

Abstract
Though compromised blood-brain barrier (BBB) is a pathological hallmark of WNV- associated neurological sequelae, underlying mechanisms are unclear. We characterized the expression of matrix metalloproteinases (MMP) in WNV-infected human brain-microvascular endothelial cells (HBMVE) and -cortical astrocytes (HBCA), components of BBB and their role in BBB disruption. Expression of multiple MMPs was significantly induced in WNV-infected HBCA cells. Naïve HBMVE cells incubated with the supernatant from WNV-infected HBCA cells demonstrated loss of tight junction proteins, which was rescued in the presence of MMP inhibitor, GM6001. Further, supernatant from WNV-infected HBCA cells compromised the in-vitro BBB models integrity. Our data suggests astrocytes as one of the sources of MMP in the brain, which mediates BBB disruption allowing unrestricted entry of immune cells into the brain, thereby contributing to WNV-neuropathogenesis. Because of the unavailability of WNV antivirals and vaccines, use of MMP inhibitors as an adjunct therapy to ameliorate WNV disease progression is warranted.
Keywords: MMP, TIMP, tight junction proteins, ZO-1, Claudin-1, meningoencephalitis, BBB, astrocytes, human brain microvascular endothelial cells, GM6001
Introduction
Although immune surveillance in the brain is a normal physiological response, increased traversing of activated immune cells and pathogens across the blood-brain barrier (BBB) leads to pathophysiological changes in neuroinflammatory diseases such as multiple sclerosis, cerebral ischemia (Persidsky et al., 2006; Petty and Lo, 2002), bacterial meningitis (BM) and infections with viruses such as HIV (Kanmogne et al., 2007), human T cell leukemia virus (Afonso et al., 2007) and West Nile virus (WNV) (Arjona et al., 2007a; Wang et al., 2004). Matrix metalloproteinases (MMP), a large family of endopeptidases, play a major role in leukocyte migration, modulating cytokine activity and disruption of the BBB (Gearing et al., 1995; Johnatty et al., 1997; Leppert et al., 2001; Mun-Bryce et al., 2002; Yong et al., 2007). Excess MMP production and activation is the key pathophysiological hallmark of several neurodegenerative diseases (Cunningham, Wetzel, and Rosenberg, 2005; Hu et al., 2007; Rosenberg, 1995; Rosenberg, 2002). Physiologically, MMP activity is tightly regulated at the level of gene transcription, pro-enzyme activation and by the action of tissue inhibitors of MMP (TIMP). Though the basal levels of MMP expression in the brain are low, under pathological conditions, MMP are synthesized by most of the resident CNS cells such as endothelial cells, astrocytes, microglia and neurons as well as the infiltrating immune cells (Hummel et al., 2001; Mandal et al., 2003; Rosenberg, 2002).
WNV, an enveloped, single-stranded positive-sense, neurotropic flavivirus, is an important human pathogen that targets neurons to cause potentially lethal encephalitis (Diamond et al., 2003; Hayes and Gubler, 2006). Neurological complications such as inflammation, failure of the BBB, and neuronal death contribute to the mortality and morbidity associated with WNV-induced meningitis. Increased production of pro-inflammatory mediators facilitate WNV neuroinvasion by compromising the BBB integrity and are associated with high virus titers in the brain and increased mortality in WNV mouse models (Arjona et al., 2007b; Wang et al., 2008; Wang et al., 2004). Recent studies using MMP-9 knock out mouse model provides important evidence for the role of WNV-induced MMP-9 in opening of the BBB (Wang et al., 2008). The BBB primarily consists of microvascular endothelial cells and perivascular astrocytes, separated by the basement membrane (Persidsky et al., 2006). The tight junction proteins (TJP) such as zona occludens (ZO), claudins and occludin, the main structural basis of BBB integrity, play a key role in the physiology of the BBB (Persidsky et al., 2006).
Though neurons are the prime target of WNV infection, we and others have previously characterized WNV infection in the BBB cells, human brain microvascular endothelial (HBMVE) cells and brain astrocytes (Cheeran et al., 2005; van Marle et al., 2007; Verma et al., 2009). Recent report documenting the presence of WNV antigen in the astrocytes of WNV-infected human brain tissue further suggests the role of these cells in WNV neuropathogenesis (van Marle et al., 2007). However, our understanding of the cellular mechanisms associated with WNV-induced BBB disruption, specifically the contribution of BBB-associated cells is limited. Therefore, the aim of this study was to examine the effect of WNV infection on the expression profile of MMP family genes in the BBB cells, characterize their role in disrupting the tight junctions of the BBB and assess the ability of MMP inhibitor GM6001 in reversing the disruption of an in-vitro BBB model.
Results
Human brain cortical astrocytes cells are susceptible to WNV infection
Brain endothelial cells and astrocytes are the two main components of the BBB. In this report we demonstrate that the WNV titer in infected human brain cortical astrocytes (HBCA) cells peaks (log 7–8 PFU/mL) at day 3 after infection, followed by a sharp decline (Fig. 1A). Similar to our previous study, we further confirmed that WNV can efficiently replicate in HBMVE cells (Fig. 1A) (Verma et al., 2009). The specificity of WNV infection in HBCA cells was further characterized by immunostaining of HBCA cells with astrocyte specific marker, glial fibrillary acidic protein (GFAP), and WNV antigen (Fig 1B). Almost 100% cells were found to be GFAP positive, thereby confirming the purity of these primary HBCA cells (Fig. 1B, i). Robust staining of WNV antigen was detected in the cytoplasm of HBCA cells at day 2 after infection (Fig. 1B, ii). Infected HBCA cells stained with only secondary antibody against both, anti-GFAP (data not shown) and WNV antigen (Fig. 1B, iii) did not show any immunostaining.
Fig. 1.

WNV replication kinetics in HBCA cells. (A) WNV titers in infected HBCA and HBMVE cells supernatants collected at various time points after infection were determined by plaque assay using Vero cells. Viral titers, expressed as plaque forming units (PFU)/mL of supernatant represent mean ± SD of data obtained from at least three independent experiments. (B) HBCA cells grown and fixed on coverslips at day 2 after infection were stained with anti-GFAP antibody or anti-WNV envelope antibody, and counterstained with DAPI (blue). (i) mock-infected HBCA cells demonstrate GFAP staining in the cytoplasm, (ii) WNV-infected HBCA cells demonstrate robust virus staining in the cytoplasm, and (iii) WNV-infected HBCA cells stained with secondary antibody, negative control, did not show any immunostaining.
