

DNA copy number aberrations (CNAs) are a characteristic feature of cancer genomes. In this work, Rebecka Jörnsten, Sven Nelander and colleagues combine network modeling and experimental methods to analyze the systems-level effects of CNAs in glioblastoma.
Keywords: cancer biology, cancer genomics, glioblastoma
Abstract
DNA copy number aberrations (CNAs) are a hallmark of cancer genomes. However, little is known about how such changes affect global gene expression. We develop a modeling framework, EPoC (Endogenous Perturbation analysis of Cancer), to (1) detect disease-driving CNAs and their effect on target mRNA expression, and to (2) stratify cancer patients into long- and short-term survivors. Our method constructs causal network models of gene expression by combining genome-wide DNA- and RNA-level data. Prognostic scores are obtained from a singular value decomposition of the networks. By applying EPoC to glioblastoma data from The Cancer Genome Atlas consortium, we demonstrate that the resulting network models contain known disease-relevant hub genes, reveal interesting candidate hubs, and uncover predictors of patient survival. Targeted validations in four glioblastoma cell lines support selected predictions, and implicate the p53-interacting protein Necdin in suppressing glioblastoma cell growth. We conclude that large-scale network modeling of the effects of CNAs on gene expression may provide insights into the biology of human cancer. Free software in MATLAB and R is provided.
Introduction
Gains and losses of chromosomal material that alter DNA copy number are a hallmark of cancer genomes. At the level of a single locus, the effects of a copy number aberration (CNA) are well known: on average, increased copy number (gene amplification) leads to increased gene expression, decreased copy number (gene deletion) leads to decreased gene expression (Pollack et al, 2002; Lee et al, 2008; Nilsson et al, 2008). However, CNAs also affect the expression of genes located outside the amplified/deleted region itself via indirect mechanisms. For example, deletion of a transcriptional repressor may increase the expression of its targets, amplification of a kinase may drive a signaling cascade, and so on. Our knowledge of how CNAs affect gene expression at a genome-wide level is limited.
Global network modeling of expression and copy number changes can elucidate such causal connections, and prove helpful in the study of several key problems in cancer biology. Specifically, such models may (1) identify functionally important genes whose perturbations have a significant and dispersed impact on transcription; (2) facilitate the discovery of possible therapeutic targets by matching model-identified key regulators or their targets to pharmacological databases; and (3) assist in the identification of distinct CNA and mRNA features that are predictive of patient prognosis or therapeutic response.
Current methods for transcriptional network modeling seemingly fall into three main categories. One common approach is to derive models from mRNA profiles only, using, e.g., gene–gene (partial) correlations, Bayesian networks, ordinary differential equations or mutual information (Friedman et al, 2000; Schäfer and Strimmer, 2005; Margolin et al, 2006; Bansal et al, 2007; Opgen-Rhein and Strimmer, 2007). A second common technique is to construct models of mRNA expression from targeted perturbation experiments, as controlled perturbations strongly facilitate causal inference (Yeung et al, 2002; Tegner et al, 2003; di Bernardo et al, 2005; Bansal et al, 2007; Bonneau et al, 2007; Lehár et al, 2007; Nelander et al, 2008; Lauria et al, 2009). A third alternative is to use the naturally occurring genetic variation in a separating population to study the relationship between genotype and expression phenotype (Jansen, 2003; Lee et al, 2006, 2009; Rockman, 2008; Suthram et al, 2008; Zhu et al, 2008; see also Discussion). Here, we focus on the role of acquired genetic variations in tumors, specifically CNAs, and ask how these can be used to derive transcriptional networks. CNAs are prevalent in several human cancers, and tend to appear in a patient-specific and multifactorial manner in the tumors, which resembles an optimal experimental design to derive causality (Fisher, 1926).
We present a global model of CNA-driven transcription in the brain tumor glioblastoma. The model is derived using EPoC (Endogenous Perturbation analysis of Cancer), a computational method that constructs network models of mRNA expression, viewing CNAs as informative system perturbations introduced endogenously during the evolution of the tumor, and the corresponding mRNA profiles as the steady-state response to that perturbation. We apply EPoC to glioblastoma data from The Cancer Genome Atlas (TCGA) consortium. Previous analyses of glioblastoma have revealed altered pathways and disease subtypes (Pollack et al, 2002; Freije et al, 2004; Phillips et al, 2006; Tso et al, 2006; Lee et al, 2008; TCGA-Consortium, 2008; Dahlback et al, 2009; Verhaak et al, 2010; Cerami et al, 2010) and networks of correlating transcripts (Carro et al, 2010) (ARACNE). Key examples of CNA/mRNA analyses for other tumors include clustering and modular network modeling, leading to the discovery of regulators such as MITF, RAB27A and TBC1D16 in malignant melanoma (Garraway et al, 2005; Akavia et al, 2010), and linkage analysis to reveal the association of cMYC amplification to wound healing signatures in breast cancer (Adler et al, 2006). Network analysis of 654 selected breast cancer transcripts and 384 genomic regions has identified a candidate regulatory region on chromosome 17 (Peng et al, 2008). Canonical correlation analysis (CCA) has also been put forth as an alternative non-network approach to integrating DNA/mRNA data (Waaijenborg et al, 2008; Witten et al, 2009).
We use EPoC to construct a gene-level model, which encompasses 10 672 genes, causally connecting CNAs to expression changes in glioblastoma. First, we establish that the parameters of the EPoC network model can be robustly estimated from paired genome-wide DNA- and RNA-level data from a set of tumors, using a combination of lasso regression and bootstrap. Second, we show that a novel score, based on a sparse singular value decomposition of the derived CNA–mRNA network model, identifies prognostic biomarkers capable of clinical stratification into short-term and long-term survivors. Third, EPoC identifies key mechanisms (disease-driving CNAs), which we assess by chemoinformatic analyses and comparisons to known biological pathways, revealing the likely existence of short regulatory paths between EPoC hubs and targets, as well as 15 candidate drug targets. We confirm a candidate hub, the p53-interacting protein Necdin, NDN, in U87MG, U373MG, U343MG and T98 glioma-derived cell lines by experimentally testing a small transcriptional network around NDN, receptor tyrosine kinases EGF receptor (EGFR) and platelet-derived growth factor receptor alpha (PDGFRA). Finally, we demonstrate rapid and consistent performance of EPoC in comparison with mRNA-only methods, standard expression quantitative locus (eQTL) methods and two recent multivariate methods for genotype–mRNA coupling (Peng et al, 2008; Lee et al, 2009).
Results
Modeling copy number-dependent transcription in tumors
Transcriptional and CNA-driven networks
To connect mRNA levels with DNA copy number in glioblastoma tumors, we adapt a common model for mRNA transcription regulation and turnover. This model formulation, related to the so-called S-system (Savageau, 1969, 1976; Crampin et al, 2004), takes the form of sets of differential equations:
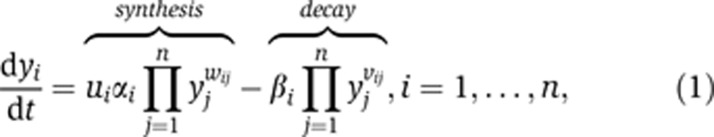 |
1 |
where n is the number of genes, dyi/dt and yi, i=1,2, …, n denote the change rate and average mRNA concentrations in a tumor respectively, and ui⩾0, i=1,2, …, n the average number of gene copies corresponding to a particular transcript (Figure 1B). Equation (1) states that the change rate of transcript yi is the difference between its synthesis rate and its decay rate. The synthesis rate is determined by the number of copies of the gene's DNA, ui, the regulatory effects of other genes, wij and a gene-specific synthesis constant, αi. Similarly, the decay rate is determined by the regulatory effects of other genes, vij and a gene-specific decay constant, βi. Obviously, the assumption of proportionality on ui is a simplification and unlikely to hold for all genes in the genome (e.g., gene copies may generate transcripts at different rates due to epigenetic differences). Nevertheless, recent data indicate that it is a reasonable approximation for a large proportion of genes in the genome (Nilsson et al, 2008).
Figure 1.

Overview of the EPoC modeling framework. (A) Using genome-wide, paired mRNA- and DNA-level data as input, EPoC generates a quantitative causal network model of the global effects of copy number aberrations on mRNA expression. The resulting model is subsequently used to predict disease-driving genes and prognostic indicators. (B) EPoC is based on systems of differential equations that take into account that the transcription of a gene is determined both by its own DNA copy number (straight arrows) and the product of other genes (bent arrows). (C) Our method generates two mutually complementary networks denoted as A and G. The A network captures transcript–transcript interactions (left), whereas the G network contains the direct and indirect effects of CNA perturbations on transcription (middle). The singular value decomposition of G can be used to identify the CNAs whose perturbations are maximally amplified by the network (i.e., they have a strong overall transcriptional effect; yellow nodes), and the mRNA transcripts whose expression are most altered by these perturbations (green nodes; right panel).
The procedure used to estimate the model parameters in Equation (1) is described in detail in Materials and methods. In short, assuming steady-state conditions, the log-transformed and zero-centered mRNA and CNA profiles of glioblastoma can be summarized by two mutually complementing linear systems. The first of these represents the transcriptional network (A):
| 2 |
where ΔY and ΔU are stack matrices of log-transformed and zero-centered mRNA and CNA profiles of glioblastoma, respectively, and R (defined by the α's and β's of the original model, Materials and methods) is a matrix that captures the effects on transcription of non-CNA perturbations in individual tumors (e.g., SNPs, sequence mutations or environmental effects). The transcriptional network A={aij} relates to the original model by aij=wij−vij, meaning its elements aij represents the net influence from transcript j to transcript i; aij>0 indicates activation of transcription i by transcript j, aij<0 inhibition, and the magnitude aij the strength of the interaction.
