

Abstract
Amongst the pathologies associated with infection by Kaposi’s sarcoma-associated herpesvirus (KSHV), multicentric Castleman’s disease is distinctive for involvement of the lytic phase of the virus replication cycle. This B cell lymphoproliferative disorder has shown clinical responsiveness not only to generalized immunotherapy and cytotoxic chemotherapy, but also to inhibitors of herpesvirus DNA replication, consistent with the involvement of lytic phase of replication. These findings suggest that selective killing of virus-producing cells might represent a novel therapeutic strategy. We designed an immunotoxin, YC15-PE38, containing a single chain variable region fragment of a monoclonal antibody against KSHV glycoprotein H (gH) linked to the effector domains of Pseudomonas aeruginosa exotoxin A. Purified YC15-PE38 displayed highly selective and potent killing of a gH-expressing transfectant cell line (subnanomolar IC50). The immunotoxin also strongly inhibited production of infectious KSHV virions from an induced chronically infected cell line, by virtue of selective killing of the virus-producing cells. Combination treatment studies indicated complementary activities between YC15-PE38 and the herpesviral DNA replication inhibitor ganciclovir. These results provide support for the development of anti-KSHV strategies based on targeted killing of infected cells expressing lytic phase genes.
Keywords: Kaposi’s sarcoma-associated herpesvirus, human herpesvirus-8, immunotoxin, glycoprotein H, multicentric Castleman’s disease, lytic infection, ganciclovir
1. Introduction
Kaposi’s sarcoma-associated herpesvirus (KSHV, human herpesvirus 8) is a gammaherpesvirus initially discovered by DNA analysis of Kaposi’s sarcoma (KS) biopsy tissues from AIDS patients (Chang et al., 1994). Beyond its initial etiological implication in KS, the virus was subsequently linked to two B-cell lymphoproliferative disorders (Cesarman and Knowles, 1997; Chang and Moore, 1996): primary effusion lymphoma (PEL) (Cesarman et al., 1995) and multicentric Castleman’s disease (MCD) (Soulier et al., 1995). Typical of the Herpesviridae (Pellett and Roizman, 2007), KSHV infected cells can exist in both latent and lytic phases of the infection cycle, with marked variations observed in different KSHV-associated pathologies. Thus in KS and PEL, the virus persists extensively in the latent phase, whereas in MCD, lytic replication is prominent (Burbelo et al., 2010; Marcelin et al., 2007). The likely differences in gene regulation patterns and associated cellular signaling pathways for these distinct pathologies suggest that effective treatment strategies might differ (Casper, 2008; Schulz, 2006; Sullivan et al., 2008).
Castleman’s disease is a relatively rare lymphoproliferative syndrome first described over a half century ago (Castleman and Towne, 1954). It can occur in either the relatively benign unicentric form or the more aggressive MCD characterized by diffuse peripheral lymphadenopathy and systemic symptoms (Bower, 2010; Oksenhendler, 2009; Stebbing et al., 2008). Like the other hallmark KSHV-associated diseases, MCD is commonly observed in the context of HIV co-infection. The syndrome involves episodic remission and relapse, with active disease associated with abnormally large plasmablasts in lymph nodes as well as high levels of KSHV DNA in blood. Curiously, while antiretroviral therapy has been associated with a marked reduction of incidence of AIDS-KS as well as regression of KS lesions (Dittmer et al., 2005), recent reports have indicated an increasing incidence of HIV-associated MCD despite highly active antiretroviral therapy (HAART) and the associated improvements in CD4 cell counts (Kenyon et al., 2007; Mylona et al., 2008; Powles et al., 2009). Prognosis and median survival time remain poor, with frequent progression to non-Hodgkins lymphoma (Mylona et al., 2008; Stebbing et al., 2008).
At present there are no standardized treatment regimens for MCD. Beneficial clinical effects have been reported with several classes of agents, both nonherpesvirus-specific and herpes-directed (Bower, 2010; Oksenhendler, 2009; Stebbing et al., 2008). The former include anticancer treatments such as steroids and cytotoxic chemotherapy (single agent or combination) (Herrada et al., 1998), as well as immunotherapy with monoclonal antibodies (mAbs) such as rituximab against the B-cell antigen CD20 as reported in cohort studies (Bower et al., 2007; Gerard et al., 2007) and Tociliizumab against the IL6 receptor (Matsuyama et al., 2007). Aggressive combination treatment with chemotherapy and rituximab has shown efficacy in recent case reports of advanced MCD (Bestawros et al., 2008; Schmidt et al., 2008). Herpes-directed treatments have been based on the in vitro activities of several inhibitors of herpesvirus DNA polymerase against KSHV (Oksenhendler, 2009; Stebbing et al., 2008). Promising findings reported in humans include a case study indicating that ganciclovir promoted symptomatic relief and reduction of KSHV DNA load in plasma of MCD subjects (Casper et al., 2004) followed by a randomized double-blind placebo controlled crossover trial demonstrating that oral valganciclovir inhibited KSHV replication in the oral mucosa of asymptomatic subjects as judged by reduced frequency and quantity of KSHV shedding (Casper et al., 2008). However the dose-limiting toxicities of these drugs (Andrei et al., 2008) may compromise their long-term use for management of MCD.
