

Abstract
Reversible protein phosphorylation is one of the most pervasive post-translational modifications, regulating diverse cellular processes in various organisms. High throughput experimental studies using mass spectrometry have identified many phosphorylation sites, primarily from eukaryotes. However, the vast majority of phosphorylation sites remain undiscovered, even in well studied systems. Because mass spectrometry-based experimental approaches for identifying phosphorylation events are costly, time-consuming, and biased toward abundant proteins and proteotypic peptides, in silico prediction of phosphorylation sites is potentially a useful alternative strategy for whole proteome annotation. Because of various limitations, current phosphorylation site prediction tools were not well designed for comprehensive assessment of proteomes. Here, we present a novel software tool, Musite, specifically designed for large scale predictions of both general and kinase-specific phosphorylation sites. We collected phosphoproteomics data in multiple organisms from several reliable sources and used them to train prediction models by a comprehensive machine-learning approach that integrates local sequence similarities to known phosphorylation sites, protein disorder scores, and amino acid frequencies. Application of Musite on several proteomes yielded tens of thousands of phosphorylation site predictions at a high stringency level. Cross-validation tests show that Musite achieves some improvement over existing tools in predicting general phosphorylation sites, and it is at least comparable with those for predicting kinase-specific phosphorylation sites. In Musite V1.0, we have trained general prediction models for six organisms and kinase-specific prediction models for 13 kinases or kinase families. Although the current pretrained models were not correlated with any particular cellular conditions, Musite provides a unique functionality for training customized prediction models (including condition-specific models) from users' own data. In addition, with its easily extensible open source application programming interface, Musite is aimed at being an open platform for community-based development of machine learning-based phosphorylation site prediction applications. Musite is available at http://musite.sourceforge.net/.
With many genomes being sequenced at an increasingly fast pace, a key and challenging issue is inferring protein function and downstream regulatory networks. As a pervasive regulatory mechanism, reversible protein phosphorylation plays an important role in signaling networks (1). Annotation of phosphorylation and other modification sites in proteomes is a critical first step toward decoding such signaling networks.
In recent years, protein phosphorylation data have accumulated rapidly due to large scale mass spectrometry studies of protein phosphorylation in different organisms (2–9) and development of associated web resources (10–18). In particular, there are currently about 100,000 annotated phosphorylation sites in all organisms in UniProt/Swiss-Prot (V57.8). About 27,000 of these sites are from human. Nevertheless, our knowledge of protein phosphorylation is still limited. The majority of proteins are estimated to be phosphorylated at multiple sites (>100,000 sites in the human proteome alone) (19). Furthermore, our understanding of phosphorylation events in signaling networks is even more lacking, largely due to the lag in elucidating kinase-substrate interactions. For example, fewer than 5,000 (5%) of reported phosphorylation sites in UniProt/Swiss-Prot are annotated for their cognate protein kinases.
Despite improvements in phosphopeptide enrichment and mass spectrometry analysis, experimental identification of phosphorylation sites in a global manner is still a difficult, expensive, and time-consuming task. In addition, high throughput proteomics techniques have some limitations. Because only proteotypic peptides are observed, mass spectrometry tends to provide fractional sequence coverage for proteins. Detection of low abundance proteins is also problematic. Consequently, a significant portion of phosphorylation sites are missed by current techniques. Moreover, it is even harder to characterize kinase-substrate interactions experimentally. Hence, in silico prediction of phosphorylation events can be highly valuable in many cases. As genome and proteome data in various organisms have been increasing dramatically, comprehensive and accurate prediction of protein phosphorylation sites is becoming more advantageous for proteome annotation and large scale experimental design. For example, in hypothesis-driven experiments, the researchers may want to use prediction tools to focus on putative phosphorylation sites above a high stringency level.
More than a dozen phosphorylation site prediction tools have been developed; they can be divided into two categories: tools for general phosphorylation site prediction and tools for kinase-specific phosphorylation site prediction. DISPHOS (20), NetPhos (21), and scan-x (22) fall into the first category. The latter category includes Scansite (23), NetPhosK (24), GPS (25), KinasePhos (26), Predikin (27), CRPhos (28), AutoMotif (29), pkaPS (30), PPSP (31), PhoScan (32), PredPhospho (33), and NetPhorest (34). More information about these tools is given in supplemental Table S1. Although kinase-specific prediction is of interest because of its essential role in constructing signaling networks, general prediction is also important because the majority of phosphorylation sites remain undiscovered, and the kinase-specific predictors may only be able to unveil a small fraction of them.
Despite the availability of various phosphorylation site prediction tools, they have limitations when applied to whole proteomes. The most important issue of phosphorylation site prediction is accuracy. Because different training data and techniques were used with these programs, prediction performance varies greatly among them as discussed later. Another notable issue is that most tools were only released as web servers and have restrictions for the data uploaded by users (see supplemental Table S1). This makes large scale predictions a laborious or impossible task. Besides web servers, GPS 2.1 (25) and PhoScan (32) were also released as stand-alone tools, capable of handling large data sets, but both tools only support kinase-specific predictions. NetPhos 2.0 and NetPhosK 1.0 were also released as both web servers and stand-alone applications under Unix/Linux, but prediction performance could be improved as we demonstrate in this study. In Schwartz et al. (22), proteome scale scans on human, mouse, fly, and yeast were performed using motif-x (35) and scan-x, and the prediction results were accessible. However, the tool scan-x has not been publicly released (as of May 26, 2010), and hence “on-the-fly” predictions for user-uploaded sequences are not possible. Another concern regarding the current tools is the stringency control of predictions. User control on the prediction stringency is important, especially for large scale predictions, because typically a user is interested only in predictions above a certain confidence threshold, and different users may have different requirements on the threshold. However, current tools either preset the threshold and do not support stringency adjustment or only support several predefined stringency levels from which a user can choose that may not meet every user's requirement.
To address the limitations of existing tools and to take advantage of the large magnitude of experimentally verified phosphorylation sites, we developed a bioinformatics tool, Musite, specifically designed for large scale prediction of both general and kinase-specific phosphorylation sites. As a stand-alone application, Musite can be easily used to perform large scale phosphorylation site prediction in an automated fashion. We modeled phosphorylation site prediction as an unbalanced binary classification problem and solve it with a comprehensive machine-learning approach. Reliable experimental phosphoproteomics data in multiple organisms were collected from several sources and utilized to train phosphorylation site prediction models by a comprehensive machine-learning procedure termed bootstrap aggregating. Three sets of features (k nearest neighbor (KNN)1 scores, disorder scores, and amino acid frequencies) were extracted from the collected data and combined using support vector machine (SVM) to make predictions. KNN scores capture local sequence similarity around sites phosphorylated by the same kinase or kinase family whether or not the kinase-substrate interactions are known. Disorder scores reflect the higher probability of phosphorylated residues to be in disordered regions, which are segments of proteins that lack a stable tertiary structure. Phosphorylation sites have been shown to be preferentially located within disordered regions (20, 36); this was confirmed on phosphoproteomics data in six organisms by this study.
Applications of Musite on several proteomes yielded tens of thousands of putative phosphorylation sites with high stringency. Cross-validation tests and comparisons with other tools show that Musite performs better on general predictions and at least comparably with existing methods on kinase-specific predictions. In Musite V1.0, we have trained general prediction models for six organisms and kinase-specific prediction models for 13 kinases or kinase families. It is noted, however, that using the current pretrained models users cannot correlate prediction results with any particular cellular condition. To do so, users can utilize a unique functionality in Musite for training customized prediction models from their own condition-specific phosphorylation data. In addition, Musite supports continuous stringency adjustment to meet different confidence requirements for users. Taken together, Musite provides a valuable tool for biologists to predict phosphorylation sites up to the whole proteome level. In addition, with its open source, well designed, and easily extensible application programming interface (API), Musite is also beneficial to bioinformaticians as a platform to build their own machine learning-based applications for phosphorylation site prediction.
MATERIALS AND METHODS
Data Collection
Phosphorylation data for six model organisms, Homo sapiens, Mus musculus, Drosophila melanogaster, Caenorhabditis elegans, Saccharomyces cerevisiae, and Arabidopsis thaliana, from several sources including UniProt/Swiss-Prot (11) (version 57.8, September 22, 2009), Phospho.ELM (15) (version 8.2, April 2009), PhosphoPep (as of October 2, 2009), and PhosPhAt (18) (V3.0, October 2009) were collected. Table I lists the phosphorylation data sources and statistics for different phosphorylation types of each organism. Because a serine/threonine-specific kinase can often phosphorylate both serine and threonine residues (38), phosphoserines and phosphothreonines in each organism were combined when training prediction models in Musite.
