

Abstract
Purpose
To identify the prevalence of rhodopsin (RHO) mutations in French patients with autosomal dominant rod-cone dystrophies (adRP).
Methods
Detailed phenotypic characterization was performed including precise family history, best corrected visual acuity using the ETDRS chart, slit lamp examination, kinetic and static perimetry, full field and multifocal electroretinography (ERG), fundus autofluorescence imaging (FAF) and optical coherence tomography (OCT). For genetic diagnosis, genomic DNA of seventy-nine families was isolated by standard methods. The coding exons and flanking intronic regions of RHO were PCR amplified, purified and sequenced in the index patient.
Results
Among this French adRP cohort, 16.5% revealed a RHO mutation. While three unrelated families showed each a novel missense mutation (p.Leu88Pro, p.Met207Lys and p.Gln344Pro), ten unrelated families showed recurrent previously published mutations (p.Asn15Ser, p.Leu131Pro, p.Arg135Trp, p.Ser334GlyfsX20 and p.Pro347Leu). All mutations co-segregated with the phenotype within a family and the novel mutations were not identified in a control population.
Conclusion
Our studies revealed that the prevalence of RHO mutations in French adRP patients is in accordance with other studies from Europe. Most of the changes identified herein reflect recurrent mutations within which p.Pro347Leu substitution is the most prevalent. Nevertheless, almost a quarter of the changes are novel indicating that, although RHO is the first gene implicated and probably the most studied gene in RP, it is still relevant to perform mutation analysis in the coding exons of RHO to detect novel changes. Our detailed phenotype-genotype analyses in all family members available deliver the basis for therapeutic approaches in those families.
Keywords: Adolescent; Adult; Child; DNA Mutational Analysis; Electroretinography; European Continental Ancestry Group; genetics; Female; Fluorescein Angiography; France; epidemiology; Genes, Dominant; Genotype; Humans; Male; Middle Aged; Mutation; Pedigree; Phenotype; Photoreceptor Cells, Vertebrate; pathology; Polymerase Chain Reaction; Prevalence; Retinitis Pigmentosa; diagnosis; genetics; Rhodopsin; genetics; Tomography, Optical Coherence; Visual Acuity; Young Adult
Rod-cone dystrophies, also called retinitis pigmentosa (RP), are a clinically and genetically heterogeneous group of inherited retinal disorders primarily affecting rods with secondary cone degeneration 1. RP patients initially often complain of night blindness. This is attributed to the primarily affected rods and clinical sign of the impaired rod function. Later on, when the secondary cone dysfunctions manifests, progressive visual field constriction, abnormal color vision and loss of central vision can be observed – signs of decreasing cone function. As the disease progresses and retinal dysfunction decreases, visual impairment increases: in some cases the disease may eventually result in very severe visual impairment or even blindness. RP is the most common inherited form of severe retinal degeneration, with a frequency of about 1 in 4000 births and more than 1 million affected individuals over the world. The mode of inheritance can be X-linked (5–15%) autosomal dominant (30–40%) or autosomal recessive (50–60%) The remaining patients represent isolated cases of which the inheritance trait can not be established 1.
To date, 20 autosomal dominant RP (adRP) genes have been reported (http://www.sph.uth.tmc.edu/Retnet/). One of the major genes underlying this disorder is rhodopsin (RHO) coding for the light absorbing molecule that initiates the signal transmission cascade in rod photoreceptors. According to the literature, RHO mutation prevalence ranges from 0 to 50% cases of adRP in cohorts from various geographical origins, with higher numbers reported in the United States 2–18.
The genetic and phenotypic heterogeneity is not only found in RP in general but also specifically reflected in adRP with RHO mutations: Over 120 mutations have been identified in different sites of the gene including specific hot spots (http://www.sph.uth.tmc.edu/Retnet/, http://www.hgmd.cf.ac.uk/ac/all.php, http://www.retina-international.org/sci-news/rhomut.htm) 19.
Certain mutations in RHO lead to diffuse rod-cone dysfunction whereas other cases are implicated in a more restricted disease that may predominate in the inferior part of the retina such as in sector RP 20. Phenotypic classifications have been proposed to reflect this variability. In particular, Cideciyan and co-workers have distinguished two classes of disease expression with allele-specificity 21: class A mutants show severely generalized abnormal rod function early in life with a constant rate of cone disease progression across the retina with time. Class B mutants show more restricted disease and absent or lateonset night blindness.