We have been pursuing antiviral strategies directed at targeted killing of infected cells based on their surface expression of virus-encoded gene products. This concept is analogous to approaches under active development in the cancer field, using antibodies or ligands to target cytotoxic payloads to selectively kill cells expressing tumor-associated antigens (Sharkey and Goldenberg, 2008). In particular, several groups including ours initially developed recombinant protein immunotoxins generated from bacterial and plant protein toxins that selectively kill HIV productively infected cells based on their surface expression of the HIV Env glycoprotein (Aullo et al., 1992; Chaudhary et al., 1988; Pincus et al., 1989; Till et al., 1988). We have argued that improved versions of such agents might provide a critical complement to suppressive antiretroviral therapy in efforts to deplete persisting infected cell reservoirs (Berger and Pastan, 2010). In the present report, we describe an immunotoxin directed against glycoprotein H (gH) of KSHV, for possible use in treatment of pathologies such as MCD in which cells in the lytic phase of the viral replication cycle play a significant role. Combination treatment of KSHV-infected cells with the immunotoxin plus a herpesvirus replication inhibitor is also examined.
2. Materials and Methods
2.1 Cells
293 cells (derived from human embryonic kidney, Freestyle 293F subclone from Invitrogen) were cultured in DMEM with 10% fetal bovine serum (FBS), 2 mM L-glutamine and amino acid supplement. Vero cells (derived African green monkey kidney) harboring recombinant KSHV rKSHV.219 (Vieira and O’Hearn, 2004) (herein referred to as Vero-219, generously donated by Jeffrey Vieira, University of Washington) were cultured in DMEM supplemented with 10% (FBS), 2mM L-glutamine and 5ng/ml puromicin.
2.2 Immunotoxin plasmid construction
Immunotoxins composed of single chain variable fragments (scFvs) of mAbs linked to the translocation and ADP-ribosylation domains of Pseudomonas aeruginosa exotoxin A were designed by standard methods (Pastan et al., 2003). Total cellular RNAs from 107 YC15 hybridoma cells were isolated by the Qiagen RNA isolation kit as described in the product manual. 2.5 μg of total RNA and isotype-specific VH hinge primer (MG2a-Hinge: 5′-TCT GGG CTC AAT TTT CTT GTC CAC C-3′) or VL hinge primer (MK-Edge: 5′-CTC ATT CTT GTT GAA GCT CTT GAC AAT-3′) were used to set up the reaction for cDNA synthesis as described in the SMART RACE cDNA amplification kit (Clonetech, Palo Alto, CA). The prepared cDNAs were used as the template to set up the 5′RACE PCR with 10 pmol of isotype specific primers (MG2a-PCR: 5′-AGG GGC CAG TGG ATA GAC CGA TGG GGC TGT-3′, MK-PCR: 5′-GGA TGG TGG GAA GAT GGA TAC AGT TGG TGC AGC-3′). The PCR products were cloned into pCR2.1-TOPO vector through TOPO TA cloning kit (Invitrogen). At least 10 clones for each chain were sequenced. The sequences were analyzed and aligned according to the Ig BLAST program using the Kabat database (http://www.ncbi.nlm.nih.gov/igblast/).
The primers used to synthesize VH and VL fragments were designed according to the nucleotide sequences alignment. The primer pair used to amplify the heavy chain Fv region were 5′ H15-NdeI (5′-AAA CAT ATG GAG GTT CAG CTC CAG CAG TCT-3′) and 3′ H15-Linker (5′-TCC AGA TCC GCC ACC ACC TGA TCC GCC TCC GCC TGA GGA GAC GGT GAC TGA GGT-3′). The primer pair used to amplify the light chain Fv region were 5′L15-Linker (5′-TCA GGT GGT GGC GGA TCT GGA GGT GGC GGA AGC GAC ATC CAA ATG ACA CAA TCT -3′) and 3′ L15-HindIII (5′-GGA AGC TTT CCG TTT GAT TTC CAG CCT GGT -3′). The PCR products encoding the VH and VL domains of mAb YC15 connected by a 15 amino acid linker (Gly4Ser)3 were generated by fusion PCR using the purified individual VH and VL PCR fragments through the primers 5′ H15-NdeI and 3′ L15-HindIII. The PCR product was then digested with Nde I and Hind III and the resulting fragment was used to replace the Nde I - Hind III fragment from expression plasmid pTK21.8 encoding 3B3(Fv)-PE38 (Bera et al., 1998). The resulting YC15-PE38 expression plasmid is designated pYC51. We also produced an expression plasmid designated pYC52 encoding a negative control protein YC15- PE38E553D, a variant of YC15-PE38 containing the E553D mutation in the PE38 domain that inactivates its ADP ribosylation activity (Douglas and Collier, 1987). A control immunotoxin, BL22-PE38 directed against the CD22 antigen (Kreitman et al., 2001), was generously provided by Ira Pastan (NCI, NIH).
2.3 Immunotoxin expression and purification
Immunotoxin proteins were expressed and purified as described previously (Pastan et al., 2003). Briefly, the immunotoxin expression plasmids were transformed into E. Coli BL21 (DE3). The bacterial cultures were induced with isopropyl-1-thio-β-d-galactopyranoside (IPTG, 1 mM final concentration) for 2 hr to express the recombinant proteins. The insoluble inclusion bodies were isolated by centrifugation steps, solubilized in 6M guanidine HCl and subsequently reduced by dithioerythritol. The solubilized reduced proteins were refolded in the refolding solution containing oxidized glutathione and L-arginine. After the refolding step the protein was dialyzed to remove guanidine hydrochloride and purified by anion exchange chromatography using Q-Sepharose and Mono-Q (Amersham Pharmacia Biotech), and by size exclusion chromatography (TSK3000; TOSOH, Tokyo, Japan). The proteins were analyzed by SDS-PAGE under reducing conditions. Protein concentrations of the purified immunotoxins were determined by a protein assay (Bio-Rad) with BSA as the standard.
