

Abstract
Background
The goal of metabolomics analyses is a comprehensive and systematic understanding of all metabolites in biological samples. Many useful platforms have been developed to achieve this goal. Gas chromatography coupled to mass spectrometry (GC/MS) is a well-established analytical method in metabolomics study, and 200 to 500 peaks are routinely observed with one biological sample. However, only ~100 metabolites can be identified, and the remaining peaks are left as "unknowns".
Result
We present an algorithm that acquires more extensive metabolite information. Pearson's product-moment correlation coefficient and the Soft Independent Modeling of Class Analogy (SIMCA) method were combined to automatically identify and annotate unknown peaks, which tend to be missed in routine studies that employ manual processing.
Conclusions
Our data mining system can offer a wealth of metabolite information quickly and easily, and it provides new insights, particularly into food quality evaluation and prediction.
Background
Metabolomics is based on biology, analytical chemistry, and information science, and it has become an important tool in many research areas [1-5]. The metabolome information can be used to extrapolate novel biological knowledge [1,6-8]. The main platforms in metabolomics studies are based on hybrid systems such as GC/MS, liquid chromatography (LC)/MS, and capillary electrophoresis (CE)/MS, all of which have been applied in many fields - including biomarker studies in medical diagnosis and quality evaluation and prediction in food science [2,3,5,9-11]. Among these platforms, GC/MS is a relatively mature method because the reproducible measurement is possible and many peaks (200 to 500) can be reliably obtained from a biological sample [1,3,12]. In addition, peak identification is straightforward when retention time (RT) and mass spectra data are compared to those of accumulated compound information in a laboratory (reference library). For these reasons, GC/MS is generally recognized as one of the most versatile and applicable platform in metabolomics.
Since GC/MS is mature enough to run a batch of analyses and to easily identify metabolite peaks, the development of a fast data analysis tool is essential [6,7]. Currently, peak identification and annotation is time-consuming when these processes are performed manually. Moreover, manual analysis results in serious problems in the accuracy of peak identification and annotation depending on the knowledge and expertise of individual researchers. Peak annotation is especially difficult because the extensive knowledge of fragmentation patterns by electron ionization (EI) is required. Therefore, it is an important challenge to develop data processing tools that identify and annotate metabolites easily, accurately, and rapidly.
Previous software platforms for peak identification utilize retention indexes that depend on an n-alkane mix (AMDIS [13], BinBase [14], MetaQuant [15], TagFinder [16], MetaboliteDetector [17]). But the retention index method requires some complicated procedures such as sample preparation and data analysis due to the n-alkane mix of the exogenous compounds. Moreover, the obtained metabolite information is limited to identifiable peaks because these platforms treat the ambiguous peak as "unknown". Therefore, many potentially interesting biomarkers tend to be disregarded.
There are several reasons why extracted peaks are left unidentified. First, peaks with a low signal-to-noise ratio, i.e., those with a large amount of noise, decrease the degree of coincidence (DOC) when compared to a reference library. Second, de-convolution may be unsuccessful because of co-elution (i.e., simultaneous elution of multiple compounds). Last and most importantly, no reference library is complete or covers information on all possible metabolites. If a certain metabolite is known to exist in a biological sample, a standard compound can be analyzed to resolve one unknown peak. However, if there is no information for a large number of unknown peaks, the cost of collecting standard compounds is prohibitively expensive; moreover, if a compound is not commercially available, the compound must be synthesized. For these reasons, it is important to deduce any kind of chemical information about unknown peaks.
We developed a data mining system to easily obtain metabolite information by using two mathematical methods. The first method is a Pearson's product-moment correlation coefficient for identification that we based on retention time and weighted mass spectrum [18,19]. Using 1) a retention time correction based on pseudo-internal standard and 2) a relaxed mass fitting to a reference library resulted in an identification process that was less dependent on column aging, column cuts, or column lot. In spectral comparison, higher masses are given more weight to reduce false positives and false negatives.
The second method is the Soft Independent Modeling of Class Analogy (SIMCA) [20] for the annotation of unknown peaks, and some techniques of SIMCA utilizing mass spectra have been developed, especially in toxic studies [21-25]. SIMCA is a supervised classification technique that is based on principal component analysis (PCA) [26], and it is useful for building multiple class models. New measurements are projected in each principle component (PC) space that describes a specific class, and the F-test is used to evaluate the Euclidean distances of the objects toward the model. We constructed the five chemical class models including amine, organic acid, fatty acid, sugar, and sugar phosphate groups as initiative. Using this method, we developed an annotation algorithm for unidentified peaks.
We utilized the free software MetAlign [27] for baseline correction, peak detection, and peak alignment. MetAlign has been a powerful tool for data preprocessing of GC/MS-based metabolomics [28,29]. The CSV format file exported from MetAlign can be analyzed by program written in Visual Basic, which software name is AIoutput. Our system and manual is given as additional files 1, 2, 3, and 4.
For validation, we performed two experiments. The first experiment included the standard mixtures: fifteen samples each mixed with 99 well-known standard compounds. In the standard-mix experiment, we demonstrated that the identification and annotation algorithms were robust and resulted in very few false positives or false negatives. The second experiment was a re-analysis of our published data. This experiment demonstrated that the required time for data processing was much shorter and that the novel system produced superior results. The proposed algorithm can be a powerful tool for quality evaluation and prediction, particularly in food science.
Methods
1. Theoretical aspect
Retention time correction
Retention times provide important information for identifying metabolites. A common problem in accurate identification is chromatographic shift resulting from column aging or lot differences. To adjust such shifts, retention indexes based on an n-alkane mix are usually calculated. However, retention index correction has some disadvantages. First, the requirements for sample preparation, such as density adjustment between metabolites and an n-alkane mix, are complicated. Moreover, if the type or number of n-alkane mix used in each laboratory is different, results may not be compatible among laboratories. Therefore, we used stable metabolite peaks derived from biological samples as indexes in order to reduce the problem of chromatographic shift. Retention times from the reference library were updated by several pseudo-internal standards. The update method was as follows.
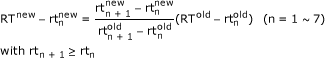 |
RTnew represents the retention time after update in the reference library, RTold represents that of original data (See also additional file 4), rtnew and rtold represent the retention time of the updated pseudo-internal standard and that of original one, respectively.
In an actual implementation, a user can choose up to eight compounds as pseudo-internal standards. The selection of standards is user-dependent, but the use of standards that result in early and late peaks is recommended for more accurate adjustment.
