

Abstract
We report a new strategy based on mercury cation exchange in nonpolar solvents to prepare bright and compact alloyed quantum dots (QDs) (HgxCd1-xE, where E = Te, Se, or S) with equalized particle size and broadly tunable absorption and fluorescence emission in the near infrared. In particular, we focus on HgxCd1-xTe nanocrystals because cubic CdTe and HgTe have nearly identical lattice constants, but very different bandgap energies and electron/hole masses. Thus, replacement of Cd2+ by Hg2+ in CdTe nanocrystals does not change the particle size, but it greatly alters the bandgap energy. After capping with a multilayer shell and solubilization with a multidentate ligand, this class of cation-exchanged QDs are compact (6.5 nm nanocrystal size and 10 nm hydrodynamic diameter) and very bright (60–80% quantum yield), with narrow and symmetric fluorescence spectra tunable across the wavelength range from 700 nm to 1150 nm.
Semiconductor nanocrystals with near-infrared (NIR) absorption and emission spectra are currently the subject of intensive research and development for biomedical imaging and diagnostics applications.1,2 In comparison with visible quantum dots (QDs), NIR nanocrystals with emission spectra in the 700–1700 nm spectral range offer several advantages such as improved tissue penetration, lower background interference, and reduced photochemical damage. To date, several strategies have been explored to synthesize NIR QDs, including the use of narrow-bandgap materials (CdSe1-xTex, InAs, PbS, Cd3P2, and CuInS2),3 charge carrier separation by staggering band offsets (type-II QDs),2,4 and lattice-mismatch strain tuning.5,6 However, the resulting NIR nanocrystals have found only limited applications in biology and medicine due to their large sizes, broad emission spectra, low quantum yields, or poor photostability. In addition, traditional multicolor QDs have very different physical dimensions (by a factor of 3–4 in diameter, and by a factor of 27–64 in volume), leading to large variations in signal brightness, steric hindrance, and binding kinetics in biological environments.7
Here we report a strategy based on mercury cation exchange to prepare bright and compact HgxCd1-xTe and related II–VI QDs with equalized particle size and tunable NIR fluorescence emission. The central rationale for this approach is that cubic CdTe and HgTe have nearly identical lattice constants (aCdTe = 6.48 Å, aHgTe = 6.46 Å), but very different bandgap energies and electron/hole masses. The bulk bandgap energy of CdTe is 1.5 eV whereas that of HgTe is −0.15 eV (a near-zero bandgap in which the bottom of the conduction band is almost aligned with the top of the valence band). Thus, starting with CdTe seed nanocrystals, substitution of Cd2+ by Hg2+ does not change the lattice parameter or the particle size, but this cation substitution reaction greatly alters the bandgap energy. As shown schematically in Figure 1, this strategy leads to constant-sized QDs with optical absorption and emission spectra that are tunable by the extent of cation exchange. Due to the large exciton Bohr diameter (90 nm) of binary HgTe and the strong quantum confinement for the alloyed HgxCd1-xTe QDs, the mercury exchanged nanocrystals have substantially wider bandgaps than their bulk counterparts. As a result, their electronic absorption and fluorescence emission spectra are not infinitely shifted to the infrared (as might be expected for nanocrystals with a nearly zero bandgap). We further show that this new class of cation-exchanged QDs can be capped with a multilayer shell for high quantum yield and photostability, and also can be solubilized in water by using low-molecular-weight multidentate ligands. The capped and solubilized QDs are compact (6.5 nm in core/shell crystal size and 10 nm in hydrodynamic diameter) and very bright (fluorescence quantum yield of 60–80%), with narrow and symmetric fluorescence spectra tunable between 700–1150 nm. This result is a significant improvement from the ill-defined NIR absorption and emission spectra of HgCdTe nanocrystals reported previously.9
Figure 1.
