

Abstract
An expedient enantioselective total synthesis of epicoccin G and related dithiodiketopiperazines through a strategy featuring direct two-directional sulfenylation, photooxygenation and Kornblum–DeLaMare rearrangement is described.
Diketopiperazines are an important class of natural products whose molecular structures are as varied as their biological properties.1 Those that contain sulfur atoms within their structures are particularly interesting due to the challenge they present to synthesis and their potent activities against viruses, bacteria and cancer cells.2 Combined with their scarcity, these properties inspire studies toward their synthesis as a means to develop new chemistry and render them readily available for further biological investigations.3 Epicoccin G [1, Figure 1, isolated from endophytic fungus Epicoccum nigrum; exhibits anti-HIV activity in C8166 cells (IC50 = 13.5 μM)],4 rostratin B [2, Figure 1, isolated from the marine-derived fungus Exserohilum rostratum; exhibits cytotoxicity against human colon carcinoma (HTC-116, IC50 = 1.9 μM)],5 and exserohilone [3, Figure 1, isolated from endophytic fungus Exserohilum holmii; suspected of antibacterial and antifungal activity]6 are three examples representing this class of compounds whose structural motifs are situated on a 6-5-6-5-6 diketopiperazine framework (I, Figure 1). In this communication we report a total synthesis of epicoccin G (1) and 8,8′-epi-ent-rostratin B (4) featuring a direct and improved procedure for the introduction of the sulfur atoms in diketopiperazines and a novel singlet oxygen/DeLaMare rearrangement cascade sequence for the attachment of the oxygen functionalities of the target molecules.
Figure 1.
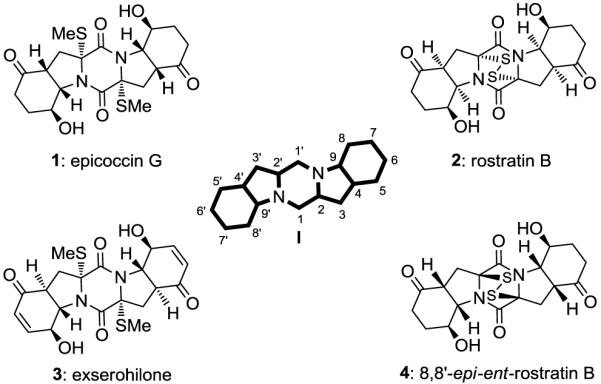
Molecular structures of selected epidithiodiketopiperazines: epicoccin G (1), rostratin B (2), exserohilone (3) and 8,8′-epi-ent-rostratin B (4).
Figure 2 shows, in retrosynthetic format, the evolution of the synthetic strategy toward epicoccin G (1). The C2-symmetry of 1 allowed for a general two-directional strategy for all three epidithiodiketopiperazine natural products shown in Figure 1 (1–3), their diastereoisomers (e.g. 4) and their analogues. Given the special reactivity of the sulfur moieties, the timing of their introduction was crucial. Thus, while early introduction of sulfur may have thwarted subsequent steps required for pending functionalizations, their late installation was excluded by the higher reactivity of the ketone groups (as compared to the diketopiperazine moiety) under the basic conditions needed for the sulfenylation reaction. On balance, it was decided to explore the possibility of introducing the sulfur atoms at the bis-diene 7 stage and attempt to navigate the growing molecule through selective endoperoxide formation effected by a photooxygenation reaction, followed by the rarely employed Kornblum–DeLaMare rearrangement7 and reduction of the remaining olefinic bonds. The requisite bis-diene system 7 was to be formed from N-Boc tyrosine (9) through intermediate 8 via appropriate functional group manipulations and dimerization.
Figure 2.
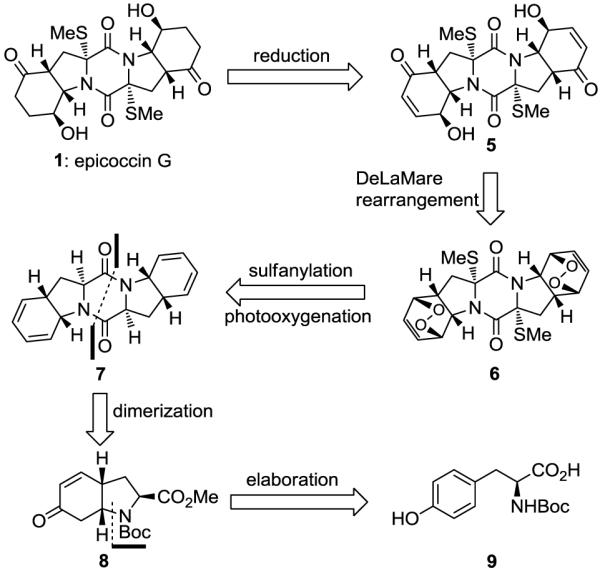
Retrosynthetic analysis of epicoccin G (1).