The second representation is termed the CNA-driven network (G):
| 3 |
G={gij} consists of CNA–mRNA couplings: gij>0 indicates CNA-driven transcriptional activation (i.e., transcription of gene i is increased because the copy number of gene j has been altered), gij<0 CNA-driven transcriptional inhibition, and the magnitude of gij the strength of the interaction. This network is related to the first as G=−A−1 and the topologies of the two networks are thus related (Figure 1C). However, while A reflects direct transcriptional interaction, corrected for the impact of a transcript's own CNA, G models how the effects of CNA perturbations propagate through the system to produce their steady-state responses and should contain key disease-driving CNAs as hubs, as well as their downstream targets (Figure 1C).
To identify the transcriptional interactions (non-zero elements in A) and the CNA–mRNA couplings (non-zero elements in G), we need to solve the large linear equation systems ((2) and (3)). We use a gene-level lasso regression approach paired with cross-validation and bootstrap to robustly identify these network parameters (Materials and methods).
Survival scores derived from network decompositions
We next describe how survival scores can be derived from the EPoC model, based on a particular interpretation of the CNA-driven network as a signal amplifier. From a systems perspective, it is natural to view the copy number profile as the input to the system G, whereas the gene expression profile is the corresponding output. One common way to summarize a system's input–output behavior is to compute the main axes of signal gain, defined as the singular value decomposition G=CΛDT (Golub and Loan, 1996; Skogestad and Postlethwaite, 2005; Nordling and Jacobsen, 2009) (Materials and methods). When applied to the CNA-driven network G, this decomposition should reveal CNA perturbations that are strongly amplified by the system (in the leading columns of D), as well as the transcripts which are most affected by CNA perturbations (in the leading columns of C) (Figure 1C). We use sparse SVD (Zou et al, 2006), which ensures that only a small subset of perturbations and transcripts are present in the leading columns (Materials and methods). Once estimates of C and D have been obtained, EPoC computes the level of signal amplification in each tumor by the scalar projection scores Zy=CTΔY and Zu=DTΔU (Materials and methods). Concisely put, these scores summarize the total burden of molecular changes consistent with the CNA-driven network, and should therefore correlate with clinical survival. Below, we confirm this conjecture for the patients in the TCGA glioblastoma cohort.
Global CNA-driven networks of glioblastoma
EPoC finds 512 robust associations between CNAs and mRNAs in glioblastoma
We proceed to estimate EPoC networks for human glioblastoma. We use CNA- and mRNA-level data (10 672 matched genes, 186 patients) provided by the TCGA consortium (TCGA-Consortium, 2008). Before estimating the network, EPoC applies a filter to select possible CNA regulators in the data (defined as genes that are recurrently amplified or deleted across the patients; Materials and methods). In total, we keep 2640 genes as possible CNA regulators, whereas we keep all 10 672 genes as possible targets and/or transcriptional regulators. On the basis of these data, network modeling proceeds as follows. First, EPoC determines a suitable model size, i.e., the number of interactions in the network. For this, we have developed a customized procedure utilizing random data splits (Figure 2A). In brief, the tumors (i.e., the 186 glioma cases) are split into two random groups. We estimate a network for each group, and compare the two networks using Kendall's W (akin to rank correlation of detected network interactions, Materials and methods). This procedure is repeated for different network sizes, and we select the network size that optimizes W. The optimal network size for the TCGA glioblastoma data is estimated to 200–500 interactions (Figure 2A). We then construct a robust final network of the optimal network size using bootstrap: On each of 1000 bootstrap data sets (resampling from the 186 tumors; Friedman et al, 2000), we generate a network of size around 400 (as obtained in Figure 2A). We retain interactions that appear in at least 20% of the 1000 bootstrap networks, a cutoff set well above appearance frequencies expected by chance (Figure 2B). This results in a final CNA-driven network with 512 interactions, of which 263 are stimulatory and 249 are inhibitory (Figure 3A).
Figure 2.

Derivation of robust and optimally sized network models for glioblastoma. (A) To select the network size, we use a customized validation technique in which networks generated in random data splits are compared using a rank correlation metric (one minus Kendall's W). Upper panel: Using this approach, we find glioblastoma networks with 200–500 interactions to be the structurally most consistent. The preferred network size is indicated by an asterisk (*) (details in Materials and methods). Lower panel: To assess the ability of a model to predict mRNA levels from CNAs, we estimate the normalized sum-of-squares prediction error by 10-fold cross-validation. This cross-validation identifies optimal networks of about 10 000 interactions. (B) We infer a robust CNA-driven network of size 512 from 186 paired gene expression and gene copy number profiles provided by The Cancer Genome Atlas (TCGA) consortium. For each of 1000 pseudo-bootstrap data sets, we generate a network of size around 400 (as obtained in Figure 2A). The final network retains interactions that appear in at least a fraction f of the bootstrap networks (frequency distribution shown as red curve). As a negative control, we permute the patients in the CNA data set (but not in the mRNA data) and repeat the estimation procedure, producing low frequencies for all individual interactions (dashed black curve and *). On the basis of these results, we here use f=20% (black line) as a frequency cutoff to generate our network model (Figure 3), which is well above frequencies expected by chance.
Figure 3.

CNA-driven network of glioblastoma. (A) Overall structure of the resulting glioblastoma network, defined as the set of interactions detected in at least 20% of the bootstrap networks (Figure 2B). Red arrows represent stimulatory interactions, blue arrows indicate inhibitory interactions. (B) Close-up of a network region containing early neural differentiation markers, including glioblastoma stem cell marker CD133/PROM1, under the control of hub genes CHIC2 and PDGFRA. The main hubs of the full network are listed in Table I. Note hub gene NDN, further analyzed in Figure 4. (C) Close-up of a network region containing genes that are targets of pharmaceutical compounds (as determined by searching the Ingenuity and Drugbank databases). Examples of compounds involved in links include simvastatin, SDX-102 (selectively active in MTAP-deficient tumors), PD173074 (a FGFR3 inhibitor), and cyclooxygenase 1 (COX1) inhibitors (PTGS1 encodes the COX1 protein).
CNA hubs that best explain mRNA variability in glioblastoma
The identified CNA-driven network G contains a number of copy number-altered genes that control multiple downstream genes (Figure 3A, Table I). Among these highly connected hub genes, we find well-known oncogenes and tumor supressors that are frequently deleted or amplified in glioblastoma, including EGFR, PDGFRA, CDKN2A and CDKN2B (Figure 3A), confirming a clear association between these alterations and transcriptional variability of glioblastoma. In addition, EPoC identifies a number of interesting hub genes that have not previously been associated with glioblastoma, e.g., MTAP and SEC61G. MTAP is located close to CDKN2A/B on chromosome 9, and SEC61G is located close to EGFR on chromosome 7, but both MTAP and SEC61G have unique and robustly identified targets in our network model. This may suggest that they are not mere innocent bystanders (passenger mutations), but may have tumorigenic effects of their own. Recent work has shown that amplification of SEC61G leads to a more than 10-fold transcriptional induction of this gene (Bralten et al, 2010); deletion of MTAP is believed to confer sensitivity to purine synthesis inhibitors such as SDX-102 (Kindler et al, 2009). Additional hubs include interferon alpha 1 (IFNA1), myeloid/lymphoid or mixed-lineage leukemia translocated to 10 (MLLT10, a well-known leukemia gene), glutamate decarboxylase 2 (GAD2), a postulated glutamate receptor GPR158 and Necdin (NDN, pursued below). As expected, the model does not contain hubs to represent copy number neutral glioma oncogenes/tumor suppressors altered by missense, nonsense or frameshift mutations (TP53, ERBB2, NF1, RBI, PIK3R1, PIK3CA; Parsons et al, 2008; TCGA-Consortium, 2008). To account for the effects of additional types of mutations, we would require a model for the non-CNA perturbation term, R, in Equation (2), which is reserved for future work.
Table 1. Hubs in the CNA-driven glioblastoma network model–based on 10 672 genes and 186 patients.