WNV induces mRNA expression of MMP family genes in HBCA cells
The global response of HBMVE and HBCA cells infected with WNV at multiplicity of infection-5 (MOI-5) was determined at days 1 and 3 after infection by cDNA microarray analysis. Though WNV infection was robust and comparable in both the cell types, WNV infection did not alter the expression profile of MMP and TIMP genes in HBMVE cells at days 1 and 3 after infection (data not shown). Whereas WNV infection did not induce any MMP family genes in HBCA cells at day 1 after infection, a significant increase in the expression of MMP-1 (34-fold) and -3 (26-fold) genes was observed in these cells at day 3 after infection. Increase in the expression of MMP and TIMP in HBCA cells was further validated by qRT-PCR at different time points after infection. In concurrence with the microarray data, as seen in Figure 2A, MMP-1 and -3 genes expression increased at day 2, and was significantly up-regulated, 20- to 40-fold, at days 3 and 4 after infection which coincided with the peak in the WNV titers. In addition, MMP-9 expression demonstrated significant increase (9- to 30-fold) at day 3 and 4 after infection. A 2- to 3-fold decrease in TIMP-2 and -3 transcripts were observed only at days 3 and 4 after infection (Fig. 2B). Infection of HBCA cells with UV-WNV did not induce any change in the expression profile of these genes (data not shown), further indicating that these alterations were a result of WNV replication, rather than just virus entry into the cells.
Fig. 2.
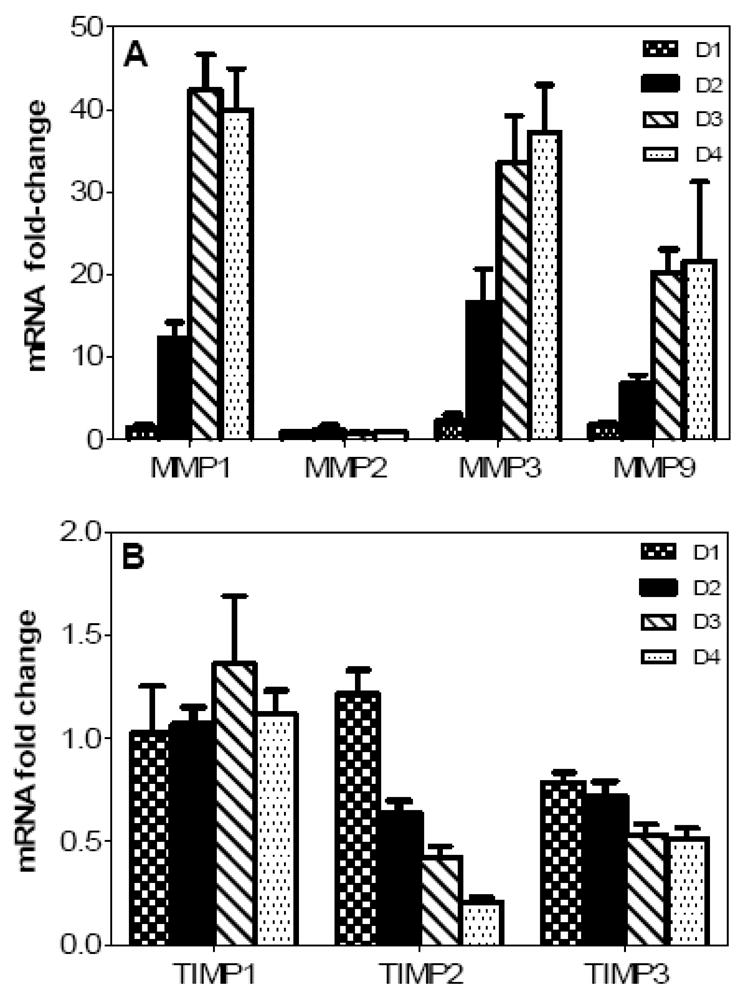
WNV differentially modulates expression of MMP family genes in HBCA cells. qRT-PCR was conducted on cDNA templates from WNV- and mock-infected HBCA cells harvested from days 1 to 4 after infection to determine the fold-change of (A) MMP-1, -2, -3 and -9 and (B) TIMP-1, -2 and -3 genes expression. Change in the levels of each gene was first normalized to the GAPDH gene and then the fold-change in infected cells as compared to corresponding control cells was calculated. Data represents mean of at least four independent experiments conducted in duplicate.
Increase in MMP protein expression
Immunocytochemical analysis did not exhibit increase in MMP-9 immunostaining in mock-infected HBCA cells (Fig. 3A, i), however, a strong signal of MMP-9 expression was observed at day 3 after infection in both, WNV-infected as well as neighboring un-infected HBCA cells (Fig. 3A, iii and iv). Further, as depicted in Figure 3B and C, a significant increase of 50% in the MMP-1 protein expression was first evident at day 2 after infection, and was consistently high at days 3 and 4 after infection. On the other hand, increase in MMP-3 and -9 proteins expression was mostly observed at days 3 and 4 after infection, thus coinciding with the increase in their mRNA transcripts.
Fig. 3.
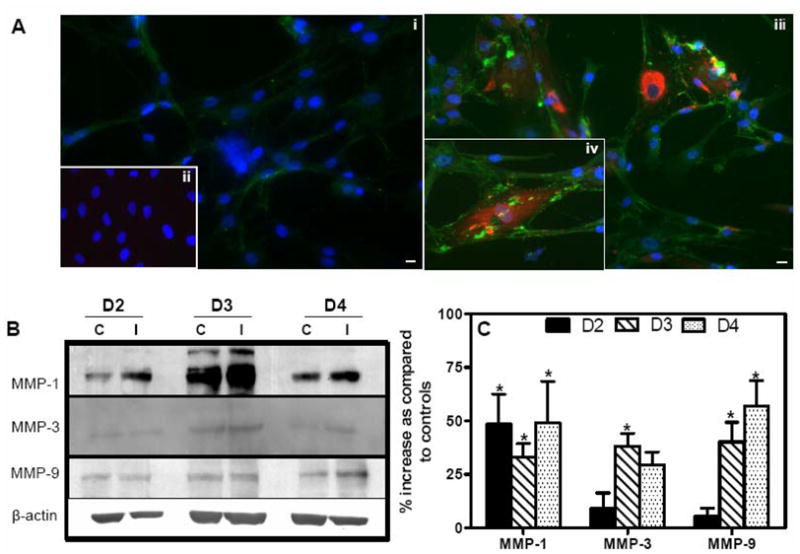
WNV induces protein expression of MMPs in HBCA cells. (A) Confluent primary HBCA cells were infected with WNV at MOI-5 and MMP-9 expression was determined using immunofluorescence. (i) Mock-infected HBCA cells demonstrated very faint and diffused MMP-9 immunoreactivity (green), and (ii) Mock-infected cells incubated without MMP-9 antibody did not show any staining. (iii and iv) marked increase in MMP-9 staining (green) was observed at day 3 in WNV-infected HBCA cells. MMP-9 increase was evident in both, WNV-infected (red) as well as neighboring un-infected HBCA cells. The images shown are representative results of three independent experiments. Scale bar represents 10 μm at 20× magnification. (B) Western blot analysis of MMP-1, -3 and -9. Cellular proteins extracted from WNV- and mock-infected HBCA cells were separated on PAGE, transferred onto PVDF membranes and immunoblotted with antibodies specific to MMP-1, -3 and -9. The membranes were stripped and re-probed with anti-β-actin and the bands were detected by the chemiluminescence method. (C) Quantitative analysis of western blots represented as percentage increase in MMPs compared to respective control HBCA cells at the corresponding time point. Mean comparisons were based upon extrapolated CI for three data points. *p<0.05, compared to corresponding controls. C, mock- and I, WNV-infected HBCA cells.