Other classifications have been proposed based on the underlying pathogenic mechanism involved in adRP due to RHO mutations. Mendes and co-workers classified the different types of mutations in 6 groups. Class I refers exclusively to rhodopsin mutations that fold correctly but are not transported to the outer segment. Class II, refers to mutations that misfold, are retained in the endopasmic reticulum (ER) and cannot easily reconstitute with 11-cis-retinal. Class III refers to mutations that affect endocytosis. Class IV mutations do not affect folding per se but might affect rhodopsin stability and posttranslational modification. Similarly, Class V mutations have no obvious folding defect but show an increased activation rate for transducin. Mutants that appear to fold correctly but lead to the constitutive activation of opsin in the absence of the chromophore and in the dark constitute Class VI. Other mutations with unclear biochemical or cellular defect, or uninvestigated defect were not classified 19.
Our comprehensive study presented herein aim to investigate in detail a French adRP cohort coming from 2 different clinical centers, namely Quinze-Vingts hospital in Paris and the Centre Hospitalier Régional in Montpellier located in the south of France. We will present the prevalence of rhodopsin mutations in this cohort and show precise phenotype-genotype correlations. Novel mutations will be analyzed on its predicted pathogenic mechanism as well as frequently mutated sites will be presented as putative candidates for therapeutic approaches.
METHODS
Clinic
Seventy-nine families with a provisional diagnosis of autosomal dominant rod-cone dystrophy, (adRP) were ascertained in the CIC of the Quinze-Vingts hospital, Paris (67 families) and in Montpellier (12 families). Informed consent was obtained from each patient and normal individual controls after explanation of the study and its potential outcome. The study protocol adhered to the tenets of the Declaration of Helsinki and was approved by the local ethics committees. Each patient underwent full ophthalmic examination with assessment of best corrected visual acuity using ETDRS chart, kinetic and static perimetry and colour vision using the desaturated Farnsworth Panel D-15. Fullfield and multifocal electroretinography (ERG and mfERG) were performed with DTL recording electrodes and incorporated the ISCEV Standards (Espion2 Diagnosys® for full field ERG and Veris II for Multifocal ERG) 22, 23. Severe rod-cone dysfunction was considered when no detectable responses where recorded. Clinical assessment was completed with Fundus Autofluorescence Imaging (FAF) and Optical Coherence Tomography (OCT) (HRAII® and Spectralis® OCT, Heidelberg Engineering, Dossenheim, Germany). At the end of clinical evaluation, patients and family members were asked to donate a blood sample for further genetic studies.
Mutation analysis
Total genomic DNA was extracted from peripheral leucocytes in blood samples by standard salting out procedures 24 or according to manufacturer recommendation (Puregen Kit, Qiagen, Courtaboeuf, France). Subsequently, either genotyping or direct sequencing of RHO was performed. For genotyping 2 to 3 polymorphic microsatellite markers within or contiguous to known adRP genes (RHO, RDS, PRPF31, RP1, PRPF8, IMPDH1, PRPF3, NRL, CA4, CRX, TOPORS, PAP1, NR2E3) was used. Results were analysed with GeneMapper software (version 4.0, Applied Biosystems). The coding 5 exons of rhodopsin (RHO RefSeq NM000539.2) and the flanking intronic regions were amplified with oligonucleotides previously described 25 At least 125 commercially available control samples were used to validate the pathogenicity of the novel sequence variants (Human random control panel 1–3, Health Protection Agency Culture Collections, Salisbury, United Kingdom).
RESULTS
Mutation analysis
Thirteen index patients of the investigated 79 French autosomal dominant RP patients revealed a RHO mutation (Table 1). These mutations co-segregated with the phenotype when tested in family members available. Three index patients showed each a novel missense mutation, while ten index patients revealed previously described mutations in RHO (Table 1 and Figure 2a).
Table 1.
Novel and known RHO mutations in the French cohort.
| Index (family) | Exon | Nucleotide Exchange | Protein Effect | Publication |
|---|---|---|---|---|
| 2810 (PB41) 2923 (PB42) |
1 | c.44A>G | p.Asn15Ser | 36 |
| CIC00218 (F155) | 1 | c.263T>C | p.Leu88Pro | novel |
| CIC00123 (F172/96) | 2 | c.392T>C | p.Leu131Pro | 26 |
|
CIC00364 (F247) CIC00974 (F610) |
2 | c.403C>T | p.Arg135Trp | 4 |
| CIC00716 (F475) | 3 | c. 620T>A | p.Met207Lys | novel (phenotypegenotype correlation published Audo et al., in press) |
| 2296 (RP827) | 5 | c.998_999insAGGC | p.Ser334GlyfsX20 | 13 |
| CIC00590 (F394) | 5 | c.1031A>C | p.Gln344Pro | novel |
|
CIC00161 (F119) CIC00841 (F546) CIC00944 (F598) CIC01125 (F681) |
5 | c.1040C>T | p.Pro347Leu | 29 |
Figure 2.