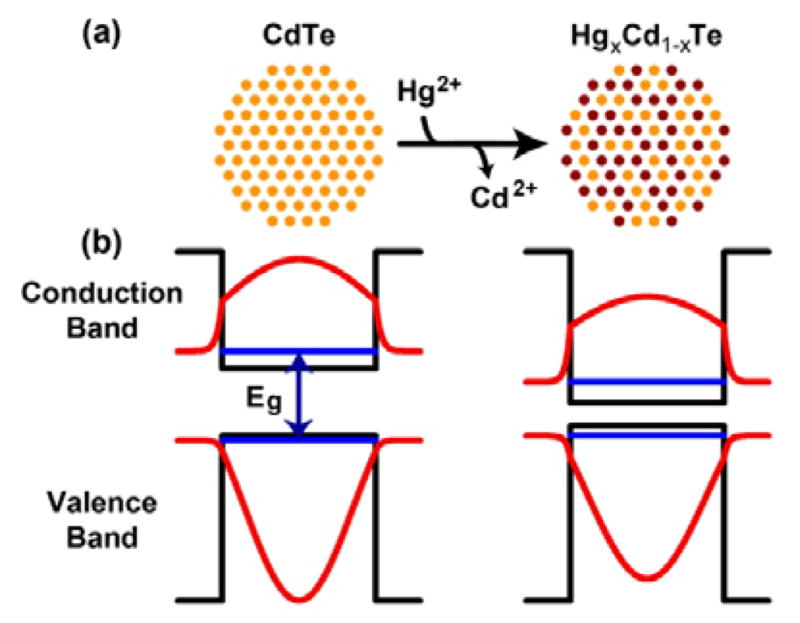
Structure and energy band diagrams of CdTe and cation-exchanged HgxCd1-xTe QDs. (a) Schematic illustration of Cd2+ replacement by Hg2+. (b) Diagrams depicting the potential energy wells (black lines), quantum-confined kinetic energy levels (blue lines), and wavefunctions (red) of electrons and holes in CdTe and HgxCd1-xTe QDs, as calculated using the effective mass approximation.8
In contrast to the “wet” aqueous synthesis method used by Rogach, Weller, and coworkers,9 we have developed a new method to synthesize CdTe seed nanocrystals and HgxCd1-xTe alloyed QDs in entirely nonpolar organic solvents. We find that this nonpolar-solvent approach permits unmatched control over the nanocrystal growth process (with narrow size distribution and high crystallinity) as well as the overgrowth of crystalline and coherent capping shells. Brightly fluorescent and monodisperse CdTe core nanocrystals were first synthesized by using cadmium oxide and tellurium in a high temperature mixture consisting of oleylamine, trioctylphosphine, tetradecylphosphonic acid, and octadecene.5,10 After purification, the nanoparticles were subject to Hg2+ cation exchange in chloroform (see Supporting Information). This resulted in a partial exchange of Cd2+ in the nanocrystal lattice by Hg2+, leaving the anion lattice unchanged. A critical factor for the success of this mercury-exchange reaction in nonpolar solvents is the use of mercury octanethiolate as a precursor. We have studied various mercury complexes including carboxylates, phosphonates, and other thiolates, and have found that the octanethiolate precursor provides the best results for controlling the extent of mercury exchange and for ensuring that the exchange reaction is homogeneous across the nanocrystal populations. After cation exchange, transmission electron microscopy (TEM) reveals that the particle size indeed does not change (see Figure 2), while energy dispersive X-ray spectroscopy confirms the incorporation of mercury and depletion of cadmium in the nanocrystal lattice (see Supporting Information). Significantly, the cadmium-to-mercury exchange strategy is not limited to HgxCd1-xTe, but can be readily extended to other mercury-containing nanomaterials such as HgxCd1-xSe and HgxCd1-xS, as described in more details in Supporting Information. Previous work by Alivisatos and coworkers11 has exploited the use of cation exchange to synthesize nanocrystals of various shapes and compositions but mercury cation exchange was not demonstrated, most likely due to the difficulties in controlling the chemical reactivity of mercury precursors. The reaction kinetics of mercury compounds can be better controlled in aqueous solution, but the resulting core-shell and alloyed QDs have poor optical properties as judged by their broad and ill-defined absorption and emission spectra.9
Figure 2.