Table I. Phosphorylation data collected in this study.
| Organism | Data source | Number of proteinsa | Number of phosphoproteins | Residue type | Number of residuesa | Number of phosphositesa |
|---|---|---|---|---|---|---|
| H. sapiens (human) | UniProt/Swiss-Prot | 20,642 | 6,993 | Serine | 964,479 (747,779) | 22,693 (20,060) |
| Phospho.ELM | (15,355) | (5,729) | Threonine | 618,337 (472,154) | 5,082 (4,470) | |
| Tyrosine | 306,980 (227,240) | 3,110 (2,572) | ||||
| M. musculus (mouse) | UniProt/Swiss-Prot | 16,429 | 5,959 | Serine | 767,185 (642,486) | 17,694 (16,085) |
| Phospho.ELM | (13,147) | (5,070) | Threonine | 488,903 (402,799) | 3,738 (3,336) | |
| Tyrosine | 245,940 (196,880) | 2,617 (2,219) | ||||
| D. melanogaster (fruit fly) | UniProt/Swiss-Prot | 17,011 | 3,314 | Serine | 781,887 (571,333) | 8,960 (8,828) |
| PhophoPepb | (12,928) | (3,209) | Threonine | 531,531 (387,518) | 2,463 (2,407) | |
| C. elegans (worm) | UniProt/Swiss-Prot | 23,090 | 2,099 | Serine | 813,784 (590,442) | 3,910 (2,792) |
| PhophoPepb | (16,758) | (1,565) | Threonine | 591,985 (424,206) | 562 (396) | |
| S. cerevisiae (bakers' yeast) | UniProt/Swiss-Prot | 13,205 | 2,556 | Serine | 550,611 (244,689) | 8,817 (8,409) |
| (6,092) | (2,379) | Threonine | 360,928 (155,870) | 2,053 (1,932) | ||
| A. thaliana (thale cress) | PhosPhAtb | 33,410 | 2,268 | Serine | 1,223,257 (666,523) | 3,748 (3,159) |
| TAIR | (17,732) | (1,864) | Threonine | 683,707 (356,487) | 580 (504) |
a Numbers in parentheses represent unique proteins or residues after running the non-redundant data set construction procedure.
b For phosphorylation data in PhosphoPep and PhosPhAt, only unambiguously determined sites were included.
We used the same type of residues (serine/threonine or tyrosine), excluding known phosphorylation sites as the negative training data (non-phosphorylation sites). Although not all these sites are necessarily true negatives, it is reasonable to believe that a large majority of them are. The data were extracted from organism-wise complete proteomes annotated in UniProt (as of October 2, 2009) for H. sapiens, D. melanogaster, C. elegans, and S. cerevisiae. Because the complete proteome for M. musculus was not provided in UniProt, Swiss-Prot protein entries of M. musculus were used. For A. thaliana, TAIR9 gene models and annotations were used (39).
Non-redundant Data Set Construction
For each of the six organisms, after combining the positive and negative data, protein sequences with high similarities were removed to build a non-redundant (NR) protein data set using BLASTClust in BLAST (40) package version 2.2.19 with a sequence identity threshold of 50%. Proteins with similar sequences were first clustered into groups by BLASTClust. Within each group, we selected the protein with the largest number of known phosphorylation sites into the NR data set; if there were no phosphorylation sites in any of the proteins in a group, we selected the longest protein. In this way, the maximum number of NR phosphorylation sites was kept in the NR data set.
Machine Learning
Phosphorylation site prediction can be formulated as a binary classification problem; namely, each serine/threonine or tyrosine can be classified as either a phosphorylation site or a non-phosphorylation site. As with all general binary classification problems, there are three key issues: (i) having a well collected and curated data set including positive and negative data, (ii) having a set of effective features to characterize the commonalities in each category and the difference between the two categories, and (iii) developing a classifier trained from the known data and capable of making reliable predictions for new data.
In this study, for different types of phosphorylation (i.e. phosphoserine/threonine and phosphotyrosine) in each organism, separate prediction models were trained from the NR data sets as summarized in Fig. 1. Output from a protein disorder predictor, KNN scores, and amino acid frequencies around the phosphorylation sites were taken as features. More features as listed in supplemental Table S2 will be evaluated and incorporated into our prediction scheme if they improve the prediction accuracy and are computationally feasible. We used the aggregation of multiple SVMs (41) as the classifier.
Fig. 1.

Overall work flow of Musite.
Feature Extraction
KNN Features
Local sequence clusters often exist around phosphorylation sites because substrate sites of the same kinase or kinase family usually share similar patterns in local sequences (42). To take advantage of such cluster information of local sequences for predicting phosphorylation sites, we took the local sequence around a possible phosphorylation site in a query protein and extracted features from its similar sequences in both positive and negative sets by a KNN algorithm as follows.
- For a query site (possible phosphorylation site), find its k nearest neighbors in positive and negative sets, respectively, according to local sequence similarity. For two local sequences, s1 = {s1(−w), s1(−w+ 1), …, s1(0), …, s1(w − 1), s1(w)} and s2 = {s2(−w), s2(−w + 1), …, s2(0), …, s2(w − 1), s2(w)} define the distance Dist(s1, s2) between s1 and s2 as
where w is the number of residues included in the window in each side, and hence, the window size is 2w + 1 (in Musite, w = 6 by default; “*,” which represents gaps in the BLOSUM matrix, will be added to the termini if the upstream or downstream regions of the sites have less than w residues) and Sim, the amino acid similarity matrix, is derived from the normalized amino acid substitution matrix (BLOSUM62 by default) (43) as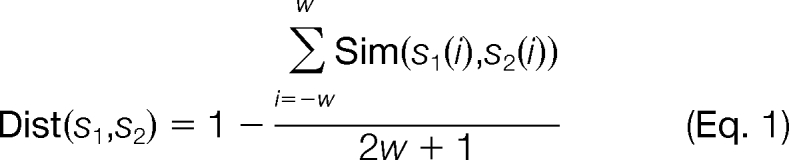
where a and b are two amino acids, M is the substitution matrix, and max/min{M} represent the largest/smallest number in the matrix, respectively.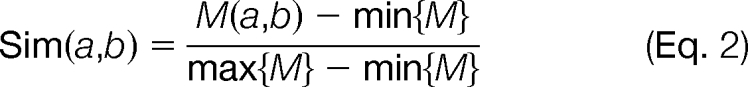
- The corresponding KNN feature is then extracted as follows.
- Form a set of neighbors by combining the positive and negative sets.
- Calculate the average distances from the query sequence s to all neighbors.
- Sort the neighbors by the distances and pick the k nearest neighbors.
- Calculate the KNN score, the percentage of positive neighbors (phosphorylation sites) in its k nearest neighbors.
To take advantage of different properties of neighbors with various similarities, steps I and II were repeated for different k values to obtain multiple features for the phosphorylation predictor. In Musite, by default, k was chosen to be 0.25, 0.5, 1, 2, and 4% of the size of the bootstrapped training data set, and thus, five KNN scores were extracted as features for phosphorylation prediction.
It is worthwhile mentioning that the nearest neighbors represent only local sequence similarity as global sequence similarities are removed in NR data sets.
Disorder Features
Phosphorylation sites have been observed to have a strong tendency to be located in disordered regions (20, 36). In Iakoucheva et al. (20), predicted disorder scores for phosphorylation sites were used as features in the phosphorylation predictor DISPHOS. In this study, we extracted the disorder information for surrounding residues of each possible phosphorylation site in the query protein and combined them to form a set of disorder features in the following procedure.
Predict the disordered scores for the query protein sequence by means of a widely used disorder prediction tool, VSL2B (44).
Extract the disorder prediction scores for the residues around each possible phosphorylation site.
Take the average scores surrounding each site with different window sizes as features for the phosphorylation predictor. In Musite, by default, the window sizes are chosen to be 1, 5, and 13 (i.e. with 0, 2, and 6 surrounding residues from each side). Therefore, three disorder scores were extracted as features for phosphorylation site prediction. If there are not enough residues in either side, we use a truncated window starting from or ending at the terminus. For example, if the phosphorylation site is at the fourth position in the protein, we averaged the disorder scores over positions 1–10 for the window size of 13.