The construction of the C2-symmetric bis-diene diketopiperazine 7 from N-Boc tyrosine 9 is summarized in Scheme 1. Thus, 9 was converted to bicyclic hydroxy enone 10 through a known two-step procedure.8 Deoxygenation of the latter was achieved through a three-step sequence involving acetylation, zinc reduction and base-induced isomerization of the resulting ß,γ-unsaturated ketone to afford the desired bicyclic enone (8) in 51% overall yield. Luche reduction9 of this enone led to hydroxy N-Boc methyl ester 11 (92% yield), which was advanced through acid (TFA) and base (LiOH) treatment to intermediates hydroxy amine 12 and hydroxy acid 13, respectively. The dimerization step was realized through BOP-Cl facilitated coupling of 12 and 13 to afford N-Boc methyl ester amide 14 (86% yield). Deprotection of the amine and Et3N-induced ring closure gave pentacyclic bis-allylic system 15 in 77% overall yield for the two steps. The desired bis-diene 7 was generated from 15, through intermediate bis-trifluoroacetate 16, by treatment with (CF3CO)2O and Et3N (69% yield), followed by exposure to Pd(PPh3)4 (cat.) (90% yield).10
Scheme 1. Synthesis of Diketopiperazine bis-Diene 7a.

aReagents and conditions. a) Ac2O (2.0 equiv), Et3N (3.0 equiv), DMAP (0.2 equiv), CH2Cl2, 0→25 °C, 4 h; b) Zn (8.0 equiv), AcOH (2.0 equiv), MeOH, 65 °C, 0.5 h; c) DBU (5.0 equiv), PhMe, 65 °C, 3 h, 51% for the three steps; d) NaBH4 (1.1 equiv), CeCl3·7H2O (1.0 equiv), MeOH, −78→0 °C, 1 h, 92%; e) TFA/CH2Cl2 (1:1), 0→25 °C, 0.5 h, 99%; f) aq. LiOH (1.0 M)/THF (4:1), 0→25 °C, 3 h, 99%; g) 12, 13 (1.0 equiv each), BOP-Cl (1.1 equiv), Et3N (3.0 equiv), CH2Cl2, 0→25 °C, 15 h, 86%; h) TFA (32 equiv), CH2Cl2, 0→25 °C, 1.5 h; then Et3N (5.0 equiv), CH2Cl2, 0→25 °C, 15 h, 77% for the two steps; i) (CF3CO)2O (4.0 equiv), Et3N (6.0 equiv), DMAP (0.3 equiv), MeCN, −40→25 °C, 1 h, 69%; j) Pd(PPh3)4 (0.1 equiv), K2CO3 (2.1 equiv), dioxane, 65 °C, 0.5 h, 90%.
With diketopiperazine bis-diene 7 in hand, the installation of the sulfur atoms became the next task. Initial attempts to accomplish bis-sulfenylation of 7 by the classical method11 of treatment of the diketopiperazine substrate with base followed by addition of S8 failed; at best, only trace amounts of bis-sulfenylated products were obtained. Upon extensive experimentation, we discovered that pre-treatment of S8 with three equiv of NaHMDS at ambient temperature, followed by sequential addition of the diketopiperazine substrate 7 and further two equiv of NaHMDS, provided a mixture of oligosulfenylated compounds (17, Scheme 2). bis-Methylthio derivative 18 [together with its chromatographically separable 2,2′-epi-diastereoisomer (2,2′-epi-18, not shown)]12 was obtained as the major product upon reduction of oligosuflide 17 with NaBH4 and subsequent quenching of the resulting dianion with MeI (58% yield, ca. 1.4:1 dr). Oxidation of the dianion resulting from NaBH4 reduction of 17 with KI3 led to the corresponding epidithiodiketopiperazine (19) as the major product, formed together with its chromatographically separable 2,2′-epi- diastereoisomer (2,2′-epi-19, not shown) in 55% combined yield (ca. 1.4:1 dr). Selective synthesis of 18 could also be achieved starting from 19 by reduction with NaBH4 and quenching with MeI (65% yield). At this stage, the stereochemical configurations of 18 and 2,2′-epi-18 (and by extension 19 and 2,2′-epi-19) were assigned by NMR studies (see NOESY correlations, Figure 3) and confirmed later the successful conversion of 18 to epicoccin G (1, see below).