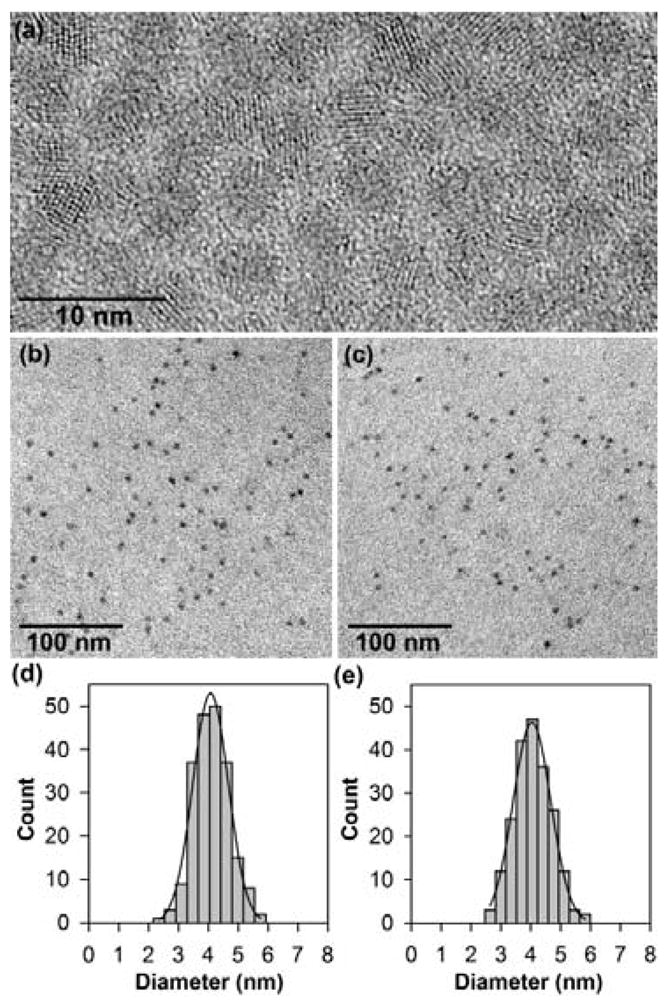
(a) High-resolution transmission electron micrograph showing the high crystallinity of 4.0 nm CdTe nanocrystals, which have a primarily cubic lattice structure.6 (b, c) TEM micrographs of CdTe (left) and Hg0.61Cd0.39Te (right) showing constant crystalline sizes before and after cation exchange. (d, e) Particle size histogram data of CdTe (left) and Hg0.61Cd0.39Te (right) showing that the particle size distributions do not change after cation exchange.
In contrast, narrow and well-defined absorption and photoluminescence spectra are obtained from HgxCd1-xTe QDs synthesized by mercury cation exchange in nonpolar solvents. As shown in Figure 3 for two size series (2.3 nm and 4.0 nm), the absorption and emission spectra are both shifted toward the NIR as the extent of mercury exchange increases (increasing x), while the particles maintain their original sizes. Thus, the particle size and the degree of cation exchange can be independently controlled to achieve broad tuning of the photoluminescence peak across the wavelength range of 500 nm to 1100 nm (Figure 3b and d). Also, the cation-exchanged QDs exhibit discrete electronic transitions and symmetric fluorescence peaks without deep trap emission. The overall shapes of the absorption spectra for CdTe and HgxCd1-xTe nanocrystals are quite similar, indicating that these particles are nearly homogenous alloys without a core/shell or gradient-type structure. This is corroborated by the absence of a high-temperature alloying point that was observed in CdSe/ZnSe nanocrystals.12
Figure 3.
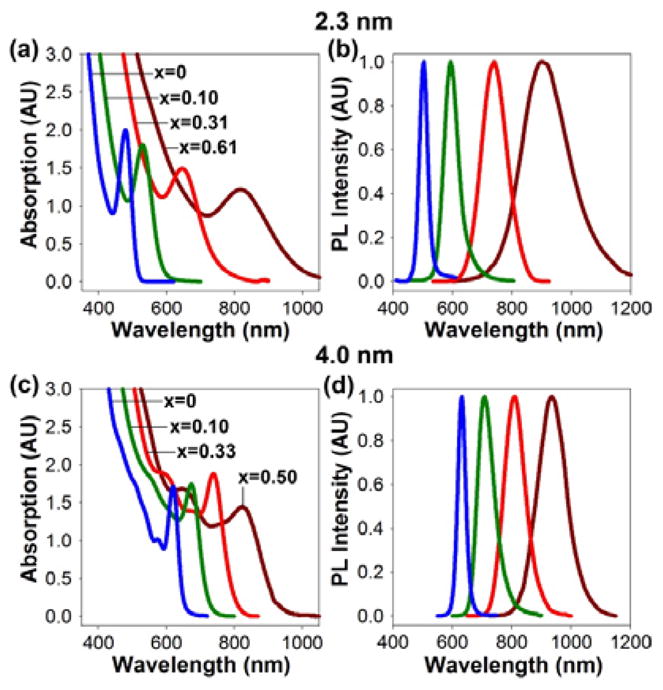
Absorption spectra (left panels) and photoluminescence (PL) spectra (right panels) of CdTe and cation-exchanged HgxCd1-xTe QDs for two nanocrystal sizes with a series of chemical compositions, indicated by the values of x in the absorption spectra. The compositions were determined quantitatively by inductively coupled plasma mass spectrometry.