| Symbol | Amp | Del | Targets | Chrom | Pos | Description |
|---|---|---|---|---|---|---|
| EGFR | 146 | 2 | 134 | 7 | 55054218 | Epidermal growth factor receptor (erythroblastic leukemia viral (v-erb-b) oncogene homolog, avian) |
| CDKN2B | 6 | 108 | 46 | 9 | 21992905 | Cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4) |
| CDKN2A | 6 | 108 | 27 | 9 | 21957751 | Cyclin-dependent kinase inhibitor 2A (melanoma, p16, inhibits CDK4) |
| MTAP | 6 | 101 | 22 | 9 | 21792634 | Methylthioadenosine phosphorylase |
| SEC61G | 134 | 2 | 21 | 7 | 54787434 | Sec61-gamma subunit |
| PDGFRA | 33 | 5 | 19 | 4 | 54790203 | Platelet-derived growth factor receptor, alpha polypeptide |
| IFNA1 | 6 | 101 | 14 | 9 | 21430439 | Interferon-alpha 1 |
| COMMD3 | 8 | 130 | 10 | 10 | 22645304 | COMM domain containing 3 |
| CHIC2 | 35 | 5 | 9 | 4 | 54570714 | Cysteine-rich hydrophobic domain 2 |
| GAD2 | 8 | 130 | 9 | 10 | 26545599 | Glutamate decarboxylase 2 (pancreatic islets and brain, 65 kDa) |
| IFNA14 | 6 | 98 | 8 | 9 | 21191233 | Interferon-alpha 14 |
| C10orf97 | 9 | 126 | 6 | 10 | 15860180 | Chromosome 10 open reading frame 97 |
| MLLT10 | 9 | 129 | 5 | 10 | 21863579 | Myeloid/lymphoid or mixed-lineage leukemia (trithorax homolog, Drosophila); translocated to 10 |
| PPYR1 | 12 | 130 | 5 | 10 | 46503539 | Pancreatic polypeptide receptor 1 |
| WIPI2 | 129 | 5 | 5 | 7 | 5220425 | WD repeat domain, phosphoinositide interacting 2 |
| NDN | 1 | 29 | 5 | 15 | 21481654 | Necdin homolog (mouse) |
| ADARB2 | 9 | 123 | 4 | 10 | 1218072 | Adenosine deaminase, RNA-specific, B2 (RED2 homolog rat) |
| ELAVL2 | 7 | 91 | 4 | 9 | 23680104 | ELAV (embryonic lethal, abnormal vision, Drosophila)-like 2 (Hu antigen B) |
| GNA12 | 128 | 5 | 4 | 7 | 2734268 | Guanine nucleotide binding protein (G protein) alpha-12 |
| ARMC4 | 8 | 132 | 3 | 10 | 28141104 | Armadillo-repeat containing 4 |
| IFNA21 | 6 | 96 | 3 | 9 | 21155635 | Interferon alpha-21 |
| LANCL2 | 120 | 2 | 3 | 7 | 55400634 | LanC lantibiotic synthetase component C-like 2 (bacterial) |
| MAD1L1 | 129 | 5 | 3 | 7 | 1821953 | MAD1 mitotic arrest deficient-like 1 (yeast) |
| MGAM | 133 | 7 | 3 | 7 | 141342147 | Maltase-glucoamylase (alpha-glucosidase) |
| MOBKL2B | 10 | 71 | 3 | 9 | 27315207 | MOB1, Mps One Binder kinase activator-like 2B (yeast) |
| ACTR3B | 136 | 1 | 2 | 7 | 152087783 | ARP3 actin-related protein 3 homolog B (yeast) |
| C1GALT1 | 130 | 5 | 2 | 7 | 7240413 | Core 1 synthase, glycoprotein-N-acetylgalactosamine 3-beta-galactosyltransferase 1 |
| CAMK1D | 8 | 126 | 2 | 10 | 12431588 | Calcium/calmodulin-dependent protein kinase ID |
| CENTA1 | 129 | 6 | 2 | 7 | 904065 | Centaurin-alpha 1 |
| FBXL18 | 130 | 5 | 2 | 7 | 5481953 | F-box and leucine-rich repeat protein 18 |
| FTSJ2 | 128 | 5 | 2 | 7 | 2240453 | FtsJ homolog 2 (E. coli) |
Besides nominating disease drivers, the derived network itself contains additional useful information. For instance, we detect robust CNA–mRNA links between the hubs EGFR, PDGFRA, and CHIC2 and target genes that are markers of early neural development, such as the glioblastoma stem cell marker CD133 (PROM1) and the transcription factors SOX10, SOX11, NR2E1(TLX) and NKX2.2 (Figure 3B) (Shi et al, 2008; Haslinger et al, 2009; Piccirillo et al, 2009). For instance, neural stem cell renewal is under epistatic control of both SOX10 and NR2E1(TLX) (Shi et al, 2008), and our model may suggest that PDGFRA and EGFR may act as complementary drivers of these two regulators (Discussion). Comparing the CNA-driven network with the two compound-target databases Drugbank and Ingenuity, we nominate a set of compounds with known activities that could, in principle, counteract endogenous perturbations in our model (Figure 3C).
Phenotypic and transcriptional consequences of hub gene perturbation in glioblastoma cell lines
To assess the biological relevance of a hub gene in the G network that has not been previously associated with glioblastoma, we have chosen to perform directed validation experiments on NDN. This gene has five downstream targets in the G network and shares a common target, fibroblast growth factor 9 (FGF9; also known as glia-activating factor), with PDGFRA which is frequently amplified in glioblastoma (Figure 3B). NDN is maternally imprinted, located on chromosome 15, and encodes a p53-interacting protein that belongs to the melanoma antigen family (Taniura et al, 1998). In the TCGA data, NDN is deleted in 29/186 patients. We introduce perturbations of NDN by overexpressing the gene in four glioblastoma cell lines (T98G, U-87MG, U-343MG and U-373MG), leading to decreased cell cycling time in all cell lines, except T98G (Figure 4A–C). Using the U-343MG cell line, we measure the expression of a set of downstream targets of NDN and PDGFRA by qPCR to assess the transcriptional response of NDN overexpression and inhibition/stimulation of PDGFRA, respectively. The results confirm a set of EPoC predictions, including induction of CPNE8 by NDN, induction of KCNH8 by PDGF-AA protein dimers (i.e., a PDGFRA agonist) and suppression of KCNH8 by Imatinib (a selective inhibitor of PDGFRA and other tyrosine kinases). Further, when NDN is overexpressed, FGF9 does not respond to PDGFRA perturbation. This is not only consistent with the prediction that NDN and PDGFRA regulate the transcription of FGF9 in opposite directions, but may also suggest a more complicated mechanism that is not captured by our model because NDN perturbation by itself did not suppress FGF9 levels (Figure 4E). For the other two of the tested transcripts, GALNT13 and ALK, which are both expressed at very low levels in U-343MG cells, we did not detect any significant changes. Further, we perturbed the activity of the EGFR by activating it using one of its ligands (EGF) and inhibiting it with a selective EGFR inhibitor (Gefitinib). As readout, we measured the transcriptional effect on SOCS2 (a modulator of STAT signaling), NR2E1 (also known as TLX, a transcription factor believed to be important for neural stem cell renewal), yielding results compatible with a coupling between hub perturbation and transcriptional response (Figure 4F).
Figure 4.

Experimental perturbations of a network region controlled by NDN and PDGFRA. (A–D) NDN overexpression slows the growth of glioblastoma cell lines. (A) Interactions in the network around EGFR, NDN and PDGFRA. (B) Perturbation of NDN by stable overexpression in two separate U343-derived cell lines, denoted as NDN+ (moderate overexpression) and NDN++ (high overexpression). (C) Growth curves collected during 6 days showed that NDN overexpression inhibits growth of U343 cells. Error bars indicate 95% confidence intervals. (D) Single-time point (7 days) measurement of cell number in NDN-overexpressing cells. Error bars indicate s.e.m. (E) Perturbation of PDGFRA by PDGF-AA protein (ligand) and imatinib (STI-571; Gleevec™; inhibits PDGFRA and certain other tyrosine kinases), respectively, produces opposite responses in target genes KCNH8 and FGF9, which were identified as downstream targets of PDGFRA in the model. NDN overexpression induces CPNE8 target genes and modulates FGF9 response to PDGFRA. Error bars indicate 95% confidence intervals of mRNA expression log2-relative to untreated controls. (F) Perturbation of EGFR by its ligand EGF and gefitinib (ZD-1839 Iressa™; inhibits EGFR) produces opposite responses in the predicted EGFR target genes SOCS2 and NR2E1.
Taken together, these observations support that the estimated CNA-driven network is mechanistically informative, and that CNA–mRNA links identified by EPoC broadly agree with data from relevant validation experiments. Our approach operates at the gene level and our data also support that individual hub genes can be identified in practice (e.g., NDN). Very large aberrations, however, cannot be fully resolved by the current modeling strategy (e.g., EPoC identifies a small set of candidate hubs in a 7 Mb region on the short arm of chromosome 10, which is often lost in its entirety in glioblastoma; see Discussion, Table I).
CNA-driven networks contain prognostic information
A crucial test for any disease network model is to ask if it produces clinically relevant predictions. While it is well established that CNA and mRNA patterns may predict survival and response to therapy using a range of supervised or unsupervised techniques (Broëtet al, 2009; Zhang et al, 2009; Verhaak et al, 2010), less work has been done in deriving prognostic scores from actual network models. We thus proceed to test the patient prognostic value of the CNA-driven network G, using the derived survival scores Zy and Zu (above).
As predicted, we find that these scores achieve a significant degree of prognostic separation (Figure 5A). In contrast, when we examine the prognostic properties of the transcriptional network, A, we find no evident survival stratification when separating patients along the leading components of the SVD of A. We also demonstrate that a standard singular value decomposition (SVD) calculated from mRNA profiles or CNA profiles fails to detect survival differences in the data. We further calculate survival curves for the first six components of both mRNA and CNA data in the G, A and data SVD cases, revealing that survival differences are only seen in the first SVD component of G (Table II).
Figure 5.

Derivation of prognostic scores from the network model. (A) Kaplan–Meier curves to assess prognostic scores extracted from the CNA-driven network. Prognostic scores are computed by a sparse singular value decomposition of the CNA-driven network G (Materials and methods). Patients are divided into two groups by projecting their CNA profiles and mRNA profiles onto the main left and right axis of the singular vectors of G, respectively. This separates patients with favorable and poor prognosis (upper panels). By contrast, the corresponding analysis of the transcriptional network A (middle panels) does not produce any significant separation of patients in terms of survival, nor does a standard singular value decomposition (SVD) of the mRNA profiles or CNA profiles (lower panels). The panels show the results obtained by projection onto the first SVD components. The results obtained when projecting onto additional components are given in Table II. (B) The sparse singular value decomposition of the CNA-driven network G identifies genes with strong scores for signal amplification, i.e., genes whose perturbations are highly amplified by the network system (here illustrated as yellow nodes, e.g., PDGFRA), as well as mRNA transcripts that are most affected by these perturbations (green nodes, e.g., GRIK1; Figure 1C).
Table 2. Survival differences.
| Network and data type | Comp 1 | Comp 2 | Comp 3 | Comp 4 | Comp 5 | Comp 6 |
|---|---|---|---|---|---|---|
| (A) log-rank test P-values of survival difference between patient with a positive versus negative loading on SVD components 1–6 | ||||||
| EPoC G, CNA | *0.0014 | 0.1129 | 0.0216 | 0.1157 | 0.0853 | 0.0147 |
| EPoC G, RNA | *0.0004 | 0.1560 | 0.0759 | 0.0818 | 0.0516 | 0.0412 |
| EPoC A, CNA | 0.3109 | 0.1468 | 0.1100 | 0.0393 | 0.2817 | 0.1069 |
| EPoC A, RNA | 0.2198 | 0.3526 | 0.0479 | 0.2402 | 0.1570 | 0.3621 |
| SVD, CNA | 0.0505 | 0.1266 | 0.0261 | 0.0042 | 0.4677 | 0.4251 |
| SVD, RNA | 0.0869 | 0.0963 | 0.2822 | 0.1198 | 0.0225 | 0.4091 |
| Subtype | Classical | Proneural | Neural | Mesenchymal |
|---|---|---|---|---|
| (B) log-rank test P-values of survival difference between patients in different molecular subclasses, as defined in (Verhaak et al, 2010) | ||||
| Classical | — | |||
| Proneural | 0.4517 | — | ||
| Neural | 0.6744 | 0.5598 | — | |
| Mesenchymal | 0.3600 | 0.7371 | 0.5220 | — |
To visualize the contribution of individual genes to the survival scores, we color-code the CNA-driven network G (Figure 5B). As an example, from the leading singular vectors of G, we note that CNAs in EGFR and PDGFRA are highly amplified by the network system model (yellow nodes) and identify the leading mRNA responders to these perturbations (green nodes), which include, e.g., growth factor PDGFA, glutamate receptor GRIK1, the transcription factor SOX11, and the STAT pathway modulator SOCS2.