Peak identification
The most important information for peak identification is the mass spectrum of a compound. Pearson's product-moment correlation coefficient was used to measure the similarity of two mass spectra, which were represented as vectors of intensity for each integer mass unit. Because the EI ionization method is a hard ionization method, recorded mass spectra generally show larger intensities for lower masses than for higher masses. Because higher masses provide more reliable information for compound identification, higher masses were given larger weights in comparing two mass spectra. The identification method was as follows.
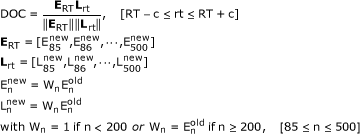 |
ERT and Lrt represent the totally-weighted vectors of an extracted peak and of a reference compound, respectively. The parameter c presents the time width for a reference search. Eold and Enew represent the original intensity and the weighted intensity of the extracted spectrum, respectively. Lnew and Lold represent the original intensity and the weighted intensity of a reference compound. For example, if an extracted peak, A, is eluted at 600 sec and the time width parameter c is set to 2 sec, the compounds from 598 to 602 sec in a reference library are selected as candidate matches. The compound from the reference library with the highest DOC when fitted to peak A is further selected as the match. If no candidate match is found, a prediction algorithm, described in the next section, is applied.
It should be noted that the time width was set by a user. Although pseudo-internal standard correction may impair accuracy compared to retention index correction, this relaxed mass fitting may have reduced the number of false negatives. This assertion is based on the assumption that mass spectra are more consistent and reliable than retention time for peak identification. In addition, although a few compounds have high similarity, the weighted mass spectra may have reduced false positives because the difference of the intensity in high masses was emphasized.
Peak prediction
SIMCA is a well-known pattern recognition method that distinguishes each class separately in a principal component (PC) space. SIMCA can also evaluate whether new objects belong to a specific model or not.
A training matrix, X, contains objects of different known classes. The sub-matrix, XK, (m × p) contains m training objects belonging to class K that were measured at p variables. Each class training set is modeled separately by PCA. XK is described with a score matrix, TK, and loading matrix, VK, as follows.
The number of important PCs, r, to describe the class, K, is usually determined by cross-validation [30,31]. EK is the matrix containing the residuals. XK is divided into two parts. One part 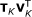 is described by r PCs, and the other EK is the residuals of the PC space. The standard deviation of EK, i.e., the residual standard deviation (RSD), and the RSD of new objects fitted to class K model are first compared, and then new objects are evaluated to determine whether they belong to class K. The RSD of EK is, in fact, a measure for the Euclidean distance of the class K objects toward the r PC space.
is described by r PCs, and the other EK is the residuals of the PC space. The standard deviation of EK, i.e., the residual standard deviation (RSD), and the RSD of new objects fitted to class K model are first compared, and then new objects are evaluated to determine whether they belong to class K. The RSD of EK is, in fact, a measure for the Euclidean distance of the class K objects toward the r PC space.
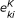 represents the residual of object, k, of the class K training set at variable i.
represents the residual of object, k, of the class K training set at variable i.
To predict whether an object, 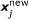 , belongs to the class K, it is projected on the space defined by the selected PCs of the class K training set.
, belongs to the class K, it is projected on the space defined by the selected PCs of the class K training set.
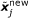 represents the predicted object, , in the space of the class K training set. The residual vector
represents the predicted object, , in the space of the class K training set. The residual vector 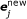 of object is calculated as follows.
of object is calculated as follows.
And the RSD, sj i.e., a Euclidean distance taking into account the degree of freedom, is obtained as follows.
One determines whether the residual variances 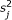 and
and 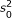 are significantly different by calculating the F-value compared to the tabulated critical F-crit for (m - r -1) and (m - r -1)2 degree of freedom.
are significantly different by calculating the F-value compared to the tabulated critical F-crit for (m - r -1) and (m - r -1)2 degree of freedom.
If the residual variances and are significantly different, the new object will not be classified into the class K. On the other hand, if the residual variances are not significantly different, the new object will be classified into class K. The test is performed under all classes.
In the AIoutput software, SIMCA is applied to unidentified peaks to classify them into a metabolite group (sugar, sugar phosphate, organic acid, amine, or fatty acid). If an unidentified peak could be classified into multiple groups, the group associated with the largest p-value is chosen. In this study, however, unknown peaks were rarely classified into multiple groups (3 out of 84 cases in re-analysis). If an unidentified peak is not classified into any class, the peak is ultimately reported as unknown. But the AIoutput software creates an organized data matrix that includes the unknown peak information. This type of output represents the ultimate goal of metabolomics studies, which is a comprehensive analysis of all metabolites in the biological samples.
2. Practical workflow
Construction of the SIMCA model
We prepared five metabolite groups for annotation: sugar, sugar phosphate, organic acid, fatty acid, and amine, and 12, 10, 12, 9, and 13 compounds, respectively, were prepared for the training matrix (Table 1). We used the relative intensities of each mass value ranging m/z 85 to 500 as variables in the SIMCA model.
Table 1.