2.4 Establishment of KSHV gH/gL stable transfectant cell line
Full length KSHV gH and gL sequence were PCR amplified from plasmids pJK2-gH and pJK3-gL (Kaleeba and Berger, 2006) with the following primers: 5′gH-BamHI (5′-ATT GGA TCC ACC ATG CAG GGT CTA GCC TTC TTG-3′), 3′gH-HindIII (5′-AAT AAG CTT CTA ATA AAG GAT GGA AAA CAG-3′), 5′gL-NotI (5′-AAT GCG GCC GCC ATG GGG ATC TTT GCG CTA TTT-3′) and 3′gL-XhoI (5′-AAT CTC GAG TTA TTT TCC CTT TTG ACC TGT GTG-3′). The gL fragment was cloned into the Not I and Xho I sites of pBudCE4.1 vector (Invitrogen) under the EF-1α promoter. The gH fragment was subsequently cloned into the Hind III and BamH I sites of the same construct under CMV promoter. The resulting gH/gL expression plasmid is designated pYC50. 293 cells were transfected with pYC50 and selected by Zeocine antibiotics for at least two weeks. Cell lines stably expressing KSHV gH/gL were established by cell cloning; the cloned 293-gH/gL transfectant cell line used in this report is designated YC-293-gH/gL-clone 5.
2.5 Flow cytometry analysis
YC15-PE38 or 3B3(Fv)-PE38 immunotoxins were labeled with Alexa fluor 488 according to the manufacturer instruction (Alexa Fluor 488 microscale protein labeling kit, Invitrogen). 5×105 cells per sample were washed with PBS and blocked with 3%BSA in PBS at 4°C for 30 mins. The cells were incubated with different concentrations of labeled immunotoxins at 4°C for 1h. After washing, each sample was fixed with a 2% paraformaldehyde and flow cytometry analysis was performed with a FACS Scan (Becton Dickinson Co.).
2.6 Direct Cytotoxicity assay
WST-8 assay cell counting kit-8 (Dojindo Molecular Technologies, Gaithersburg, MD) was used to measure the cytotoxicity activities of the immunotoxins against the designated cells. Briefly, 5×103 cells/well were incubated with various concentrations of immunotoxins in a 96-well plate for 3 days. CCK-8 reagent (10 μl) was added to each well and the plate was incubated at 37°C incubator for 2 hours. Absorbance at 450nm was read. Cycloheximide (10 μg/ml) was used as a control for complete killing. All values were normalized to untreated controls.
2.7 Inhibition of infectious virus production assay
Vero-219 cells were seeded at 2×105 cells/well in a 24-well plate format. The cells were infected with recombinant baculovirus BacK50 (Vieira and O’Hearn, 2004) (from Jeffrey Vieira) at M.O.I. 100 for 3 hrs and washed one time with PBS. Then complete medium supplemented with the 1.25 mM Sodium Butyrate and different concentration of immunotoxin was added into the cells. 24hrs later, the medium containing sodium butyrate was removed and replaced with complete medium containing the same immunotoxin concentrations. At 3 days post induction, the supernatants were harvested and assayed for infectious KSHV virions. Aliquots (0.2 ml) of supernatants were spin-inoculated (1500 RPM, 30 min) onto fresh 293 cells in 24 well plates (2×105 cells/well, 0.5 ml total volume); after incubation at 37°C for 3 hours, the inoculum was replaced with 0.5 ml fresh medium and the plates were incubated at 37°C for 2 days. The number of infected target cells was then determined by flow cytometry; infectivity is expressed as percentage of target cells that scored GFP-positive.
3. Results
3.1 Design and production of KSHV gH-targeted immunotoxin
We chose a recently developed murine hybridoma designated YC15 for immunotoxin production, since our analyses indicated that the corresponding mAb binds to gH expressed on the surface of gH transfectant cell lines and neutralizes infection by KSHV virions (Y. Cai and E. A. Berger, unpublished). From cDNA clones of the variable light and heavy chain regions, we designed an scFv and linked the sequences to those encoding PE38, a Pseudomonas aeruginosa exotoxin A construct lacking the native N-terminal cell binding domain but containing the domains involved in internalization and cell killing by inhibition of protein synthesis via ADP-ribosylation of elongation factor 2. The single chain recombinant immunotoxin, designated YC15-PE38, is shown schematically in Fig. 1A. The protein was expressed in E. coli and purified from inclusion bodies (see Materials and Methods). SDS-PAGE coupled with Coomassie Blue staining (Fig. 1B) indicated that YC15-PE38 was readily detectable in the total cell extract (compare lanes 2 and 3) and was the major component in the inclusion body preparation (lane 4). The purity of the final product (lane 5) was ≥ 95%. We also expressed and purified the negative control protein YC15-PE38E553D, which lacks cytotoxic activity due to an inactivating mutation in the PE38 moiety.
Fig. 1.

Expression and purification of recombinant YC15-PE38 immunotoxin direct against the KSHV gH glycoprotein (A) Schematic of construction of the YC15-PE38 immunotoxin expression plasmid and the relative positions of the PCR primers used for the amplification and cloning procedure (see Materials and Methods). The VH and VL segments of anti-KSHV gH mAb YC15 were linked by a 15 amino acid linker (G4S)3 and fused in frame to PE38. (B) SDS-PAGE analysis of steps in purification of YC15-PE38. Lane 1, Molecular weight markers (kDa values shown at the left). Lane 2, uninduced bacterial cell lysate. Lane 3, IPTG-induced bacterial cell lysate. Lane 4, inclusion body preparation from induced cells. Lane 5, purified YC15-PE38 immunotoxin. Similar results were obtained for YC15-PE38E553D containing the inactivating mutation in the PE38 moiety.
3.2 Binding of YC15-PE38 to cell surface gH
We used flow cytometry to test the ability of the immunotoxin to bind to gH expressed on the cell surface (Fig. 2). We observed dose-dependent binding of YC15-PE38 to a KSHV gH/gL stable transfectant of 293 (panel A). Specificity was demonstrated by the lack of binding of this immunotoxin to parental 293 cells lacking gH (panel B), and of an irrelevant immunotoxin [BL22-PE38, which targets CD22] to either cell type (panels C and D).