Scheme 2. Synthesis of Dithiodiketopiperazines 18 and 19a.
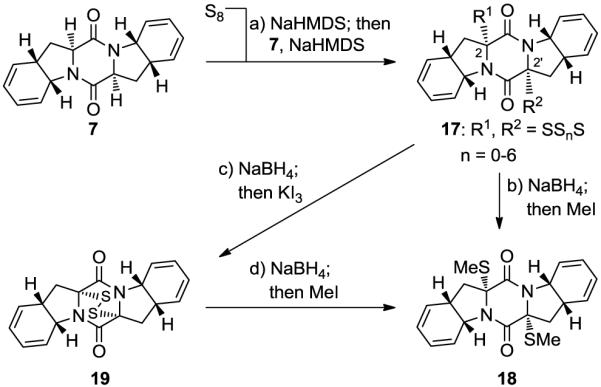
aReagents and conditions. a) NaHMDS (0.6 M in PhMe, 3.0 equiv), S8 (1.0 equiv), THF, 25 °C, 1 min; then 7 (1 M in THF, 1.0 equiv) 1 min; then NaHMDS (0.6 M in PhMe, 2.0 equiv), 25 °C, 0.5 h; b) NaBH4 (25 equiv), THF/MeOH (1:1), 0→25 °C, 0.75 h; then MeI (50 equiv), 25 °C, 15 h, 58% over three steps from 7 (18:2,2′-epi-18, ca. 1.4:1 dr); c) NaBH4 (25 equiv), THF/MeOH (1:1), 0→25 °C, 0.75 h; then aq. KI3 (1.4 M), 25 °C, 10 min, 55% over three steps from 7 (19: 2,2′-epi-19, ca. 1.4:1 dr); d) NaBH4 (25 equiv), THF/MeOH (1:1), 0→25 °C, 0.75 h; then MeI (50 equiv), 25 °C, 15 h, 65% from 19.
Figure 3.
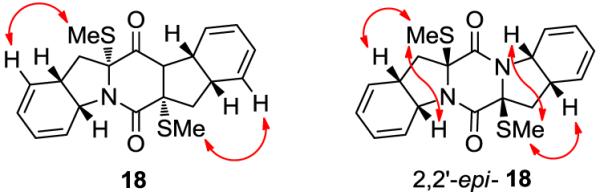
Stereochemical assignments of 18 and 2,2′-epi-18 by NOESY studies. Arrows designate NOESY correlations.
With the challenging sulfenylation successfully completed through the newly developed procedure, the next hurdle, that of regio- and stereoselective oxygenation of the diene systems of 18 in the presence of the sulfur atoms, was addressed. Reaction of 18 with singlet oxygen, generated from triplet oxygen and light in the presence of TPP as a sensitizer13 (CH2Cl2, 0 °C, 15 min), gave bis-endoperoxide 6 (Scheme 3). The latter underwent regioselective Kornblum–DeLaMare rearrangement7 upon in situ treatment with DBU to afford bis-hydroxy enone 20 in 52% overall yield. Finally, catalytic hydrogenation of the highly functionalized pentacyclic precursor 20 (H2, 20% Pd(OH)2/C) furnished epicoccin G (1, 86% yield). The physical properties of synthetic 1 (i.e. 1H and 13C NMR, mass spec data and optical rotation) matched those reported for the natural substance.4b
Scheme 3. Completion of the Total Synthesis of Epicoccin G (1)a.
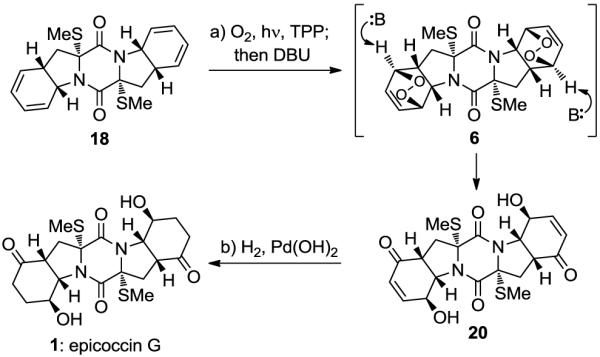
aReagents and conditions. a) O2, TPP (0.02 equiv), CH2Cl2, 400W Philips-MH400/U sunlamp, −45 °C, 0.7 h; then DBU (10.0 equiv), −45→0 °C; 1 h, 52% from 18; b) H2, Pd(OH)2/C (20% w/w, 0.4 equiv), MeOH, 25 °C, 1 h, 86%.