MMP-3 and -9 activities are increased in the supernatant of WNV-infected HBCA cells
Since MMPs are secretory proteins, their increase was further determined in the supernatant of WNV-infected HBCA cells by assessing the gelatinolytic activity of MMPs by zymography as well as ELISA. As depicted in Figure 4A, the supernatant from mock-infected HBCA cells gave a faint gelatinolytic band of 92-kDa, which became intense and strong in the supernatant from WNV-infected HBCA cells. Densitometric analysis of the intensity of the bands demonstrated a 90% and 208% increase in MMP-9 activity as compared to controls at days 3 and 4 after infection, respectively (p<0.005, Fig. 4B). Similarly, MMP-3 enzyme activity determined by casein zymography demonstrated an increase in its activity by 48% and 56% in WNV-infected HBCA cells at the same time points (Fig. 4A and B). Interestingly, MMP-2 band at 72-kDa demonstrated decreased gelatinolytic activity in WNV-infected HBCA cells at both time points. In addition, total MMP-9 protein measured by ELISA also increased significantly in the supernatant from WNV-infected HBCA cells at day 3 after infection (p<0.05, Fig. 4C).
Fig. 4.

WNV infection induces enzymatic activities of MMP-3 and -9 in HBCA cells. (A) The conditioned media from WNV- and mock-infected HBCA cells were analyzed for MMP -2/9 and -3 enzymes activities by gelatin and casein zymography, respectively. (B) The densitometric analysis demonstrates significant increase in MMP-3 and -9 activities in infected HBCA cells. (C) Total MMP-9 levels were measured by ELISA in the supernatant from mock- and WNV-infected HBCA cells at day 3 after infection. Mean comparisons were based upon extrapolated CI for three data points. Values represent mean ± S.D. *p<0.005, **p<0.05 compared to corresponding control cells.
The MMPs released in the supernatant of WNV-infected HBCA cells can degrade TJP of HBMVE cells
We previously demonstrated that WNV infection of HBMVE cells has no effect on the key TJPs (Verma et al., 2009). To determine the ability of HBCA-derived MMPs to digest TJP, naïve HBMVE cells were either infected directly with WNV or incubated with supernatant media from mock- or WNV-infected HBCA cells. As observed in Figure 5A, naïve HBMVE cells demonstrated distinct ZO-1 staining at the cell borders (i and ii), which did not change in response to direct WNV infection, at days 3 and 4 after infection (iii and iv). Further, ZO-1 immunostaining of naïve HBMVE cells treated for six hours with supernatant from mock-infected HBCA cells (Fig. 5A, v and vi) was similar to naïve HBMVE cells (i and ii), however HBMVE cells treated with supernatant from WNV-infected HBCA cells, collected at day 3 after infection, demonstrated distinct loss of ZO-1 protein, which became even more prominent when incubated with supernatant from WNV-infected HBCA cells collected at day 4 after infection (Fig. 5A, vii and viii). To further confirm the specificity of MMPs in the degradation of TJP, the supernatant from mock- and WNV-infected HBCA cells were treated with broad-spectrum MMP inhibitor GM6001 for 15 min before incubation with naïve HBMVE cells. While the incubation of naïve HBMVE cells with supernatant from mock-infected HBCA cells treated with GM6001 did not affect ZO-1 staining (Fig.5Aix and x), loss of ZO-1 was noticeably reversed in the naïve HBMVE cells incubated with GM6001-treated supernatant from WNV-infected HBCA cells collected at both, days 3 and 4 after infection (Fig. 5Axi and xii). Gelatin zymography of GM6001-treated supernatant from WNV-infected HBCA cells demonstrated a distinct loss of MMP-2 and -9 enzymes activities as compared to the untreated supernatant, thereby confirming the ability of GM6001 to block MMP-9 enzyme activity (Fig.5A, inset).
Fig. 5.

WNV-induced MMPs lead to degradation of TJP of HBMVE cells. Confluent primary HBMVE cells were either infected with WNV or incubated with the supernatant from mock- or WNV-infected HBCA cells for six hours and TJPs expression was visualized using confocal immunofluorescence microscopy. (A) ZO-1 staining in (i and ii) naïve HBMVE cells characterized by staining at cell borders, did not change as a result of (iii and iv) WNV infection or (v and vi) after incubation of naïve HBMVE cells with supernatant media from mock-infected HBCA cells collected at days 3 and 4 after infection. Whereas there was a distinct loss of ZO-1 staining in naïve HBMVE cells incubated with supernatant media from WNV-infected HBCA cells at days 3 and 4 after infection (vii and viii). The treatment of supernatant media from mock-infected HBCA cells with GM6001 had no effect on ZO-1 staining (ix and x), however disrupted ZO-1 staining was markedly reversed in naïve HBMVE cells incubated with GM6001-treated supernatant from WNV-infected HBCA cells at days 3 and 4 after infection (xi and xii). The inset depicts, MMP-9 activity in (a) supernatant of infected HBCA cells and (b) in the presence of GM6001. (B) Immunostaining of claudin-1 in HBMVE cells by WNV-induced MMPs. Claudin-1 immunoreactivity which was characterized by intracellular and junctional pattern in (i and ii) naïve HBMVE cells, increased at days 3 and 4 after (iii and iv) WNV infection. (v and vi) incubation of naïve HBMVE cells with supernatant from mock-infected HBCA cells for six hours did not alter the claudin-1 staining pattern, however (vii and viii) incubation with supernatant from WNV-infected HBCA cells at days 3 and 4 after infection, disrupted the claudin-1 staining. Effect of GM6001 treatment on supernatant from (ix and x) mock-infected and (xi and xii) WNV-infected HBCA cells on the claudin-1 immunostaining. The images shown are representative results of at least three independent experiments in duplicate. Scale bar represents 20 μm at 63× magnification.
Expression of claudin-1 is also critical for maintaining the BBB integrity. Therefore, in addition to ZO-1 we also investigated the experssion of claudin-1 in HBMVE cells by immunostaining to further understand the effect of MMPs on BBB integrity parameters. As compared to naïve HBMVE cells (Fig 5B, i and ii), expression of claudin-1 increased at days 3 and 4 after WNV infection of HBMVE cells (Fig. 5Biii and iv), which concurs with our previous observation (Verma et al., 2009). However, similar to ZO-1 (Fig. 5A), claudin-1 expression which did not change in the presence of supernatant from mock-infected HBCA cells (Fig. 5Bv and vi), decreased considerably in the presence of supernatant from WNV-infected HBCA cells collected at days 3 and 4 after infection (Fig. 5B, vii and viii) and the treatment of supernatant with GM6001 markedly reversed the degradation of claudin-1 induced by supernatant from WNV-infected HBCA cells (Fig. 5B, xi- xii). To further differentiate between MMP-induced degradation of TJP and cytotoxicity triggered by HBCA supernatant, the cytotoxicity assays were performed on HBMVE cells after similar treatments and no significant change in the cell viability of HBMVE cells was observed (data not shown).