Compounds used in the training set for the SIMCA method
| Class | Name | IUPAC | CAS | KEGG |
|---|---|---|---|---|
| Sugar | Fructose | (3S,4R,5R)-2-(hydroxymethyl)oxane-2,3,4,5-tetrol | 57-48-7 | C00095 |
| Galactose | (3R,4S,5R,6R)-6-(hydroxymethyl)oxane-2,3,4,5-tetrol | 59-23-4 | C00124 | |
| Glucose | (3R,4S,5S,6R)-6-(hydroxymethyl)oxane-2,3,4,5-tetrol | 50-99-7 | C00031 | |
| Glycerol | propane-1,2,3-triol | 56-81-5 | C00116 | |
| Maltose | (2R,3S,4S,5R,6R)-2-(hydroxymethyl)-6-[(2R,3S,4R,5R)-4,5,6-trih ydroxy-2-(hydroxymethyl)oxan-3-yl]oxyoxane-3,4,5-triol | 69-79-4 | C00208 | |
| Sucrose | (2R,3R,4S,5S,6R)-2-[(2S,3S,4S,5R)-3,4-dihydroxy-2,5-bis(hydrox ymethyl)oxolan-2-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol | 57-50-1 | C00089 | |
| Trehalose | (2R,3S,4S,5R,6R)-2-(hydroxymethyl)-6-[(2R,3R,4S,5S,6R)-3,4,5-t rihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoxane-3,4,5-triol | 99-20-7 | C01083 | |
| Xylitol | (2R,4S)-pentane-1,2,3,4,5-pentol | 83-99-0 | C00379 | |
| Inositol | cyclohexane-1,2,3,4,5,6-hexol | 87-89-8 | C00137 | |
| Sorbitol | (2R,3R,4R,5S)-hexane-1,2,3,4,5,6-hexol | 50-70-4 | C00794 | |
| Ribose | (3R,4S,5R)-5-(hydroxymethyl)oxolane-2,3,4-triol | 50-69-1 | C00121 | |
| Maltitol | (2S,3R,4R,5R)-4-[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxyl methyl)oxan-2-yl]oxyhexane-1,2,3,5,6-pentol | 81025-03-8 | C13542 | |
| Sugar phosphate | Fructose-6-phosphate | [(2R,3R,4S)-2,3,4,6-tetrahydroxy-5-oxohexyl] dihydrogen phosphate | 643-13-0 | C00085 |
| Glucosamine-6-phosphate | [(2R,3S,4R,5R)-5-amino-2,3,4-trihydroxy-6-oxohexyl] dihydrogen phosphate | 3616-42-0 | C00352 | |
| Glycerol-2-phosphate | 1,3-dihydroxypropan-2-yl phosphate | 17181-54-3 | C02979 | |
| Arabinose-5-phosphate | [(2R,3R,4S)-2,3,4-trihydroxy-5-oxopentyl] phosphate | 13137-52-5 | C01112 | |
| Ribulose-5-phosphate | [(2R,3R)-2,3,5-trihydroxy-4-oxopentyl] phosphate | 551-85-9 | C00199 | |
| Sorbitol-6-phosphate | 2,3,4,5,6-pentahydroxyhexyl phosphate | 20479-58-7 | C01096 | |
| Phosphoenolpyruvic acid | 2-phosphonooxyprop-2-enoic acid | 138-08-9 | C00074 | |
| Deoxyribose-5'-phosphate | [(2R,3S)-3-hydroxyoxolan-2-yl]methyl hydrogenphosphate | 7685-50-9 | C00673 | |
| Glucose-6-phosphate | [(2R,3S,4S,5R)-3,4,5,6-tetrahydroxyoxan-2-yl]methyl dihydrogen phosphate | 56-73-5 | C00092 | |
| Ribulose-1,5-bisphosphate | (2,3-dihydroxy-4-oxo-5-phosphonatooxypentyl) | 24218-00-6 | C01182 | |
| Organic acid | Oxalic acid | oxalic acid | 144-62-7 | C00209 |
| Isocitric acid | 1-hydroxypropane-1,2,3-tricarboxylic acid | 320-77-4 | C00311 | |
| 2-Isopropylmalic acid | 2-hydroxy-2-propan-2-ylbutanedioic acid | 3237-44-3 | C02504 | |
| Succinic acid | butanedioic acid | 110-15-6 | C00042 | |
| Maleic acid | (Z)-but-2-enedioic acid | 110-16-7 | C01384 | |
| Malic acid | 2-hydroxybutanedioic acid | 617-48-1 | C00711 | |
| Malonic acid | propanedioic acid | 141-82-2 | C00383 | |
| Glutaric acid | pentanedioic acid | 110-94-1 | C00489 | |
| Glycolic acid | 2-hydroxyacetic acid | 79-14-1 | C00160 | |
| Citramalic acid | 2-hydroxy-2-methylbutanedioic acid | 2306-22-1 | C00815 | |
| Citric acid | 2-hydroxypropane-1,2,3-tricarboxylic acid | 77-92-9 | C00158 | |
| Methylmalonic acid | 2-methylpropanedioic acid | 516-05-2 | C02170 | |
| Fatty acid | Elaidic acid | (E)-octadec-9-enoic acid | 112-79-8 | C01712 |
| Heptadecanoic acid | heptadecanoic acid | 506-12-7 | Not found | |
| Icosanoic acid | icosanoic acid | 506-30-9 | C06425 | |
| Lauric acid | dodecanoic acid | 143-07-7 | C02679 | |
| Lignoceric acid | tetracosanoic acid | 557-59-5 | C08320 | |
| n-Caprylic acid | octanoic acid | 124-07-2 | C06423 | |
| Nonanoic acid | nonanoic acid | 112-05-0 | C01601 | |
| Octacosanoic acid | octacosanoic acid | 506-48-9 | Not found | |
| Palmitoleic acid | (E)-hexadec-9-enoic acid | 373-49-9 | C08362 | |
| Amine | Dopamine | 4-(2-aminoethyl)benzene-1,2-diol | 51-61-6 | C03758 |
| Cadaverine | pentane-1,5-diamine | 462-94-2 | C01672 | |
| n-Butylamine | butan-1-amine | 109-73-9 | C18706 | |
| Putrescine | butane-1,4-diamine | 110-60-1 | C00134 | |
| Tyramine | 4-(2-aminoethyl)phenol | 51-67-2 | C00483 | |
| Isobutylamine | 2-methylpropan-1-amine | 78-81-9 | C02787 | |
| 2-Aminoethanol | 2-aminoethanol | 141-43-5 | C00189 | |
| 1,3-Propanediamine | N',N'-dimethylpropane-1,3-diamine | 109-76-2 | C00986 | |
| n-Propylamine | propan-1-amine | 107-10-8 | Not found | |
| Tryptamine | 2-(1H-indol-3-yl)ethanamine | 61-54-1 | C00398 | |
| Histamine | 2-(1H-imidazol-5-yl)ethanamine | 51-45-6 | C00388 | |
| 1-Methylhistamine | 2-(1-methylimidazol-4-yl)ethanamine | 501-75-7 | C05127 | |
| Serotonin | 3-(2-aminoethyl)-1H-indol-5-ol | 50-67-9 | C00780 | |
Compounds in each metabolite group were randomly selected from our reference library based on the metabolite feature. The popular name, IUPAC name, CAS registry number, and KEGG ID were described, respectively.
Standard mixture experiment
In order to validate the accuracy of our identification and annotation algorithms, we performed the following verification experiment. Standard compounds (99 total, see Table 2 and 3) were dispensed into 2 ml eppendorf tubes at three concentrations (5 μl, 10 μl, or 15 μl each standard solution of 10 mM). For each pattern, five tubes were prepared (15 standard mixtures in total). Any methanol in the mixtures was evaporated in a vacuum centrifuge dryer for 1 hour, and the mixtures were freeze-dried overnight.
Table 2.