As a demonstration of the power of the developed methodology to construct complex epidithiodiketopiperazine systems, we successfully completed the total synthesis of the epidithiodiketopiperazine 8,8′-epi-ent-rostratin B (4) as shown in Scheme 4. Thus, photooxygenation of 2,2′-epi-19 followed by in situ treatment of the so-generated bis-endoperoxide (21) with Et3N furnished, regioselectively, bis-hydroxy enone 22 in 55% overall yield. Reduction of the olefinic bonds within 22 was achieved through the use of Stryker’s reagent,14 followed by treatment with KI3, furnished 8,8′-epi-ent-rostratin B (4) in 82% yield.
Scheme 4. Completion of the Total Synthesis of 8,8′-epi-ent-Rostratin B (4)a.
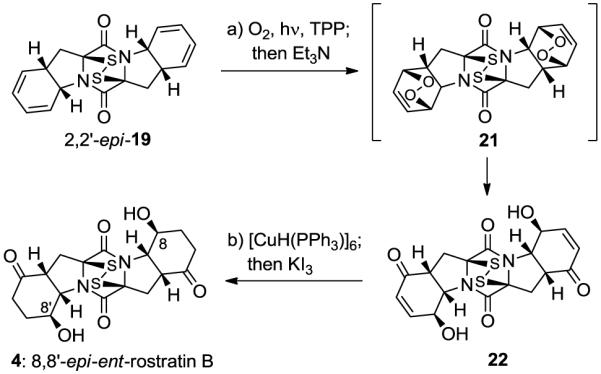
aReagents and conditions. a) O2, TPP (0.02 equiv), CH2Cl2, 400W Philips-MH400/U sunlamp, 0 °C, 2 h; then Et3N (5.0 equiv), 0→25 °C, 3 h, 55% for the two steps; b) [CuH(PPh3)]6 (10.0 equiv), benzene, 25 °C, 0.5 h; then aq. KI3 (1.4 M), 82%.
Described above is an improved direct procedure for sulfenylation of diketopiperazines and its application to the synthesis of the bis-methylthio- and epidithiodiketopiperazine structural motifs as exemplified by the total synthesis of epicoccin G (1) and 8,8′-epi-ent-rostratin B (4). Employing endoperoxide intermediates, the described chemistry also demonstrates the applicability of the Kornblum–DeLaMare rearrangement in total synthesis, and should facilitate the construction of other members of the dithiodiketopiperazine class of compounds, natural or designed, for biological investigations.
Supplementary Material
ACKNOWLEDGMENT
We thank Drs. D. H. Huang and L. Pasternack for NMR spectroscopic assistance and Dr. Gary Siuzdak for mass spectrometric assistance. This work was supported by the Skaggs Institute for Research and grants from the National Institutes of Health (USA) (AI055475-09, CA100101-05), as well as fellowships from Fonds de Recherche Québec (to D.G.) and Bristol-Myers Squibb (to D.S.).
Footnotes
Supporting Information. Experimental procedures and characterization data for key compounds. This material is available from the author or free of charge via the Internet at http://pubs.acs.org/.
