normalized to the peak I6.0 in the absence of PcTX1 and SPM. Each point represents an average of four to seven cells. **p < 0.01, unpaired t test. B, Dose–response of SPM for the potentiation of I6.0 in the absence (square; derived from Fig. 4C) or presence (triangle) of 5 nm PcTX1. All responses are normalized to the peak I6.0 in the presence of 10 mm SPM. Each point represents an average of four to six cells. C, Representative current traces and statistical data showing that ASIC1a currents were very small under the Ca2+-free condition and were dramatically increased by SPM (n = 4). ***p < 0.001, paired t test. Error bars indicate SEM.
Interestingly, such a competitive mode of action with PcTX1 has been suggested for extracellular Ca2+ (Chen et al., 2005), which also alters the desensitization of ASIC1a channels (Babini et al., 2002) in a similar fashion as spermine. Our standard extracellular solution contains 2 mm Ca2+, which is sufficient to regulate the desensitization of ASIC1a and enhance channel currents. Therefore, to learn how extracellular Ca2+ affects the role of spermine on ASIC1a channel regulation, the effect of spermine was tested using a Ca2+-free bath solution. In the absence of extracellular Ca2+ and spermine, I6.0 was very small, consistent with notion that extracellular Ca2+ is important to maintain ASIC1a activity (Babini et al., 2002). Addition of spermine caused a dramatic increase (>20-fold) in the current evoked by pH 6.0 (Fig. 6C), suggesting that spermine and Ca2+ may share a similar mechanism in modulating ASIC1a channels and thus further argue for a role of the PcTX1 binding site(s) in such a modulation, as has been implicated for Ca2+ (Chen et al., 2005).
Next, we attempted to identify the critical site(s) of spermine modulation by testing a series of mutants at the acidic amino acids of the extracellular pocket shown to be involved in PcTX1 binding based on previous docking studies using 3D models of human ASIC1 (Pietra, 2009; Qadri et al., 2009). These include E97, D126, E177, E219, D237, E238, E242, D347, D351, F352, and E355 located at the interface between two neighboring subunits. We used human ASIC1a because of the larger and more clear-cut shift in pH50 in steady-state desensitization in response to spermine (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Recording of I6.0 in CHO cells expressing these mutated channels showed that all mutants were functional except for ASIC1aD347A, which did not respond to pH 6.0 solution and was excluded from additional analysis. Notably, spermine failed to enhance I6.0 in E219A and E242D (but enhanced E242A) (data not shown) mutants. By contrast, spermine increased I6.0 in E355A to the extent similar as the WT channel and induced even larger enhancement of I6.0 in E97A, D126A, E177A, D237A, D238A, D351A, and F352L mutants when compared with the WT (Fig. 7A). The different results in E242D and E242A were interesting. Given the structural similarity of Asp and Glu, we tested two additional mutants, E242C and E242K, and found that spermine did not enhance I6.0 in these mutants either (supplemental Fig. 5C, available at www.jneurosci.org as supplemental material). We also found that E219A or E242D substitution completely eliminated the steady-state desensitization of ASIC1a at the holding pH of 7.2 (supplemental Fig. 5B, available at www.jneurosci.org as supplemental material). By contrast, mutations at E97, D126, E177, D237, E238, D351, F352, and E355 all enhanced desensitization (supplemental Fig. 5A,B, available at www.jneurosci.org as supplemental material). Furthermore, pH50 values derived from the steady-state desensitization curves were significantly shifted to acidic values in both E219A and E242D mutants [E219A, 6.85 ± 0.07 (supplemental Fig. 5D, available at www.jneurosci.org as supplemental material); E242D, 7.01 ± 0.05 (Fig. 7B); WT, 7.22 ± 0.01 (supplemental Fig. 4, available at www.jneurosci.org as supplemental material)]. More interestingly, the shift of the pH50 values in response to spermine was mostly attenuated in the E219A and E242D mutants [E219A, 6.79 ± 0.02 (supplemental Fig. 5D, available at www.jneurosci.org as supplemental material); E242D, 6.97 ± 0.04 (Fig. 7B); WT, 7.00 ± 0.01 (supplemental Fig. 4, available at www.jneurosci.org as supplemental material)]. Together, these results are consistent with the notion that residues of putative PcTX1 binding sites, especially E219 and E242, are important for spermine interaction with ASIC1a, regulating the channel desensitization.
Spermine enhances ASIC1a-mediated Ca2+ elevation and membrane depolarization
Activation of homomeric ASIC1a channels leads to elevation of intracellular Ca2+ concentration ([Ca2+]i), which activates ischemic injury-related kinases such as CaMKII (Colbran, 1992; Zha et al., 2006; Vila-Petroff et al., 2007) and contributes to ischemic neuronal injury (Xiong et al., 2004; Yermolaieva et al., 2004; Gao et al., 2005). We next examined whether spermine affects acid-induced [Ca2+]i elevation by Ca2+ imaging of cultured rat hippocampal neurons. In the presence of the blockers for Na+ channels (1 μm TTX), L-type Ca2+ channels (5 μm nimodipine), and ionotropic glutamate receptors (20 μm AP5 and 20 μm CNQX), reduction of extracellular pH from 7.2 to 6.0 induced a transient elevation of [Ca2+]i (Fig. 8A). The effect was abolished by the ASIC antagonist amiloride (100 μm) (data not shown), suggesting that the acid-induced [Ca2+]i elevation could be attributed to Ca2+ influx mediated by ASIC1a channels. Spermine significantly enhanced the acid-induced [Ca2+]i elevation in a dose-dependent manner (Fig. 8A–C), but by itself the polyamine had no effect on the basal [Ca2+]i, consistent with the notion that spermine enhances ASIC1a channel function.