43 out of 99 compounds included in the five classes
| Class | Name | IUPAC | Predicted Name |
|---|---|---|---|
| Organic acid | Citramalic acid | 2-hydroxy-2-methylbutanedioic acid | Organic acid |
| Citric acid | 2-hydroxypropane-1,2,3-tricarboxylic acid | Organic acid | |
| Fumaric acid | (E)-but-2-enedioic acid | Organic acid | |
| Glycolic acid | 2-hydroxyacetic acid | Organic acid* and Sugar | |
| Maleic acid | (Z)-but-2-enedioic acid | Organic acid | |
| Malic acid | 2-hydroxybutanedioic acid | Organic acid | |
| Malonic acid | propanedioic acid | Organic acid | |
| Mandelic acid | 2-hydroxy-2-phenylacetic acid | Organic acid | |
| Oxalic acid | oxalic acid | Organic acid | |
| Oxamic acid | oxamic acid | Organic acid | |
| Shikimic acid | (3R,4S,5R)-3,4,5-trihydroxycyclohexene-1-carboxylic acid | No annotation | |
| Succinic acid | butanedioic acid | Organic acid | |
| Sugar | Arabinose | (2S,3R,4R)-2,3,4,5-tetrahydroxypentanal | Sugar |
| Arabitol | (2R,4R)-pentane-1,2,3,4,5-pentol | Sugar | |
| Fructose | (3S,4R,5R)-2-(hydroxymethyl)oxane-2,3,4,5-tetrol | Sugar | |
| Galactose | (3R,4S,5R,6R)-6-(hydroxymethyl)oxane-2,3,4,5-tetrol | Sugar | |
| Glucose | (3R,4S,5S,6R)-6-(hydroxymethyl)oxane-2,3,4,5-tetrol | Sugar | |
| Inositol | cyclohexane-1,2,3,4,5,6-hexol | Sugar* and Organic acid | |
| Maltose | (2R,3S,4S,5R,6R)-2-(hydroxymethyl)-6-[(2R,3S,4R,5R)-4,5,6-trihydrox y-2-(hydroxymethyl)oxan-3-yl]oxyoxane-3,4,5-triol | Sugar | |
| Mannose | (3S,4S,5S,6R)-6-(hydroxymethyl)oxane-2,3,4,5-tetrol (2R,3R,4S,5S,6R)-2-[(2S,3S,4R,5R)-4-hydroxy-2,5-bis(hydroxymethyl) | Sugar | |
| Melezitose | -2-[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]ox yoxolan-3-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol | Sugar | |
| Ribitol | pentane-1,2,3,4,5-pentol | Sugar | |
| Ribose | (3R,4S,5R)-5-(hydroxymethyl)oxolane-2,3,4-triol | Sugar | |
| Sucrose | (2R,3R,4S,5S,6R)-2-[(2S,3S,4S,5R)-3,4-dihydroxy-2,5-bis(hydroxymet hyl)oxolan-2-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol | Sugar | |
| Threitol | (2R,3R)-butane-1,2,3,4-tetrol | Sugar | |
| Trehalose | (2R,3S,4S,5R,6R)-2-(hydroxymethyl)-6-[(2R,3R,4S,5S,6R)-3,4,5-trihyd roxy-6-(hydroxymethyl)oxan-2-yl]oxyoxane-3,4,5-triol | Sugar | |
| Xylose | (2S,3R,4S,5R)-oxane-2,3,4,5-tetrol | Sugar | |
| Glycerol | propane-1,2,3-triol | Sugar | |
| Sugar phosphate | Ribulose-5-phosphate | [(2R,3R)-2,3,5-trihydroxy-4-oxopentyl] dihydrogen phosphate | Sugar phosphate |
| Amine | Cadaverine | pentane-1,5-diamine | Amine |
| Dopamine | 4-(2-aminoethyl)benzene-1,2-diol | Amine | |
| Isobutylamine | 2-methylpropan-1-amine | Amine | |
| n-Butylamine | butan-1-amine | Amine | |
| n-Propylamine | propan-1-amine | Amine | |
| Putrescine | butane-1,4-diamine | Amine | |
| Spermidine | N'-(3-aminopropyl)butane-1,4-diamine | No annotation | |
| Spermine | N,N'-bis(3-aminopropyl)butane-1,4-diamine | No annotation | |
| Tyramine | 4-(2-aminoethyl)phenol | Amine | |
| Histamine | 2-(1H-imidazol-5-yl)ethanamine | Amine | |
| Serotonin | 3-(2-aminoethyl)-1H-indol-5-ol | Amine | |
| Tryptamine | 2-(1H-indol-3-yl)ethanamine | Amine | |
| Fatty acid | Heptadecanoic acid | heptadecanoic acid | Fatty acid |
| Octadecanoic acid | octadecanoic acid | Fatty acid | |
Table 2 shows 43 standard compounds classified to the five metabolite groups constituting the SIMCA method. Table 3 shows the remaining 56 standard compounds. Table 2 and 3 also show the predicted name of each compound by the SIMCA algorithm. If a compound was classified into some groups, the groups were fastened by "and". The asterisk (*) indicates the group with higher p-value. If a compound was not classified into any groups, the predicted name was described as "No annotation".
Table 3.