We thus conclude that the estimated CNA-driven EPoC model correlates with a clinically relevant phenotype. This is in general support of model validity, and suggests that integrative models may help to identify clinically useful glioblastoma biomarkers.
Technical comparison with mRNA-based and eQTL-type methods
We compare the performance of EPoC to a set of alternative network construction methods. We include a set of mRNA-only methods based on information theory (ARACNE), partial correlations (GeneNet) and sparse estimation of the inverse covariance (precision) matrix (glasso). We also consider methods based on combinations of genotype and expression data. These include (i) a univariate SNP-eQTL (Stranger et al, 2007a, 2007b), here using CNAs in place of SNPs; (ii) a recent network method termed remMap (Peng et al, 2008); and (iii) the SNP-eQTL module network solver LirNet (Lee et al, 2009), here using CNAs genotypes in place of SNPs. remMap and LirNet, similar to EPoC, use variants of lasso for model fitting and are thus preferred points of comparison. remMap was recently proposed to relate genomic-region variations to select genes in breast cancer, and LirNet was of late advantageously compared with a set of eQTL-type methods, including Geronemo (Lee et al, 2006) and Bayesian networks with eQTL priors (Zhu et al, 2008). Details of the comparison are given in Materials and methods.
Model consistency between independent glioblastoma data sets
We identify the subset of 146 patients (out of the 186 patients analyzed above), for which two independent CNA and mRNA data sets have been produced at different institutes in the TCGA consortium. These technically independent data sets provide an ideal setting for an unbiased comparison of the methods. We thus apply each method to the two data sets, and use Kendall's W to investigate the consistency between the two solutions (Materials and methods). This analysis shows stronger performance by EPoC CNA-driven networks G over all other methods for all but the largest network sizes (Figure 6A), i.e., EPoC G network solutions from two technically independent data sets largely agree both in terms of detection and estimated strength of network interactions.
Figure 6.

Method comparisons: network consistency and pathway interactions. (A) We compare network models derived from two full replicate glioblastoma data sets (146 identical tumors; same patients and samples) but processed at different centers with slightly different technological setups (Affymetrix and Agilent technologies, run at MSKCC, Harvard Medical School and Broad Institute, Materials and methods). This test measures each method's reliability, i.e., its robustness to noise and technological factors. EPoC estimation of the CNA-driven network G is the best-performing method on the TCGA data (1−W lower, arrow ↗). Glasso is second best, followed by sparse estimation of the transcriptional network A (EPoC A), and remMap. LirNet, eQTL, GeneNet and ARACNE all exhibit less robust performance compared with EPoC G. (B) We map interactions found by EPoC and other methods to molecular links in the pathway repositories HPRD, Reactome, Intact and NCI-nature. Each interaction is characterized by the number of steps minimally needed to ‘walk’ between the network gene and its target (i.e., the shortest path). We argue that a well-estimated network should be comprised of identified interactions that either match known interactions in the databases or are enriched for shorter paths. The figure depicts the enrichment (relative proportion of interactions that correspond to a shortest path length of 1 or 2 interactions in a pooled network based on the four different pathway databases). EPoC G interactions are clearly enriched for short or direct paths in the databases, followed by glasso and EPoC A.
Apart from glasso, methods that incorporate combined genomic/transcriptional data perform better than mRNA-based networks (ARACNE and GeneNet). We also derive transcriptional EPoC A networks (solving Equation (2); Materials and methods). EPoC A corrects transcripts for their own CNAs prior to network construction and performs quite well, but clearly worse than EPoC G. This is best explained by the stronger correlations among mRNAs compared with CNAs (predictor variables in EPoC A and EPoC G, respectively), as it is well known that regression modeling with highly correlated predictors is subject to instability (Breiman, 1996; Skogestad and Postlethwaite, 2005; Nordling and Jacobsen, 2009; and references therein). While CNAs may exhibit strong correlation within a genomic region, CNA correlation between genomic regions is globally much lower than between mRNAs (data not shown). As expected, EPoC G, remMap and LirNet perform better than standard eQTL, which likely reflects the benefit of a multivariate modeling approach, using regularization (L1 in EPoC G, and L1+L2 in remMap and LirNet) over a univariate approach (eQTL).
Pathway overlap and prediction error
We proceed to determine the overlap between the derived networks and two protein–protein interaction (PPI) and two pathway databases. Results from this analysis demonstrate a similar ranking of methods as in the robustness tests above (Figure 6B, Materials and methods). That is, EPoC G captures the most known direct and short-path interactions. In absolute numbers, 65 interactions in the CNA-driven network correspond to known short-range interactions (shortest path between the genes is one or two steps in at least one of the databases). Among the other methods, EPoC A and glasso also exhibit a good overlap with known pathways.
We emphasize robustness in the above analysis, but EPoC can also be used to generate network models for mRNA prediction from CNA data (Materials and methods). We assess the prediction accuracy by computing cross-validation errors for EPoC G, remMap and LirNet, which all produce natural prediction models. We conclude that EPoC G results in the smallest prediction errors on the glioblastoma data set (Supplementary Figure 2).
Qualitative differences and speed
Additional comparisons (Supplementary information) show that the genotype-driven networks and mRNA-based networks capture complementary genomic process information in terms of structure and gene content: the EPoC CNA-driven network has a more hub-oriented structure, with several development-associated genes as responders, whereas the transcriptional network captures inflammatory and cell cycle processes (Supplementary Figure 1). The distinction between CNA-driven and transcriptional networks is well illustrated by a hierarchical clustering summary, structurally comparing networks produced by all the above methods using Kendall's W. We see a clear structural separation between mRNA-based and genome-type-driven networks (Supplementary Figure 1). As a practical consideration, we also demonstrate that the scalability and algorithmic speed of EPoC is highly competitive (Supplementary information).
From these comparisons, we conclude that EPoC exhibits excellent performance in terms of model reproducibility, validity and algorithmic speed (see Materials and methods and Supplementary information). EPoC is primarily of interest for derivation of large networks at the single gene level and thus complements methods that use different levels of description, such as grouping CNAs into whole regions, integrating CNA and mRNA events over PPI networks, or by condensing transcripts into modules or linear superpositions (below). In Materials and methods and Supplementary information, we discuss the relative performance of the methods in more detail, including plausible explanations for performance differences.
Discussion
We have demonstrated that combined network modeling of CNAs and mRNA expression levels in tumors generates mechanistically and prognostically informative results. The network-based survival scores introduced here serve to identify molecular features useful for predicting the outcome of individual patients, adding to our understanding of survival differences in the TCGA cohort (Verhaak et al, 2010). Extensive computational and experimental assessments confirm EPoC as an efficient and robust methodology to interpret CNA–mRNA profiles of glioblastoma. Applying our method to other tumors is facilitated by an efficient solver in R and MATLAB packages (Materials and methods).
Possible limitations
CNAs often span multiple genes in large chromosomal regions (sometimes a whole chromosome or chromosome arm), introducing copy number correlations between genes in the affected region. This may lead to erroneously identified CNA–mRNA couplings in network construction. EPoC tries to address this in two ways: first, each transcript's mRNA signal is corrected by its own CNA value (Materials and methods), which largely de-correlates the transcript's signal with neighboring CNAs, thus reducing the risk of including false couplings in such regions; second, the bootstrap procedure will work against CNA–mRNA associations that cannot be resolved at the single gene level. Empirically, this seems to work well in glioblastoma, as known oncogenes and tumor suppressors are recovered as single gene hubs by the algorithm. However, we also note cases where EPoC does not resolve a single regulator (e.g., Chromosome 10, see Results section). While our tests show good support for pursuing a gene-level approach for glioblastoma, optimally modeling larger genomic regions is an interesting future research problem; possible approaches to explore are clustering of CNA profiles into regions (Peng et al, 2008), adapting statistical techniques from linkage analysis (Jiang and Zeng, 1995) or using annotation features as regulatory priors (Lee et al, 2009).
Our analysis focuses on the full set of glioblastoma patients in the TCGA compendium. We also considered subtype-specific models, using the recent classification of the TCGA glioblastoma cohort into Classical, Mesenchymal, Proneural and Neural subtypes (Verhaak et al, 2010), but found that the number of patients within each subtype is too small to produce a robust network model (as indicated by the bootstrap procedure, data not shown). We reserve work on specific cancer subtypes for when more patients become available, and suggest a rule of thumb that EPoC be applied to 100 patients or more. Future data sets may also involve finer anatomical sampling, helping to model distinct sub-populations of cells in glioblastoma tumors.
Other approaches
In this article we have explored and compared several methods for the analysis of CNA and mRNA data. We briefly discuss some additional methods here.
CCA is a traditional multivariate technique, and has recently been extended and applied to integrate CNA and mRNA data (Waaijenborg et al, 2008; Witten et al, 2009). CCA links modules of CNA to modules of mRNA, i.e., identifies a sequence of linear combinations of subsets of CNAs maximally correlated with linear combinations of subsets of mRNA. Thus, CCA does not provide a gene-level network model, but opts to summarize CNA–mRNA interactions at a module level. CCA treats CNA and mRNA data symmetrically. Therefore, CCA components (module–module links) do not generally agree with the survival score decomposition of the CNA-driven network G, except under very unrealistic assumptions (Materials and methods and Supplementary information). An investigation to further compare module-level and gene-level methods, and alternative decomposition techniques is reserved for future work.