Figure 8.

SPM enhanced acid-induced elevation in [Ca2+]i and membrane depolarization in cultured rat hippocampal neurons. A, Representative plot of 340/380 ratio changes (corresponding to [Ca2+]i changes) induced by pH decrease in the absence or presence of 0.25 mm SPM. Example ratio images before (CTRL) and during pH drop in the absence, pH 6.0, and presence of SPM (SPM + pH 6.0) are shown on the top. To block secondary activation of glutamate receptors and voltage-gated Na+ and Ca2+ channels, 20 μm AP5, 20 μm CNQX, 1 μm TTX, and 5 μm nimodipine were included in the external solution. B, C, Average 340/380 ratios showing SPM dose-dependently enhanced peak (B) or average integrated Ca2+ responses (C) evoked by pH 6.0 solution (n = 27). ***p < 0.001, paired t test. D, Representative traces showing membrane depolarization (Vm) induced by a pH reduction from 7.2 to 6.0 in the absence or presence of 0.25 mm SPM. Nimodipine (5 μm), AP5 (20 μm), CNQX (20 μm), and TTX (1 μm) were included in the external solutions. E, F, Statistical data indicating SPM dose-dependently enhanced peak (E) or average integrated membrane depolarization (F) (relative Vm) induced by pH 6.0 solution (n = 5–8). *p < 0.05, ***p < 0.001, paired t test. The dashed lines indicate control values without SPM. Error bars indicate SEM.
In addition to increasing [Ca2+]i, membrane depolarization resulting from ASIC activity is known to facilitate glutamate receptor-mediated excitatory synaptic transmission (Wemmie et al., 2002), contributing to excitotoxicity associated with ischemic neuronal injury. To examine the effect of spermine on ASIC-mediated membrane potential changes, current-clamp experiments were performed in the presence of pharmacological blockers for Na+ and Ca2+ channels as well as ionotropic glutamate receptors as indicated above. As shown in Figure 8D–F, reducing extracellular pH from 7.2 to 6.0 evoked a robust depolarization in rat hippocampal neurons and spermine markedly enhanced this effect.
Spermine exacerbates ASIC1a-mediated neuronal damage in vitro
To further confirm the role of ASIC1a channels in spermine-induced neuronal damage, we performed cell toxicity assays in cortical neurons cultured from ASIC1+/+ and ASIC1−/− mice. In the presence of nimodipine, MK-801, and CNQX (see Materials and Methods), spermine treatment significantly enhanced acidosis-induced LDH release from cultured cortical neurons prepared from WT mice (Fig. 9A) but not those from ASIC1−/− mice (Fig. 9B). Moreover, in a model of ischemia induced by OGD, a condition involving both NMDA receptor (NMDAR)-dependent excitotoxicity and ASIC-dependent acidotoxicity (Goldberg and Choi, 1993; Xiong et al., 2004), spermine markedly enhanced neuronal damage in cultured rat hippocampal neurons (Fig. 9C), thus highlighting the relevance of spermine–ASIC1a interaction in ischemic neuronal cell damage. Interestingly, whereas blockade of ASICs with amiloride (100 μm) produced similar neuroprotection as the blockade of NMDARs with AP5 (20 μm) in OGD cultures in the absence of spermine, amiloride produced significantly greater neuroprotection than AP5 in spermine-treated OGD cultures (Fig. 9C). This result is consistent with the in vivo findings that spermine enhanced ischemic injury, and deleting ASIC1 (ASIC1−/− mice) mostly prevented spermine-induced increase in cortical infarct volume (Fig. 1A,B). Together, these data strongly argue for a major role of ASIC1a channels in spermine-induced cytotoxicity in both cortical and hippocampal neurons under ischemic conditions.
Endogenous spermine exacerbates ASIC1a-mediated ischemic damage
The relevance of spermine-evoked enhancement of ASIC1a channel activation to ischemic neuronal damage most likely lies in the release of endogenous spermine from affected brain regions. To test this possibility, we blocked spermine biosynthesis using a selective ODC inhibitor, DFMO (Wallace and Fraser, 2004). In the OGD model, DFMO (50 μm; added 30 min before OGD and remained during OGD and reperfusion for 24 h) treatment significantly attenuated the neuronal damage, as assessed by LDH release, in cultured hippocampal neurons from WT mice (Fig. 10A, left panel). By contrast, DFMO was ineffective in protecting OGD-induced neuronal death in cultured hippocampal neurons from ASIC1−/− mice (Fig. 10A, right panel). These results demonstrate that endogenously released spermine is involved in inducing neuronal damage during ischemia and this effect is mediated primarily through ASIC1a channels. In contrast to the finding that DFMO occluded the effect of ASIC1a inhibition, blockade of NMDARs by AP5 still significantly attenuated OGD-induced neuronal death (Fig. 10A), suggesting that inhibition of spermine synthesis did not influence the NMDAR-dependent ischemic damage in neurons. Thus, the endogenous spermine contributes mainly to acidotoxicity rather than NMDAR-mediated excitotoxicity.