Integrity of the BBB model compromised by MMPs secreted by WNV-infected HBCA cells can be reversed in presence of MMP inhibitor
Disruption of the BBB is mostly associated with degradation or redistribution of TJP. Using BBB model, we previously demonstrated that direct WNV infection of HBMVE cells per se does not compromise the BBB integrity (Verma et al., 2009). In this study, we employed similar model to investigate if MMPs derived from infected HBCA cells can degrade ZO-1 and claudin-1 of HBMVE cells and compromise the BBB integrity. At day 8 after seeding, the integrity of the BBB models demonstrated high TEER (650 Ω/cm2) and low dextran transmigration (2–3%) as compared to inserts with no cells. The BBB models incubated with synthetic MMP-9 (10 ng/mL media) as positive controls for six hours resulted in a significant decrease in the TEER (Fig 6A). The BBB models were then incubated for six hours with cell supernatants from HBCA cells collected at day 4 after infection. While the TEER of the BBB models incubated with the supernatant from mock-infected HBCA cells did not change as compared to control models (only HBMVE cell media), it decreased significantly, (~400 Ω/cm2, p<0.005), in the BBB models incubated with supernatant from WNV-infected HBCA cells (Fig. 6A). Treatment of WNV-infected HBCA cells supernatant with GM6001 prior to the incubation restored the TEER values (p<0.05). Similarly, there was no significant change in the FITC-dextran transmigrated across the BBB models incubated with supernatant from mock-infected HBCA cells as compared to control BBB models with only CSC media (Fig. 6B). However, BBB models incubated with supernatant from WNV-infected HBCA cells demonstrated a significant increase in the percent FITC-dextran transmigration (190%, p<0.005) as compared to controls, and the treatment of supernatant with GM6001 significantly inhibited FITC-dextran transmigration from 190% to 140% (p<0.05).
Fig. 6.
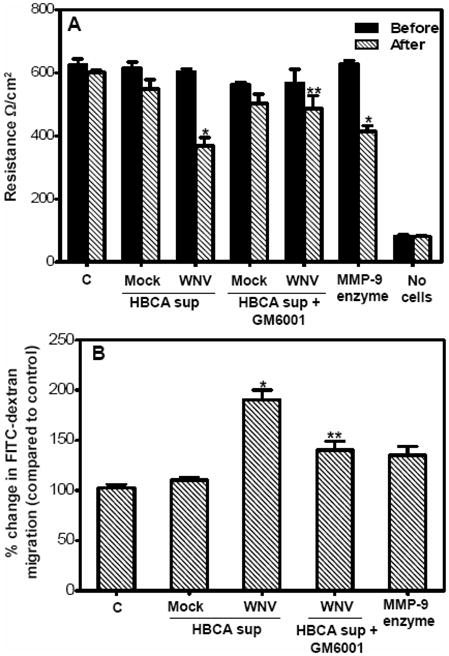
Supernatant from WNV-infected HBCA cells augment the paracellular permeability of the BBB model. At day 8 post seeding, the BBB models were incubated with supernatant from mock- and WNV-infected HBCA cells or synthetic MMP-9 enzyme as positive control for six hours and (A) The integrity of the BBB models was determined by measuring the TEER before and after the incubation. TEER values, presented as Ω/cm2, demonstrated a significant decrease only in the BBB model incubated with either supernatant from WNV-infected HBCA cells or synthetic MMP-9 enzyme. Presence of GM6001 rescued the BBB permeability. (B) FITC-dextran permeability assay of the BBB models. After six hours of incubation, the percentage of FITC-dextran that crossed the BBB models as compared to control BBB models did not vary significantly in BBB models incubated with supernatant from mock-infected HBCA cells. BBB models incubated with supernatant from WNV-infected HBCA cells demonstrated significant increase in the FITC-dextran transmigration and this increase was reversed in presence of GM6001. The data is representative of at least three independent experiments in duplicate. *p<0.005 percent change as compared to control inserts and **p<0.05 percent change of GM6001-treated supernatants as compared to corresponding BBB models without GM6001 treatment.
Discussion
The tight junctions of the BBB play a critical role in preserving the integrity of the BBB and restricting the entry of immune cells as well as pathogens into the CNS. Increased permeability of the BBB is shown to be associated with increased WNV titers in the brain (Diamond and Klein, 2004). This study characterizing the acute MMP gene and protein changes as a result of WNV infection of BBB cells highlights that astrocytes are one of the potential source of multiple MMPs, which degrade key TJPs of HBMVE cells and compromise the integrity of the BBB model thereby contributing to WNV-associated neuropathogenesis.
Recently, non-neuronal brain cells such as astrocytes, microglia and brain endothelial cells have also been shown to be susceptible to WNV infection (Cheeran et al., 2005; van Marle et al., 2007). Presence of WNV antigen has been recently demonstrated in the astrocytes of WNV-infected human brain tissue (van Marle et al., 2007). In our study, the WNV infection kinetics seen in HBCA cells is similar to the results observed in the previous studies of WNV infection in astrocytes (Cheeran et al., 2005; van Marle et al., 2007). Increased MMP-9 activity has been recently reported in the CSF of WNV-infected patients as well as in mice brain (Wang et al., 2008). However, the question remains, what is the specific cell source of MMPs in the brain and their precise role in modulating BBB tight junction properties. In other neuroinflammatory diseases such as MS, cerebral ischemia, Alzheimer’s disease and virus infections such as HIV, human herpesvirus-8 (HHV-8), and HTLV, increase in MMP-9 has been reported in brain endothelial cells (Chaudhuri et al., 2008; Conant et al., 1999; Cunningham, Wetzel, and Rosenberg, 2005; Ottino et al., 2004; Qian et al., 2007; Rosenberg, 1995; Rosenberg and Yang, 2007; Sellner et al., 2006; Shigemori et al., 2006; Sporer et al., 2000). As compared to these disease scenarios, our data demonstrate no alterations in the expression of MMPs or TIMPs in WNV-infected HBMVE cells and neuronal cells (data not shown). This observation, in context with our previous data indicating either no degradation of any TJP (Verma et al., 2009) or no significant induction of inflammatory cytokines (data not shown) in WNV-infected HBMVE cells suggest that specific host defense response of HBMVE cells to WNV infection most likely is protective in-vivo and might provide a barrier to the WNV-CNS entry, which fails in-vivo due to the neuroinflammatory mediators secreted by other cells.