A second alternative approach to integrate CNA and mRNA data would be to adapt the electrical circuit-inspired model, eQED (Suthram et al, 2008; Kim et al, 2010), which links genetic perturbations to transcriptional responses over a predefined network from molecular interaction databases. Clearly, the key distinction between this approach and EPoC is the use of PPI and other data as a scaffold on which to construct the model. In work by Zhu et al (2004, 2007, 2008), it is suggested to use SNP-eQTL priors to guide the construction of mRNA-based transcriptional networks (for a performance test against a lasso-based method, see Lee et al, 2009).
Future directions
For biological follow-up work, there are several predicted couplings between CNA hubs and stem cell markers that may be interesting to investigate further (Figure 3B). For instance, autocrine signaling loops are known to suppress differentiation in glioblastoma (Erlandsson et al, 2006; Dai et al, 2001); it is therefore interesting that our model connects the PDGFRA and EGFR hubs to at least seven known growth factors (including PDGFA, PDGFD, FGF9) and five known feedback regulators of cell signaling (including SPRY1, SPRY2 and SOCS2). Exploring such links by targeted experiments may lead to new insight regarding signaling networks in glioblastoma. In addition, the compound predictions (Figure 3C) can be explored in several ways, for instance, by assessing the level of synergism between these compounds and standard glioblastoma therapies such as Temozolomide.
Future methodological work should be aimed at incorporating other data types, including miRNA expression, DNA methylation and sequence mutations, to model the nonspecific perturbation term, Γ in Equation (3), currently treated as noise in the network model estimation procedure. Both modeling and model testing might benefit from additional molecular network reference data. For instance, TF-target links could also be included as a prior for network construction or to guide method development. Extending biological-mechanistic models to encompass all levels of the regulatory process will require careful consideration and important choices of model representations. Of particular interest will also be to further develop prognostic scoring methods by linking network decomposition techniques to more advanced survival modeling, which may be an interesting extension to methods that relate principal components in tumor molecular profiles to relative hazard (Nguyen and Rocke, 2002a, 2002b).
Ambitious efforts are currently being undertaken to acquire comprehensive genome-scale data sets for several cancer types, including the Cancer Genome Atlas, and the International Cancer Genome Consortium. The recent data deluge presents a challenging opportunity to develop mechanistically and clinically relevant models of the data. EPoC is one step in this direction, and helps to set the stage for the continued modeling efforts in the context of human cancer genome programs.
Materials and methods
Glioblastoma data preparation
We obtained DNA and mRNA molecular profiles from the Cancer Genome Atlas (TCGA) data repository (http://tcga.org and TCGA-Consortium, 2008). We use level 3 data (discrete copy number estimates and mRNA levels for known protein-coding genes) generated using Agilent 244 k DNA and Agilent 44 k mRNA, and Affymetrix U133 mRNA arrays. For model construction, mRNA and Affymetrix mRNA signals are averaged for more stable signal (Verhaak et al, 2010). Sex chromosomes are removed from the analysis.
Prior to analysis with EPoC or other network estimation methods, we standardize the amplitude of the mRNA levels, i.e., center each gene around its mean expression level and divide by its standard deviation. All methods have also been compared on unstandardized data, but standardization substantially improves the consistency of models between replicate data sets, as well as facilitates the search for an optimal regularization parameter in the sparse regression modeling, as, after standardization, a single parameter value can be used for all transcripts.
Network parameter estimation
After centering and standardization, and assuming steady state, Equation (1) is rewritten as
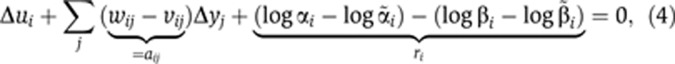 |
4 |
(where 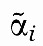 and
and 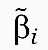 refer to mean gene-specific constants across all tumors). Collected for all transcripts i, the above translates to the linear equation systems in Equation (2)
AΔY+ΔU+R=0. Equation (2) can be transformed into Equation (3), where we have ΔY=GΔU+Γ, G=−A−1 and Γ=−A−1R. ΔY is the n × T matrix of mRNA expression levels for the n transcripts across the T tumors, similarly for the CNAs ΔU, and G is the n × n CNA-driven network matrix. Below, we outline how to solve for parameters of interest in Equation (3), although this is easily recast to estimate parameters in Equation (2) instead.
refer to mean gene-specific constants across all tumors). Collected for all transcripts i, the above translates to the linear equation systems in Equation (2)
AΔY+ΔU+R=0. Equation (2) can be transformed into Equation (3), where we have ΔY=GΔU+Γ, G=−A−1 and Γ=−A−1R. ΔY is the n × T matrix of mRNA expression levels for the n transcripts across the T tumors, similarly for the CNAs ΔU, and G is the n × n CNA-driven network matrix. Below, we outline how to solve for parameters of interest in Equation (3), although this is easily recast to estimate parameters in Equation (2) instead.
Prior to solving for parameters of interests, EPoC uses, by default, an optional filter to distinguish candidate (CNAs) from inherited (germ line) copy number variations (CNVs). For each gene, we calculate the number of patients with amplification of the gene (N1), and the number of patients with deletion of the gene (N2). Given a total number of changes of N1+N2, we evaluate P as the binomial cumulative probability function for N1 successes in N1+N2 attempts at 0.5 probability. Candidate hubs are selected as genes, for which P<δ (bias toward deletion) or P>1−δ (bias toward amplification). For our analysis, we set δ=10−8, thus including 25% of the genes as possible regulators. The key difference when the filter is not applied is that the CNV gene GSTT1 is selected as a hub; this gene has both gains and losses, indicating no selection by the tumor, and was also seen in ovarian cancer data from TCGA (data not shown), and is located within a known CNV. We expect that the recurrent CNA detection programs RAE or GISTIC could also be used in this step with good results (Beroukhim et al, 2007; Taylor et al, 2008).
For each gene i we first estimate the direct effect of the gene's own CNA by the positive truncated least-squares estimate: d=max(0,ΔUiT ΔYi), where ΔYi is the T × 1 vector transpose of the i-th row of ΔY, and similarly for ΔUi. From the CNA filter operation above, we obtain the set of H candidate hubs and denote the corresponding CNA values by ΔUH, where ΔUH is a H × T matrix. We then solve the n L1 regularized regression problems (one for each gene), treating Γ terms as noise in the estimation:
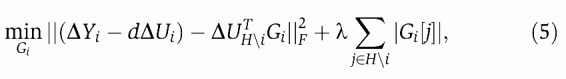 |
5 |
where ΔUHi=ΔUH[\i,\i], i.e., the ΔUH matrix excluding gene i, and Gi denotes the H × 1 vector transpose of the i-th row in G with elements corresponding to the hub set H, but excluding gene i, and λ is the regularization parameter that controls the degree of sparsity (number of non-zeroes) in G. Gi[j] denotes the j-th element in vector Gi. We solve Equation (5) using the cyclic coordinate descent algorithm (Fu, 1998; Friedman et al, 2007). Following Friedman et al (2007), we speed up the computation using the fact that λ>ΔUHi∣∣ΔYi−dΔUi∣∣∞ implies that Gi=0 (Osborne et al, 2000), meaning that we need not search for a model for transcripts that meet this criterion as the optimal model will be empty. As a global upper limit for λ, we define λmax=maxiΔUHi ∣∣ΔYi−dΔUi∣∣∞, for which all elements of G will be zero. The EPoC algorithm is summarized below (Box 1). Note, the same algorithm can be used to estimate the transcriptional network A by simply replacing ΔUHi with the mRNA data ΔYi.
EPoC flowchart.
1. Data preparation
Center the mRNA and CNA data, constructing the two n × T data matrices ΔY and ΔU.
Reduce the CNAs to candidate hubs using the germline binomial filter described above. Collect the candidate CNA hubs in the H × T data matrix ΔUH.
2. Estimate the direct effect of each transcript's own CNA
For each gene i,
estimate the direct effect of the gene's own CNA by d=max(0,ΔUiTΔYi), where ΔYi is the T × 1 vector transpose of the i-th row of ΔY, and similarly for ΔUi .
3. Estimate elements of G using lasso
For genes i=1, … ,n, solve the lasso problem
 |
where Gi denotes the vector transpose of i-th row in the gain matrix G with elements corresponding to the hub set H, but excluding gene i.
4. Optimal network size
Randomly split the data into two groups. Apply steps 2 and 3 for different values of λ to each group, and use Kendall's W to compute network agreement. Select the optimal λ that maximizes W (average W over 1000 random splits) and record the corresponding network size S. (Alternatively, here optimize for mRNA prediction.)
5. Final network
Generate B=1000 pseudo-bootstrap data sets. For selected λ in step 4—find the corresponding λ′ that generates networks of size S on the bootstrap data set (applying steps 2 and 3 above). The bootstrap networks are denoted Gb, b=1, … ,B. Collect interaction frequencies f{i,j}=∑b=1B1(Gb{i,j}≠0)/B. The final network consists of interactions with f{i,j}>f* (here with f*=0.2).
EPoC has been implemented in both R and MATLAB (with C) and the software is available for download at http://sysbio.med.gu.se/epoc.html
Optimizing the size of the network
The size of the estimated network is controlled by the lasso penalty parameter λ. To determine an appropriate value for λ between 0 (fully connected model) and λmax (smallest, non-empty model) we consider two different validation criteria.
We first compare network consistency using Kendall's W (Kendall and Smith, 1939), commonly used to assess agreement among rank-order lists and lately applied for network inference (Vacher et al, 2008; Milns et al, 2010). Here, we rank the network edges (presence and magnitude of an interaction) from different network estimates. If the rank lists agree completely, W is 1, and if the rank orders exhibit a random overlap, W is 0. Kendall's W is analogous to rank correlation but, as it can compare several rank lists instead of only two, we chose to use this measure instead of Spearman's rank correlation for future scalability. Here, we randomly split the data set into two non-overlapping groups, independently estimating a network for each group, and there are thus two rank lists to compare. For more robust inference, the above validation procedure is repeated 1000 times. The final choice of λ, or network size, is based on the mean value of Kendall's W across the 1000 random splits (Figure 2A, upper panel).