Fig. 2.

Flow cytometry analysis of the YC15-PE38 immunotoxin binding to KSHV gH-expressing transfectant cell line. YC15-PE38 and the control immunotoxin BL22-PE38 were labeled with Alexa fluor 488 and incubated at the indicated concentrations with the YC-293-gH/gL stable transfectant cell line or parental cells. After washing, the cellbound immunotoxins were detected by flow cytometry.
3.3 Selective killing of KSHV gH-expressing cell lines by YC15-PE38
We tested the cytotoxic activity of YC15-PE38 against various cell lines. As shown in Fig. 3, the immunotoxin promoted dose-dependent killing of a 293 transfectant cell line expressing KSHV gH/gL. The cytotoxicity was quite potent, with an IC50 of 10 ng/ml (corresponding to 150 pM); this compares favorably with reported potencies of PE-based immunotoxins directed at antigens over-expressed on tumor cells (Wolf and Elsasser-Beile, 2009). Specificity was demonstrated in two independent control assays involving immunotoxin treatment over the same concentration range: YC15-PE38 showed negligible cytotoxicity for parental 293 cells (gH-negative), and an irrelevant PEbased immunotoxin (BL22-PE38 targeting CD22, which is not expressed on 293 cells) showed negligible cytotoxicity for both the parental 293 cells and the gH/gL-expressing 293 transfectant. The observed killing of the gH/gL transfectant cell line by YC15-PE38 was dependent on the cytotoxic activity of the PE38 moiety, as demonstrated by the lack of effect of YC15-PE38E553D, which contains a point mutation that abolishes the ADP-ribosylation activity of the PE moiety.
Fig. 3.
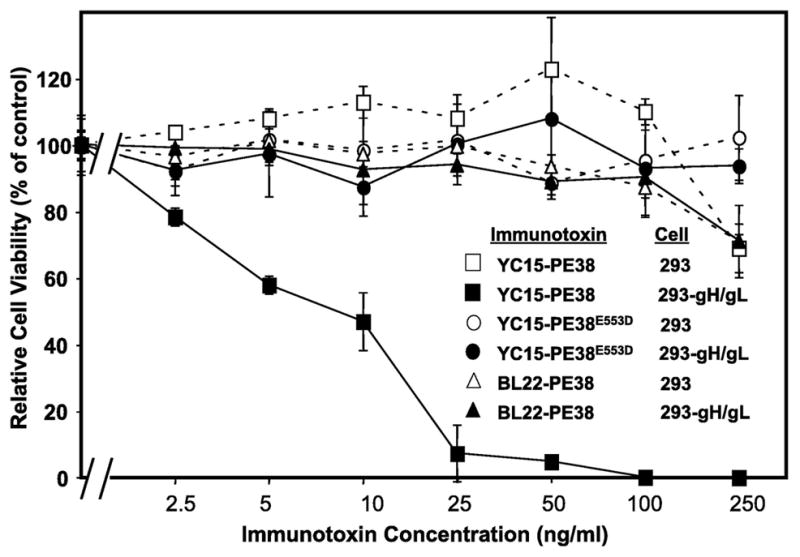
Direct cytotoxicity assay of YC15-PE38 against cell line expressing KSHV gH. 293 gH/gL stable transfectant cells (designated 293-gH/gL) or parental 293 cells were treated for 3 days with the indicated concentrations of YC15-PE38 or the negative control proteins YC15-PE38E553D or BL22-PE38. Cell viability was measured by the WST-8 assay as describe in Materials and Methods. Data points indicate the means of triplicate samples; error bars represent standard deviations.
3.4 Selective inhibition of infectious virus production by YC15-PE38
A prerequisite for the potential utility of YC15-PE38 is that it inhibit production of infectious virions from a producer cell; such an effect would reflect the ability of the immunotoxin to kill the infected cell before it produces the amount of virus that it would normally releases before succumbing to any cytopathic effect associated with the lytic phase of infection. In the experiments shown in Fig. 4, we examined the effects of immunotoxins on release of infectious virions from the Vero-219 cell line, which carries an episomal recombinant KSHV genome encoding infectious virions containing the eGFP gene linked to a strong constitutive cellular promoter. Virion release was stimulated by treatment for 3 days with sodium butyrate plus BacK50, a recombinant baculovirus encoding KSHV ORF 50 (RTA transcription factor) (Vieira and O’Hearn, 2004); the amounts of released infectious virus were quantitated by incubating the culture supernatants with susceptible 293 target cells and scoring the number of eGFP-positive cells after 2 days. Fig. 4 A shows that YC15-PE38 produced dose-dependent inhibition of infectious virus release, with potency much greater than that of the control BL22-PE38 immunotoxin. This inhibition was dependent on the cytotoxic activity of YC15-PE38 as demonstrated by the ineffectiveness of YC15-PE38E553D, thus proving that the observed inhibition was due to specific killing of the producer cells rather than to direct neutralization by the YC15 scFv moiety. This latter possibility was unlikely based on other studies (Y. Cai and E. A. Berger, unpublished) with the YC15 mAb, which demonstrated that neutralization required much higher concentrations of the divalent antibody than those used in this experiment with the monovalent immunotoxin. The experiment shown in Fig. 4B demonstrates that the specificity of inhibition of infectious KSHV release by YC15-PE38 was observed at all time points over a week-long period of induction of the Vero-219 cells.
Fig. 4.