56 out of 99 compounds not included in the five classes
| Class | Name | IUPAC | Predicted Name |
|---|---|---|---|
| Benzene | 4-Aminobenzoic acid | 4-aminobenzoic acid | No annotation |
| Benzoic acid | benzoic acid | No annotation | |
| o-Toluic acid | 2-methylbenzoate | No annotation | |
| Phenylalanine | (2S)-2-amino-3-phenylpropanoic acid | No annotation | |
| Tyrosine | (2S)-2-amino-3-(4-hydroxyphenyl)propanoic acid | No annotation | |
| Ferulic acid | (E)-3-(4-hydroxy-3-methoxyphenyl)prop-2-enoic acid | No annotation | |
| Dopa | (2S)-2-amino-3-(3,4-dihydroxyphenyl)propanoic acid | No annotation | |
| Alpha-Keto acid | 2-Oxoglutaric acid | 2-oxopentanedioic acid | No annotation |
| Pyruvic acid | 2-oxopropanoic acid | Amine | |
| Indole, Imidazole | Histidine | (2S)-2-amino-3-(1H-imidazol-5-yl)propanoic acid | No annotation |
| Histidinol | 2-amino-3-(1H-imidazol-5-yl)propan-1-ol | No annotation | |
| Tryptophan | (2S)-2-amino-3-(1H-indol-3-yl)propanoic acid | No annotation | |
| Purine, Pyrimidine | Adenine | 7H-purin-6-amine | No annotation |
| Caffeine | 1,3,7-trimethylpurine-2,6-dione | No annotation | |
| Cytosine | 6-amino-1H-pyrimidin-2-one | No annotation | |
| Guanine | 2-amino-3,7-dihydropurin-6-one | No annotation | |
| Inosine | 9-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-3H-p urin-6-one | No annotation | |
| Thymine | 5-methyl-1H-pyrimidine-2,4-dione | No annotation | |
| Uracil | 1H-pyrimidine-2,4-dione | No annotation | |
| Xanthine | 3,7-dihydropurine-2,6-dione | No annotation | |
| Amino acid | 2-Aminobutyric acid | 2-aminobutanoic acid | No annotation |
| 2-Aminoisobutyric acid | 2-amino-2-methylpropanoic acid | No annotation | |
| 4-Aminobutyric acid | 4-aminobutanoic acid | Amine | |
| Alanine | (2S)-2-aminopropanoic acid | No annotation | |
| Allothreonine | (2S,3S)-2-amino-3-hydroxybutanoic acid | No annotation | |
| Asparagine | (2S)-2,4-diamino-4-oxobutanoic acid | No annotation | |
| Aspartic acid | (2S)-2-aminobutanedioic acid | No annotation | |
| Citrulline | (2S)-2-amino-5-(carbamoylamino)pentanoic acid | No annotation | |
| Cysteine | (2R)-2-amino-3-sulfanylpropanoic acid | No annotation | |
| Glutamic acid | (2S)-2-aminopentanedioic acid | No annotation | |
| Glutamine | (2S)-2,5-diamino-5-oxopentanoic acid | No annotation | |
| Glycine | 2-aminoacetic acid | Amine | |
| Glycyl-glycine | 2-[(2-aminoacetyl)amino]acetic acid | No annotation | |
| Homoserine | 2-amino-4-hydroxybutanoic acid | No annotation | |
| Isoleucine | (2S,3S)-2-amino-3-methylpentanoic acid | No annotation | |
| Leucine | (2S)-2-amino-4-methylpentanoic acid | No annotation | |
| Lysine | (2S)-2,6-diaminohexanoic acid | No annotation | |
| Methionine | (2S)-2-amino-4-methylsulfanylbutanoic acid | No annotation | |
| N-Acetyl-DL-valine | 2-acetamido-3-methylbutanoic acid | No annotation | |
| Ornithine | (2S)-2,5-diaminopentanoic acid | No annotation | |
| Proline | (2S)-pyrrolidine-2-carboxylic acid | No annotation | |
| Sarcosine | 2-(methylamino)acetic acid | No annotation | |
| Serine | (2S)-2-amino-3-hydroxypropanoic acid | No annotation | |
| Threonine | (2S,3R)-2-amino-3-hydroxybutanoic acid | No annotation | |
| Valine | (2S)-2-amino-3-methylbutanoic acid | No annotation | |
| β-Alanine | 3-aminopropanoic acid | No annotation | |
| Other | 2-Hydroxypyridine | 1H-pyridin-2-one | No annotation |
| 4-Hydroxypyridine | 1H-pyridin-4-one | No annotation | |
| Phosphoric acid | phosphate | Sugar phosphate | |
| Kojic acid | 5-hydroxy-2-(hydroxymethyl)pyran-4-one | No annotation | |
| Nicotinic acid | pyridine-3-carboxylic acid | No annotation | |
| Quinic acid | (3R,5R)-1,3,4,5-tetrahydroxycyclohexane-1-carboxylic acid | No annotation | |
| Propyleneglycol | propane-1,2-diol | No annotation | |
| Creatinine | 2-amino-3-methyl-4H-imidazol-5-one | No annotation | |
| Urea | urea | Organic acid | |
| Ascorbic acid | (2R)-2-[(1S)-1,2-dihydroxyethyl]-4,5-dihydroxyfuran-3-one | No annotation | |
The detail is shown in Table 2.
Sample derivatization procedures were followed previously [5]. In brief, methoxyamine hydrochloride in pyridine was added for oximation, and N-methyl-N-(trimethylsilyl) trifluoroacetamide (MSTFA) was added for silylation, and 1 μl of each mixture was injected in the split mode (25:1, v/v). Auto-sampler was a 7683B series injector (Agilent Co., Palo Alto, CA), and gas chromatograph was a 6890N (Agilent Co., Palo Alto, CA), and mass spectrometer was a Pegasus III TOF (LECO, St. Joseph, MI). The column was a 30 m × 0.25 mm i.d. fused silica capillary column coated with 0.25 μm CP-SIL 8 CB low bleed/MS (Varian Inc., Palo Alto, CA). The front inlet temperature was 230°C. The helium gas flow rate through the column was 1 ml/min. The column temperature was held at 80°C for 2 min isothermally and then was raised by 15°;C/min to 330°C and was held there for 6 min isothermally. The transfer line and ion source temperatures were 250°C and 200°C, respectively. 20 scans per second were recorded over the mass range 85-500 m/z.
MS data were exported in the netCDF format (See additional file 5). Fifteen chromatograms were peak-detected and aligned using the MetAlign software (Wageningen UR, The Netherlands, freely available at http://www.pri.wur.nl/UK/products/MetAlign/). The resulting data was exported in the CSV-format file (See additional file 6). After updating retention times of our reference library by the pseudo-internal standard correction method (see above), peak identification and annotation were executed in the AIoutput software.
Published data experiment
In order to verify the utility of our system, we re-analyzed data from our previous work that is reported in Pongsuwan W et al. [5]. The analytical method used for this experiment was exactly the same as that used for the standard mixture experiment.
Result and Discussion
Validation and optimization of the SIMCA model
It was important to evaluate independence of five class models. We performed PCA toward the data matrix (56 × 416), i.e., spectral vectors of 56 compounds used in the SIMCA model (Figure 1a and 1b). The metabolite groups were clearly separated by the first and second PCs, and the amine and fatty acid groups were especially independent. As shown in Figure 1b, the loading plot shows that the m/z 86 and 174 contributed to the discrimination of amine group, and the m/z 117, 129, and 132 contributed to the discrimination of fatty acid group. To investigate the features of organic acid, sugar, and sugar phosphate groups in detail, we applied PCA to the data matrix (34 × 416) including only the three groups. As shown in Figure 1c and 1d, the m/z 299 clearly discriminated the sugar phosphate group, and the m/z 147 was a characteristic mass to the organic acid group.
Figure 1.

Model evaluation. (a), (b) The PCA score and loading plot including all compound groups. (c), (d) The score and loading plot including organic acid, sugar, and sugar phosphate groups. Mean centering was used in the data preprocessing. The legend shows each metabolite group. X-axis and Y-axis describe the first and second PCs, respectively.
After we applied PCA to five metabolite groups individually, we optimized each model using interclass distance as follows.