It is also worth noting that that CdTe cores with wider size distributions result in a proportional increase in peak width after mercury alloying, apparently due to inhomogeneous broadening in both particle size and composition. In addition, the line widths are strongly dependent on the nanocrystal size, with larger particles consistently yielding narrower peaks (compare Figure 3b and 3d). The 4.0 nm cation-exchanged QDs show spectral widths of 40–50 nm full-width at half-maximum (FWHM) in the wavelength range of 700–800 nm (after capping), which are much narrower than that of other NIR QDs such as CdTe/CdSe nanocrystals (70–80 nm FWHM), InAs (80–90 nm), Cd3P2 (75–100 nm) and I-III-VI materials such as CuInS2 (100–140 nm).3,4,9
For potential biomedical applications, we have developed a method to cap the cation-exchanged QDs with a multilayer shell (see Figure 4a). A monolayer of CdTe is first deposited on the nanocrystal using dimethylcadmium and trioctylphosphine telluride, followed by three monolayers of CdxZn1-xS using dimethylcadmium, diethylzinc, and sulfur. An important finding is that a CdTe interim layer is critical for stabilizing the optical properties; without this layer, direct deposition of CdS or ZnS results in a dramatic red-shift in the photoluminescence peak. This is likely caused by tensile strain in the CdxZn1-xS shell, which decreases the conduction band energy and results in the formation of a type-II interface, as observed in CdTe/CdS and CdTe/ZnS materials.5 With the inclusion of a CdTe interim layer, carrier electrons are insulated from the CdxZn1-xS shell, and only a small red shift is observed during shell growth.
Figure 4.
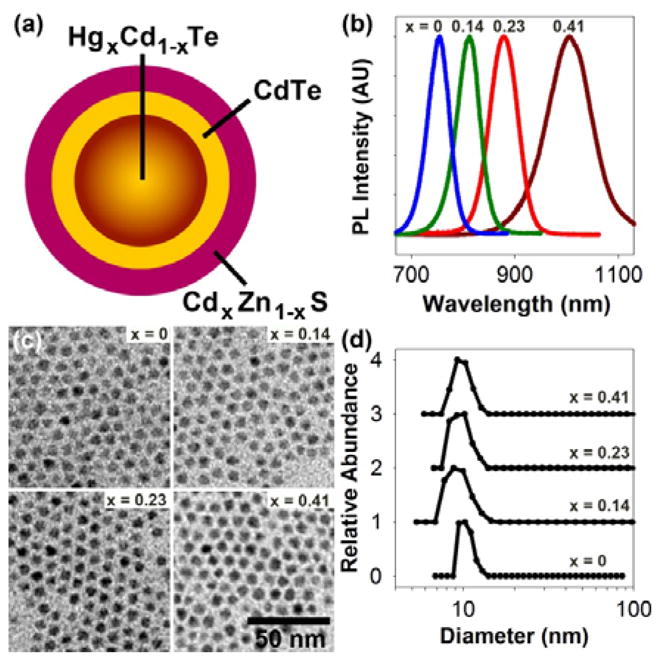
(a) Schematic diagram of a cation-exchanged HgxCd1-xTe QD capped with a multilayer shell. (b) Photoluminescence spectra of 3.5 nm HgxCd1-xTe QDs capped with CdTe and CdxZn1-xS with tunable emission between 700 nm and 1100 nm. (c) Electron micrographs of the capped QDs with the same particle size. (d) Dynamic light scattering data showing that the multicolor QDs have similar hydrodynamic sizes (approximately 10 nm) after solubilization by using multidentate polymer ligands. Chemical compositions (x) of the HgxCd1-xTe cores are indicated in (b), (c), and (d).