Figure 10.

Blockade of endogenous SPM biosynthesis significantly attenuated ASIC1a-dependent ischemic neuronal damage. A, DFMO (50 μm) significantly attenuated OGD-induced neuronal damage in cultured hippocampal neurons from WT mice (n = 12). ***p < 0.001, unpaired t test. However, in neurons from ASIC1−/− mice, DFMO had no protective effect (n = 7; NS, no statistical significance, unpaired t test). AP5 (20 μm) caused additional neuroprotection in DFMO-treated neurons in both ASIC1+/+ (n = 12) and ASIC1−/− (n = 9) groups. ###p < 0.001, unpaired t test. All results are normalized to the averaged LDH value of OGD-treated ASIC+/+ neurons (“CTRL”; dashed line). B, In a severe ischemia model (60 min MCAO), PcTX1 (1 μm) and DFMO (75 μg/μl) injection reduced the infarct volume in brains from WT mice in an additive manner (n = 4–6). **p < 0.01, ***p < 0.001, unpaired t test, compared with aCSF group from WT mice. #p < 0.05, ###p < 0.001, unpaired t test, compared with PcTX1 and DFMO cotreatment group. Coinjection of SPM (2.5 mm) with DFMO reversed the neuroprotective effect of DFMO. ##p < 0.01, unpaired t test, compared with DFMO alone. ASIC1 deletion also reduced the infarct volume but DFMO was ineffective in ASIC1−/− mice (n = 6). +++p < 0.001, unpaired t test, compared with aCSF group from WT mice. The images on the top show examples of TTC-stained brain sections and infarct areas for the corresponding treatment groups. Error bars indicate SEM.
As shown in Figure 1B, a short duration of MCAO (40 min) caused similar infarct volumes (∼10%) in both aCSF-injected WT ASIC1+/+ and ASIC1−/− mice, indicating that ASIC1a channels alone do not cause significant damage under mild ischemia. It is possible that, under this condition, the level of extracellular spermine, presumably released from damaged cells as a result of ischemia, may be insufficient to potentiate the ASIC1a channels to such an extent that substantial neuronal damage emerges. This possibility is particularly important if spermine plays a more decisive role than acidosis alone in ischemic brain damage. Since the severity of neuronal injury is correlated with the length of ischemic treatment, we reasoned that, under severe ischemic conditions, more spermine may be released and thereby intensify the effect of the endogenous polyamine. With a 60 min MCAO, a treatment that causes significant acidosis-dependent neuronal death (Xiong et al., 2004; Pignataro et al., 2007), the infarct volume was increased to 48.8 ± 2.3% in WT mice injected with aCSF intracerebroventricularly. This was significantly decreased to 23.5 ± 5.2% in mice injected with PcTX1 or to 21.0 ± 1.0% in mice injected with DFMO (75 μg in 1 μl, 30 min before MCAO). Furthermore, exogenous administration of spermine with DFMO reversed the neuroprotective action by DFMO alone in WT mice (Fig. 10B). Our results, therefore, support the idea that both endogenously generated spermine and activation of ASIC1a channels are involved in ischemic neuronal damage in vivo. Consistent with this notion, the infarct volume in ASIC1−/− mice induced by 60 min MCAO (12.4 ± 3.9%) was not significantly different from that induced by the 40 min MCAO (Fig. 1B) in the aCSF group and injection of DFMO did not cause any protection (Fig. 10B), demonstrating that the protective effect of DFMO (and hence the inhibition of spermine biosynthesis) occurs through ASIC1a channels. Together, these results strongly support a functional link between elevated spermine and ASIC1a-mediated neuronal injury during ischemia in vivo.
Discussion
In this study, we have demonstrated that spermine, one of the endogenous polyamines, exacerbates ischemic neuronal injury through sensitization of ASIC1a channels. This unique modulation is attributable to the reduced rate of desensitization of ASIC1a channels from the open state and a shift of the steady-state desensitization to lower pH. These properties allow for prolonged channel activation by acid and better recovery of the channel at more moderate alkalinization during repeated acidosis.