We also assess the prediction power of our method. When mRNA prediction is the goal, we need to optimize network size with this in mind. We thus compute the prediction errors for each mRNA transcript using cross-validation. Leaving out one 10th fraction of the data, we estimate the gain matrix: 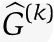 . For the left-out portion of the data, we can now predict the mRNA transcript by
. For the left-out portion of the data, we can now predict the mRNA transcript by 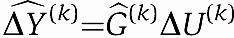 and compare with the true observed mRNA expression levels ΔY(k). Note, ΔY(k), ΔU(k) refer to the left-out data, whereas the estimate
and compare with the true observed mRNA expression levels ΔY(k). Note, ΔY(k), ΔU(k) refer to the left-out data, whereas the estimate 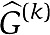 refers to the estimate based on all data, except the left-out portion. The cross-validation prediction error is defined as
refers to the estimate based on all data, except the left-out portion. The cross-validation prediction error is defined as
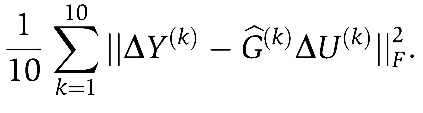 |
The validation procedure is repeated 1000 times. The final choice of λ is based on the mean prediction error across 10-fold random splits of the data (Figure 2A, lower panel). We note that networks optimized for prediction are larger than networks optimized for robustness.
For robust inference, we produce a final network model by repeating the estimation and validation procedures several times using so-called pseudo-bootstrap (Friedman et al, 2000). We generate bootstrap samples as follows: for each bootstrap simulation b=1, … ,B, we create pseudo-bootstrap mRNA data as ΔYb=(1−c)ΔY+cΔY* (here B=1000 bootstrap simulations). ΔY* is a n × T data matrix, where each column (tumor sample vector across genes) has been randomly sampled from the columns in ΔY. That is, in ΔYb each column represents a weighted combination of one tumor sample vector with another sample. The constant c is small, here set to 0.01. The pseudo-boostrap CNA data, ΔUb, is obtained in exactly the same way. We then estimate the boostrap network Gb by applying EPoC to the data set (ΔUb, ΔYb). We collect frequency information for each interaction as f{i,j}=∑b=1B1(Gb{i,j}≠0)/B, i.e., f{i,j} is the proportion of bootstrap samples for which the interaction i←j is present in the network. Large values of f{i,j} suggest that the interaction is real, whereas small values suggest it is detected just by chance (numerical instabilities in the estimation procedure). We compute a cutoff for f using permutations of the data (Figure 2B) and pick a frequency threshold of 20%, which is well above interaction frequencies expected by chance. We visualize the obtained networks using Cytoscape (Shannon et al, 2003).
Network-based survival score
The singular value decomposition of G is G=CΛDT (where CCT=I, DDT=I and Λ is diagonal). The right singular vectors (columns) in D represent the leading directions of CNA perturbations that are amplified by the system G. The left singular vectors (columns) in C represent for which mRNA transcripts these perturbation signals result in the largest effects. To aid in interpretation and identify a small set of potential prognostic biomarkers, we here construct the SVD of G using a sparse PCA method (Zou et al, 2006). This is based on a regression formulation of PCA, and employs a combined L1 and L2 penalty (a.k.a elastic net) to identify the non-zero loadings of the principal components. The sparse SVD component D is obtained through a sparse PCA of GGT, and the sparse component C is obtained through a sparse PCA of GTG. The level of sparsity is chosen such that (i) we obtain a reasonable set of biomarkers for possible therapeutic follow-up, and (ii) the solution is stable across neighboring values of the sparsity penalty.
The mRNA profiles of individual patients are projected onto the singular vector space by Zy=CTΔY (rows of Zy will be components, columns will be patients); and CNA profiles are projected by Zu=DTΔU (rows of Zu will be components, columns will be patients). For the different components of Zy and Zu we thus compare patients z>0 and z<0 in terms of clinical survival (from date of surgery to date of death); survival difference P-values are obtained by the log-rank test.
One could consider constructing alternative biomarker modules to the above using sparse canonical correlation (CCA; Waaijenborg et al, 2008; Witten et al, 2009). In the Supplementary information, we discuss this alternative in more detail, but note here that sparse SVD of G can disagree substantially from CCA. The SVD of G focuses on CNA as the input or driver of mRNA changes, whereas CCA treats CNA and mRNA symmetrically. In our toy example, SVD of G correctly identifies CNA biomarkers and mRNA responders (Figure 1C). In contrast, CCA is susceptive to, and reflective of, the structure of the noise term of Equation (3). This is a major concern in our glioblastoma data set, where we know that the noise structure is non-negligible, capturing all the mRNA–mRNA dependencies that are non-CNA related.
Method comparisons
A detailed description of the methods is found in the Supplementary information.
Structural consistency tests
We first construct two replicate versions of the TCGA data set, A and B. A comprised array-CGH and Agilent array measurements from MSKCC; B comprised Agilent array-CGH profiles and Affymetrix U133A mRNA profiles generated at Harvard and Broad Institute, with both A and B consisting of 146 individually matched samples. For 100 iterations, we select a mixture of the 250 genes with the highest mRNA variance in one of the data sets, plus an additional random 250 genes from the 10 672 genes studied. This way, we get a set of genes that can be analyzed also by the slowest methods (remMap, glasso, ARACNE), and which introduces a bias in favor of the methods that uses mRNA data only. We subsequently run each method for each of a series of parameter values corresponding to stringency, resulting in a series of networks of different sizes. The parameters tuned are glasso ρ, ARACNE dpi, GeneNet significance threshold, EPoC λ, remMap L1 penalty, LirNet L1 penalty and eQTL P-value cutoff. remMap has an additional L2 parameter, which is tuned for optimal performance (W). Similarly, LirNet is optimized to perform well by (i) using the same set of initial clusters/modules in the A and B data; (ii) by optimizing the number of modules and the L2 penalty. For all methods, we compute network agreement between data sets A and B using Kendall's W (Figure 6A).
Pathway comparisons
We download Reactome, IntAct and HPRD from Pathwaycommons.org, and map identifiers to the 10 672 genes in our data set. We subsequently calculate the undirected shortest path Rij for all gene pairs (i,j) in these databases using Johnson's algorithm (Johnson, 1977). For a given network G, we then calculate the enrichment (relative proportion) as:
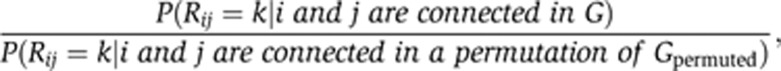 |
calculated across non-diagonal elements (i≠j) and where Gpermuted is generated by random permutation of the non-diagonal G elements (1000 simulations; Figure 6B). For Figure 6B, k=2 is used.
Prediction of mRNA levels
We calculate the 10-fold cross-validation error for the remMap, LirNet and EPoC methods on random sets of 500 genes (above) (due to speed constraint of remMap). Each method is tuned to perform well by adjusting L1 and (when applicable) L2, and module number.
Experimental methods
Cell culture and perturbation of NDN, PDGFRA and EGFR
Human U-343MG glioblastoma cells were cultured in Dulbeccos modified Eagles medium (DMEM; Lonza) supplemented with heat-inactivated 10% fetal bovine serum, 100 units/ml penicillin and 0.1 mg/ml streptomycin. Cells were kept in a 37°C humidified incubator containing 5% CO2. For transfection, 1.5 × 105 cells were seeded in a six-well plate, and the FuGENE 6 transfection Reagent (Roche) was used. The cells were transfected by the FLAGC-NecF plasmid containing Flag-tagged NDN and the neomycin selection cassette. For generating stable transfectants, 500 μg/ml neomycin was used, whereas for continuous growth 250 μg/ml was added. Two different cell lines (NDN+ and NDN++) were generated that expressed NDN at different levels. Negative control lines were generated by the same protocol, but with an insert-free plasmid. In addition to this experiment, three other cell lines, Human U-87MG, T98G and U-373MG (Cell Lines Service, Germany), were transfected with the same plasmid. U-343MG was re-transfected and previous results were confirmed. Conditions and procedures were similar, except for the transfection procedure, where cells were seeded in six replicates in six-well plates (0.7 to 4.3 × 105 cells/well) and transfected with OPTI-MEM, lipofectamine and PLUSreagent (Invitrogen Corp.). To perturb the PDGFRA node, PDGF-AA was added at 30 ng/ml. To inhibit cytoplasmic tyrosine kinases, we used STI-571 (Gleevec) at 1 μM. Cells of 50–70% of confluence were grown in a six-well plate and in serum-free DMEM medium for 1 day before PDGF-AA and STI-571 treatments. The duration of PDGF-AA and STI-571 treatment was 14 h. To perturb the EGFR node in U-87MG, U-343MG, U-373MG, T98G and A172, EGFR inhibitor Gefitinib (Selleck Chemicals) and EGFR ligand EGF (supplied by Peprotech) was added at 5 μM and 50 ng/ml, respectively. For the Gefitinib-treated cells, controls were treated with equal concentration of DMSO. Cells were seeded in triplicates at 5 × 105 cells/well in a six-well plate 24 h before addition of Gefitinib and EGF. Cells were treated for 6 h at 37°C.
Comparison of growth rates
For U-343MG, cell growth of NDN-expressing and cells transfected with insert-free control plasmid was measured using the MTT colorimetric assay. Initially, 1500 cells were seeded in 96-well plates, with 11 replicates for each time point and treatment. At the respective time point, cells were incubated with 20 μl of 5 mg/ml−1 MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) dissolved in PBS. After 6 h at 37°C, media were removed and formazan crystals were dissolved in DMSO, and absorbance was measured at 570 nm. For U-87MG, 75 000 cells/well were seeded in six-well plates and counted after 24 days. For U-373MG and T98G, 200 000 cells/well were seeded in six-well plates and counted after 7 days. Cell number was measured using a Chemometec cell counter.