Effect of YC15-PE38 on infectious KSHV production from activated Vero-219 cells. (A) Vero-219 cells were activated by recombinant baculovirus BacK50 plus sodium butyrate, and treated with different concentration of YC15-PE38, YC15-PE38E553D or control BL22-PE38 for 3 days. The supernatants were harvested and assayed for infection of fresh 293 cells to measure the infectious KSHV-r219 been produced from activated cells. The amount of infectious virus been produced from activated Vero-219 cells in the absence of immunotoxin was set as 100%. (B) Activated Vero-219 cells were treated with 100ng/ml of YC15-PE38 (black) or control BL22-PE38 (gray) or without immunotoxin (white). The infectious virus titer was measured at different time points post induction. Each data point represents the mean of triplicate culture wells, with error bars indicating standard deviations.
3.5 Effects of combinations treatment with YC15-PE38 and ganciclovir
As noted in the Introduction, the herpesviral DNA polymerase inhibitors ganciclovir and its prodrug valganciclovir have been shown to block KSHV replication in vitro and in vivo. These reports led us consider possible effects of combining such inhibitors with YC15-PE38. On the one hand, the two agents might show complementary anti-KSHV activity, since the former acts by blocking viral replication and the latter acts by killing cells that are already infected. On the other hand, since KSHV lytic gene expression is dependent on viral DNA synthesis (Lu et al., 2004), it is possible that a DNA polymerase inhibitor might compromise the activity of YC15-PE38 by blocking synthesis of gH, the target protein for the immunotoxin. We therefore compared the effects of each agent alone, and in combination over a range of concentrations of each agent, using inhibition of infectious virus production from induced Vero-219 cells as the assay. Fig. 5A shows dose-response curves of YC15-PE38 inhibition, alone and in combination with increasing concentrations of ganciclovir. The immunotoxin displayed activity over the entire range of ganciclovir; importantly as shown in the inset, the potency the immunotoxin was at least as great throughout the range of ganciclovir concentrations as it was when tested alone, as judged by the absence of increase in its IC50. Fig. 5B shows the reciprocal experiment, i.e. dose-response curves of ganciclovir in the presence of increasing concentrations of YC15-PE38. A comparable effect was observed, i.e. ganciclovir was at least as potent in the presence of increasing doses of YC15-PE38 as it was alone, based on IC50. Thus the results indicate that the two anti- KSHV agents show complementary rather than antagonistic activities.
Fig. 5.
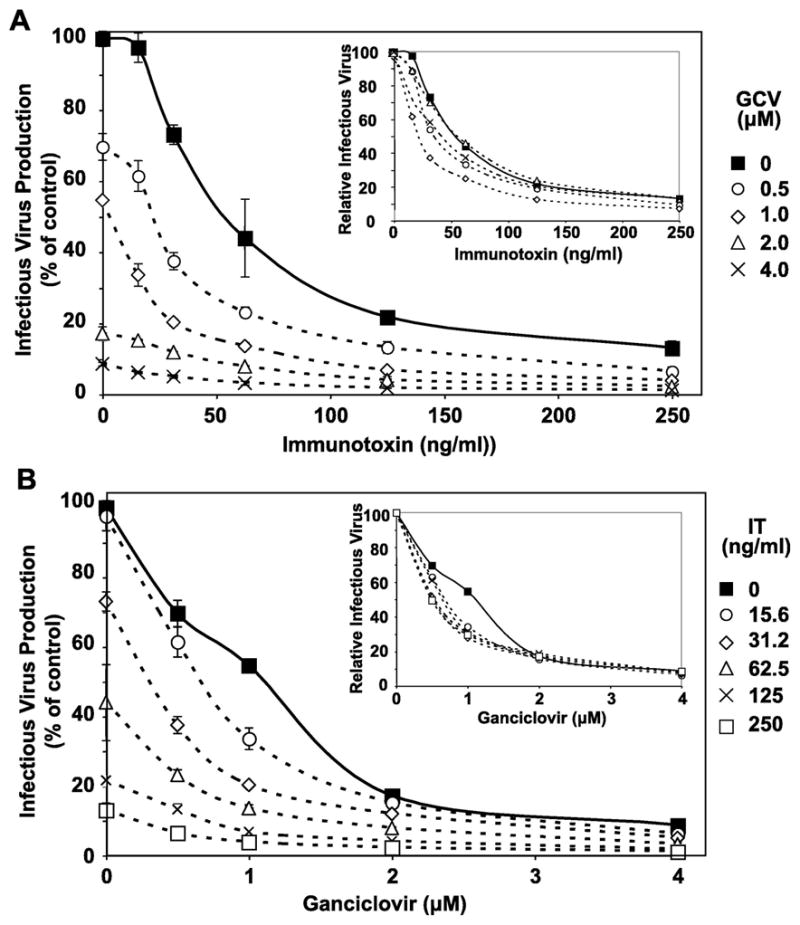
Effects of combination treatment with immunotoxin (IT) YC15-PE38 and ganciclovir on infectious KSHV production. Activated Vero-219 cells were treated for 3 days with combinations of YC15-PE38 and ganciclovir over the indicated concentration ranges for each agent. The amount of infectious virus produced was determined, with the value obtained in the absence of either agent set as 100%. Each data point represents the mean of triplicate culture wells, with error bars indicating standard deviation. A. Data are plotted as dose-response curves of YC15-PE38 in the presence of varying concentrations of ganciclovir; B. Data are plotted as dose-response curves of ganciclovir in the presence of varying concentrations of YC15-PE38. In each panel, the insets display the same data sets, with the values obtained in the absence of the agent on the X axis set as 100% for each dose-response curve.