Amino Acid Frequency Features
Iakoucheva et al. (20) analyzed the amino acid composition of the surrounding sequences of phosphorylation sites and found that rigid, buried, neutral amino acids (Trp, Cys, Phe, Ile, Tyr, Val, and Leu) were significantly depleted, whereas flexible, surface-exposed amino acids (Ser, Pro, Glu, and Lys) were significantly enriched. This conclusion was confirmed in the current study as illustrated under “Results.” Hence, the amino acid frequencies can be useful features for phosphorylation site prediction. The procedure to extract amino acid frequency features is as follows. We calculated the amino acid frequencies in the sequence surrounding the query site (the site itself is not counted). There are 20 types of amino acids, and thus 20 frequencies are calculated, the sum of which is 1. In Musite, by default, the window size is 13; i.e. 6 residues at each side were included to calculate the frequencies. If there are not enough residues in either side, a truncated window starting from or ending at the terminus is used in the same manner as described above in the disorder feature extraction procedure.
Model Training
We used the extracted features to train phosphorylation prediction models. As illustrated in Fig. 1, the training procedure consisted of two subprocedures: bootstrap aggregation and specificity estimation. Note that the training data and control data were randomly separated with no overlaps.
Bootstrap Aggregating
The sizes of positive and negative data sets in this study were highly unbalanced. The size of negative data sets was 2 orders of magnitude larger than the positive data sets as shown in Table I. To handle this problem, we used an ensemble meta-algorithm in machine learning called bootstrap aggregating or bagging (45). Given the features extracted from positive and negative data sets, the bootstrap aggregation procedure is as follows.
Bootstrap: generate a training set by sampling with replacement from positive and negative data sets randomly. This training set is called one bootstrap sample. By default, in one bootstrap sample, 2,000 data points were sampled from the positive data set, and another 2,000 were sampled from the negative data set. Therefore, the training set was balanced after bootstrapping.
Classifier training: take the bootstrap sample as a training data set to train an SVM classifier. We used the package SVMlight V6.02 (46) in this study.
Aggregating: when training, we repeated steps I and II for m times to get m training classifiers (m = 5 by default). When predicting a query site, we submitted extracted features to all trained classifiers and combined the outputs from the classifiers by averaging. This averaged output was taken as the prediction score for the query site.
Performance Evaluation and Specificity Estimation
To evaluate the prediction performance of Musite, cross-validation tests were performed. Receiver operating characteristic (ROC) curves were calculated and plotted based on specificities (Equation 3) and sensitivities (Equation 4) by taking different thresholds.
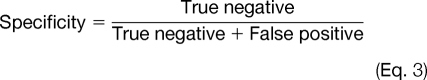 |
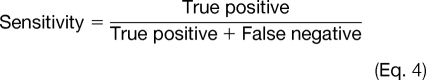 |
Areas under ROC curves (AUCs) were also calculated based on the trapezoidal approximation. As part of the training procedure, a portion of non-phosphorylation sites (10,000 by default) was randomly selected for specificity estimation as shown in Fig. 1. When making predictions, a user can choose stringency levels based on estimated specificities.
RESULTS AND DISCUSSION
KNN Scores as Features
A KNN score measures whether the local sequence surrounding a query site is more similar to the sequences containing phosphorylation sites in the positive set or those with non-phosphorylation sites in the negative set. A score greater than 0.5 means the query site is more similar to the positive set; a score smaller than 0.5 means it is more similar to the negative set. The larger the KNN score, the more similar the site is to some known phosphorylation sites, and thus, the more likely it is a phosphorylation site. Fig. 2 compares the KNN scores of phosphorylation sites with those of non-phosphorylation sites. Overall, phosphorylation sites have larger KNN scores than non-phosphorylation sites. For phosphoserines/threonines, the average KNN scores with different sizes of nearest neighbors are within 0.6–0.8 for all six organisms; for phosphotyrosines, the average KNN scores are around 0.6. Therefore, the local sequences surrounding known phosphorylation sites are more similar to their nearest neighbors in the positive set (excluding self-matches) on average as expected. Note that such similarities are not due to protein homology as the global sequence similarity between any two proteins in our NR data sets is either insignificant or low. This finding confirms that phosphorylation-related clusters exist in local sequences around phosphorylation sites. For non-phosphorylation sites, the average KNN scores are around 0.5, which means overall that the sequences in the negative set are not predominantly more similar to nearest neighbors in either the positive or negative set. This is not surprising because phosphorylation-related sequence clusters are unlikely to exist in the negative set, and thus, the sequences in the negative set have a similar chance to find close neighbors in either the positive or negative set. In short, the KNN scores capture the cluster information in the local sequence around phosphorylation sites and hence distinguish them from the background. Therefore, KNN scores are suitable to be used as features for phosphorylation site prediction.
Fig. 2.

Comparison of KNN scores between phosphorylation sites and non-phosphorylation sites. KNN scores of 1,000 phosphorylation sites and 1,000 non-phosphorylation sites randomly selected from each non-redundant data sets for six organisms were plotted. A, box plots of KNN scores (H. sapiens serine/threonine data only) for phosphorylation sites (red) and non-phosphorylation sites (blue). The horizontal axis represents the size of nearest neighbors (in percentage of the bootstrapped data set size). The vertical axis represents the KNN score. The bottom and top of the box are the 25th and 75th percentiles, respectively; the central band is the median; the whiskers extend to the most extreme data points that are not considered outliers; and the outliers are plotted individually as plus marks (+). B, comparison of mean KNN scores between phosphorylation sites (pentagrams) and non-phosphorylation sites (circles) in six organisms.
KNN scores are very effective features when used for predicting both general and kinase-specific phosphorylation sites. For general phosphorylation site predictions, KNN scores can automatically capture the local sequence similarity between substrates of a common kinase or kinase family without relying on knowledge of kinase-substrate interactions or kinase-binding sequence motifs. This means that although most known phosphorylation sites have no annotation about their corresponding kinases KNN scores can still utilize the inherent cluster information in them. KNN scores are also useful for predicting kinase-specific sites. Oftentimes, one kinase corresponds to multiple local sequence motifs, and using a single sequence profile may not be as effective as KNN, which better handles diverse sequence clusters.
Protein Phosphorylation and Protein Disorder
In this section, we will demonstrate the effectiveness of disorder scores as features for phosphorylation site prediction by studying the preference of phosphorylation sites in protein disordered regions. Fig. 3, A and C and B and D, plot the histograms of the disorder scores for the surrounding residues of phosphoserines/threonines and non-phosphoserines/threonines, respectively. For both H. sapiens (Fig. 3A) and A. thaliana (Fig. 3C), the number of phosphoserines/threonines increases exponentially when the disorder score increases from 0 to 1; the phosphoserines/threonines with disorder scores larger than 0.9 are dominant in the data set. In contrast, Fig. 3, B and D, show a different pattern for non-phosphoserines/threonines. The number of non-phosphorylation sites with disorder scores larger than 0.9 is still higher than those in the other subranges. This may be due to the fact that many of the non-phosphorylation sites are actually unassigned phosphorylation sites. Alternatively, this could also reflect a general preference of serine/threonine to be located in disordered regions. A third possibility is that these serines/threonines are actually not phosphorylated because they lack a surrounding kinase-specific motif in which case their non-phosphorylation status should be indicated by the KNN feature. In any case, it is clear that phosphoserines/threonines in H. sapiens and A. thaliana are much more overrepresented in disordered regions than non-phosphoserines/threonines. In fact, the majority (91.0% in Fig. 3A and 87.6% in Fig. 3C) of the phosphorylation sites in the example have disorder scores larger than 0.5 (note that VSL2B predicts a residue to be in the disordered region when its predicted value is larger than 0.5), whereas the corresponding percentages are only 54.9 and 50.5% for non-phosphoserines/threonines in Fig. 3, B and D, respectively. Fig. 3, E and F, plot the histograms of the disorder scores for the surrounding residues of phosphotyrosines and non-phosphotyrosines in H. sapiens, respectively. Although the phosphotyrosines are not predominantly distributed in disordered regions, there is a clear shift toward disordered regions in comparison with non-phosphotyrosines. This pattern is consistent in all six organisms studied (supplemental Fig. S1). In summary, there is a clear preference of known phosphorylation sites to be within disordered regions, which justifies the use of disorder scores as features for phosphorylation site prediction.
Fig. 3.

Preference of phosphorylation sites in disordered regions. Disorder scores for the H. sapiens NR data set and the A. thaliana NR data set are shown as examples. All phosphorylation sites and non-phosphorylation sites that have 6 or more residues at both sides were used. A, histogram of disorder scores of residues around phosphoserines/threonines (23,907 in total) in the H. sapiens NR data set. The horizontal axis represents the disorder score predicted by VSL2B, divided evenly into 10 subranges from 0 to 1; the vertical axis represents the occurrence (the number of sites) in the corresponding disorder subrange. Different colors from blue to red in each bar stand for 13 different residue positions in the window from the upstream −6 to downstream +6 residues as indicated in the color bar on the right. B, histogram of disorder scores of residues around non-phosphoserines/threonines (1,171,139 in total) in the H. sapiens NR data set. C, histogram of disorder scores of residues around phosphoserine/threonine sites (3,512 in total) in the A. thaliana NR data set. D, histogram of disorder scores of residues around non-phosphoserine/threonine sites (986,481 in total) in the A. thaliana NR data set. E, histogram of disorder scores of residues around phosphotyrosine sites (2,504 in total) in the H. sapiens NR data set. F, histogram of disorder scores of residues around non-phosphotyrosine sites (221,322 in total) in the H. sapiens NR data set.