Spermine exacerbates ischemic injury through enhancing ASIC1a activity
Spermine toxicity in the CNS has been extensively studied. Spermine can induce neuronal depolarization and cytoplasmic Ca2+ overload, which might lead to mitochondria-related injury and neuronal damage (Toninello et al., 2004). More importantly, spermine regulates many ion channels and receptors contributing to neuronal injury, particularly with regard to its role in NMDAR-mediated excitotoxicity. Spermine can modulate NMDAR function by a direct interaction on NMDARs (Benveniste and Mayer, 1993; Rock and Macdonald, 1995). Thus, many studies have attempted to link polyamine neurotoxicity to a positive modulatory action at NMDARs. However, these studies have generated inconsistent results (Johnson, 1998; Li et al., 2007). To date, very little research has focused on alternatives to NMDAR-mediated mechanisms of spermine-induced neurotoxicity. After ischemia, pH is subjected to long-term and dynamic change compared with glutamate release, suggesting a more efficient neuroprotective role of the blockade of ASIC1a channels (Gao et al., 2005; Pignataro et al., 2007). In our study, the effect of spermine on OGD-induced cell damage was more sensitive to ASIC blockade than to NMDAR antagonism (Fig. 9C). This is demonstrated by the finding from ASIC1−/− mice that spermine-induced cell damage was primarily mediated by ASIC1a channels. Consistent with this notion, blocking endogenous biosynthesis of polyamines by DFMO did not influence the NMDAR-dependent cell damage in cultured neurons (Fig. 10A). However, we cannot exclude additional effects of polyamines on neuronal death, including the contribution of oxidative products of polyamines, such as aldehydes and hydrogen peroxide, to cell death (Bolkenius and Seiler, 1986; Zimmerman et al., 1995; Cockroft et al., 1996; Ivanova et al., 2002). Because acidosis-induced activity of ASIC1a channels also enhanced the production of reactive oxygen species (ROS) (Liu et al., 2009), and there was no significant difference between aCSF- and DFMO-treated groups in terms of ischemic infarct size in ASIC1−/− mice (Fig. 10B), the two ischemic cell death pathways—ASIC1a-dependent and ASIC1a-independent pathways—seem to be intrinsically related at the downstream ROS pathway.
Physiological relevance of spermine modulation of ASIC1a channels
Spermine is synthesized and released in the setting of ischemia (Kindy et al., 1994). The concentration of spermine in secretory granules and nerve terminals from ox neurohypophyses has been estimated to be 0.26 and 0.52 mm, respectively (Krøigaard et al., 1992). It has been suggested that more polyamines could be released from necrotic neurons into the extracellular space and cause multiple effects (Li et al., 2007). This may also explain the observed lack of change (Carter et al., 1995; Baskaya et al., 1997) or decrease (Paschen et al., 1992) of spermine after cerebral ischemia in some studies, as polyamines released into the extracellular space may be cleared by the blood circulation. It is thus likely that sufficient spermine could influence the activation of ASICs during ischemia. Despite the consistently observed increase of spermidine, and especially putrescine, in ischemic brain (Paschen et al., 1992), these two polyamines only exerted a very weak effect on ASICs. It is possible that other, less obvious, polyamines may function similarly to spermine and contribute to ischemic brain damage through ASIC1a, whereas spermidine and putrescine might regulate neuronal function through other mechanisms.
The potential effects of increased ASIC1a channel activity on a neuron because of “a few second” increase in the duration and a faster recovery from desensitization caused by spermine should not be considered trivial as the enhanced Na+ conductance affects neuronal excitability and in turn Ca2+ influx through voltage-gated Ca2+ channels. This would be on top of the Ca2+ influx mediated directly through the ASIC1a channels themselves. More importantly, the paradox concerning the role of ASIC1a channels in ischemic neural damage is not how high the activity is in amplitude but how long it can last. This is because the damage requires tenth of minutes of acidosis and the severity is related to the duration of the acidosis, whereas without other modulators, the ASIC1a activity only lasts for a few seconds during continuous acidosis. We believe that our findings that spermine prolongs the activity of ASIC1a and facilitates the recovery of the channel from desensitization precisely address this issue concerning the duration of ASIC1a activation, especially since previous studies have documented extracellular pH fluctuations during brain ischemia (Obrenovitch, 1995). Furthermore, the known effects of arachidonic acid and lactate (Immke and McCleskey, 2001; Allen and Attwell, 2002) on ASIC1a channels can further complicate the issue. However, during ischemic brain damage, these factors could also work together with spermine in either additive or synergistic manners to further prolong the activation of the ASIC1a channels and contribute to neuronal damage.
Moreover, homomeric ASIC1a channels are predominantly expressed in human brain neurons and the pH50 for steady-state inactivation of ASICs in human cortical neurons is more acidic than that in mouse cortical neurons (Li et al., 2010). Clearly, spermine affects steady-state desensitization of human ASIC1a channels (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), suggesting a new mechanism underlying neuronal damage in stroke patients. Such interplay between spermine and ASIC1a becomes amplified when extracellular Ca2+ concentration was reduced (Fig. 6C), a condition that may occur during ischemia or traumatic brain injury (Harris et al., 1981; Silver and Erecinska, 1990; Nilsson et al., 1996). Accordingly, we propose that spermine and other factors known to regulate desensitization of the ASIC1a channels play contributory roles in ischemic neuronal injury, although many other contributors are also involved as the ischemic period prolongs (Moskowitz et al., 2010). Because both elevated spermine and acidosis are also involved in neurodegenerative diseases (Yatin et al., 2001; Friese et al., 2007; Uribe-San Martín et al., 2009) and chronic pain (Tan-No et al., 2000; Duan et al., 2007; Xu and Duan, 2009), the mechanism uncovered in the present study is likely of general importance for neuronal dysfunction and not just specific to ischemic stroke.