Activation of glial cells is a key pathogenic feature of WNV-associated meningoencephalitis (Cheeran et al., 2005; Diniz et al., 2006; van Marle et al., 2007). Recent data by Marle et al. provide direct evidence for the role of neurotoxic mediators released by WNV-infected astrocytes in causing neuronal cell death (van Marle et al., 2007). Both, in-vitro and in-vivo, resident astrocytes have been demonstrated to respond to neuropathological injuries and undergo functional changes including increased production of MMPs (Anthony et al., 1997; Chaudhuri et al., 2008; Leppert et al., 2001; Rosenberg, 2002). In this report, the peak increase in both mRNA expression as well as enzyme activities detected between days 2 to 4 after infection when the virus titers were also high strongly suggest that MMP production is triggered by virus replication, not merely by virus entry. The activities of MMPs are tightly controlled at the level of mRNA expression, zymogen activation and an imbalance in the ratio between TIMP and MMP which determines the extent of the degradation of the extra cellular matrix (Cunningham, Wetzel, and Rosenberg, 2005). Our data demonstrating elevated levels of MMP-1, -3 and -9 along with reduced level of TIMP-2 in WNV-infected HBCA cells indicate a clear imbalance in their ratios.
Another observation in our data was that WNV induced multiple MMPs in HBCA cells. Even though the exact role of individual MMPs is unclear, it is likely that MMP-1 and -3 also contribute to the BBB injury either by directly digesting TJP or by activating pro-MMP-9 to active MMP-9. Several novel roles of MMP-1 and -3 have been described recently such as triggering neuroinflammation (Mun-Bryce et al., 2002; Suzuki et al., 2007), apoptosis (Kim et al., 2005) and direct activation of pro-MMP-9 (Conant et al., 2002). Based on our data demonstrating increase in MMP-3 and decrease MMP-2 activity, we speculate that the activation of MMP-9 in WNV-infected astrocytes is via MMP-3.
The kinetics of TJP expression during WNV infection has not been characterized. In this report, we for the first time demonstrate that degradation of TJP of HBMVE cells is mediated by MMPs derived from WNV-infected astrocytes. In addition, we have used a BBB model to demonstrate that the loss of TJP by MMPs directly results in decreased TEER and increased FITC-dextran transmigration representing loss of membrane integrity. In-vitro BBB models are routinely used to asses the effect of virus infections or transmigration of virus-infected immune cells on the BBB permeability (Kanmogne et al., 2007; Mahajan et al., 2008; Persidsky et al., 1997). Such degradation of TJP by MMPs released by infected macrophages and glial cells has been shown in infections with other neurotropic viruses such as HIV (Johnston et al., 2001; Sporer et al., 2000). Similarly, decreased degradation of collagen IV, an important component of the basement membrane in WNV-infected MMP-9−/− mice as compared to wild type mice was observed by Wang et al. (Wang et al., 2008). Based on these observations, we believe that in vivo, these WNV-induced MMPs derived from astrocytes contribute to BBB disruption by degrading components of both basement membranes as well as tight junctions ultimately resulting in infiltration of virus and immune cells into the CNS.
TNF-α is another well known mediator of BBB disruption and responds significantly to WNV infection (Cheng, King, and Kesson, 2004; Wang et al., 2004). In HBCA cells too, WNV infection significantly induces TNF-α from days 1 to 4 after infection (data not shown). However, our data suggesting the role of HBCA-derived MMPs in directly disrupting the tight junctions is further strengthened by the fact that GM6001 can efficiently block the TJP degradation and BBB disruption. GM6001 is a broad-spectrum MMP inhibitor and is routinely used in-vitro as well as in-vivo models to understand their pathogenic roles (Chen et al., 2006; Zozulya et al., 2007). Treatment of mice with GM6001 (Ilomastat) has shown to reduce/delay BBB disruption and neuroinflammation resulting in improved disease outcome of several neurodegenerative disease models such as cerebral ischemia and infection with HIV and Semliki forest virus (Amantea et al., 2007; Johnston et al., 2001; Keogh et al., 2003). In addition, LPS-induced disruption of the BBB is reported to be reversed significantly in the presence of MMP inhibitors including GM6001 (Mun-Bryce and Rosenberg, 1998; Rosenberg, Estrada, and Mobashery, 2007). Due to the ability of MMP inhibitors to improve disease symptoms, specific synthetic MMP inhibitors have been used or proposed as short term supporting therapies for acute inflammatory diseases such as MS, BM and vascular inflammatory diseases (Leppert et al., 2001; Mandal et al., 2003; Yong et al., 2007).
In summary, our results emphasize that complex neuroinflammatory responses associated with WNV infection in the CNS are mediated by astrocytes as one of the key players, and involves up-regulation of several MMPs. Though WNV-induced TJP degradation remains to be validated in-vivo mouse model, we conclude that MMPs-induced perturbations in the TJP expression might be an important pathway in WNV-induced BBB injury in-vivo, resulting in complex downstream pathological events such as the unrestricted entry of infected and/or activated inflammatory and immune cells as ‘Trojan horse’ into the CNS. The ability of GM6001 to rescue the BBB permeability is encouraging and the therapeutic use of MMP inhibitors to block WNV-associated BBB injury, inflammation and CNS-infiltration of infected or activated immune cells in-vivo is warranted.
Materials and methods
Cells, virus and plaque assay
Primary HBCA cells were obtained from ACBRI (Kirkland, WA) and propagated in the astrocyte media (ATCC) while HBMVE cells were grown as described previously (Chapagain et al., 2007). Only early passage primary HBCA and HBMVE cells (passage 6 to 9) were used for all experiments. WNV strain NY99 was used for all infection experiments at MOI-5 as described previously (Verma et al., 2009; Verma et al., 2008). Production of infectious virus in the supernatant was determined by plaque assay (Verma et al., 2008).
qRT-PCR analysis
cDNA prepared from RNA extracted from WNV-, UV-WNV-, and mock-infected HBCA and HBMVE cells were used for qRT-PCR as described previously (Verma et al., 2009; Verma et al., 2008; Verma et al., 2006). Primer sequences and annealing temperatures used for amplification of MMPs and TIMP are described in Table 1.
Table 1.