Amino Acid Composition surrounding Phosphorylation Sites
In this section, we will study the difference between the amino acid composition surrounding phosphorylation sites and that surrounding non-phosphorylation sites. In Fig. 4, from left to right, the amino acids vary from being enriched to being depleted in the surrounding sequences of phosphorylation sites. With slight variations among different organisms, the overall trends are similar. For phosphoserine/threonine sites (Fig. 4A), amino acids Pro, Arg, Asp, Glu, Ser, Lys, and Gly are enriched in the surrounding sequences, whereas Cys, Trp, Tyr, Phe, Ile, Met, Leu, His, Thr, and Val are depleted. For phosphotyrosine sites (Fig. 4B), Asp, Glu, Pro, Ser, and Gly are enriched, whereas Trp, Cys, Phe, Leu, His, Met, and Ile are depleted. The different compositions of amino acids surrounding phosphorylation sites and non-phosphorylation sites justify the use of amino acid frequencies as features for phosphorylation site prediction.
Fig. 4.

Comparisons of amino acid compositions in positive and negative data sets. A, comparisons between phosphoserines/threonines and non-phosphoserines/threonines in six organisms. The vertical axis represents the log2 ratio between amino acid frequencies surrounding phosphoserines/threonines and those surrounding non-phosphoserines/threonines. A value larger than 0 means the corresponding amino acid is enriched surrounding phosphoserines/threonines. The horizontal axis represents the 20 amino acids sorted in descending order by the mean log2 ratio. B, similarly, comparisons between phosphotyrosines and non-phosphotyrosines in H. sapiens and M. musculus (phosphotyrosine data in the other four organisms are too sparse to derive meaningful statistics).
General and Kinase-specific Prediction for Multiple Organisms
One of the unique features of Musite is that it can be used to perform both general and kinase-specific phosphorylation site predictions. Both types of predictions use the same process except that kinase-specific phosphorylation site predictions use phosphorylation sites corresponding to a specific kinase or a kinase family as the positive training data. For general predictions, we have trained six phosphoserine/threonine prediction models for six organisms (H. sapiens, M. musculus, D. melanogaster, C. elegans, S. cerevisiae, and A. thaliana), one combined phosphoserine/threonine prediction model for general eukaryotes, and two phosphotyrosine prediction models for H. sapiens and M. musculus, respectively. We did not train phosphotyrosine prediction models for the other four organisms because there were not enough data for training in those organisms. Kinase-specific prediction models were trained from phosphorylation data in H. sapiens for 13 kinases or kinase families, including ataxia telangiectasia mutated (ATM) kinase, cyclin-dependent kinase (CDK) family, CDK1, CDK2, casein kinase 1 (CK1), CK2, mitogen-activated protein kinase (MAPK) family, MAPK1, MAPK3, protein kinase A (PKA) family, protein kinase B (PKB) family, protein kinase C (PKC) family, and proto-oncogenic tyrosine kinases (Src). Supplemental Table S3 lists the detailed information about the prediction models we trained so far. All the prediction models are downloadable at http://musite.sourceforge.net/.
Evaluation of General Phosphorylation Site Prediction Performance of Musite
To evaluate the performance of Musite for general phosphorylation site prediction, we carried out a 10-fold cross-validation test on each type of phosphorylation in each organism as follows. Taking phosphoserine/threonine in H. sapiens for example, the phosphoserines/threonines in the H. sapiens NR data set were randomly divided into 10 groups. Each group was then combined with the same number of non-phosphoserines/threonines randomly selected from the H. sapiens NR data set to form 10 sub-data sets. A single sub-data set was retained as validation data, and all the remaining positive and negative data in the NR data set was used as training data to train a prediction model. The validation data were then submitted to this trained model for prediction. The cross-validation process was repeated 10 times with each sub-data set used exactly once as the validation data. Sensitivities at different specificity levels in each cross-validation run were calculated according to Equations 3 and 4. Average sensitivities and specificities over 10 cross-validations were calculated. By taking different thresholds, we then calculated the ROC curves as plotted in Fig. 5 and the AUCs as shown in supplemental Table S4.
Fig. 5.

ROC curves of Musite predictions on NR data sets of H. sapiens, M. musculus, D. melanogaster, C. elegans, S. cerevisiae, and A. thaliana. Each curve represents the average sensitivities and specificities for difference thresholds over 10 cross-validation runs. The bottom right figure is the zoomed-in region with high prediction specificities (0.9–1).
The most confident predictions are those with high specificities. From the ROC curves, for phosphoserines/threonines, at 95% specificity, the prediction sensitivities vary from 36 to 62%; at 99% specificity, most predictions have sensitivity around 10%, whereas predictions for A. thaliana and C. elegans achieve 20 and 32% sensitivity, respectively. Interestingly, from the ROC curves, the predictions for C. elegans perform significantly better than those for the other five organisms. This can be explained by the fact that both KNN and amino acid frequency features in C. elegans show stronger patterns in distinguishing between positive and negative data as shown in Figs. 2 and 4. For phosphotyrosines, we tested our results only on H. sapiens and M. musculus. The performances for both organisms achieve around 10 and 25% in sensitivity at the 99 and 95% specificity levels, respectively.
The prediction performance using each of the three sets of features (KNN, amino acid frequency, and disorder) was also evaluated as shown in supplemental Fig. S2. The result shows that the combined features yield more accurate predictions as expected. When testing the features separately, KNN features performed the best in ROC.
Proteome-wide Phosphorylation Site Prediction
The general phosphorylation site prediction model for each organism was used to scan the corresponding proteome. Nearly 100,000 phosphorylation sites were predicted at the 99% specificity level for all six organisms combined as shown in Table II. Prediction results are available for download at http://musite.sourceforge.net/. These predictions provide useful hypotheses for experimental validations.
Table II. Proteome-wide phosphorylation site predictions at 99 and 95% specificity levels.
| Organism | Residue type | Number of predicted phosphorylation sites |
|
|---|---|---|---|
| 99% specificity | 95% specificity | ||
| H. sapiens | Serine/threonine | 20,084 | 82,748 |
| Tyrosine | 2,621 | 14,339 | |
| M. musculus | Serine/threonine | 13,658 | 61,378 |
| Tyrosine | 3,273 | 12,134 | |
| D. melanogaster | Serine/threonine | 13,931 | 68,420 |
| C. elegans | Serine/threonine | 14,287 | 74,520 |
| S. cerevisiae | Serine/threonine | 12,895 | 52,306 |
| A. thaliana | Serine/threonine | 17,877 | 92,118 |
Cross-species Prediction of General Phosphorylation Sites
The performance of cross-species predictions using Musite was evaluated against six organisms. A test data set was built by randomly selecting 100 proteins that contain phosphoserines/threonines and 100 non-phosphoproteins from the NR data set of each organism. The remaining proteins in each NR data set formed the training data set to build a prediction model for every organism. We also built a combined prediction model using the data by combining the six training data sets and running the non-redundant data set building procedure with an identity threshold of 30%. Note that there was no overlap between training and test data sets. Pairwise tests were then performed by submitting all serines/threonines in each of the six test data sets to each of the seven prediction models. The specificities, sensitivities, and AUCs were then calculated as shown in Table III. For all test data sets, prediction results from the model trained using data in the same organism performed the best (considering only AUC). For each of the six test data sets, the performances did not have large variations using different models trained on data from different organisms. As a possible explanation, although kinases and their substrates vary in different species, the biophysical mechanism of enzyme-substrate binding remained the same, and the generic features utilized by Musite (i.e. disorder scores and amino acid frequencies) could have captured such a mechanism. Supplemental Fig. S3 and supplemental Table S5 provide the scatter plots and correlation coefficients of prediction scores among the seven models for all 15,980 serines/threonines in the H. sapiens test data set, which show high positive associations among predictions from different models. The results suggest that Musite and the associated prediction models can be used for cross-species predictions of general phosphoserines/threonines, which is especially useful when phosphorylation data in species of interest is not enough for training a prediction model. Interestingly, for cross-species predictions, there is no apparent evidence that using model trained on data from an evolutionarily closer species would perform better. As an example, for the H. sapiens test data set, the predictions from the A. thaliana model performed better than others except the combined model and the H. sapiens model itself. Given the small test data size, this may not be statistically significant. The performance variations in various models may be partially due to different quantities and qualities of phosphorylation data in different organisms.