4. Discussion
Amongst the pathologies associated with KSHV infection, MCD stands out virologically for its association with the lytic phase of the KSHV replication cycle, and clinically for its poor prognosis and increasing incidence in the HIV-infected population despite HAART. The frontline therapeutic regimens for MCD, antineoplastic chemotherapy and rituximab, have shown some efficacy, but are not curative and are complicated by significant toxicities due to their generalized modes of action. Curiously rituximab was associated with not only remission of MCD symptoms, but also with flare-up of KS lesions in HIV-positive patients with prior KS (Bower et al., 2007; Gerard et al., 2007) and very recently with initial presentation of aggressive KS in an HIV-negative MCD patient (Law et al., 2010). Based on analysis of KSHV viral loads, it has been concluded that MCD relapse after a positive rituximab response was due not to the outgrowth of resistant variants, but instead to the failure to completely eliminate virus from B cells, thereby allowing resumption of lytic infection (Powles et al., 2007). These results, coupled with the promising clinical findings with herpesvirus DNA polymerase inhibitors described in the Introduction, highlight the need for therapeutic agents that act directly and specifically against KSHV, particularly in the lytic phase.
The results presented herein demonstrate that the YC15-PE38 immunotoxin displays highly potent and selective cytotoxicity against cells expressing surface KSHV gH. This selective killing activity resulted in strong suppression of infectious virus release from chronically infected cells induced into lytic phase. Moreover, the immunotoxin showed complementary activity with the herpesvirus DNA replication inhibitor ganciclovir; while more detailed studies will be required to distinguish between additivity versus formal synergy, the coordinate antiviral action of these distinct classes of agents speaks to their potential clinical value in combination.
The case for combination therapy with immunotoxin treatment plus replication inhibitors has been promoted for HIV-1, for which formal synergy between an anti-HIV targeted toxin and reverse transcriptase inhibitors was demonstrated (Ashorn et al., 1990), and combined treatment with an immunotoxin plus replication inhibitors in a murine model resulted in nearly complete suppression of viral rebound after cessation of treatment (Goldstein et al., 2000). These dramatic combination effects provided the basis for the proposal to test immunotoxin complementation of HAART as an approach to deplete persisting HIV infected cell reservoirs (Berger and Pastan, 2010). Parallels have been reported in the herpesvirus field; immunotoxins generated from mAbs against murine cytomegalovirus have been shown to display complementary activities with ganciclovir both in cell culture and in a murine model (Smee et al., 1996). These findings support continued investigation of immunotoxin treatment for pathologies associated with KSHV lytic phase infection.
Acknowledgments
We thank Deboeeta Chatterjee (NIAID, NIH) for helpful discussions and Virgilio Bundoc (NIAID, NIH) for excellent technical assistance. We extend our thanks to Joseph Newland (NIAID, NIH), Tapan Bera, John Weldon, Dawn Walker, and Ira Pastan (NCI, NIH) for technical advice and donation of PE-based immunotoxins. Jeffrey Vieira (U. Washington) kindly provided Vero-219 cells and the BacK-50 baculovirus. This research was funded in part by the Intramural Program of the NIH, NIAID. Y. C. and E. A. B. are co-inventors on an NIH-owned patent application on the YC15 hybridoma/mAb and the YC15-PE38 immunotoxin.
Abbreviations
- KSHV
Kaposi’s sarcoma-associated herpesvirus
- KS
Kaposi’s sarcoma
- MCD
multicentric Castleman’s disease
- PEL
primary effusion lymphoma
- HAART
highly active antiretroviral therapy
- mAb
monoclonal antibody
- FBS
fetal bovine serum
- scFv
single chain variable fragment
- IPTG
isopropyl-1-thio-β-d-galactopyranoside
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: targeted drug delivery for cancer. Current Opinion in Chemical Biology. 2010;14:529–537. doi: 10.1016/j.cbpa.2010.06.170. [DOI] [PubMed] [Google Scholar]
- Andrei G, De Clercq E, Snoeck R. Novel inhibitors of human CMV. Curr Opin Invest Drugs. 2008;9:132–145. [PubMed] [Google Scholar]
- Ashorn P, Moss B, Weinstein JN, Chaudhary VK, FitzGerald DJ, Pastan I, Berger EA. Elimination of infectious human immunodeficiency virus from human T-cell cultures by synergistic action of CD4-Pseudomonas exotoxin and reverse transcriptase inhibitors. Proc Natl Acad Sci USA. 1990;87:8889–8893. doi: 10.1073/pnas.87.22.8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aullo P, Alcami J, Popoff MR, Klatzmann DR, Murphy JR, Boquet P. A recombinant diphtheria toxin related human CD4 fusion protein specifically kills HIV infected cells which express gp120 but selects fusion toxin resistant cells which carry HIV. EMBO J. 1992;11:575–583. doi: 10.1002/j.1460-2075.1992.tb05089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bera TK, Kennedy PE, Berger EA, Barbas CFI, Pastan I. Specific killing of HIV-infected lymphocytes by a recombinant immunotoxin directed against the HIV-1 envelope glycoprotein. Mol Med. 1998;4:384–391. [PMC free article] [PubMed] [Google Scholar]