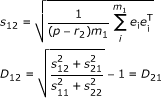 |
s12 denotes the interclass residual when Class 1 objects were projected into the PC space of Class 2. r2 and m1 represent the factor number of Class 2 and the number of training objects for Class 1, respectively. It should be noted that the interclass residual of Class 1 described by Class 2 space was different from that of Class 2 described by Class 1 space (s12 ≠ s21). For this reason, we used an interclass distance D12 as the distance between class models, and the values larger than one indicate real differences [20]. Higher distances indicate that models are more independent of one another. If two models are not independent, the interclass distance is close to zero. Table 4 shows the interclass distance, PC number, and the important m/z used in the SIMCA model. The classes were largely independent of one another. In addition, because only one PC was used as the latent variable for all metabolite groups, the model should be robust and less over-fitted. In the cross validation, the misclassifications were nothing (Table 5). This result shows that a good model can be constructed for annotating metabolites from mass spectra.
Table 4.
Interclass distance resulting from SIMCA
| Class name | Sugar phosphate | Organic acid | Sugar | Amine | Fatty acid | PC number | Important m/z |
|---|---|---|---|---|---|---|---|
| Sugar phosphate | 0.00 | 1.21 | 1.05 | 1.85 | 1.79 | 1 | 89, 147, 217, 299 |
| Organic acid | 1.21 | 0.00 | 1.46 | 3.81 | 4.38 | 1 | 101, 133, 147 |
| Sugar | 1.05 | 1.46 | 0.00 | 2.72 | 2.53 | 1 | 89, 103, 147, 217 |
| Amine | 1.85 | 3.81 | 2.72 | 0.00 | 4.32 | 1 | 86, 100, 174 |
| Fatty acid | 1.79 | 4.38 | 2.53 | 4.32 | 0.00 | 1 | 117, 129, 132, 145 |
We used only one PC for all groups in order to make a robust model without over-fit. A distance close to zero indicates that the two classes are virtually identical, and the value above 1.0 indicates real differences. The important m/z contributed to a model was indicated, and the most important m/z was shown by bold type.
Table 5.
Cross validation of SIMCA model
| Actuals\Prediction | phosphate Sugar | Organic acid | Sugar | Amine | Fatty acid |
|---|---|---|---|---|---|
| Sugar phosphate | 10 | 0 | 0 | 0 | 0 |
| Organic acid | 0 | 12 | 0 | 0 | 0 |
| Sugar | 0 | 0 | 12 | 0 | 0 |
| Amine | 0 | 0 | 0 | 13 | 0 |
| Fatty acid | 0 | 0 | 0 | 0 | 9 |
Cross validation was automatically performed by Pirouetto 4.0 software (InfoMetrix).
Identification and annotation accuracies by the standard-mix experiment
Table 6 shows the result of peak identification by Manual, ChromaTOF software, and the AIoutput software, respectively. Our system required only two minutes for analyzing the CSV-format file, and all 99 compounds in 15 samples were unmistakably identified. Several amino acids generate two peaks due to different degrees of silylation at primary amines, and sugars generate several peaks due to their geometric isomers derived from in the oxime reaction [32-34]. Such peaks were also identified accurately. Although there were the ten false positives, some of these false positive might have been generated by additional reactions in the derivatization process and by the pyrolysis reaction in the front inlet and capillary column [33,34]. The formation of TMS-pyroglutamate from TMS-glutamate is a characteristic example of an additional reaction in the derivatization process [34]. Moreover, we also confirmed the accuracy of annotation algorithm (see Table 2 and 3). Some compounds of organic acid and sugar groups were classified into two groups. Although the organic acid and sugar groups were relatively similar as shown in Figure 1 and Table 4, the end result by p-value was correct. Some compounds including an amino functional group were classified to amine group. Despite some misclassifications, however, the result suggests that our annotation algorism is acceptable because the mass fragmentation is not always dependent to the functional groups. In the fragmentation pattern, pyruvic acid, phosphoric acid, and urea have m/z 174, m/z 299, and m/z 147 as high intensity mass, respectively. Spermidine and spermine have the unique mass fragmentation patterns different in amine group (See additional file 2).
Table 6.
Peak identification results by manual, ChromaTOF and the AIoutput software
| Analysis time | False negatives | False positives | |
|---|---|---|---|
| Manual | 39 ± 15 h | 12 ± 6 | 5 ± 2 |
| ChromaTOF | 20 sec | 70 | 5 |
| AIoutput | 2 min | 0 | 10 |
Manual analysis was performed by six skilled people in our laboratory. ChromaTOF software identified the compounds based on the NIST library. The AIoutput software identified compounds based on our reference library.
System evaluation by the data re-analysis
We re-analyzed the published data in order to show the utility of our system. The biological samples used were Japanese green teas that had been ranked in an agricultural fair [5]. Our system recognized 231 peaks in these chromatograms, and offered an organized data matrix without any missing values (See additional file 7). Out of 231 peaks, 112 were matched with compounds from our reference library, and 83 peaks were classified into a predicted metabolite groups; organic acid, sugar, sugar phosphate, amine, and fatty acid groups included 56, 18, 3, 6 and 0 peaks, respectively. We applied the organized data matrix to PCA (Figure 2). Figure 2a and 2b represent the PCA score plots from the data matrix obtained by the previous analysis [5] and the new analysis, respectively. Our new system produced better classification, and the second PC space closely correlated with tea grades. Moreover, the required time for data processing was about 30 min.
Figure 2.

Result comparison. (a) The PCA score plot made by our previous method. (b) The PCA score plot made by our new system. The legend shows the ranking of the Japanese green tea samples. The variations in each group were relatively small, and each tea grade was clearly better separated in the second PC with the new system.
Because the second PC correlated with tea quality, we examined the loading of the second PC (data not shown). In addition to some identified metabolites, two annotated metabolites (Figure 3a and 3b) positively contributed to the second PC, and one annotated metabolite (Figure 3c) contributed negatively (we also confirmed the mass spectra of these annotated peaks by manual). The amounts of three metabolites clearly differed among tea grades. Note here that the second PC was insensitive to the analytical order because the tea samples had been randomly analyzed by GC-TOF/MS, also note that ribitol could be reliably used as the internal standard (Figure 3d). Of these three annotated peaks, we identified one metabolite as xylonic acid by our additional investigation (Figure 4). Xylonic acid is a minor sugar acid, and this is new insight into Japanese green tea. We also examined standard compounds of xylitol and xylose in order to confirm whether xylonic acid was generated from these compounds because of additional reaction in the derivatization process (data not shown).
Figure 3.

Annotated peaks information. (a), (b), (c) The peak height of three important metabolites for describing the tea grade in the second PC space. (d) The peak height of ribitol. The peaks of the annotated metabolites were scaled relative to the ribitol peak. The graph title indicates their annotated names and their respective retention times. These three peaks clearly varied with tea quality.