Table III. Cross-species prediction performance of Musite.
| Training | Test |
|||||
|---|---|---|---|---|---|---|
| H. sapiens | M. musculus | D. melanogaster | C. elegans | S. cerevisiae | A. thaliana | |
| H. sapiens | 0.858/43.7/15.4a | 0.817/35.1/9.9 | 0.782/28.7/8.2 | 0.873/43/6/13.4 | 0.813/33.5/9.3 | 0.834/38.9/16.8 |
| M. musculus | 0.856/44.8/13.7 | 0.825/36.9/11.6 | 0.784/28.4/8.4 | 0.877/41.1/11.9 | 0.815/33.5/9.6 | 0.837/40.0/14.6 |
| D. melanogaster | 0.850/41.3/12.2 | 0.810/30.7/9.2 | 0.815/32.6/11.3 | 0.892/47.5/18.8 | 0.815/35.3/12.3 | 0.849/45.4/15.1 |
| C. elegans | 0.845/40.5/7.5 | 0.800/33.9/9.4 | 0.795/32.1/12.1 | 0.908/53.0/20.8 | 0.809/33.5/10.8 | 0.839/40.5/20.0 |
| S. cerevisiae | 0.828/28.9/5.6 | 0.800/32.2/5.9 | 0.767/23.2/6.1 | 0.849/32.2/7.9 | 0.825/34.0/8.6 | 0.822/29.7/5.9 |
| A. thaliana | 0.857/41.8/12.4 | 0.817/34.4/12.6 | 0.781/28.7/9.2 | 0.872/41.6/13.9 | 0.809/35.5/11.3 | 0.861/47.0/16.8 |
| Combined | 0.858/44.1/14.3 | 0.819/35.9/9.4 | 0.799/31.3/10.5 | 0.886/48.5/14.9 | 0.822/36.0/11.1 | 0.839/41.1/16.8 |
a The three numbers in each cell represent the AUC, sensitivity (%) at 95% specificity, and sensitivity (%) at 99% specificity. The training model with the highest AUC for each test data set is highlighted in bold.
Comparison with Other General Prediction Tools
To further evaluate the performance of general phosphorylation site prediction by Musite, we compared it with three existing tools, NetPhos, DISPHOS, and scan-x. We applied the same H. sapiens test data set as that used in cross-species prediction evaluation containing 9,943 serines (with 390 known phosphoserines) and 6,037 threonines (with 77 known phosphothreonines). Sequences of these 100 phosphoproteins and 100 non-phosphoproteins were submitted to NetPhos, scan-x, and DISPHOS for prediction. To compare the result, we trained a model for Musite using the remaining proteins in the H. sapiens NR data set and predicted the phosphorylation sites in the test data set. The ROC curves comparing the predictions of different tools are shown in Fig. 6 and supplemental Table S6.
Fig. 6.

Comparison of phosphoserine/threonine prediction performances of NetPhos, DISPHOS, scan-x, and Musite. For NetPhos, DISPHOS, and Musite, the phosphoserine/threonine prediction scores were extracted, and the corresponding ROC curves were calculated and plotted. For scan-x, only specificities/sensitivities at the two supported stringency levels were plotted. The bottom right graph is the zoomed-in region with high prediction specificities (0.9–1).
NetPhos is an artificial neural network-based general phosphorylation site predictor for eukaryotic proteins (21). We submitted the H. sapiens test data set to the web server of NetPhos 2.0 (http://www.cbs.dtu.dk/services/NetPhos/). The default predictions of NetPhos achieved sensitivity and specificity of 77.7 and 61.9%, respectively. In addition to the default predictions, the program also provided the prediction scores for all serines/threonines in query sequences. By taking different thresholds on the scores, NetPhos achieved sensitivities of 27.4 and 6.4% at 95.2 and 99.4% specificity levels, respectively. To compare, when predicting phosphoserines/threonines on the same data set using the models trained from the H. sapiens NR data set excluding the test data set, Musite achieved sensitivities of 88.7, 42.8, and 11.3% at specificities of 61.9, 95.2, and 99.4%, respectively.
DISPHOS was the first phosphorylation site predictor that made use of protein disorder information (20). The web server DISPHOS 1.3 (http://core.ist.temple.edu/pred/) successfully predicted 190 of 200 protein sequences in the H. sapiens test data set, whereas the remaining 10 sequences failed after multiple trials. We did not include these 10 sequences when evaluating the performance of DISPHOS. The default predictions of DISPHOS reached a sensitivity of 56.1% at a specificity of 84.0%. DISPHOS also provided prediction scores. By taking different thresholds on the scores, DISPHOS achieved sensitivities of 27.0 and 10.4% at 95.0 and 99.0% specificity levels, respectively. In contrast, Musite achieved sensitivities of 72.2, 43.7, and 15.4% at specificities of 84.0, 95.0, and 99.0%, respectively.
scan-x is a tool to predict phosphorylation sites in different organisms using phosphorylation motifs determined by motif-x (22, 35). Proteome scale results on H. sapiens, M. musculus, D. melanogaster, and S. cerevisiae using scan-x are available for searching on the scan-x web site (http://scan-x.med.harvard.edu/). By searching the protein sequences in the H. sapiens test data set, the predictions of scan-x achieved a sensitivity of 34.5% at a specificity of 95.2% and a sensitivity of 15.8% at a specificity of 98.7%. To compare, Musite achieved sensitivities of 42.8 and 18.2% at specificities of 95.2 and 98.7%, respectively.
Note that, when performing the comparisons, we used a prediction model that trained from a data set excluding the protein sequences in the test data set. However, for NetPhos, DISPHOS, and scan-x, some of the test proteins might have been included in their training processes, and thus, the sensitivity performances are biased favorably toward these tools in the comparisons. This means that the performance improvement of Musite over these tools could be underestimated.
The consistency among different tools is shown in Fig. 7. At a specificity of 95%, each tool predicted about 900 phosphorylation sites. 109 total predicted sites are common among all four tools. About one-third to one-half of the predictions from each tool have no overlap with any other tools. 37 (34%) of the 109 commonly predicted sites are known phosphorylation sites, whereas the percentage for each separate tool is lower (Musite, 21%; DISPHOS, 9%; NetPhos, 9%; and scan-x, 18%). This suggests that a metapredictor combining the multiple tools to perform a consensus prediction might boost the prediction accuracy, although the data presented here are too sparse to be conclusive. An alternative explanation could be that the training data used by different tools were complementary.
Fig. 7.

Prediction consistency among different tools at specificity around 95% on same test results as in Fig. 6. Different colors indicate different tools. Blocks with edges of different colors represent overlapping predictions from corresponding tools. The numbers in each block represent the number of true positives and the number of predicted phosphorylation sites separated by a slash. The numbers in the parentheses following each tool name have a similar meaning for all the predicted sites by the tool.
Comparison with Other Kinase-specific Prediction Tools
We also evaluated the performance of kinase-specific predictions using Musite by comparison with four widely used tools, Scansite 2.0 (23), NetPhosK 1.0 (24), GPS 2.1 (25), and pkaPS (30). The well known PKA family, CK2, and MAPK family were used for comparison. The substrate proteins of each kinase or kinase family were extracted from the H. sapiens NR data set. Another 200 non-phosphoproteins were randomly selected from the H. sapiens NR data set. Combining these 200 non-phosphoproteins and the substrate proteins forms a test data set for each kinase or kinase family. The test data set for PKA was submitted to Scansite, NetPhosK, GPS, and pkaPS. The test data set of CK2 was submitted to Scansite, NetPhosK, and GPS. The test data set for MAPK was submitted to GPS.
To evaluate the performance of Musite, for each of the four kinases, a modified leave-one-out cross-validation test was performed. Each time, one phosphorylation site and the non-phosphorylation sites in the corresponding test data set formed the validation set, and the remaining phosphorylation and non-phosphorylation sites in the H. sapiens NR data set formed the training set. A prediction model was trained from the training set using Musite (1,000 data points from the positive data set and 1,000 data points from the negative data set were sampled when bootstrapping; k was chosen to be 2.5, 5, 10, and 20% of the bootstrapped sample size, i.e. 2,000, for KNN features; other parameters were by default). The model was used to predict phosphorylation sites in the validation set. The specificity level for the validation phosphorylation site was then calculated by counting the percentage of correctly classified non-phosphorylation sites in the validation set by setting the threshold as the prediction score of the validation phosphorylation site. This was repeated such that each phosphorylation site was used once in the validation set, and hence, each had a specificity level. Combining all validations, sensitivities were calculated by counting the percentage of phosphorylation sites that had specificities above certain levels. The leave-one-out cross-validation results of Musite were then compared with the prediction results of other tools. Like the comparisons of general phosphorylation site predictions, some of the test proteins might have been trained in other tools, and thus the performance is biased favorably to them when comparing the results. Therefore, we also performed self-consistency tests; i.e. we trained prediction models using all available sites for each kinase and predicted the sequences in the corresponding test data set. Both results of leave-one-out cross-validation tests and self-consistency tests by Musite were compared with the results of other tools.