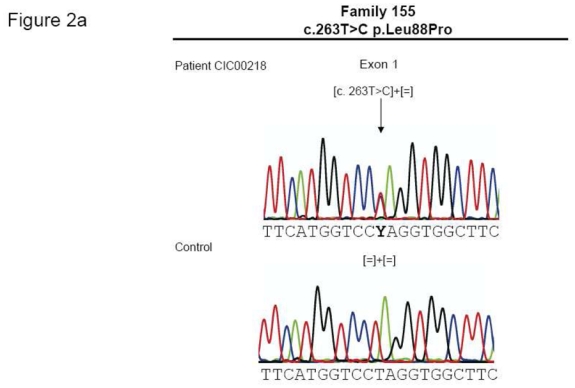
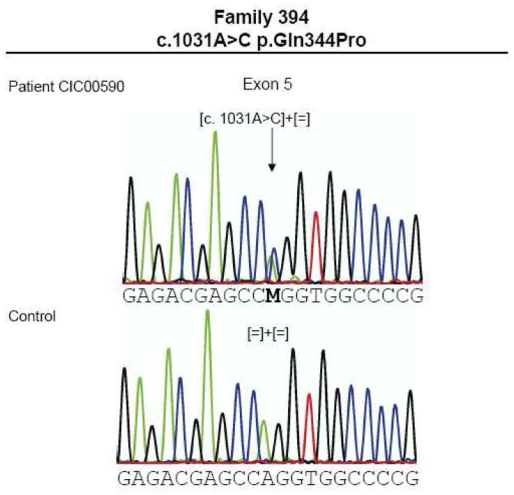

(a). Electropherograms of novel RHO mutations highlighted by an arrow. (b) Multiple amino acid sequence alignments of different species of novel mutated residues (depicted in green). Amino acid substitutions are highlighted in red. The position of the respective amino acids is shown in black numbers.
-
1
Patient CIC00218 from family 155, originating from the Southwest of France, had a novel c.263T>C mutation on exon 1 leading to a p.Leu88Pro substitution (Figures 1A and 2A)
-
2
Patient CIC00716 of family 475, from Northern France revealed a novel mutation c.620T>A in exon 3 leading to a p.Met207Arg substitution, which segregates with an unusual restricted chorioretinal atrophy phenotype (Figure 1b) (Audo et al., 2010, in press).
-
3
Patient CIC00590 from family 394, with Sephardim Jewish origins, revealed a novel mutation, c.1031A>C in exon 5, leading to a p.Gln344Pro substitution (Figure 1c and 2a).
-
4 and 5
Two patients from two unrelated families (PB41 and 42) from a similar region in France revealed the known c.44A>G mutation in exon 1 leading to a p.Asn15Ser exchange (Figure 1d).
-
6
Patient CIC00123 from family 172/96 originating from Martinique, within the French West Indies, showed a previously described heterozygous c.392T>C mutation in exon 2 leading to a p.Leu131Pro substitution (Figure 1e).
-
7 and 8
Two index patients CIC00364 and CIC00974 from two unrelated families 247 and 610 respectively revealed the known mutation c.403C>T in exon 2 leading to a p.Arg135Trp substitution, which co-segregated with the disease (Figure 1f).
-
9
Index patient 2296 from family RP827 revealed the earlier described c.998_999insAGGC insertion leading to a predicted frameshift mutation (p.Ser334GlyfsX20), which is assumed to change the open reading frame and elongates the altered protein (Figure 1g).
-
10–13
Four index patients CIC00161, CIC00841, CIC00944 and CIC01125 from 4 unrelated families with origins in 4 distinct regions of France (family 119, family 546, family 598 and family 681, respectively) revealed the c.1040C>T mutation in exon 5 leading to the p.Pro347Leu substitution, which co-segregated in family members available for genetic testing (Figure 1h).
Figure 1.