Primer sequences used for qRT-PCR
| Gene | Primer Sequence (5′-3′) | Amplicon | |
|---|---|---|---|
| GenBank No. | (bp) | Tm (°C) | |
| MMP1 | |||
| NM_002421 | |||
| Forward | ATGAAGCAGCCCAGATGTG | 98 | 55 |
| Reverse | TCAATCCTGTAGGTCAGATGTG | ||
|
| |||
| MMP2 | |||
| NM_004530 | |||
| Forward | TGCTGAAGGACACACTAAAGAAG | 147 | 55 |
| Reverse | CTTGCGAGGGAAGAAGTTGTAG | ||
|
| |||
| MMP3 | |||
| NM_002422 | |||
| Forward | TCCTGGCATCCCGAAGTG | 146 | 57 |
| Reverse | AGCCTGGAGAATGTGAGTGG | ||
|
| |||
| MMP9 | |||
| NM_004994 | |||
| Forward | GCCACTTCCCCTTCATCTTCG | 168 | 56 |
| Reverse | ATTGCCGTCCTGGGTGTAGAG | ||
|
| |||
| TIMP-1 | |||
| M12670 | |||
| Forward | TGTTGGCTGTGAGGAATGCA | 84 | 55 |
| Reverse | GGTCCGTCCACAAGCAATG | ||
|
| |||
| TIMP-2 | |||
| NM_003255 | |||
| Forward | CGTTGGAGGAAAGAAGGAATATC | 86 | 55 |
| Reverse | GCACGATGAAGTCACAGAGG | ||
|
| |||
| TIMP-3 | |||
| U67195 | |||
| Forward | GCGTCTATGATGGCAAGATG | 148 | 55 |
| Reverse | AAGCAAGGCAGGTAGTAGC | ||
Western immunoblot analysis
Total 40–50 μg of cellular protein extracted from mock- and infected-HBCA cells at days 2 to 4 after infection was used for western blotting (Verma et al., 2009; Verma et al., 2008). The membranes were incubated with antibodies against rabbit polyclonal anti-MMP-1 (1 mg/mL, Chemicon), mouse polyclonal anti-MMP-3 (1:1000, Calbiochem) and rabbit polyclonal anti-MMP-9 (1:1000, Abcam) followed by HRP-conjugated secondary antibodies, and were developed using ECL detection kit and analyzed by Bio-Rad Quantity one Program.
MMP-3 and -9 enzymes activity analysis by zymography and ELISA
At days 3 and 4 after infection, the supernatant medium from HBCA cells was collected for zymography and ELISA assays. For zymography, the supernatant was concentrated 10-fold using Microcon centrifugal filter units (Millipore), electrophoresed on 10% SDS-polyacrylamide gels containing 1 mg/mL gelatin or casein as a protease substrate and the gelatinolytic bands were determined as described previously (Chua et al., 2003). ELISA for total MMP-9 in the supernatant from mock- and WNV-infected HBCA cells at day 3 after infection was performed using Quantikine human MMP-9 ELISA kit according to the manufacturers’ instructions (R&D systems), and a standard curve was generated in the range of 0 to 20 ng/mL. The plates were read using a Victor 3 microtiter reader equipped with Workout 1.5 software (Perkin Elmer).
Treatment of HBMVE cells with supernatant of HBCA cells and cytotoxicity assays
HBMVE cells grown on coverslips were incubated for six hours with supernatant from aforementioned mock- and infected HBCA cells. Briefly 250 μL of the supernatant from HBCA cells was mixed with 250 μL of the CSC media of HBMVE cells and applied to naïve HBMVE cells. HBCA media collected on days 3 and 4 after infection were treated with broad-spectrum MMP inhibitor GM6001 (25 μM/mL, Calbiochem) for 15 min at room temperature before applying to naïve HBMVE cells. Following incubation, the cells were washed twice with 1X PBS and fixed with 4% PFA. Similar treatments were performed on HBMVE cells grown in 96-well plate and cell viability was assayed as described previously (Verma et al., 2008).
Immunocytochemical analysis
Fixed HBCA cells were immunostained using monoclonal anti-WNV envelope (1:800), polyclonal anti- GFAP, (1:100, Chemicon) or anti-MMP-9 (1:100) antibodies followed by secondary antibodies conjugated with Alexa Fluor 546 (GFAP and WNV) or FITC (MMP-9). HBMVE cells with or without treatment with supernatant from HBCA cells were immunostained with monoclonal mouse anti-ZO-1 or polyclonal rabbit anti-claudin-1 (Verma et al., 2009).
Development of BBB model and WNV infection
An in-vitro monolayer BBB model comprised of HBMVE cells was developed on a BioCoat® Cell Environment™ Human Fibronectin PET (polyethylene terephthalate) insert with 3.0 μm pore size in a 24-well plate (BD Bioscience, Bedford, MA) as described previously (Verma et al., 2009). The integrity of the in-vitro BBB model was determined every day, as described previously (Verma et al., 2009). The FITC-dextran (4-KDa MW) transmigration across the inserts was calculated as percentage of the total amount added in the upper well (10 μg/100 μL) and the TEER was measured using EVOMX and was expressed as Ω/cm2. At day 8 after seeding, upper and lower chambers of the BBB models were incubated with 400 μL of the supernatant from HBCA cells collected at day 4 after infection in the presence or absence of GM6001 (25 nM/mL). After six hours of incubation, the models were washed twice with 1X PBS and the BBB integrity was assayed by TEER and FITC-dextran transmigration assay. For positive control, the inserts were treated with synthetic MMP-2/9 enzyme (10 μg/membrane).
Statistical analysis
Data are reported as mean ± standard error of mean of at least three independent experiments. Unpaired student t-test using GraphPad prism 5.0 (GraphPad software) was used to compare the values of arbitrary units of densitometry scans and permeability assays.
Acknowledgments
This work was partially supported by grants from the Hawaii Community Foundation (20050405), Research Centers in Minority Institutions Program (G12RR003061) and Centers of Biomedical Research Excellence (P20RR018727), National Center for Research Resources, National Institutes of Health. We thank Dr. Duane J. Gubler for the generous gift of the WNV strain NY99 and the monoclonal human anti-WNV envelope antibody. We also thank Dr. Kathy Conant for valuable discussions related to MMPs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afonso PV, Ozden S, Prevost MC, Schmitt C, Seilhean D, Weksler B, Couraud PO, Gessain A, Romero IA, Ceccaldi PE. Human blood-brain barrier disruption by retroviral-infected lymphocytes: role of myosin light chain kinase in endothelial tight-junction disorganization. J Immunol. 2007;179(4):2576–83. doi: 10.4049/jimmunol.179.4.2576. [DOI] [PubMed] [Google Scholar]
- Amantea D, Russo R, Gliozzi M, Fratto V, Berliocchi L, Bagetta G, Bernardi G, Corasaniti MT. Early upregulation of matrix metalloproteinases following reperfusion triggers neuroinflammatory mediators in brain ischemia in rat. Int Rev Neurobiol. 2007;82:149–69. doi: 10.1016/S0074-7742(07)82008-3. [DOI] [PubMed] [Google Scholar]
- Anthony DC, Ferguson B, Matyzak MK, Miller KM, Esiri MM, Perry VH. Differential matrix metalloproteinase expression in cases of multiple sclerosis and stroke. Neuropathol Appl Neurobiol. 1997;23(5):406–15. [PubMed] [Google Scholar]