All of the four other tools that we compared have predefined several stringency levels, and all except Scansite also provided prediction scores for all potential sites. Therefore, for Scansite, we calculated the specificity and sensitivity at its predefined three stringency levels, but for the other three tools and Musite, we adjusted the prediction thresholds to set the specificity levels as close as possible to 99.99, 99.9, 99.8, 99.5, 98.0, 97.0, 95.0, and 90.0% and compared the corresponding sensitivities. Table IV shows that Musite self-consistency tests performed superiorly to Scansite, NetPhosK, and pkaPS. Musite leave-one-out tests performed better than Scansite and NetPhosK in most cases except the results of NetPhosK at sensitivities of 99.99 and 99.90%. Musite leave-one-out tests performed comparably with pkaPS. GPS performed slightly better than the Musite leave-one-out tests; however, in most cases, Musite self-consistency tests outperformed the results of GPS, especially at high stringency levels. Note that because GPS is a new and well maintained tool it is likely that most, if not all, of our training phosphorylation sites have been included in their training process. In any case, the prediction performance of Musite is at least comparable with other kinase-specific prediction tools.
Table IV. Performance comparison of Musite with existing kinase-specific prediction tools.
Sensitivities (Sn) at different specificities (Sp) were compared. Different specificity levels were taken as similar as possible (in each column) among different tools. The best performed result in each specificity level (column) for each kinase or kinase family is highlighted in bold. LOO and Self stand for “leave-one-out cross-validation test” and “self-consistency test,” respectively.
| PKA | |||||||||
| ScanSite 1.0 | |||||||||
| Sp (%) | —a | — | 99.83 | — | 99.08 | — | 97.03 | — | — |
| Sn (%) | — | — | 16.67 | — | 41.88 | — | 61.54 | — | — |
| GPS 2.1 | |||||||||
| Sp (%) | 99.99 | 99.90 | 99.80 | 99.50 | 99.00 | 98.00 | 97.00 | 95.00 | 90.00 |
| Sn (%) | 0.85 | 8.12 | 19.66 | 32.91 | 49.57 | 58.97 | 67.52 | 72.65 | 83.76 |
| NetPhosK 1.0 | |||||||||
| Sp (%) | 99.99 | 99.90 | 99.79 | 99.46 | 99.02 | 97.98 | 97.06 | 95.16 | 89.86 |
| Sn (%) | 1.59 | 8.47 | 13.76 | 23.81 | 28.04 | 38.62 | 48.68 | 56.08 | 72.49 |
| pkaPS | |||||||||
| Sp (%) | 99.99 | 99.90 | 99.80 | 99.50 | 99.00 | 98.00 | 97.00 | 95.05 | 90.06 |
| Sn (%) | 1.71 | 8.12 | 15.38 | 31.62 | 42.73 | 58.12 | 66.67 | 73.93 | 85.04 |
| Musite 1.0LOO | |||||||||
| Sp (%) | 99.99 | 99.90 | 99.80 | 99.50 | 99.00 | 98.02 | 97.10 | 95.08 | 90.03 |
| Sn (%) | 0.43 | 6.84 | 16.67 | 31.62 | 44.87 | 57.26 | 63.68 | 72.79 | 81.62 |
| Musite 1.0Self | |||||||||
| Sp (%) | 99.99 | 99.90 | 99.80 | 99.50 | 99.00 | 98.00 | 97.00 | 95.00 | 90.00 |
| Sn (%) | 1.71 | 8.97 | 18.80 | 34.62 | 47.01 | 58.55 | 69.23 | 74.36 | 85.47 |
| CK2 | |||||||||
| ScanSite 1.0 | |||||||||
| Sp (%) | — | 99.87 | — | — | 99.14 | — | 96.8 | — | — |
| Sn (%) | — | 14.22 | — | — | 36.44 | — | 59.6 | — | — |
| GPS 2.1 | |||||||||
| Sp (%) | 99.99 | 99.90 | 99.80 | 99.50 | 99.00 | 98.00 | 97.00 | 95.00 | 90.00 |
| Sn (%) | 4.87 | 13.72 | 20.35 | 36.73 | 49.56 | 61.95 | 68.58 | 74.34 | 82.74 |
| NetPhosK 1.0 | |||||||||
| Sp (%) | 99.99 | 99.89 | 99.71 | 99.47 | 98.91 | 97.80 | 97.06 | 95.02 | 89.83 |
| Sn (%) | 3.66 | 13.61 | 20.42 | 29.32 | 37.70 | 51.31 | 55.50 | 62.30 | 74.35 |
| Musite 1.0LOO | |||||||||
| Sp (%) | 99.99 | 99.90 | 99.80 | 99.49 | 99.00 | 98.04 | 97.02 | 95.04 | 89.99 |
| Sn (%) | 5.58 | 14.60 | 22.57 | 34.51 | 49.12 | 60.62 | 66.81 | 72.12 | 81.42 |
| Musite 1.0Self | |||||||||
| Sp (%) | 99.99 | 99.90 | 99.80 | 99.50 | 99.00 | 98.00 | 97.00 | 95.00 | 90.00 |
| Sn (%) | 6.19 | 15.04 | 23.45 | 37.61 | 48.67 | 60.62 | 68.58 | 75.66 | 83.63 |
| MAPK | |||||||||
| GPS 2.1 | |||||||||
| Sp (%) | 99.99 | 99.90 | 99.80 | 99.50 | 99.00 | 98.00 | 97.00 | 95.00 | 90.00 |
| Sn (%) | 0.00 | 2.26 | 11.76 | 18.10 | 24.89 | 40.27 | 52.04 | 71.04 | 81.00 |
| Musite 1.0LOO | |||||||||
| Sp (%) | 99.99 | 99.90 | 99.80 | 99.50 | 99.00 | 98.00 | 96.99 | 95.02 | 90.93 |
| Sn (%) | 0.90 | 2.26 | 5.43 | 18.10 | 27.15 | 37.10 | 44.80 | 62.90 | 81.90 |
| Musite 1.0Self | |||||||||
| Sp (%) | 99.99 | 99.90 | 99.80 | 99.50 | 99.00 | 98.00 | 97.00 | 95.00 | 90.00 |
| Sn (%) | 0.91 | 4.52 | 4.98 | 15.38 | 27.15 | 38.46 | 47.06 | 63.35 | 81.90 |
a —, the prediction result at this specificity level was not available for this tool.
Software Implementation
We developed a stand-alone software system, Musite, to implement the described phosphorylation site prediction method. Currently, Musite V1.0 has been released for Windows, Mac OS X, and Linux/Unix platforms. Written in Java and released under a GNU general public license open source license, the Musite project provides an open platform for development of machine learning-based applications in predicting protein phosphorylation sites. With well designed API, Musite can be easily extended by programming. For example, other sequence features, such as protein secondary structure and solvent accessibility, can be easily incorporated in phosphorylation prediction by extending Musite API. Furthermore, Musite can be extended to train models and make predictions for other types of post-translational modifications. Musite, together with its source code, is available at http://musite.sourceforge.net/.
To make Musite user-friendly, we have implemented an easy-to-use graphical user interface. The most important utility of Musite is phosphorylation prediction, and therefore, in the main dialog, a user can submit protein sequences, a FASTA file, or a Musite XML file (Musite XML is a customized XML file format used by Musite for storing phosphorylation data in a compact yet comprehensive way). Fig. 8 shows an example of the result panel after submitting the human p53 protein sequence for phosphoserine/threonine prediction. Musite supports continuous adjustment of specificity cutoff from 0 to 1, rather than predefined confidence levels, to meet all stringency requirements of different studies. The predicted phosphorylation sites above the cutoff are rendered in different colors according to their stringency levels. The tab “Predicted Sites” contains a table with detailed information about each predicted site, such as its position, prediction score, and specificity level. The predicted result can be saved for future analysis or exported as a tab-delimited text file.
Fig. 8.

Screenshot of Musite V1. 0 graphical user interface. As an example, the phosphoserine/threonine prediction result of human p53 is displayed.