REFERENCES
- (1).(a) Bull SD, Davies SG, Parkin RM, Sancho FS. J. Chem. Soc., Perkin Trans. 1. 1998:2313–2319. [Google Scholar]; (b) Ding G, Jiang L, Guo L, Chen X, Zhang H, Che Y. J. Nat. Prod. 2008;71:1861–1865. doi: 10.1021/np800357g. [DOI] [PubMed] [Google Scholar]; (c) Kamei H, Oka M, Hamagishi Y, Tomita K, Komishi M, Oki T. J. Antibiot. 1990;43:1018–1020. doi: 10.7164/antibiotics.43.1018. [DOI] [PubMed] [Google Scholar]; (d) Huang R, Zhou X, Xu T, Yang X, Liu Y. Chem.Biodiversity. 2010;7:2809–2829. doi: 10.1002/cbdv.200900211. [DOI] [PubMed] [Google Scholar]
- (2).(a) Gardiner DM, Waring P, Howlett BJ. Microbiology. 2005;151:1021–1032. doi: 10.1099/mic.0.27847-0. [DOI] [PubMed] [Google Scholar]; (b) Rezanka T, Sobotka M, Spizek J, Sigler K. Anti-infect. Agents Med. Chem. 2006;5:187–224. [Google Scholar]
- (3).For selected epidithiodiketopiperazine total syntheses, see: Kishi Y, Fukuyama T, Nakatsuka S. J. Am. Chem. Soc. 1973;95:6492–6493. doi: 10.1021/ja00800a078. Kishi Y, Nakatsuka S, Fukuyama T, Havel M. J. Am. Chem. Soc. 1973;95:6493–6495. doi: 10.1021/ja00800a079. Fukuyama T, Kishi Y, Nakatsuka S. Tetrahedron Lett. 1974;16:1549–1552. Fukuyama T, Kishi Y. J. Am. Chem. Soc. 1976;21:6723–6724. doi: 10.1021/ja00437a063. Williams RM, Rastetter WH. J. Org. Chem. 1980;45:2625–2631. Miknis F, Williams R,M. J. Am. Chem. Soc. 1993;115:536–547. Kim J, Ashenhurst JA, Movassaghi M. Science. 2009;324:238–241. doi: 10.1126/science.1170777. Iwasa E, Hamashima Y, Fujishiro S, Higuchi E, Ito A, Yoshida M, Sodeoka M. J. Am. Chem. Soc. 2010;132:4078–4079. doi: 10.1021/ja101280p. Kim J, Movassaghi M. J. Am. Chem. Soc. 2010;132:14376–14378. doi: 10.1021/ja106869s. and references cited therein. DeLorbe JE, Salman YJ, Mennen SM, Overman LE, Zhang F. J. Am. Chem. Soc. 2011;133:6549–6552. doi: 10.1021/ja201789v.
- (4).Guo H, Sun B, Gao H, Chen X, Liu S, Yao X, Liu X, Che Y. J. Nat. Prod. 2009;72:2115–2119. doi: 10.1021/np900654a. Wang J-M, Ding G-Z, Fang L, Dai J-G, Yu S-S, Wang Y-H, Chen X-G, Ma S-G, Qu J, Xu S, Du D. J. Nat. Prod. 2010;73:1240–1249. doi: 10.1021/np1000895. These two reports claim the isolation and structural characterization of epicoccin G and ent-epicoccin G, respectively, from the same species. In the latter, the authors employed X-ray crystallographic and Mosher ester techniques to assign the absolute stereochemistry shown in 1, a compound they called ent-epicoccin G. Our synthetic material matched the enantiomer depicted in structure 1 reported in ref. 4b. We thank Professor Y. Che for assuring us that epicoccin G and ent-epicoccin G are one and the same, now called epicoccin G.
- (5).Tan RX, Jensen PR, Williams PG, Fenical W. J. Nat. Prod. 2004;67:1374–1382. doi: 10.1021/np049920b. [DOI] [PubMed] [Google Scholar]
- (6).Sugawara K, Sugawara M, Strobel GA, Fu Y, Cun-Heng H, Clardy J. J. Org. Chem. 1985;50:5631–5633. [Google Scholar]
- (7).(a) Kornblum N, DeLaMare HE. J. Am. Chem. Soc. 1951;73:880–881. [Google Scholar]; (b) Staben ST, Linghu X, Toste FD. J. Am. Chem. Soc. 2006;128:12658–12659. doi: 10.1021/ja065464x. [DOI] [PubMed] [Google Scholar]
- (8).Wipf P, Kim Y. Tetrahedron Lett. 1992;33:5477–5480. [Google Scholar]
- (9).Luche JL. J. Am. Chem. Soc. 1978;100:2226–2227. [Google Scholar]
- (10).Barrero A,F, Arseniyadis S, Quilez del Moral J, Mar Herrador M, Valdivia M, Jeminez D. J. Org. Chem. 2002;67:2501–2508. doi: 10.1021/jo0161882. [DOI] [PubMed] [Google Scholar]
- (11).Ohler E, Poisel H, Tataruch F, Schmidt U. Chem. Ber. 1972;105:635–641. doi: 10.1002/cber.19721050229. (b) For an insightful review on sulfenylation reaction of carbonyl compounds, see: Wladislaw B, Marzoati L, Di Vitta C. Org. React. Proc. Int. 2007;39:447–494.
- (12).No trans products corresponding to 18 and 2,2′-epi-18 were isolated, suggesting intramolecular delivery of the second sulfur atom. The generality, scope and mechanism of this sulfenylation reaction are currently under investigation and will be reported in due course.
- (13).For a review on synthetic applications of bicyclic endoperoxides, see: Balci M. Chem. Rev. 1981;81:91–108.
- (14).Mahoney WS, Brestensky DM, Stryker JM. J. Am. Chem. Soc. 1988;110:291–293. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.