- Arjona A, Foellmer HG, Town T, Leng L, McDonald C, Wang T, Wong SJ, Montgomery RR, Fikrig E, Bucala R. Abrogation of macrophage migration inhibitory factor decreases West Nile virus lethality by limiting viral neuroinvasion. J Clin Invest. 2007a;117(10):3059–66. doi: 10.1172/JCI32218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arjona A, Ledizet M, Anthony K, Bonafe N, Modis Y, Town T, Fikrig E. West Nile Virus Envelope Protein Inhibits dsRNA-Induced Innate Immune Responses. J Immunol. 2007b;179(12):8403–9. doi: 10.4049/jimmunol.179.12.8403. [DOI] [PubMed] [Google Scholar]
- Chapagain ML, Verma S, Mercier F, Yanagihara R, Nerurkar VR. Polyomavirus JC infects human brain microvascular endothelial cells independent of serotonin receptor 2A. Virology. 2007;364(1):55–63. doi: 10.1016/j.virol.2007.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri A, Yang B, Gendelman HE, Persidsky Y, Kanmogne GD. STAT1 signaling modulates HIV-1-induced inflammatory responses and leukocyte transmigration across the blood-brain barrier. Blood. 2008;111(4):2062–72. doi: 10.1182/blood-2007-05-091207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeran MC, Hu S, Sheng WS, Rashid A, Peterson PK, Lokensgard JR. Differential responses of human brain cells to West Nile virus infection. J Neurovirol. 2005;11(6):512–24. doi: 10.1080/13550280500384982. [DOI] [PubMed] [Google Scholar]
- Chen KM, Liu JY, Lai SC, Hsu LS, Lee HH. Association of plasminogen activators and matrix metalloproteinase-9 proteolytic cascade with blood-CNS barrier damage of angiostrongyliasis. Int J Exp Pathol. 2006;87(2):113–9. doi: 10.1111/j.0959-9673.2006.00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, King NJ, Kesson AM. The role of tumor necrosis factor in modulating responses of murine embryo fibroblasts by flavivirus, West Nile. Virology. 2004;329(2):361–70. doi: 10.1016/j.virol.2004.06.050. [DOI] [PubMed] [Google Scholar]
- Chua PK, Melish ME, Yu Q, Yanagihara R, Yamamoto KS, Nerurkar VR. Elevated levels of matrix metalloproteinase 9 and tissue inhibitor of metalloproteinase 1 during the acute phase of Kawasaki disease. Clin Diagn Lab Immunol. 2003;10(2):308–14. doi: 10.1128/CDLI.10.2.308-314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant K, Haughey N, Nath A, St Hillaire C, Gary DS, Pardo CA, Wahl LM, Bilak M, Milward E, Mattson MP. Matrix metalloproteinase-1 activates a pertussis toxin-sensitive signaling pathway that stimulates the release of matrix metalloproteinase-9. J Neurochem. 2002;82(4):885–93. doi: 10.1046/j.1471-4159.2002.01038.x. [DOI] [PubMed] [Google Scholar]
- Conant K, McArthur JC, Griffin DE, Sjulson L, Wahl LM, Irani DN. Cerebrospinal fluid levels of MMP-2, 7, and 9 are elevated in association with human immunodeficiency virus dementia. Ann Neurol. 1999;46(3):391–8. doi: 10.1002/1531-8249(199909)46:3<391::aid-ana15>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Cunningham LA, Wetzel M, Rosenberg GA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia. 2005;50(4):329–39. doi: 10.1002/glia.20169. [DOI] [PubMed] [Google Scholar]
- Diamond MS, Klein RS. West Nile virus: crossing the blood-brain barrier. Nat Med. 2004;10(12):1294–5. doi: 10.1038/nm1204-1294. [DOI] [PubMed] [Google Scholar]
- Diamond MS, Shrestha B, Mehlhop E, Sitati E, Engle M. Innate and adaptive immune responses determine protection against disseminated infection by West Nile encephalitis virus. Viral Immunol. 2003;16(3):259–78. doi: 10.1089/088282403322396082. [DOI] [PubMed] [Google Scholar]
- Diniz JA, Da Rosa AP, Guzman H, Xu F, Xiao SY, Popov VL, Vasconcelos PF, Tesh RB. West Nile virus infection of primary mouse neuronal and neuroglial cells: the role of astrocytes in chronic infection. Am J Trop Med Hyg. 2006;75(4):691–6. [PubMed] [Google Scholar]
- Gearing AJ, Beckett P, Christodoulou M, Churchill M, Clements JM, Crimmin M, Davidson AH, Drummond AH, Galloway WA, Gilbert R, et al. Matrix metalloproteinases and processing of pro-TNF-alpha. J Leukoc Biol. 1995;57(5):774–7. doi: 10.1002/jlb.57.5.774. [DOI] [PubMed] [Google Scholar]
- Hayes EB, Gubler DJ. West Nile virus: epidemiology and clinical features of an emerging epidemic in the United States. Annu Rev Med. 2006;57:181–94. doi: 10.1146/annurev.med.57.121304.131418. [DOI] [PubMed] [Google Scholar]
- Hu J, Van den Steen PE, Sang QX, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov. 2007;6(6):480–98. doi: 10.1038/nrd2308. [DOI] [PubMed] [Google Scholar]
- Hummel V, Kallmann BA, Wagner S, Fuller T, Bayas A, Tonn JC, Benveniste EN, Toyka KV, Rieckmann P. Production of MMPs in human cerebral endothelial cells and their role in shedding adhesion molecules. J Neuropathol Exp Neurol. 2001;60(4):320–7. doi: 10.1093/jnen/60.4.320. [DOI] [PubMed] [Google Scholar]
- Johnatty RN, Taub DD, Reeder SP, Turcovski-Corrales SM, Cottam DW, Stephenson TJ, Rees RC. Cytokine and chemokine regulation of proMMP-9 and TIMP-1 production by human peripheral blood lymphocytes. J Immunol. 1997;158(5):2327–33. [PubMed] [Google Scholar]
- Johnston JB, Zhang K, Silva C, Shalinsky DR, Conant K, Ni W, Corbett D, Yong VW, Power C. HIV-1 Tat neurotoxicity is prevented by matrix metalloproteinase inhibitors. Ann Neurol. 2001;49(2):230–41. doi: 10.1002/1531-8249(20010201)49:2<230::aid-ana43>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Kanmogne GD, Schall K, Leibhart J, Knipe B, Gendelman HE, Persidsky Y. HIV-1 gp120 compromises blood-brain barrier integrity and enhances monocyte migration across blood-brain barrier: implication for viral neuropathogenesis. J Cereb Blood Flow Metab. 2007;27(1):123–34. doi: 10.1038/sj.jcbfm.9600330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keogh B, Sheahan BJ, Atkins GJ, Mills KH. Inhibition of matrix metalloproteinases ameliorates blood-brain barrier disruption and neuropathological lesions caused by avirulent Semliki Forest virus infection. Vet Immunol Immunopathol. 2003;94(3–4):185–90. doi: 10.1016/s0165-2427(03)00082-5. [DOI] [PubMed] [Google Scholar]
- Kim YS, Kim SS, Cho JJ, Choi DH, Hwang O, Shin DH, Chun HS, Beal MF, Joh TH. Matrix metalloproteinase-3: a novel signaling proteinase from apoptotic neuronal cells that activates microglia. J Neurosci. 2005;25 (14):3701–11. doi: 10.1523/JNEUROSCI.4346-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppert D, Lindberg RL, Kappos L, Leib SL. Matrix metalloproteinases: multifunctional effectors of inflammation in multiple sclerosis and bacterial meningitis. Brain Res Brain Res Rev. 2001;36(2–3):249–57. doi: 10.1016/s0165-0173(01)00101-1. [DOI] [PubMed] [Google Scholar]