Mechanisms underlying ASIC1a channel desensitization
Several lines of evidence show that divalent cations such as Ca2+, Mg2+ (Babini et al., 2002), endogenous RFamide-related peptides (Sherwood and Askwith, 2008), dynorphin (Sherwood and Askwith, 2009), and spider toxin PcTX1 (Chen et al., 2005) all alter steady-state desensitization of ASIC1a channels. However, the action site(s) for divalent and polyvalent cations and whether spermine acts via the same or a different mechanism than divalent cations await additional experimentation. Negatively charged amino acids in the large extracellular loop of the ASIC1a subunit (Jasti et al., 2007) are the best candidates for forming the binding site(s) of positively charged spermine. Previous studies have shown that extracellular Ca2+ competes with H+ and reduces the apparent affinity for H+ (Babini et al., 2002; Immke and McCleskey, 2003), whereas PcTX1 competes with Ca2+ for binding to ASIC1a channels (Chen et al., 2005). Our results show that extracellular spermine also competes with PcTX1 and reduces the apparent affinity for H+ (Fig. 6A,B), similar to the effect of Ca2+ on ASIC1a channels (Fig. 6C), suggesting that spermine may share similar interaction domains with PcTX1 in the extracellular loop of ASIC1a channel. Interestingly, PcTX1 exerts an inhibitory effect by “opening” the ASIC1a channel at much higher (e.g., basic) pH (Chen et al., 2005) and therefore rendering the channel into steady-state desensitization at neutral pH. Spermine appears to do just the opposite, by shifting the steady-state desensitization to lower pH. Indeed, our mutagenesis experiments point to the importance of two acidic residues E219 and E424 of the putative PcTX1 binding sites in mediating spermine enhancement of ASIC1a activity. Dynorphin directly interacts with ASIC1a at F352, a location overlapping with the PcTX1 binding site (Sherwood and Askwith, 2009), and enhances ASIC1a activity by preventing the channel desensitization. However, the F352L mutation did not reverse spermine effect on ASIC1a channels, suggesting that spermine interaction sites could be more dynamic. Although the biological significance of ASIC1a desensitization remains mostly mysterious, this property may open a new avenue for developing modulators for ASICs.
In conclusion, we have shown that spermine sensitizes ASIC1a channels to extracellular acidosis in both heterologous expression systems and native CNS neurons, through modulation of channel desensitization. Furthermore, our results support that an ASIC1a-mediated pathway plays a major part in spermine-induced cytotoxicity during brain ischemia. The reduction in desensitization and acceleration in reactivation are particularly relevant in facilitating ASIC1 activity during repetitive acidosis. When extracellular spermine and protons are both elevated under ischemia, chronic pain, and neurodegeneration, the spermine–ASIC interaction may contribute significantly to the pathogenesis of these diseases. Identification of drugs influencing this novel interaction may lead to the discovery of new clinical therapeutics against ischemia and other neurological disorders involving elevated spermine release and acidosis.
Footnotes
This study was supported by National Natural Science Foundation of China Grants 30830035 and 30700145, National Basic Research Program of China Grant 2011CBA00408, Shanghai Municipal Government Grant 09XD1404900, and National Institutes of Health Grants R01 NS049470, R01 NS047506, R01 NS050610, and R01 GM081658. B. Duan is a postdoctoral fellow supported by the K. C. Wong Education Foundation (Hong Kong). We thank Drs. Q. Hu, Z.-Y. Ye, J. Gao, Y. Ding, and D. Branigan for technical assistance, and Drs. J. A. Wemmie, M. P. Price, and M. J. Welsh (Howard Hughes Medical Institute, University of Iowa, Iowa City, IA) for providing ASIC knock-out mice.