Musite makes it possible for a user to perform proteome scale prediction of phosphorylation sites in an automated fashion. We have performed the proteome scans in Table III on a standard work station (2.13-GHz dual core processor and 2-GB memory). Processing time was ∼18 h to predict general phosphorylation sites in all 20,319 proteins in the H. sapiens complete proteome from UniProt using a model trained with the default parameters (2,000 boots and five SVM classifiers). The running time will decrease by using a model with fewer boots and fewer SVM classifiers. If a user is only interested in predicting selected residues in the proteome, to save computing time, one can label each of those residues by appending a mark, “?,” e.g. replacing the serines (“S”) of interest by “S?”, and then Musite will make predictions only for the labeled residues.
Musite also provides other related functionalities, such as customized prediction model training, file format conversion, file statistics, and NR data set building tool integration (37). Customized prediction model training is a unique utility provided by Musite that enables users to train their own models from any phosphorylation data sets. As phosphorylation sites in various species are accumulating rapidly, it is difficult for a phosphorylation prediction tool to keep track of all available phosphorylation data. We have built phosphorylation prediction models in six model eukaryotic species, and we are in the progress of building more models in other species. Customized model training makes it possible for users, especially for the non-computational biologists, to train models using phosphorylation data specific to their own work. To train a customized prediction model based on the default parameters, a user only needs to specify the training sequence file (in the format of Musite XML or FASTA), the output model name, and amino acid types of sites. The user must annotate the phosphorylation sites in the input file, which is a simple and straightforward task. For example, if the input file is in the FASTA format, a user can specify a serine residue “S” as a phosphorylation site, appending a mark “#.” Although keeping simplicity, we also managed to maintain the flexibility of the program. The “Advanced Option” in the training dialog allows users to customize all the parameters for training models using their own data. Details on how to set the training parameters are explained in the tutorials at http://musite.sourceforge.net. It is worth mentioning that we have provided tools to convert the UniProt XML format to the Musite XML format and extract the phosphorylation data in user-defined organisms so that the users can easily make use of the latest phosphorylation annotations in UniProt/Swiss-Prot when training their own models.
Limitations and Future Work
Although Musite provides a useful alternative strategy for annotating phosphorylation events in proteomes, it has some limitations, most of which are common for all current phosphorylation site prediction tools. First of all, although computational predictions indicate the possibilities that query sites can/cannot be phosphorylated, our predicted results have not been correlated to different cell states or tissue conditions. This is one of the reasons that we provided a utility of customized model training in Musite. However, users still have to provide high quality training data related to specific cellular conditions, which are sparse in the current phosphorylation studies. Second, the phosphorylation sites used in the training data were mostly identified by mass spectrometry methods, which may have inherent bias in terms of representing the global phosphorylation events and hence affect the prediction performance. As techniques like electron transfer dissociation and alternative proteases are helping to resolve technology limitations, more complete phosphorylation data sets will be released. We will adapt our program and prediction models as the new data become available. Another limitation of the data is that we have only labeled positive data, but we do not have labeled negative data (i.e. we do not know whether the non-phosphorylation sites are truly negatives), and therefore, if some of them are predicted as phosphorylation sites, we do not know whether they are false positives. This fact makes it hardly feasible to estimate the precision (TP/(TP + FP)) where TP is true positive and FP is false positive) of any phosphorylation site prediction tool. Moreover, ignorance of the inherent prior ratio between the positive and negative data, which is hard to estimate, also created some bias in predictions. For future work, we will explore other methods, such as semisupervised learning, to address these limitations. We will include more kinases/kinase families and more organisms, extend Musite to other types of post-translational modifications, and integrate Musite in work flows of experimental phosphorylation studies.
CONCLUSION
Annotation of protein phosphorylation sites in proteomes is a crucial step to decode the signaling networks in living cells. In recent years, tens of thousands of phosphorylation sites in various species have been identified by large scale mass spectrometry-based studies. However, the vast majority of phosphorylation sites, especially in non-mammal species, still remain undiscovered. Considering limitations of mass spectrometry-based experimental studies, a more practical and efficient approach will be in silico large scale phosphorylation site prediction. In this work, we presented a new bioinformatics tool, Musite, specifically designed for large scale prediction of phosphorylation sites. Musite modeled phosphorylation site prediction as a binary classification problem with highly unbalanced data sets and solved it with a comprehensive machine-learning approach. After studying the properties of phosphorylation sites and their surrounding sequences, we adopted three sets of features to distinguish phosphorylation sites from non-phosphorylation sites: KNN scores, protein disorder scores, and amino acid frequencies. KNN scores were utilized to take advantage of the sequence cluster information around phosphorylation sites. Disorder scores and amino acid frequencies were used to characterize the generic patterns of phosphorylation sites. By combining both sequence and generic features, Musite is capable of identifying both phosphorylation sites with local sequence patterns similar to known phosphorylation sites and those beyond local sequence similarities. Combining all three sets of features, we have trained models based on a bootstrap aggregating procedure for predicting both general and kinase-specific phosphorylation sites in multiple organisms. It should be noted that the pretrained models in Musite V1.0 were not correlated to any particular cellular conditions. To perform condition-specific predictions, users can train customized models from phosphorylation data of a certain cellular condition. Proteome-wide predictions of phosphorylation sites were performed for six organisms. Cross-validation tests and comparisons with other tools show that Musite performs better on general predictions and at least comparably with existing methods on kinase-specific predictions.
Musite provides a unique application system, specifically designed for large scale prediction of both general and kinase-specific phosphorylation sites and for better utilizing the large magnitude of experimentally verified phosphorylation sites. Musite is the first tool that provides utility for training a phosphorylation site prediction model from users' own data and supports continuous adjustment of stringency levels. With its user-friendly graphic interface, Musite can be easily used by biologists to make predictions on their sequences and train prediction models from phosphorylation data of their own interest. Unlike experimental approaches, computational predictions are capable of proteome-wide predictions without inherent technical biases. Furthermore, Musite could provide an even more powerful and cost-effective approach by combining experimental and computational methods iteratively, which could be especially useful for some hypothesis-driven experiments. Alternatively, for bioinformaticians, Musite can serve as an open platform for building machine-learning applications for phosphorylation site prediction. In conclusion, Musite provides a unique tool for large scale phosphorylation site identification, and it is our hope that Musite will accelerate accumulation of our knowledge on protein phosphorylation and hence help explore the corresponding regulatory networks in living cells.
Supplementary Material
Acknowledgments
We thank Dr. Zoran Obradovic for helpful discussion, Dr. Waltraud Schulze for providing data of PhosPhAt3.0, and Dr. John Obenauer for technical assistance when testing on Scansite. We also thank the anonymous reviewers for useful suggestions.
Footnotes
* This work was supported, in whole or in part, by National Institutes of Health Grant R21/R33 GM078601 (to D. X.). This work was also supported by National Science Foundation-Plant Genome Research Program Grant DBI-0604439 (to J. J. T.).
 This article contains supplemental Figs. S1–S3 and Tables S1–S6.
This article contains supplemental Figs. S1–S3 and Tables S1–S6.
1 The abbreviations used are:
- KNN
- k nearest neighbor
- API
- application programming interface
- AUC
- area under ROC curve
- CDK
- cyclin-dependent kinase
- CK
- casein kinase
- NR
- non-redundant
- ROC
- receiver operating characteristic
- SVM
- support vector machine
- XML
- extensible markup language.