After capping, the fluorescence emission of the cation-exchanged QDs can be tuned between 700–1150 nm, with quantum yields as high as near 80% at room temperature. This high quantum efficiency is likely a result of two factors: (i) the high crystallinity of the initial QD cores, which also have fluorescence quantum yields over 80%; and (ii) the high crystallinity of the epitaxial shell, which strongly confines the charge carriers to the core materials.5 It is worth noting that immediately after cation exchange, the HgxCd1-xTe nanocrystals often have a substantially reduced quantum yield, sometimes less than 1%, but the efficiency is nearly entirely recovered after shell growth. This effect is not surprising because the cation exchange process modifies the particle surface, which has previously been shown to quench the fluorescence of semiconductor nanocrystals.13
Figure 4b shows the fluorescence spectra of a series of cation-exchanged and capped QDs prepared from 3.5 nm CdTe cores. TEM reveals that all of these particles are similar in crystal size (~ 6.5 nm) (Figure 4c). After water solubilization with a multidentate polymeric ligand,14 these nanoparticles remain bright and compact, with quantum yields over 60% and hydrodynamic sizes around 10 nm (Figure 4d). In comparison with current NIR QDs (often with elongated shapes and hydrodynamic sizes of 20–30 nm), the cation-exchanged NIR dots are 2–3-fold smaller and are expected to show improved binding kinetics for both live cell imaging and in-vitro diagnostic applications. In terms of potential toxicity, divalent mercury has been reported to have a similar or lower acute toxicity profile in comparison with divalent cadmium,15 but work is in progress to evaluate the chemical stability and cellular toxicity of cation-exchanged QDs relative to traditional core-shell particles.
In conclusion, we have reported a cation-exchange strategy in nonpolar solvents to prepare bright and compact HgxCd1-xTe and related II–VI QDs with equalized particle size and tunable NIR fluorescence emission. After capping with a multilayer shell and solubilization with multidentate polymers, the cation-exchanged QDs offer a number of attractive properties including high quantum yield, narrow spectral width, broad wavelength tunability, and an overall compact size. This class of bright and compact QDs is thus well suited for multiplexed imaging and detection in the NIR optical window with improved tissue penetration and detection sensitivity.
Supplementary Material
Acknowledgments
This work was supported by NIH grants (PN2EY018244, R01 CA108468, and U54CA119338). S.M.N is a Distinguished Scholar of the Georgia Cancer Coalition (GCC). A. M. S. acknowledges the NCI Nano-Alliance Program for a Pathway to Independence Award (1K99CA154006-01) and also the Emory-Georgia Tech Cancer Nanotechnology Center for fellowship support.
Footnotes
Supporting Information Available: Detailed methods, UV-Vis absorption and fluorescence spectra, TEM micrographs, and energy dispersive x-ray data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lim YT, Kim S, Nakayama A, Stott NE, Bawendi MG, Frangioni JV. Mol Imaging. 2003;2:50–64. doi: 10.1162/15353500200302163. [DOI] [PubMed] [Google Scholar]; Gao XH, Cui YY, Levenson RM, Chung LWK, Nie SM. Nat Biotechnol. 2004;22:969–976. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]; Smith AM, Mancini MC, Nie SM. Nat Nanotech. 2009;4:710–711. doi: 10.1038/nnano.2009.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim S, Lim YT, Soltesz EG, De Grand AM, Lee J, Nakayama A, Parker JA, Mihaljevic T, Laurence RG, Dor DM, Cohn LH, Bawendi MG, Frangioni JV. Nat Biotechnol. 