References
- Allen NJ, Attwell D. Modulation of ASIC channels in rat cerebellar Purkinje neurons by ischaemia-related signals. J Physiol. 2002;543:521–529. doi: 10.1113/jphysiol.2002.020297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askwith CC, Wemmie JA, Price MP, Rokhlina T, Welsh MJ. Acid-sensing ion channel 2 (ASIC2) modulates ASIC1 H+-activated currents in hippocampal neurons. J Biol Chem. 2004;279:18296–18305. doi: 10.1074/jbc.M312145200. [DOI] [PubMed] [Google Scholar]
- Babini E, Paukert M, Geisler HS, Grunder S. Alternative splicing and interaction with di- and polyvalent cations control the dynamic range of acid-sensing ion channel 1 (ASIC1) J Biol Chem. 2002;277:41597–41603. doi: 10.1074/jbc.M205877200. [DOI] [PubMed] [Google Scholar]
- Baron A, Waldmann R, Lazdunski M. ASIC-like, proton-activated currents in rat hippocampal neurons. J Physiol. 2002;539:485–494. doi: 10.1113/jphysiol.2001.014837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron A, Voilley N, Lazdunski M, Lingueglia E. Acid sensing ion channels in dorsal spinal cord neurons. J Neurosci. 2008;28:1498–1508. doi: 10.1523/JNEUROSCI.4975-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskaya MK, Rao AM, Dogan A, Donaldson D, Gellin G, Dempsey RJ. Regional brain polyamine levels in permanent focal cerebral ischemia. Brain Res. 1997;744:302–308. doi: 10.1016/S0006-8993(96)01116-X. [DOI] [PubMed] [Google Scholar]
- Benveniste M, Mayer ML. Multiple effects of spermine on N-methyl-d-aspartic acid receptor responses of rat cultured hippocampal neurones. J Physiol. 1993;464:131–163. doi: 10.1113/jphysiol.1993.sp019627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein HG, Müller M. The cellular localization of the l-ornithine decarboxylase/polyamine system in normal and diseased central nervous systems. Prog Neurobiol. 1999;57:485–505. doi: 10.1016/s0301-0082(98)00065-3. [DOI] [PubMed] [Google Scholar]
- Bolkenius FN, Seiler N. Developmental aspects of polyamine interconversion in rat brain. Int J Dev Neurosci. 1986;4:217–224. doi: 10.1016/0736-5748(86)90061-4. [DOI] [PubMed] [Google Scholar]
- Carter C, Poignet H, Carboni S, Fage D, Voltz C, Scatton B. Release of spermidine from the rat cortex following permanent middle cerebral artery occlusion. Fundam Clin Pharmacol. 1995;9:129–140. doi: 10.1111/j.1472-8206.1995.tb00272.x. [DOI] [PubMed] [Google Scholar]
- Chen X, Kalbacher H, Gründer S. The tarantula toxin psalmotoxin 1 inhibits acid-sensing ion channel (ASIC) 1a by increasing its apparent H+ affinity. J Gen Physiol. 2005;126:71–79. doi: 10.1085/jgp.200509303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu XP, Zhu XM, Wei WL, Li GH, Simon RP, MacDonald JF, Xiong ZG. Acidosis decreases low Ca2+-induced neuronal excitation by inhibiting the activity of calcium-sensing cation channels in cultured mouse hippocampal neurons. J Physiol. 2003;550:385–399. doi: 10.1113/jphysiol.2003.043091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu XP, Wemmie JA, Wang WZ, Zhu XM, Saugstad JA, Price MP, Simon RP, Xiong ZG. Subunit-dependent high-affinity zinc inhibition of acid-sensing ion channels. J Neurosci. 2004;24:8678–8689. doi: 10.1523/JNEUROSCI.2844-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockroft KM, Meistrell M, 3rd, Zimmerman GA, Risucci D, Bloom O, Cerami A, Tracey KJ. Cerebroprotective effects of aminoguanidine in a rodent model of stroke. Stroke. 1996;27:1393–1398. doi: 10.1161/01.str.27.8.1393. [DOI] [PubMed] [Google Scholar]
- Colbran RJ. Regulation and role of brain calcium/calmodulin-dependent protein kinase II. Neurochem Int. 1992;21:469–497. doi: 10.1016/0197-0186(92)90080-b. [DOI] [PubMed] [Google Scholar]
- DeGraba TJ, Ostrow PT, Strong RA, Earls RM, Zong ZJ, Grotta JC. Temporal relation of calcium-calmodulin binding and neuronal damage after global ischemia in rats. Stroke. 1992;23:876–882. doi: 10.1161/01.str.23.6.876. [DOI] [PubMed] [Google Scholar]
- Duan B, Wu LJ, Yu YQ, Ding Y, Jing L, Xu L, Chen J, Xu TL. Upregulation of acid-sensing ion channel ASIC1a in spinal dorsal horn neurons contributes to inflammatory pain hypersensitivity. J Neurosci. 2007;27:11139–11148. doi: 10.1523/JNEUROSCI.3364-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escoubas P, De Weille JR, Lecoq A, Diochot S, Waldmann R, Champigny G, Moinier D, Ménez A, Lazdunski M. Isolation of a tarantula toxin specific for a class of proton-gated Na+ channels. J Biol Chem. 2000;275:25116–25121. doi: 10.1074/jbc.M003643200. [DOI] [PubMed] [Google Scholar]
- Friese MA, Craner MJ, Etzensperger R, Vergo S, Wemmie JA, Welsh MJ, Vincent A, Fugger L. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat Med. 2007;13:1483–1489. doi: 10.1038/nm1668. [DOI] [PubMed] [Google Scholar]