REFERENCES
- 1. Johnson L. N. (2009) The regulation of protein phosphorylation. Biochem. Soc. Trans. 37, 627–641 [DOI] [PubMed] [Google Scholar]
- 2. Beausoleil S. A., Jedrychowski M., Schwartz D., Elias J. E., Villén J., Li J., Cohn M. A., Cantley L. C., Gygi S. P. (2004) Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U.S.A. 101, 12130–12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 4. Villén J., Beausoleil S. A., Gerber S. A., Gygi S. P. (2007) Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. U.S.A. 104, 1488–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chi A., Huttenhower C., Geer L. Y., Coon J. J., Syka J. E., Bai D. L., Shabanowitz J., Burke D. J., Troyanskaya O. G., Hunt D. F. (2007) Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 104, 2193–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Munton R. P., Tweedie-Cullen R., Livingstone-Zatchej M., Weinandy F., Waidelich M., Longo D., Gehrig P., Potthast F., Rutishauser D., Gerrits B., Panse C., Schlapbach R., Mansuy I. M. (2007) Qualitative and quantitative analyses of protein phosphorylation in naive and stimulated mouse synaptosomal preparations. Mol. Cell. Proteomics 6, 283–293 [DOI] [PubMed] [Google Scholar]
- 7. Sugiyama N., Nakagami H., Mochida K., Daudi A., Tomita M., Shirasu K., Ishihama Y. (2008) Large-scale phosphorylation mapping reveals the extent of tyrosine phosphorylation in Arabidopsis. Mol. Syst. Biol. 4, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhai B., Villén J., Beausoleil S. A., Mintseris J., Gygi S. P. (2008) Phosphoproteome analysis of Drosophila melanogaster embryos. J. Proteome Res. 7, 1675–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boersema P. J., Foong L. Y., Ding V. M., Lemeer S., van Breukelen B., Philp R., Boekhorst J., Snel B., den Hertog J., Choo A. B., Heck A. J. (2010) In depth qualitative and quantitative profiling of tyrosine phosphorylation using a combination of phosphopeptide immunoaffinity purification and stable isotope dimethyl labeling. Mol. Cell. Proteomics 9, 84–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hornbeck P. V., Chabra I., Kornhauser J. M., Skrzypek E., Zhang B. (2004) PhosphoSite: a bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics 4, 1551–1561 [DOI] [PubMed] [Google Scholar]
- 11. Farriol-Mathis N., Garavelli J. S., Boeckmann B., Duvaud S., Gasteiger E., Gateau A., Veuthey A. L., Bairoch A. (2004) Annotation of post-translational modifications in the Swiss-Prot knowledge base. Proteomics 4, 1537–1550 [DOI] [PubMed] [Google Scholar]
- 12. Gnad F., Ren S., Cox J., Olsen J. V., Macek B., Oroshi M., Mann M. (2007) PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 8, R250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Linding R., Jensen L. J., Ostheimer G. J., van Vugt M. A., Jørgensen C., Miron I. M., Diella F., Colwill K., Taylor L., Elder K., Metalnikov P., Nguyen V., Pasculescu A., Jin J., Park J. G., Samson L. D., Woodgett J. R., Russell R. B., Bork P., Yaffe M. B., Pawson T. (2007) Systematic discovery of in vivo phosphorylation networks. Cell 129, 1415–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bodenmiller B., Campbell D., Gerrits B., Lam H., Jovanovic M., Picotti P., Schlapbach R., Aebersold R. (2008) PhosphoPep—a database of protein phosphorylation sites in model organisms. Nat. Biotechnol. 26, 1339–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Diella F., Gould C. M., Chica C., Via A., Gibson T. J. (2008) Phospho.ELM: a database of phosphorylation sites—update 2008. Nucleic Acids Res. 36, D240–D244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gao J., Agrawal G. K., Thelen J. J., Xu D. (2009) P3DB: a plant protein phosphorylation database. Nucleic Acids Res. 37, D960–D962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Keshava Prasad T. S., Goel R., Kandasamy K., Keerthikumar S., Kumar S., Mathivanan S., Telikicherla D., Raju R., Shafreen B., Venugopal A., Balakrishnan L., Marimuthu A., Banerjee S., Somanathan D. S., Sebastian A., Rani S., Ray S., Harrys Kishore C. J., Kanth S., Ahmed M., Kashyap M. K., Mohmood R., Ramachandra Y. L., Krishna V., Rahiman B. A., Mohan S., Ranganathan P., Ramabadran S., Chaerkady R., Pandey A. (2009) Human Protein Reference Database—2009 update. Nucleic Acids Res. 37, D767–D772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Durek P., Schmidt R., Heazlewood J. L., Jones A., MacLean D., Nagel A., Kersten B., Schulze W. X. (2010) PhosPhAt: the Arabidopsis thaliana phosphorylation site database. An update. Nucleic Acids Res. 38, D828–D834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang H., Zha X., Tan Y., Hornbeck P. V., Mastrangelo A. J., Alessi D. R., Polakiewicz R. D., Comb M. J. (2002) Phosphoprotein analysis using antibodies broadly reactive against phosphorylated motifs. J. Biol. Chem. 277, 39379–39387 [DOI] [PubMed] [Google Scholar]
- 20. Iakoucheva L. M., Radivojac P., Brown C. J., O'Connor T. R., Sikes J. G., Obradovic Z., Dunker A. K. (2004) The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32, 1037–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Blom N., Gammeltoft S., Brunak S. (1999) Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 294, 1351–1362 [DOI] [PubMed] [Google Scholar]
- 22. Schwartz D., Chou M. F., Church G. M. (2009) Predicting protein post-translational modifications using meta-analysis of proteome scale data sets. Mol. Cell. Proteomics 8, 365–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Obenauer J. C., Cantley L. C., Yaffe M. B. (2003) Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31, 3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blom N., Sicheritz-Pontén T., Gupta R., Gammeltoft S., Brunak S. (2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–1649 [DOI] [PubMed] [Google Scholar]
- 25. Xue Y., Ren J., Gao X., Jin C., Wen L., Yao X. (2008) GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics 7, 1598–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wong Y. H., Lee T. Y., Liang H. K., Huang C. M., Wang T. Y., Yang Y. H., Chu C. H., Huang H. D., Ko M. T., Hwang J. K. (2007) KinasePhos 2.0: a web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucleic Acids Res. 35, W588–W594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saunders N. F., Brinkworth R. I., Huber T., Kemp B. E., Kobe B. (2008) Predikin and PredikinDB: a computational framework for the prediction of protein kinase peptide specificity and an associated database of phosphorylation sites. BMC Bioinformatics 9, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dang T. H., Van Leemput K., Verschoren A., Laukens K. (2008) Prediction of kinase-specific phosphorylation sites using conditional random fields. Bioinformatics 24, 2857–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Plewczynski D., Tkacz A., Wyrwicz L. S., Rychlewski L., Ginalski K. (2008) AutoMotif Server for prediction of phosphorylation sites in proteins using support vector machine: 2007 update. J. Mol. Model. 14, 69–76 [DOI] [PubMed] [Google Scholar]
- 30. Neuberger G., Schneider G., Eisenhaber F. (2007) pkaPS: prediction of protein kinase A phosphorylation sites with the simplified kinase-substrate binding model. Biol. Direct 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xue Y., Li A., Wang L., Feng H., Yao X. (2006) PPSP: prediction of PK-specific phosphorylation site with Bayesian decision theory. BMC Bioinformatics 7, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li T., Li F., Zhang X. (2008) Prediction of kinase-specific phosphorylation sites with sequence features by a log-odds ratio approach. Proteins 70, 404–414 [DOI] [PubMed] [Google Scholar]
- 33. Kim J. H., Lee J., Oh B., Kimm K., Koh I. (2004) Prediction of phosphorylation sites using SVMs. Bioinformatics 20, 3179–3184 [DOI] [PubMed] [Google Scholar]
- 34. Miller M. L., Jensen L. J., Diella F., Jørgensen C., Tinti M., Li L., Hsiung M., Parker S. A., Bordeaux J., Sicheritz-Ponten T., Olhovsky M., Pasculescu A., Alexander J., Knapp S., Blom N., Bork P., Li S., Cesareni G., Pawson T., Turk B. E., Yaffe M. B., Brunak S., Linding R. (2008) Linear motif atlas for phosphorylation-dependent signaling. Sci. Signal. 1, ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schwartz D., Gygi S. P. (2005) An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 23, 1391–1398 [DOI] [PubMed] [Google Scholar]
- 36. Dunker A. K., Oldfield C. J., Meng J., Romero P., Yang J. Y., Chen J. W., Vacic V., Obradovic Z., Uversky V. N. (2008) The unfoldomics decade: an update on intrinsically disordered proteins. BMC Genomics 9, Suppl. 2, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li W., Godzik A. (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 [DOI] [PubMed] [Google Scholar]
- 38. Shi Y. (2009) Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484 [DOI] [PubMed] [Google Scholar]
- 39. Poole R. L. (2007) The TAIR database. Methods Mol. Biol. 406, 179–212 [DOI] [PubMed] [Google Scholar]
- 40. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vapnik V. (1995) The Nature of Statistical Learning Theory, Springer-Verlag, New York [Google Scholar]
- 42. Kennelly P. J., Krebs E. G. (1991) Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J. Biol. Chem. 266, 15555–15558 [PubMed] [Google Scholar]
- 43. Henikoff S., Henikoff J. G. (1992) Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. U.S.A. 89, 10915–10919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Obradovic Z., Peng K., Vucetic S., Radivojac P., Dunker A. K. (2005) Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins 61, Suppl. 7, 176–182 [DOI] [PubMed] [Google Scholar]
- 45. Breiman L. (1996) Bagging predictors. Mach. Learn. 24, 123–140 [Google Scholar]
- 46. Thorsten J. (2002) Learning to Classify Text Using Support Vector Machines: Methods, Theory and Algorithms, Kluwer Academic Publishers, Dordrecht, The Netherlands [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.