Figure 4.

Xylonic acid mass spectra. (a) Mass spectra of an annotated metabolite in a Japanese green tea sample. This metabolite is the same as Fig. 3a. (b) Xylonic acid mass spectra.
Conclusion
The purpose of metabolomics is a comprehensive analysis of metabolites in biological samples. GC-TOF/MS offers highly reproducible information on primary metabolites. Our new data analysis tool provided the useful metabolite information and the organized data matrix accurately and rapidly. The system identified compounds by a retention time correction based on pseudo-internal standard and a relaxed mass fitting without requiring complicated sample preparation procedures, such as density control. This system can be also used to re-analyze past data if the reference library is provided. As shown by the re-analysis of our published data, novel knowledge about Japanese green tea research is available for quality evaluation and prediction in food science. Our study suggests that researchers can achieve high-quality GC/MS-based metabolomics relatively easily. However, GC-TOF/MS is comparatively expensive; therefore, we are working to develop a similar system for GC-Q/MS, which is considerably less expensive. Moreover, this method will be also used to develop the "Known" and "Known unknown" metabolite library database for non-targeted metabolomics analysis.
Authors' contributions
EF initiated and supervised the project. HT improved the initial concept and designed and implemented the program, and wrote the manuscript. YT and HT prepared the reference library of 500 compounds. MA contributed to the system performance and also contributed to writing the paper. EF and TB proposed the retention time correction method. EF and TB also contributed to manuscript brushing up. All authors read and approved the final manuscript.
Supplementary Material
Main program of the system. Excel file including the source program for peak identification and annotation.
Example reference library. Excel file of an example reference library used in the main program.
SIMCA model book. Excel file for SIMCA method used in the main program.
Manual. The manual for using our system.
Example raw data. Example of a raw data file in standard mixture experiment.
Example CSV file. Example of a CSV file from MetAlign.
Example peak table. Example of the peak table exported from the system.
Contributor Information
Hiroshi Tsugawa, Email: hiroshi_tsugawa@bio.eng.osaka-u.ac.jp.
Yuki Tsujimoto, Email: yuki_tsujimoto@bio.eng.osaka-u.ac.jp.
Masanori Arita, Email: arita@bi.s.u-tokyo.ac.jp.
Takeshi Bamba, Email: bamba@bio.eng.osaka-u.ac.jp.
Eiichiro Fukusaki, Email: fukusaki@bio.eng.osaka-u.ac.jp.
Acknowledgements
The authors thank Dr. Nakako Shibagaki for her highly constructive comments to the program. The study represents a portion of the dissertation submitted by Hiroshi Tsugawa to Osaka University in partial fulfillment of the requirement for his PhD.
References
- Fiehn O, Kopka J, Dörmann P, Altmann T, Trethewey R, Willmitzer L. Metabolite profiling for plant functional genomics. Nature biotechnology. 2000;18:1157–1161. doi: 10.1038/81137. [DOI] [PubMed] [Google Scholar]
- Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, Laxman B, Mehra R, Lonigro RJ, Li Y, Nyati MK, Ahsan A, Kalyana-Sundaram S, Han B, Cao X, Byun J, Omenn GS, Ghosh D, Pennathur S, Alexander DC, Berger A, Shuster JR, Wei JT, Varambally S, Beecher C, Chinnaiyan AM. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457:910–914. doi: 10.1038/nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Denkert C, Budczies J, Kind T, Weichert W, Tablack P, Sehouli J, Niesporek S, Könsgen D, Dietel M, Fiehn O. Mass spectrometry-based metabolic profiling reveals different metabolite patterns in invasive ovarian carcinomas and ovarian borderline tumors. Cancer research. 2006;66:10795–10804. doi: 10.1158/0008-5472.CAN-06-0755. [DOI] [PubMed] [Google Scholar]
- Yoshida R, Tamura T, Takaoka C, Harada K, Kobayashi A, Mukai Y, Fukusaki E. Metabolomics-based systematic prediction of yeast lifespan and its application for semi-rational screening of ageing-related mutants. Aging cell. 2010;9:616–625. doi: 10.1111/j.1474-9726.2010.00590.x. [DOI] [PubMed] [Google Scholar]
- Pongsuwan W, Fukusaki E, Bamba T, Tonetani T, Yamahara T, Kobayashi A. Prediction of Japanese green tea ranking by gas chromatography/mass spectrometry-based hydrophilic metabolite fingerprinting. Journal of agricultural and food chemistry. 2007;55:231–236. doi: 10.1021/jf062330u. [DOI] [PubMed] [Google Scholar]
- Bylesjö M, Eriksson D, Kusano M, Moritz T, Trygg J. Data integration in plant biology: the O2PLS method for combined modeling of transcript and metabolite data. The Plant journal: for cell and molecular biology. 2007;52:1181–1191. doi: 10.1111/j.1365-313X.2007.03293.x. [DOI] [PubMed] [Google Scholar]
- Fiehn O. Extending the breadth of metabolite profiling by gas chromatography coupled to mass spectrometry. Trends in analytical chemistry. 2008;27:261–269. doi: 10.1016/j.trac.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cevallos-Cevallos JM Reyes-De-Corcuera JI Etxeberria E Danyluk MD Rodrick GE Metabolomic analysis in food science: a review Trends in Food Science & Technology 200920557–566. 10.1016/j.tifs.2009.07.00221536537 [DOI] [Google Scholar]
- Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nature chemical biology. 2009;5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto M, Wong DT, Hirayama A, Soga T, Tomita M. Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer-specific profiles. Metabolomics. 2010;6:78–95. doi: 10.1007/s11306-009-0178-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongsuwan W, Bamba T, Harada K, Yonetani T, Kobayashi A, Fukusaki E. High-throughput technique for comprehensive analysis of Japanese green tea quality assessment using ultra-performance liquid chromatography with time-of-flight mass spectrometry (UPLC/TOF MS) Journal of agricultural and food chemistry. 2008;56:10705–10708. doi: 10.1021/jf8018003. [DOI] [PubMed] [Google Scholar]
- Fiehn O, Kopka J, Trethewey RN, Willmitzer L. Identification of uncommon plant metabolites based on calculation of elemental compositions using gas chromatography and quadrupole mass spectrometry. Analytical chemistry. 2000;72:3573–3580. doi: 10.1021/ac991142i. [DOI] [PubMed] [Google Scholar]