Statistical model to test growth rate differences
We assume an exponential model for the growth of glioblastoma cells in culture, h(t)=h02kt, in which where t=time (days), h ∝ number of cells and h0 is the value of h at t=0. Following log transformation, this is expressed by the linear equation log2(h(t))=log2(h0)+kt, which is in good agreement with experimental data (not shown). The constant k is estimated by linear least squares, with confidence interval k±sk·t0.975,n−2, where sk is the empirical standard deviation of k. The time tD, it takes to double the number of cells, is given by the inverse of k, i.e., tD=1/k, which has confidence interval
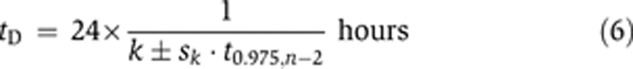 |
6 |
In our experiment, we tested the hypothesis that cells that overexpress Necdin (NDN) grow slower than cells that have been transfected with a negative control plasmid (CTRL). Thus, our null hypothesis H0 is that kNDN=kCTRL and our alternative hypothesis H1 is that kNDN<kCTRL. To compare the doubling rates kNDN and kCTRL, we calculate a T statistic:
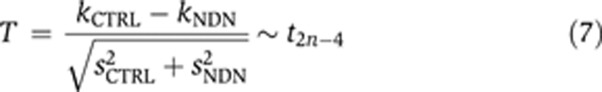 |
7 |
Detection of Necdin expression
For U-343MG, cells of 50–70% confluence were solubilized with lysis buffer (150 mM NaCl, 5 mM Tris, pH 8, 0.5% deoxycholate, 10% glycerol, 1% NP-40, 1 mM PMSF, 1 mM DTT, 1 mM aprotonin). Samples were boiled for 10 min and protein content was measured using Bio-Rad protein-assay Dye Reagent. Samples of 10 μg total protein were analyzed on a 10% SDS–PAGE. After electrophoresis, the proteins were transferred onto a nitrocellulose membrane (Amersham) with a semi-dry transfer equipment (Bio-Rad). The membrane was processed for immunoblotting using an anti-FLAG primary antibody (1:2000, Sigma-Aldrich) at 4°C over night and a secondary anti-mouse antibody (1:10 000, GE Healthcare) for 1 h. To detect signal, the membrane was developed using the ECL Advance System (GE Healthcare) according to the manufacturer's protocol and scanned using LAS-1000 Plus (Fujilm).
qPCR analysis
Cells cultured in a six-well plate were harvested and RNA isolated using the TRIzol reagent (Invitrogen) and Ethanol precipitation. We synthesized cDNA by annealing oligo-dT primers and elongating with M-MuLV reverse transcriptase (Fermentas) according to the manufacturer's protocol. To avoid DNA contamination, the TURBO DNA-free kit (Ambion, Applied Biosystems) was used. We measured levels of transcripts and estimated Ct values for GAPDH, NDN, CPNE8, FGF9 and KCHN8 (primer sequences in Supplementary information) using the StepOne Real-Time PCR system (Applied Biosystems). Primer sequences were selected using the software Primer Express 3.0 (Applied Biosystems). For qPCR analysis of SOCS2 and NR2E1, cells cultured in a six-well plate were harvested and cellular total RNA was extracted using the RNeasy Plus Mini Kit (Qiagen), according to the manufacturer's protocol. cDNAs were synthesized from total RNA (1 μg) using random primers according to the protocol (High Capacity cDNA Reverse Transcription kit, Applied Biosystems). The expression levels of human SOCS2 and NR2E1 mRNA was evaluated using quantitative real-time PCR (TaqMan Gene Expression Assays, Applied Biosystems). Each reaction was run according to manufacturer's protocol (Applied Biosystems). TaqMan Gene Expression Assays used were Hs99999903 m1 for human ACTB; Hs00919620 m1 for human SOCS2; and Hs01128417 m1 for human NR2E1/TLX. The reaction was run using an ABI PRISM 7900 HT Sequence Detection System (Applied Biosystems). Data were collected and recorded by ABI PRISM 7900 SDS Software (Applied Biosystems). Samples were run in duplicates.
Statistical analysis of qPCR experiments
Quantity levels (on an arbitrary scale, using four-point standard curves) were estimated using Applied Biosystems software. Statistical testing was conducted to test the following null hypothesis (using a four-sample, unequal variance t-test): log(quant. of test gene in treated cells)–log(quant. of ctrl gene in treated cells)=log(quant. of test gene in normal cells)–log(quant. of ctrl gene in normal cells). The validity of using a t-test was confirmed by assessing the distribution of residuals (difference between log(quantity) and the mean of log(quantity)). Residuals were compared with a normal distribution by the Kolmogorov–Smirnov test, showing no evidence (P-value=0.1793). We used both ACTB and GAPDH as housekeeping controls.
Supplementary Material
Supplementary Methods, Supplementary Figures S1–2
Acknowledgments
This project receives funding from Cancerfonden (SN), Barncancerfonden (SN), NBCNS consortium (SN), Vetenskapsradet (SN, RJ) and BioCare (SN).
Author contributions: The study was initiated by SN. Computational methodology was developed by SN, RJ, TA, TK, PG and TN. High-performance software was carried out by TA. Data integration was performed by TK, SN and CS. Biological network model assessment was by LS, LL, SN, BjN, BoN and KF. Method benchmarking tests was performed by TA, SN, RJ, TK. Experiments were designed and performed by LS, LL, KF, EJ and BoN. Experiments were analyzed by TK, LS, LL, BoN. Paper was written by SN and RJ with significant contributions from all authors.
Footnotes
The authors declare that they have no conflict of interest.
References
- Adler AS, Lin M, Horlings H, Nuyten DSA, van de Vijver MJ, Chang HY (2006) Genetic regulators of large-scale transcriptional signatures in cancer. Nat Genet 38: 421–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akavia UD, Litvin O, Kim J, Sanchez-Garcia F, Kotliar D, Causton HC, Pochanard P, Mozes E, Garraway LA, Pe'er D (2010) An integrated approach to uncover drivers of cancer. Cell 143: 1005–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal M, Belcastro V, Ambesi-Impiombato A, di Bernardo D (2007) How to infer gene networks from expression profiles. Mol Syst Biol 3: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, Vivanco I, Lee JC, Huang JH, Alexander S, Du J, Kau T, Thomas RK, Shah K, Soto H, Perner S, Prensner J, Debiasi RM, Demichelis F, Hatton C et al. (2007) Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci USA 104: 20007–20012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneau R, Facciotti MT, Reiss DJ, Schmid AK, Pan M, Kaur A, Thorsson V, Shannon P, Johnson MH, Bare JC, Longabaugh W, Vuthoori M, Whitehead K, Madar A, Suzuki L, Mori T, Chang DE, Diruggiero J, Johnson CH, Hood L et al. (2007) A predictive model for transcriptional control of physiology in a free living cell. Cell 131: 1354–1365 [DOI] [PubMed] [Google Scholar]
- Bralten LBC, Kloosterhof NK, Gravendeel LAM, Sacchetti A, Duijm EJ, Kros JM, van den Bent MJ, Hoogenraad CC, Smitt PAES, French PJ (2010) Integrated genomic profiling identifies candidate genes implicated in glioma-genesis and a novel LEO1-SLC12A1 fusion gene. Genes Chromosomes Cancer 49: 509–517 [DOI] [PubMed] [Google Scholar]
- Breiman L (1996) Heuristics of instability and stabilization in model selection. Ann Statist 24: 2350–2383 [Google Scholar]
- Broët P, Camilleri-Broët S, Zhang S, Alifano M, Bangarusamy D, Battistella M, Wu Y, Tuefferd M, Régnard JF, Lim E, Tan P, Miller LD (2009) Prediction of clinical outcome in multiple lung cancer cohorts by integrative genomics: implications for chemotherapy selection. Cancer Res 69: 1055–1062 [DOI] [PubMed] [Google Scholar]
- Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, Lasorella A, Aldape K, Califano A, Iavarone A (2010) The transcriptional network for mesenchymal transformation of brain tumours. Nature 463: 318–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Demir E, Schultz N, Taylor BS, Sander C (2010) Automated network analysis identifies core pathways in glioblastoma. PLoS ONE 5: e8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crampin EJ, Schnell S, McSharry PE (2004) Mathematical and computational techniques to deduce complex biochemical reaction mechanisms. Prog Biophys Mol Biol 86: 77–112 [DOI] [PubMed] [Google Scholar]
- Dahlback HSS, Brandal P, Meling TR, Gorunova L, Scheie D, Heim S (2009) Genomic aberrations in 80 cases of primary glioblastoma multiforme: pathogenetic heterogeneity and putative cytogenetic pathways. Genes Chromosomes Cancer 48: 908–924 [DOI] [PubMed] [Google Scholar]
- Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC (2001) PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev 15: 1913–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Bernardo D, Thompson MJ, Gardner TS, Chobot SE, Eastwood EL, Wojtovich AP, Elliott SJ, Schaus SE, Collins JJ (2005) Chemogenomic profiling on a genome-wide scale using reverse-engineered gene networks. Nat Biotechnol 23: 377–383 [DOI] [PubMed] [Google Scholar]
- Erlandsson A, Brännvall K, Gustafsdottir S, Westermark B, Forsberg-Nilsson K (2006) Autocrine/paracrine platelet-derived growth factor regulates proliferation of neural progenitor cells. Cancer Res 66: 8042–8048 [DOI] [PubMed] [Google Scholar]
- Fisher R (1926) The arrangement of field experiments. J Ministry of Agriculture of Great Britain 33: 503–515 [Google Scholar]
- Freije WA, Castro-Vargas FE, Fang Z, Horvath S, Cloughesy T, Liau LM, Mischel PS, Nelson SF (2004) Gene expression profiling of gliomas strongly predicts survival. Cancer Res 64: 6503–6510 [DOI] [PubMed] [Google Scholar]
- Friedman J, Hastie T, Höfling H, Tibshirani R (2007) Pathwise coordinate optimization. Ann Appl Stat 1: 302–332 [Google Scholar]
- Friedman N, Linial M, Nachman I, Pe'er D (2000) Using Bayesian networks to analyze expression data. J Comput Biol 7: 601–620 [DOI] [PubMed] [Google Scholar]
- Fu WJ (1998) Penalized regressions: the bridge versus the lasso. J Comput Graph Statist 7: 397–416 [Google Scholar]
- Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, Lee C, Wagner SN, Li C, Golub TR, Rimm DL, Meyerson ML, Fisher DE, Sellers WR (2005) Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436: 117–122 [DOI] [PubMed] [Google Scholar]
- Golub GH, Loan CFV (1996) Matrix computations. Baltimore, MD, USA: Johns Hopkins University Press