2004;22:93–97. doi: 10.1038/nbt920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bailey RE, Nie SM. J Am Chem Soc. 2003;125:7100–7106. doi: 10.1021/ja035000o. [DOI] [PubMed] [Google Scholar]; Aharoni A, Mokari T, Popov I, Banin U. J Am Chem Soc. 2006;128:257–264. doi: 10.1021/ja056326v. [DOI] [PubMed] [Google Scholar]; Allen PM, Liu W, Chauhan VP, Lee J, Ting AY, Fukumura D, Jain RK, Bawendi M. J Am Chem Soc. 2010;132:470–471. doi: 10.1021/ja908250r. [DOI] [PMC free article] [PubMed] [Google Scholar]; Pietryga JM, Werder DJ, Williams DJ, Casson JL, Schaller RD, Klimov VI, Hollingsworth JA. J Am Chem Soc. 2008;130:4879–4885. doi: 10.1021/ja710437r. [DOI] [PubMed] [Google Scholar]; Miao S, Hickey SG, Rellinghaus B, Waurisch C, Eychmuller A. J Am Chem Soc. 2010;132:5613–5615. doi: 10.1021/ja9105732. [DOI] [PubMed] [Google Scholar]; Allen PM, Bawendi M. J Am Chem Soc. 2008;130:9240–9241. doi: 10.1021/ja8036349. [DOI] [PMC free article] [PubMed] [Google Scholar]; Xie R, Rutherford M, Peng XG. J Am Chem Soc. 2009;131:5691–5697. doi: 10.1021/ja9005767. [DOI] [PubMed] [Google Scholar]
- 4.Kim S, Fisher B, Eisler HJ, Bawendi M. J Am Chem Soc. 2003;125:11466–11467. doi: 10.1021/ja0361749. [DOI] [PubMed] [Google Scholar]
- 5.Smith AM, Mohs AM, Nie SM. Nat Nanotech. 2009;4:56–63. doi: 10.1038/nnano.2008.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng Z, Schulz O, Lin S, Ding B, Liu X, Wei XX, Ros R, Yan H, Liu Y. J Am Chem Soc. 2010;132:5592–5593. doi: 10.1021/ja101476b. [DOI] [PubMed] [Google Scholar]
- 7.Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Ipe BI, Bawendi MG, Frangioni JV. Nat Biotechnol. 2007;25:1165–1170. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]; Smith AM, Nie SM. Nat Biotechnol. 2009;27:732–733. doi: 10.1038/nbt0809-732. [DOI] [PMC free article] [PubMed] [Google Scholar]; Choi HS, Liu W, Liu F, Nasr K, Misra P, Bawendi M, Frangioni JV. Nat Nanotech. 2010;5:42–47. doi: 10.1038/nnano.2009.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schooss D, Mews A, Eychmuller A, Weller H. Phys Rev B. 1994;49:17072–17078. doi: 10.1103/physrevb.49.17072. [DOI] [PubMed] [Google Scholar]; Haus JW, Zhou HS, Honma I, Komiyama H. Phys Rev B. 1993;47:1359–1365. doi: 10.1103/physrevb.47.1359. [DOI] [PubMed] [Google Scholar]
- 9.Kershaw SV, Burt M, Harrison M, Rogach A, Weller H, Eychmuller A. App Phys Lett. 1999;75:1694–1696. [Google Scholar]; Harrison MT, Kershaw SV, Burt MG, Eychmuller A, Weller H, Rogach AL. Mater Sci Eng B. 2000;69:355–360. [Google Scholar]; Harrison MT, Kershaw SV, Rogach AL, Kornowski A, Eychmüller A, Weller H. Adv Mat. 2000;12:123–125. [Google Scholar]; Rogach A, Kershaw S, Burt M, Harrison M, Kornowski A, Eychmüller A, Weller H. Adv Mat. 1999;11:552–555. [Google Scholar]
- 10.Yu WW, Wang YA, Peng XG. Chem Mater. 2003;15:4300–4308. [Google Scholar]
- 11.Son DH, Hughes SM, Yin YD, Alivisatos AP. Science. 2004;306:1009–1012. doi: 10.1126/science.1103755. [DOI] [PubMed] [Google Scholar]; Sadtler B, Demchenko DO, Zheng H, Hughes SM, Merkle MG, Dahmen U, Wang LW, Alivisatos AP. J Am Chem Soc. 2009;131:5285–5293. doi: 10.1021/ja809854q. [DOI] [PubMed] [Google Scholar]; Luther JM, Zheng H, Sadtler B, Alivisatos AP. J Am Chem Soc. 2009;131:16851–16857. doi: 10.1021/ja906503w. [DOI] [PubMed] [Google Scholar]
- 12.Zhong XH, Han MY, Dong Z, White TJ, Knoll W. J Am Chem Soc. 2003;125:8589–8594. doi: 10.1021/ja035096m. [DOI] [PubMed] [Google Scholar]; Regulacio MD, Han MY. Acc Chem Res. 2010;43:621–630. doi: 10.1021/ar900242r. [DOI] [PubMed] [Google Scholar]
- 13.Jain PK, Amirav L, Aloni S, Alivisatos AP. J Am Chem Soc. 2010;132:9997–9999. doi: 10.1021/ja104126u. [DOI] [PubMed] [Google Scholar]
- 14.Smith AM, Nie SM. J Am Chem Soc. 2008;130:11278–11279. doi: 10.1021/ja804306c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bucio L, Souza V, Albores A, Sierra A, Chavez E, Carabez A, Guiterrez-Ruiz MC. Toxicology. 1995;102:285–299. doi: 10.1016/0300-483x(95)03095-w. [DOI] [PubMed] [Google Scholar]; Han SG, Castranova V, Vallyathan V. J Toxicol Environ Health Part A. 2007;70:852–860. doi: 10.1080/15287390701212695. [DOI] [PubMed] [Google Scholar]; Strubelt O, Kremer J, Tilse A, Keogh J, Pentz R. J Toxicol Environ Health Part A. 1996;47:267–283. doi: 10.1080/009841096161780. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.