- Halket JM, Przyborowska A, Stein SE, Mallard WG, Down S, Chalmers RA. Deconvolution gas chromatography/mass spectrometry of urinary organic acids--potential for pattern recognition and automated identification of metabolic disorders. Rapid communications in mass spectrometry. 1999;13:279–284. doi: 10.1002/(SICI)1097-0231(19990228)13:4<279::AID-RCM478>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Fiehn O, Wohlgemuth G, Scholz M. Setup and annotation of metabolomic experiments spectrometric metadata. Lecture Notes in Computer Science. 2005;3615:224–239. doi: 10.1007/11530084_18. [DOI] [Google Scholar]
- Bunk B, Kucklick M, Jonas R, Münch R, Schobert M, Jahn D, Hiller K. MetaQuant: a tool for the automatic quantification of GC/MS-based metabolome data. Bioinformatics. 2006;22:2962–2965. doi: 10.1093/bioinformatics/btl526. [DOI] [PubMed] [Google Scholar]
- Luedemann A, Strassburg K, Erban A, Kopka J. TagFinder for the quantitative analysis of gas chromatography - mass spectrometry (GC-MS) based metabolite profiling experiments. Bioinformatics. 2008;24:732–737. doi: 10.1093/bioinformatics/btn023. [DOI] [PubMed] [Google Scholar]
- Hiller K, Hangebrauk J, Jäger C, Spura J, Schreiber K, Schomburg D. MetaboliteDetector: comprehensive analysis tool for targeted and nontargeted GC/MS based metabolome analysis. Analytical chemistry. 2009;81:3429–3439. doi: 10.1021/ac802689c. [DOI] [PubMed] [Google Scholar]
- Horai H, Arita M, Kanaya S, Nihei Y, Ikeda T, Suwa K, Ojima Y, Tanaka K, Tanaka S, Aoshima K, Oda Y, Kakazu Y, Kusano M, Tohge T, Matsuda F, Sawada Y, Hirai MY, Nakanishi H, Ikeda K, Akimoto N, Maoka T, Takahashi H, Ara T, Sakurai N, Suzuki H, Shibata D, Neumann S, Iida T, Tanaka K, Funatsu K, Matsuura F, Soga T, Taguchi R, Saito K, Nishioka T. MassBank: a public repository for sharing mass spectral data for life sciences. Journal of mass spectrometry. 2010;45:703–714. doi: 10.1002/jms.1777. [DOI] [PubMed] [Google Scholar]
- Horai H, Arita M, Nishioka T. Comparison of ESI-MS Spectra in MassBank Database. 2008 International Conference on BioMedical Engineering and Informatics. 2008;2:853–857. [Google Scholar]
- Svante W, Michael S. SIMCA: a method for analyzing chemical data in terms of similarity and analogy. ACS Symposium Series. 1977;52:243–282. [Google Scholar]
- Scott DR. Determination of chemical classes from mass spectra of toxic organic compounds by SIMCA pattern recognition and information theory. Analytical chemistry. 1986;58:881–890. doi: 10.1021/ac00295a051. [DOI] [Google Scholar]
- Scott DR. Classification of binary mass spectra of toxic compounds with an inductive expert system and comparison with SIMCA class modeling. Analytica chimica Acta. 1988;211:11–29. doi: 10.1016/S0003-2670(00)83665-X. [DOI] [Google Scholar]
- Dunn WJ III, Emery SL, Glen WG, Scott DR. Preprocessing, variable selection, and classification rules in the application of SIMCA pattern recognition to mass spectral data. Environmental Science and Technology. 1989;23:1499–1505. doi: 10.1021/es00070a008. [DOI] [Google Scholar]
- Scott DR. Classification and identification of mass spectra of toxic compounds with an inductive rule-building expert system and information theory. Analytica chimica Acta. 1989;223:105–121. [Google Scholar]
- Werther W, Lohninger H, Stancl F, Varmuza K. Classification of mass spectra: a comparison of yes/no classification methods for the recognition of simple structural properties. Chemometrics and Intelligent Laboratory Systems. 1994;22:63–76. doi: 10.1016/0169-7439(94)85018-6. [DOI] [Google Scholar]
- Jolliffe IT. Principal Component Analysis. Springer Series in Statistics. 2002.
- Lommen A. MetAlign: interface-driven, versatile metabolomics tool for hyphenated full-scan mass spectrometry data preprocessing. Analytical chemistry. 2009;81:3079–3086. doi: 10.1021/ac900036d. [DOI] [PubMed] [Google Scholar]
- Lommen A, van der Weg G, van Engelen MC, Bor G, Hoogenboom LAP, Nielen MWF. An untargeted metabolomics approach to contaminant analysis: pinpointing potential unknown compounds. Analytica chimica acta. 2007;584:43–49. doi: 10.1016/j.aca.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Tikunov Y, Lommen A, Vos CHRD, Verhoeven HA, Bino RJ, Hall RD, Bovy AG. A novel approach for nontargeted data analysis for metabolomics. Large-scale profiling of Tomato fruit volatiles1[w] Plant Physiology. 2005;139:1125–1137. doi: 10.1104/pp.105.068130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maesschalck R. Decision criteria for soft independent modeling of class analogy applied to near infrared data. Chemometrics and Intelligent Laboratory Systems. 1999;47:65–77. doi: 10.1016/S0169-7439(98)00159-2. [DOI] [Google Scholar]
- Svante W. Pattern recognition by means of disjoint principal components models. Pattern Recognition. 1976;8:127–139. doi: 10.1016/0031-3203(76)90014-5. [DOI] [Google Scholar]
- Roger AL, Charles CS. Analysis of trimethylsilyl O-methyloximes of carbohydrates by combined gas-liquid chromatography-mass spectrometry. Analytical Biochemistry. 1971;43:533–538. doi: 10.1016/0003-2697(71)90284-3. [DOI] [PubMed] [Google Scholar]
- Kenneth RL, Roy HR, Charles WG. Complete mass spectra of the per-trimethylsilylated amino acids. Journal of Chromatography. 1977;141:355–375. doi: 10.1016/S0021-9673(00)93539-8. [DOI] [Google Scholar]
- Kanani HH, Klapa MI. Data correction strategy for metabolomics analysis using gas chromatography-mass spectrometry. Metabolic engineering. 2007;9:39–51. doi: 10.1016/j.ymben.2006.08.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Main program of the system. Excel file including the source program for peak identification and annotation.
Example reference library. Excel file of an example reference library used in the main program.
SIMCA model book. Excel file for SIMCA method used in the main program.
Manual. The manual for using our system.
Example raw data. Example of a raw data file in standard mixture experiment.
Example CSV file. Example of a CSV file from MetAlign.
Example peak table. Example of the peak table exported from the system.