- Gao J, Duan B, Wang DG, Deng XH, Zhang GY, Xu L, Xu TL. Coupling between NMDA receptor and acid-sensing ion channel contributes to ischemic neuronal death. Neuron. 2005;48:635–646. doi: 10.1016/j.neuron.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RJ, Symon L, Branston NM, Bayhan M. Changes in extracellular calcium activity in cerebral ischaemia. J Cereb Blood Flow Metab. 1981;1:203–209. doi: 10.1038/jcbfm.1981.21. [DOI] [PubMed] [Google Scholar]
- Immke DC, McCleskey EW. Lactate enhances the acid-sensing Na+ channel on ischemia-sensing neurons. Nat Neurosci. 2001;4:869–870. doi: 10.1038/nn0901-869. [DOI] [PubMed] [Google Scholar]
- Immke DC, McCleskey EW. Protons open acid-sensing ion channels by catalyzing relief of Ca2+ blockade. Neuron. 2003;37:75–84. doi: 10.1016/s0896-6273(02)01130-3. [DOI] [PubMed] [Google Scholar]
- Ivanova S, Batliwalla F, Mocco J, Kiss S, Huang J, Mack W, Coon A, Eaton JW, Al-Abed Y, Gregersen PK, Shohami E, Connolly ES, Jr, Tracey KJ. Neuroprotection in cerebral ischemia by neutralization of 3-aminopropanal. Proc Natl Acad Sci U S A. 2002;99:5579–5584. doi: 10.1073/pnas.082609299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid-sensing ion channel 1 at 1.9 Å resolution and low pH. Nature. 2007;449:316–323. doi: 10.1038/nature06163. [DOI] [PubMed] [Google Scholar]
- Johnson TD. Polyamines and cerebral ischemia. Prog Drug Res. 1998;50:193–258. doi: 10.1007/978-3-0348-8833-2_5. [DOI] [PubMed] [Google Scholar]
- Kindy MS, Hu Y, Dempsey RJ. Blockade of ornithine decarboxylase enzyme protects against ischemic brain damage. J Cereb Blood Flow Metab. 1994;14:1040–1045. doi: 10.1038/jcbfm.1994.136. [DOI] [PubMed] [Google Scholar]
- Krishtal O. The ASICs: signaling molecules? Modulators? Trends Neurosci. 2003;26:477–483. doi: 10.1016/S0166-2236(03)00210-8. [DOI] [PubMed] [Google Scholar]
- Krøigaard M, Thams P, Thorn NA. Polyamines in nerve terminals and secretory granules isolated from neurohypophyses. Acta Physiol Scand. 1992;146:233–239. doi: 10.1111/j.1748-1716.1992.tb09412.x. [DOI] [PubMed] [Google Scholar]
- Li J, Doyle KM, Tatlisumak T. Polyamines in the brain: distribution, biological interactions, and their potential therapeutic role in brain ischaemia. Curr Med Chem. 2007;14:1807–1813. doi: 10.2174/092986707781058841. [DOI] [PubMed] [Google Scholar]
- Li M, Inoue K, Branigan D, Kratzer E, Hansen JC, Chen JW, Simon RP, Xiong ZG. Acid-sensing ion channels in acidosis-induced injury of human brain neurons. J Cereb Blood Flow Metab. 2010;30:1247–1260. doi: 10.1038/jcbfm.2010.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhang R, Liu K, Zhou H, Yang X, Liu X, Tang M, Su J, Dong Q. Tissue kallikrein protects cortical neurons against in vitro ischemia-acidosis/reperfusion-induced injury through the ERK1/2 pathway. Exp Neurol. 2009;219:453–465. doi: 10.1016/j.expneurol.2009.06.021. [DOI] [PubMed] [Google Scholar]
- Masuko T, Kusama-Eguchi K, Sakata K, Kusama T, Chaki S, Okuyama S, Williams K, Kashiwagi K, Igarashi K. Polyamine transport, accumulation, and release in brain. J Neurochem. 2003;84:610–617. doi: 10.1046/j.1471-4159.2003.01558.x. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson P, Laursen H, Hillered L, Hansen AJ. Calcium movements in traumatic brain injury: the role of glutamate receptor-operated ion channels. J Cereb Blood Flow Metab. 1996;16:262–270. doi: 10.1097/00004647-199603000-00011. [DOI] [PubMed] [Google Scholar]
- Obrenovitch TP. The ischaemic penumbra: twenty years on. Cerebrovasc Brain Metab Rev. 1995;7:297–323. [PubMed] [Google Scholar]
- Paschen W, Widmann R, Weber C. Changes in regional polyamine profiles in rat brains after transient cerebral ischemia (single versus repetitive ischemia): evidence for release of polyamines from injured neurons. Neurosci Lett. 1992;135:121–124. doi: 10.1016/0304-3940(92)90150-6. [DOI] [PubMed] [Google Scholar]
- Pietra F. Docking and MD simulations of the interaction of the tarantula peptide psalmotoxin-1 with ASIC1a channels using a homology model. J Chem Inf Model. 2009;49:972–977. doi: 10.1021/ci800463h. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Simon RP, Xiong ZG. Prolonged activation of ASIC1a and the time window for neuroprotection in cerebral ischaemia. Brain. 2007;130:151–158. doi: 10.1093/brain/awl325. [DOI] [PubMed] [Google Scholar]
- Qadri YJ, Berdiev BK, Song Y, Lippton HL, Fuller CM, Benos DJ. Psalmotoxin-1 docking to human acid-sensing ion channel-1. J Biol Chem. 2009;284:17625–17633. doi: 10.1074/jbc.M109.003913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock DM, Macdonald RL. Polyamine regulation of N-methyl-d-aspartate receptor channels. Annu Rev Pharmacol Toxicol. 1995;35:463–482. doi: 10.1146/annurev.pa.35.040195.002335. [DOI] [PubMed] [Google Scholar]