- Berger EA, Pastan I. Immunotoxin complementation of HAART to deplete persisting HIV-infected cell reservoirs. PloS Pathog. 2010;6 doi: 10.1371/journal.ppat.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestawros A, Miche R, Seguin C, Routy JP. Multicentric Castleman’s disease treated with combination chemotherapy and rituximab in four HIV-positive men: A case series. American Journal of Hematology. 2008;83:508–511. doi: 10.1002/ajh.21108. [DOI] [PubMed] [Google Scholar]
- Bower M. How I treat HIV-associated multicentric Castleman’s disease. Blood. 2010 doi: 10.1182/blood-2010-07-290213. onine. [DOI] [PubMed] [Google Scholar]
- Bower M, Powles T, Williams S, Davis TN, Atkins M, Montoto S, Orkin C, Webb A, Fisher M, Nelson M, et al. Rituximab in HIV-associated multicentric Castleman disease. Annals of Internal Medicine. 2007;147:836–839. doi: 10.7326/0003-4819-147-12-200712180-00003. [DOI] [PubMed] [Google Scholar]
- Burbelo PD, Issa AT, Ching KH, Wyvill KM, Little RF, Iadarola MJ, Kovacs JA, Yarchoan R. Distinct profiles of antibodies to Kaposi sarcoma-associated herpesvirus antigens in patients with Kaposi sarcoma, multicentric Castleman disease, and primary effusion lymphoma. J Infect Dis. 2010;201:1919–1922. doi: 10.1086/652869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper C. New approaches to the treatment of human herpesvirus 8- associated disease. Rev Med Virol. 2008;18:321–329. doi: 10.1002/rmv.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper C, Krantz EM, Corey L, Kuntz SR, Wang J, Selke S, Hamilton S, Huang ML, Wald A. Valganciclovir for suppression of human herpesvirus-8 replication: A randomized, double-blind, placebo-controlled, crossover trial. J Infect Dis. 2008;198:23–30. doi: 10.1086/588820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper C, Nichols WG, Huang ML, Corey L, Wald A. Remission of HHV-8 and HIV-associated multicentric Castleman disease with ganciclovir treatment. Blood. 2004;103:1632–1634. doi: 10.1182/blood-2003-05-1721. [DOI] [PubMed] [Google Scholar]
- Castleman B, Towne VW. Case records of the Massachusetts general hospital: case No. 40231. N Engl J Med. 1954;250:1001–1005. doi: 10.1056/NEJM195406102502308. [DOI] [PubMed] [Google Scholar]
- Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s sarcoma-associated herpesvirus-like DNA-sequences in AIDS-related body-cavity-based lympjomas. N Engl J Med. 1995;332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- Cesarman E, Knowles DM. Kaposi’s sarcoma-associated herpesvirus: A lymphotropic human herpesvirus associated with Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. Seminars in Diagnostic Pathology. 1997;14:54–66. [PubMed] [Google Scholar]
- Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA-sequences in AIDS-associated Kaposis-sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- Chang Y, Moore PS. Kaposi’s sarcoma (KS)-associated herpesvirus and its role in KS. Infectious Agents and Disease-Reviews Issues and Commentary. 1996;5:215–222. [PubMed] [Google Scholar]
- Chaudhary VK, Mizukami T, Fuerst TR, FitzGerald DJ, Moss B, Pastan I, Berger EA. Selective killing of HIV-infected cells by recombinant human CD4- Pseudomonas exotoxin hybrid protein. Nature. 1988;335:369–372. doi: 10.1038/335369a0. [DOI] [PubMed] [Google Scholar]
- Dadachova E, Casadevall A. Radioimmunotherapy of infectious diseases. Seminars in Nuclear Medicine. 2009;39:146–153. doi: 10.1053/j.semnuclmed.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer DP, Vahrson W, Staudt M, Hilscher C, Fakhari FD. Kaposi’s sarcoma in the era of HAART - An update on mechanisms, diagnostics and treatment. Aids Reviews. 2005;7:56–61. [PubMed] [Google Scholar]
- Douglas CM, Collier RJ. Exotoxin-A of Pseucomonas-aeruginosa - substitution of glutamic aid-553 with aspartic acid drastically reduces toxicity and enzymatic activity. J Bacteriol. 1987;169:4967–4971. doi: 10.1128/jb.169.11.4967-4971.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard L, Berezne A, Galicier L, Meignin V, Obadia M, De Castro N, Jacomet C, Verdon R, Madelaine-Chambrin I, Boulanger E, et al. Prospective study of rituximab in chemotherapy-dependent human immunodeficiency virus-associated multicentric Castleman’s disease: ANRS 117 CastlemaB trial. Journal of Clinical Oncology. 2007;25:3350–3356. doi: 10.1200/JCO.2007.10.6732. [DOI] [PubMed] [Google Scholar]
- Goldstein H, Pettoello-Mantovani M, Bera TK, Pastan IH, Berger EA. Chimeric toxins targeted to the human immunodeficiency virus type 1 envelope glycoprotein augment the in vivo activity of combination antiretroviral therapy in thy/liv-scid-hu mice. J Infect Dis. 2000;181:921–926. doi: 10.1086/315351. [DOI] [PubMed] [Google Scholar]
- Herrada J, Cabanillas F, Rice L, Manning J, Pugh W. The clinical behavior of localized and multicentric Castleman disease. Annals of Internal Medicine. 1998;128:657–662. doi: 10.7326/0003-4819-128-8-199804150-00010. [DOI] [PubMed] [Google Scholar]