- Mahajan SD, Aalinkeel R, Sykes DE, Reynolds JL, Bindukumar B, Adal A, Qi M, Toh J, Xu G, Prasad PN, Schwartz SA. Methamphetamine alters blood brain barrier permeability via the modulation of tight junction expression: Implication for HIV-1 neuropathogenesis in the context of drug abuse. Brain Res. 2008;1203:133–48. doi: 10.1016/j.brainres.2008.01.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal M, Mandal A, Das S, Chakraborti T, Sajal C. Clinical implications of matrix metalloproteinases. Mol Cell Biochem. 2003;252(1–2):305–29. doi: 10.1023/a:1025526424637. [DOI] [PubMed] [Google Scholar]
- Mun-Bryce S, Lukes A, Wallace J, Lukes-Marx M, Rosenberg GA. Stromelysin-1 and gelatinase A are upregulated before TNF-alpha in LPS-stimulated neuroinflammation. Brain Res. 2002;933(1):42–9. doi: 10.1016/s0006-8993(02)02303-x. [DOI] [PubMed] [Google Scholar]
- Mun-Bryce S, Rosenberg GA. Gelatinase B modulates selective opening of the blood-brain barrier during inflammation. Am J Physiol. 1998;274(5 Pt 2):R1203–11. doi: 10.1152/ajpregu.1998.274.5.R1203. [DOI] [PubMed] [Google Scholar]
- Ottino P, Finley J, Rojo E, Ottlecz A, Lambrou GN, Bazan HE, Bazan NG. Hypoxia activates matrix metalloproteinase expression and the VEGF system in monkey choroid-retinal endothelial cells: Involvement of cytosolic phospholipase A2 activity. Mol Vis. 2004;10:341–50. [PubMed] [Google Scholar]
- Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. 2006;1(3):223–36. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Stins M, Way D, Witte MH, Weinand M, Kim KS, Bock P, Gendelman HE, Fiala M. A model for monocyte migration through the blood-brain barrier during HIV-1 encephalitis. J Immunol. 1997;158(7):3499–510. [PubMed] [Google Scholar]
- Petty MA, Lo EH. Junctional complexes of the blood-brain barrier: permeability changes in neuroinflammation. Prog Neurobiol. 2002;68(5):311–23. doi: 10.1016/s0301-0082(02)00128-4. [DOI] [PubMed] [Google Scholar]
- Qian LW, Xie J, Ye F, Gao SJ. Kaposi’s sarcoma-associated herpesvirus infection promotes invasion of primary human umbilical vein endothelial cells by inducing matrix metalloproteinases. J Virol. 2007;81(13):7001–10. doi: 10.1128/JVI.00016-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg GA. Matrix metalloproteinases in brain injury. J Neurotrauma. 1995;12(5):833–42. doi: 10.1089/neu.1995.12.833. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39(3):279–91. doi: 10.1002/glia.10108. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Estrada EY, Mobashery S. Effect of synthetic matrix metalloproteinase inhibitors on lipopolysaccharide-induced blood-brain barrier opening in rodents: Differences in response based on strains and solvents. Brain Res. 2007;1133(1):186–92. doi: 10.1016/j.brainres.2006.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg GA, Yang Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg Focus. 2007;22 (5):E4. doi: 10.3171/foc.2007.22.5.5. [DOI] [PubMed] [Google Scholar]
- Sellner J, Simon F, Meyding-Lamade U, Leib SL. Herpes-simplex virus encephalitis is characterized by an early MMP-9 increase and collagen type IV degradation. Brain Res. 2006;1125(1):155–62. doi: 10.1016/j.brainres.2006.09.093. [DOI] [PubMed] [Google Scholar]
- Shigemori Y, Katayama Y, Mori T, Maeda T, Kawamata T. Matrix metalloproteinase-9 is associated with blood-brain barrier opening and brain edema formation after cortical contusion in rats. Acta Neurochir Suppl. 2006;96:130–3. doi: 10.1007/3-211-30714-1_29. [DOI] [PubMed] [Google Scholar]
- Sporer B, Koedel U, Paul R, Kohleisen B, Erfle V, Fontana A, Pfister HW. Human immunodeficiency virus type-1 Nef protein induces blood-brain barrier disruption in the rat: role of matrix metalloproteinase-9. J Neuroimmunol. 2000;102(2):125–30. doi: 10.1016/s0165-5728(99)00170-8. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Nagai N, Umemura K, Collen D, Lijnen HR. Stromelysin-1 (MMP-3) is critical for intracranial bleeding after t-PA treatment of stroke in mice. J Thromb Haemost. 2007;5(8):1732–9. doi: 10.1111/j.1538-7836.2007.02628.x. [DOI] [PubMed] [Google Scholar]
- van Marle G, Antony J, Ostermann H, Dunham C, Hunt T, Halliday W, Maingat F, Urbanowski MD, Hobman T, Peeling J, Power C. West Nile virus-induced neuroinflammation: glial infection and capsid protein-mediated neurovirulence. J Virol. 2007;81(20):10933–49. doi: 10.1128/JVI.02422-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, Lo Y, Chapagain M, Lum S, Kumar M, Gurjav U, Luo H, Nakatsuka A, Nerurkar VR. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: Transmigration across the in vitro blood-brain barrier. Virology. 2009;385:425–433. doi: 10.1016/j.virol.2008.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, Molina Y, Lo YY, Cropp B, Nakano C, Yanagihara R, Nerurkar VR. In vitro effects of selenium deficiency on West Nile virus replication and cytopathogenicity. Virol J. 2008;5:66. doi: 10.1186/1743-422X-5-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, Ziegler K, Ananthula P, Co JK, Frisque RJ, Yanagihara R, Nerurkar VR. JC virus induces altered patterns of cellular gene expression: interferon-inducible genes as major transcriptional targets. Virology. 2006;345(2):457–67. doi: 10.1016/j.virol.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Wang P, Dai J, Bai F, Kong KF, Wong SJ, Montgomery RR, Madri JA, Fikrig E. Matrix metalloproteinase 9 facilitates west nile virus entry into the brain. J Virol. 2008;82(18):8978–85. doi: 10.1128/JVI.00314-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10(12):1366–73. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- Yong VW, Zabad RK, Agrawal S, Goncalves Dasilva A, Metz LM. Elevation of matrix metalloproteinases (MMPs) in multiple sclerosis and impact of immunomodulators. J Neurol Sci. 2007;259(1–2):79–84. doi: 10.1016/j.jns.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Zozulya AL, Reinke E, Baiu DC, Karman J, Sandor M, Fabry Z. Dendritic cell transmigration through brain microvessel endothelium is regulated by MIP-1alpha chemokine and matrix metalloproteinases. J Immunol. 2007;178(1):520–9. doi: 10.4049/jimmunol.178.1.520. [DOI] [PMC free article] [PubMed] [Google Scholar]