- Seiler N, Delcros JG, Moulinoux JP. Polyamine transport in mammalian cells. An update. Int J Biochem Cell Biol. 1996;28:843–861. doi: 10.1016/1357-2725(96)00021-0. [DOI] [PubMed] [Google Scholar]
- Sherwood TW, Askwith CC. Endogenous arginine-phenylalanine-amide-related peptides alter steady-state desensitization of ASIC1a. J Biol Chem. 2008;283:1818–1830. doi: 10.1074/jbc.M705118200. [DOI] [PubMed] [Google Scholar]
- Sherwood TW, Askwith CC. Dynorphin opioid peptides enhance acid-sensing ion channel 1a activity and acidosis-induced neuronal death. J Neurosci. 2009;29:14371–14380. doi: 10.1523/JNEUROSCI.2186-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver IA, Erecinska M. Intracellular and extracellular changes of [Ca2+] in hypoxia and ischemia in rat brain in vivo. J Gen Physiol. 1990;95:837–866. doi: 10.1085/jgp.95.5.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan-No K, Taira A, Wako K, Niijima F, Nakagawasai O, Tadano T, Sakurada C, Sakurada T, Kisara K. Intrathecally administered spermine produces the scratching, biting and licking behaviour in mice. Pain. 2000;86:55–61. doi: 10.1016/s0304-3959(99)00312-7. [DOI] [PubMed] [Google Scholar]
- Tantini B, Fiumana E, Cetrullo S, Pignatti C, Bonavita F, Shantz LM, Giordano E, Muscari C, Flamigni F, Guarnieri C, Stefanelli C, Caldarera CM. Involvement of polyamines in apoptosis of cardiac myoblasts in a model of simulated ischemia. J Mol Cell Cardiol. 2006;40:775–782. doi: 10.1016/j.yjmcc.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Toninello A, Salvi M, Mondovì B. Interaction of biologically active amines with mitochondria and their role in the mitochondrial-mediated pathway of apoptosis. Curr Med Chem. 2004;11:2349–2374. doi: 10.2174/0929867043364559. [DOI] [PubMed] [Google Scholar]
- Uribe-San Martín R, Herrera-Molina R, Olavarría L, Ramírez G, von Bernhardi R. Reduction of beta-amyloid-induced neurotoxicity on hippocampal cell cultures by moderate acidosis is mediated by transforming growth factor beta. Neuroscience. 2009;158:1338–1347. doi: 10.1016/j.neuroscience.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Vila-Petroff M, Salas MA, Said M, Valverde CA, Sapia L, Portiansky E, Hajjar RJ, Kranias EG, Mundiña-Weilenmann C, Mattiazzi A. CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovasc Res. 2007;73:689–698. doi: 10.1016/j.cardiores.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Vukicevic M, Kellenberger S. Modulatory effects of acid-sensing ion channels on action potential generation in hippocampal neurons. Am J Physiol Cell Physiol. 2004;287:C682–C690. doi: 10.1152/ajpcell.00127.2004. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature. 1997;386:173–177. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- Wallace HM, Fraser AV. Inhibitors of polyamine metabolism: review article. Amino acids. 2004;26:353–365. doi: 10.1007/s00726-004-0092-6. [DOI] [PubMed] [Google Scholar]
- Wang W, Duan B, Xu H, Xu L, Xu TL. Calcium-permeable acid-sensing ion channel is a molecular target of the neurotoxic metal ion lead. J Biol Chem. 2006;281:2497–2505. doi: 10.1074/jbc.M507123200. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Chen J, Askwith CC, Hruska-Hageman AM, Price MP, Nolan BC, Yoder PG, Lamani E, Hoshi T, Freeman JH, Jr, Welsh MJ. The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron. 2002;34:463–477. doi: 10.1016/s0896-6273(02)00661-x. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Xu TL, Duan B. Calcium-permeable acid-sensing ion channel in nociceptive plasticity: a new target for pain control. Prog Neurobiol. 2009;87:171–180. doi: 10.1016/j.pneurobio.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Yatin SM, Yatin M, Varadarajan S, Ain KB, Butterfield DA. Role of spermine in amyloid beta-peptide-associated free radical-induced neurotoxicity. J Neurosci Res. 2001;63:395–401. doi: 10.1002/1097-4547(20010301)63:5<395::AID-JNR1034>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Yermolaieva O, Leonard AS, Schnizler MK, Abboud FM, Welsh MJ. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci U S A. 2004;101:6752–6757. doi: 10.1073/pnas.0308636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha XM, Wemmie JA, Green SH, Welsh MJ. Acid-sensing ion channel 1a is a postsynaptic proton receptor that affects the density of dendritic spines. Proc Natl Acad Sci U S A. 2006;103:16556–16561. doi: 10.1073/pnas.0608018103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman GA, Meistrell M, 3rd, Bloom O, Cockroft KM, Bianchi M, Risucci D, Broome J, Farmer P, Cerami A, Vlassara H. Neurotoxicity of advanced glycation endproducts during focal stroke and neuroprotective effects of aminoguanidine. Proc Natl Acad Sci U S A. 1995;92:3744–3748. doi: 10.1073/pnas.92.9.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]