- Kaleeba JAR, Berger EA. Broad target cell selectivity of Kaposi’s sarcoma-associated herpesvirus glycoprotein-mediated cell fusion and virion entry. Virology. 2006;354:7–14. doi: 10.1016/j.virol.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Kenyon C, Pillay K, Jacobs P. Castleman’s disease and retroviral therapy. Transfusion and Apheresis Science. 2007;37:81–84. doi: 10.1016/j.transci.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Kreitman RJ, Wilson WH, Bergeron K, Raggio M, Stetler-Stevenson M, FitzGerald DJ, Pastan I. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N Engl J Med. 2001;345:241–247. doi: 10.1056/NEJM200107263450402. [DOI] [PubMed] [Google Scholar]
- Law AB, Ryan G, Lade S, Prince HM. Development of Kaposi’s sarcoma after complete remission of multicentric Castlemans disease with rituximab therapy in a HHV8-positive, HIV-negative patient. International Journal of Hematology. 2010;91:347–348. doi: 10.1007/s12185-010-0497-9. [DOI] [PubMed] [Google Scholar]
- Lu M, Suen J, Frias C, Pfeiffer R, Tsai MH, Chuang E, Zeichner SL. Dissection of the Kaposi’s sarcoma-associated herpesvirus gene expression program by using the viral DNA replication inhibitor cidofovir. J Virol. 2004;78:13637–13652. doi: 10.1128/JVI.78.24.13637-13652.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama M, Suzuki T, Tsuboi H, Ito S, Mamura M, Goto D, Matsumoto I, Tsutsumi A, Sumida T. Anti-interleukin-6 receptor antibody (tocilizumab) treatment of multicentric Castleman’s disease. Internal Medicine. 2007;46:771–774. doi: 10.2169/internalmedicine.46.6262. [DOI] [PubMed] [Google Scholar]
- Mylona EE, Baraboutis IG, Lekakis LJ, Georgiou O, Papastamopoulos V, Skoutelis A. Multicentric Castleman’s disease in HIV infection: a systematic review of the literature. AIDS Reviews. 2008;10:25–35. [PubMed] [Google Scholar]
- Oksenhendler E. HIV-associated multicentric Castleman disease. Curr Opin HIV AIDS. 2009;4:16–21. doi: 10.1097/coh.0b013e328319bca9. [DOI] [PubMed] [Google Scholar]
- Pastan I, Beers R, Bera TK. Recombinant immunotoxins in the treatment of cancer. In: Lo BKC, editor. Meth Molec Biol. Totowa, NJ: Humana Press Inc; 2003. pp. 503–518. [DOI] [PubMed] [Google Scholar]
- Pellett PE, Roizman B. The family Herpesviridae: a brief introduction. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Philadelphia: Lipincott Williams & Wilkens; 2007. pp. 2479–2499. [Google Scholar]
- Pincus SH, Wehrly K, Chesebro B. Treatment of HIV tissue culture infection with monoclonal antibody-ricin A chain conjugates. J Immunol. 1989;142:3070–3075. [PubMed] [Google Scholar]
- Powles T, Stebbing J, Bazeos A, Hatzimichael E, Mandalia S, Nelson M, Gazzard B, Bower M. The role of immune suppression and HHV-8 in the increasing incidence of HIV-associated multicentric Castleman’s disease. Annals of Oncology. 2009;20:775–779. doi: 10.1093/annonc/mdn697. [DOI] [PubMed] [Google Scholar]
- Powles T, Stebbing J, Montoto S, Nelson M, Gazzard B, Orkin C, Webb A, Bower M. Rituximab as retreatment for rituximab pretreated HIV-associated multicentric Castleman disease. Blood. 2007;110:4132–4133. doi: 10.1182/blood-2007-08-106187. [DOI] [PubMed] [Google Scholar]
- Schmidt SM, Raible A, Kortuem F, Mayer F, Riessen R, Adam P, Gregor M, Bissinger AL. Successful treatment of multicentric Castleman’s disease with combined immunochemotherapy in an AIDS patient with multiorgan failure. Leukemia. 2008;22:1782–1785. doi: 10.1038/leu.2008.54. [DOI] [PubMed] [Google Scholar]
- Schulz TF. The pleiotropic effects of Kaposi’s sarcoma herpesvirus. Journal of Pathology. 2006;208:187–198. doi: 10.1002/path.1904. [DOI] [PubMed] [Google Scholar]
- Sharkey RM, Goldenberg DM. Use of antibodies and immunoconjugates for the therapy of more accessible cancers. Advanced Drug Delivery Reviews. 2008;60:1407–1420. doi: 10.1016/j.addr.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smee DF, Sidwell RW, Barnett BB. Combination of antiviral immunotoxin and ganciclovir or cidofovir for the treatment of murine cytomegalovirus infections. Antiviral Research. 1996;32:165–171. doi: 10.1016/s0166-3542(95)00986-8. [DOI] [PubMed] [Google Scholar]
- Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazalshatem D, Babinet P, Dagay MF, Clauvel JP, Raphael M, Degos L, et al. Kaposi’s sarcomaassociated herpesvirus-like DNA-sequence in multicentri Castleman’s disease. Blood. 1995;86:1276–1280. [PubMed] [Google Scholar]
- Stebbing J, Pantanowitz L, Dayyani F, Sullivan RJ, Bower M, Dezube BJ. HIV-associated multicentric Castleman’s disease. American Journal of Hematology. 2008;83:498–503. doi: 10.1002/ajh.21137. [DOI] [PubMed] [Google Scholar]
- Sullivan RJ, Pantanowitz L, Casper C, Stebbing J, Dezube BJ. Epidemiology, pathophysiology, and treatment of Kaposi sarcoma-associated herpesvirus disease: Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease. Clinical Infectious Diseases. 2008;47:1209–1215. doi: 10.1086/592298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till MA, Ghetie V, Gregory T, Patzer EJ, Porter JP, Uhr JW, Capon DJ, Vitetta ES. HIV-infected cells are killed by rCD4-ricin A chain. Science. 1988;242:1166–1168. doi: 10.1126/science.2847316. [DOI] [PubMed] [Google Scholar]
- Vieira J, O’Hearn PM. Use of the red fluorescent protein as a marker of Kaposi’s sarcoma-associated herpesvirus lytic gene expression. Virology. 2004;325:225–240. doi: 10.1016/j.virol.2004.03.049. [DOI] [PubMed] [Google Scholar]
- Wolf P, Elsasser-Beile U. Pseudomonas exotoxin A: From virulence factor to anti-cancer agent. Int J of Med Microbiol. 2009;299:161–176. doi: 10.1016/j.ijmm.2008.08.003. [DOI] [PubMed] [Google Scholar]