- Haslinger A, Schwarz TJ, Covic M, Lie DC (2009) Expression of Sox11 in adult neurogenic niches suggests a stage-specific role in adult neurogenesis. Eur J Neurosci 29: 2103–2114 [DOI] [PubMed] [Google Scholar]
- Jansen RC (2003) Studying complex biological systems using multifactorial perturbation. Nat Rev Genet 4: 145–151 [DOI] [PubMed] [Google Scholar]
- Jiang C, Zeng ZB (1995) Multiple trait analysis of genetic mapping for quantitative trait loci. Genetics 140: 1111–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D (1977) Efficient algorithms for shortest paths in sparse networks. J Acm 24: 1–13 [Google Scholar]
- Kendall MG, Smith BB (1939) The problem of m rankings. Ann Math Statist 10: 275–287 [Google Scholar]
- Kim YA, Wuchty S, Przytycka TM (2010) Simultaneous identification of causal genes and Dys-regulated pathways in complex disease. Res Comput Mol Biol (RECOMB) 6044: 263–280 [Google Scholar]
- Kindler HL, Burris HA, Sandler AB, Oliff IA (2009) A phase II multicenter study of L-alanosine, a potent inhibitor of adenine biosynthesis, in patients with MTAP-deficient cancer. Invest New Drugs 27: 75–81 [DOI] [PubMed] [Google Scholar]
- Lauria M, Iorio F, di Bernardo D (2009) NIRest: a tool for gene network and mode of action inference. Ann N Y Acad Sci 1158: 257–264 [DOI] [PubMed] [Google Scholar]
- Lee H, Kong SW, Park PJ (2008) Integrative analysis reveals the direct and indirect interactions between DNA copy number aberrations and gene expression changes. Bioinformatics 24: 889–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SI, Dudley AM, Drubin D, Silver PA, Krogan NJ, Pe'er D, Koller D (2009) Learning a prior on regulatory potential from eQTL data. PLoS Genet 5: e1000358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SI, Pe'er D, Dudley AM, Church GM, Koller D (2006) Identifying regulatory mechanisms using individual variation reveals key role for chromatin modification. Proc Natl Acad Sci USA 103: 14062–14067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehár J, Zimmermann GR, Krueger AS, Molnar RA, Ledell JT, Heilbut AM, Short GF, Giusti LC, Nolan GP, Magid OA, Lee MS, Borisy AA, Stockwell BR, Keith CT (2007) Chemical combination effects predict connectivity in biological systems. Mol Syst Biol 3: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Favera RD, Califano A (2006) ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 7(Suppl 1): S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milns I, Beale CM, Smith VA (2010) Revealing ecological networks using Bayesian network inference algorithms. Ecology 91: 1892–1899 [DOI] [PubMed] [Google Scholar]
- Nelander S, Wang W, Nilsson B, She QB, Pratilas C, Rosen N, Gennemark P, Sander C (2008) Models from experiments: combinatorial drug perturbations of cancer cells. Mol Syst Biol 4: 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DV, Rocke DM (2002a) Partial least squares proportional hazard regression for application to DNA microarray survival data. Bioinformatics 18: 1625–1632 [DOI] [PubMed] [Google Scholar]
- Nguyen DV, Rocke DM (2002b) Tumor classification by partial least squares using microarray gene expression data. Bioinformatics 18: 39–50 [DOI] [PubMed] [Google Scholar]
- Nilsson B, Johansson M, Heyden A, Nelander S, Fioretos T (2008) An improved method for detecting and delineating genomic regions with altered gene expression in cancer. Genome Biol 9: R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordling TEM, Jacobsen EW (2009) Interampatteness—a generic property of biochemical networks. IET Syst Biol 3: 388–403 [DOI] [PubMed] [Google Scholar]
- Opgen-Rhein R, Opgen-Rhein and Strimmer K (2007) From correlation to causation networks: a simple approximate learning algorithm and its application to high-dimensional plant gene expression data. BMC Syst Biol 1: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne MR, Presnell B, Turlach BA (2000) On the LASSO and its dual. J Comput Graph Statist 9: 319–337 [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JCH, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA, Hartigan J et al. (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321: 1807–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Zhu J, Bergamaschi A, Han W, Noh DY, Pollack JR, Wang P (2008) Regularized multivariate regression for identifying master predictors with application to integrative genomics study of breast cancer. ArXiv, stat.AP [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, Aldape K (2006) Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9: 157–173 [DOI] [PubMed] [Google Scholar]
- Piccirillo SGM, Binda E, Fiocco R, Vescovi AL, Shah K (2009) Brain cancer stem cells. J Mol Med 87: 1087–1095 [DOI] [PubMed] [Google Scholar]
- Pollack JR, Sørlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Børresen-Dale AL, Brown PO (2002) Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci USA 99: 12963–12968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman MV (2008) Reverse engineering the genotype-phenotype map with natural genetic variation. Nature 456: 738–744 [DOI] [PubMed] [Google Scholar]
- Savageau MA (1969) Biochemical systems analysis. II. The steady-state solutions for an n-pool system using a power-law approximation. J Theor Biol 25: 370–379 [DOI] [PubMed] [Google Scholar]
- Savageau MA (1976) Biochemical Systems Analysis: A Study of Function and Design in Molecular Biology; with a Foreword by Robert Rosen. Addison-Wesley Pub Co, Advanced Book Program [Google Scholar]
- Schäfer J, Strimmer K (2005) An empirical Bayes approach to inferring large-scale gene association networks. Bioinformatics 21: 754–764 [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Sun G, Zhao C, Stewart R (2008) Neural stem cell self-renewal. Crit Rev Oncol Hematol 65: 43–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skogestad S, Postlethwaite I (2005) Multivariable Feedback Control: Analysis and Design. Chichester, UK: John Wiley & Sons [Google Scholar]
- Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, Tyler-Smith C, Carter N, Scherer SW, Tavaré S, Deloukas P, Hurles ME, Dermitzakis ET (2007a) Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315: 848–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, Ingle CE, Dunning M, Flicek P, Koller D, Montgomery S, Tavaré S, Deloukas P, Dermitzakis ET (2007b) Population genomics of human gene expression. Nat Genet 39: 1217–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthram S, Beyer A, Karp RM, Eldar Y, Ideker T (2008) eQED: an efficient method for interpreting eQTL associations using protein networks. Mol Syst Biol 4: 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniura H, Taniguchi N, Hara M, Yoshikawa K (1998) Necdin, a postmitotic neuron-specific growth suppressor, interacts with viral transforming proteins and cellular transcription factor E2F1. J Biol Chem 273: 720–728 [DOI] [PubMed] [Google Scholar]
- Taylor BS, Barretina J, Socci ND, Decarolis P, Ladanyi M, Meyerson M, Singer S, Sander C (2008) Functional copy-number alterations in cancer. PLoS ONE 3: e3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCGA-Consortium (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455: 1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegner J, Yeung MKS, Hasty J, Collins JJ (2003) Reverse engineering gene networks: integrating genetic perturbations with dynamical modeling. Proc Natl Acad Sci USA 100: 5944–5949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tso CL, Freije WA, Day A, Chen Z, Merriman B, Perlina A, Lee Y, Dia EQ, Yoshimoto K, Mischel PS, Liau LM, Cloughesy TF, Nelson SF (2006) Distinct transcription profiles of primary and secondary glioblastoma subgroups. Cancer Res 66: 159–167 [DOI] [PubMed] [Google Scholar]
- Vacher C, Piou D, Desprez-Loustau ML (2008) Architecture of an antagonistic tree/fungus network: the asymmetric influence of past evolutionary history. PLoS ONE 3: e1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O′Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS et al. (2010) Integrated genomic analysis identifies clinically relevant Subtypes of glioblastoma characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17: 98–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waaijenborg S, de Witt Hamer PCV, Zwinderman AH (2008) Quantifying the association between gene expressions and DNA-markers by penalized canonical correlation analysis. Stat Appl Genet Mol Biol 7, Article3 [DOI] [PubMed] [Google Scholar]
- Witten DM, Tibshirani R, Hastie T (2009) A penalized matrix decomposition, with applications to sparse principal components and canonical correlation analysis. Biostatistics 10: 515–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung MKS, Tegnér J, Collins JJ (2002) Reverse engineering gene networks using singular value decomposition and robust regression. Proc Natl Acad Sci USA 99: 6163–6168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Martens JWM, Yu JX, Jiang J, Sieuwerts AM, Smid M, Klijn JGM, Wang Y, Foekens JA (2009) Copy number alterations that predict metastatic capability of human breast cancer. Cancer Res 69: 3795–3801 [DOI] [PubMed] [Google Scholar]
- Zhu J, Lum PY, Lamb J, GuhaThakurta D, Edwards SW, Thieringer R, Berger JP, Wu MS, Thompson J, Sachs AB, Schadt EE (2004) An integrative genomics approach to the reconstruction of gene networks in segregating populations. Cytogenet Genome Res 105: 363–374 [DOI] [PubMed] [Google Scholar]
- Zhu J, Wiener MC, Zhang C, Fridman A, Minch E, Lum PY, Sachs JR, Schadt EE (2007) Increasing the power to detect causal associations by combining genotypic and expression data in segregating populations. PLoS Comput Biol 3: e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhang B, Smith EN, Drees B, Brem RB, Kruglyak L, Bumgarner RE, Schadt EE (2008) Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat Genet 40: 854–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Hastie T, Tibshirani R (2006) Sparse principal component analysis. J Computat Graphical Statist 2: 262–286 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods, Supplementary Figures S1–2