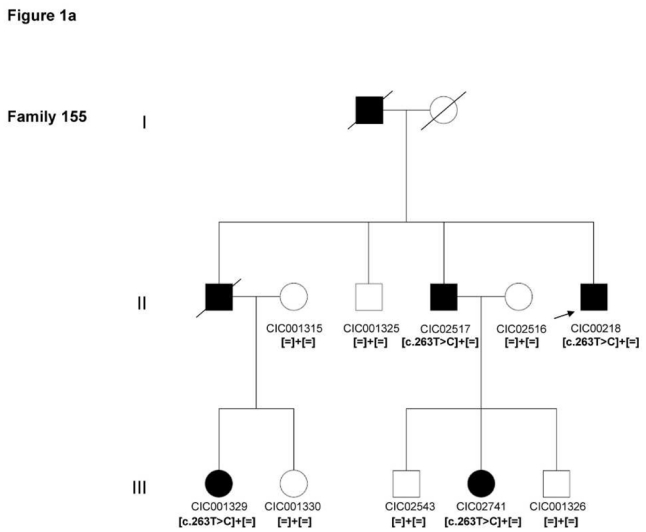
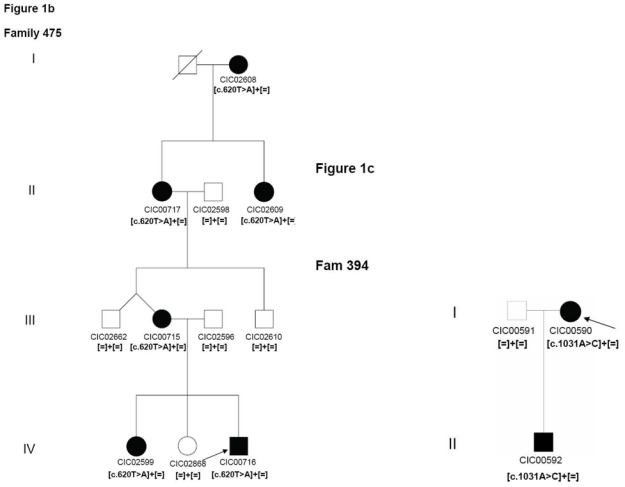
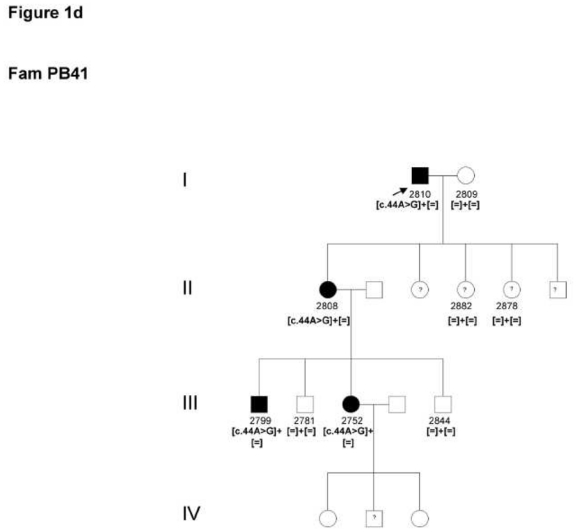
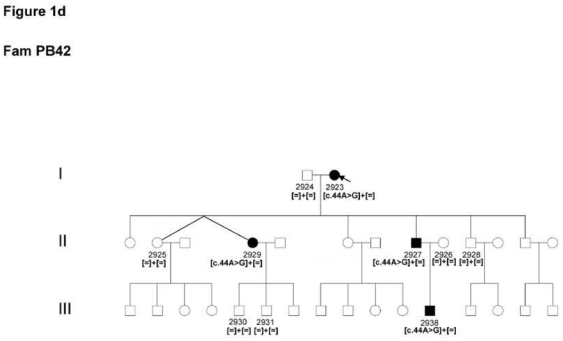
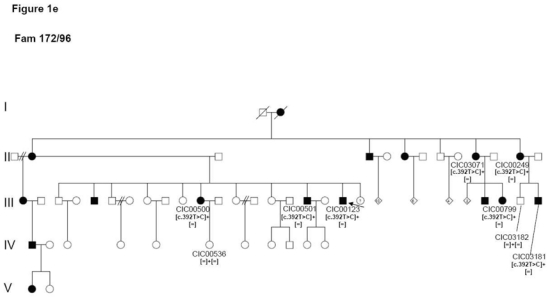
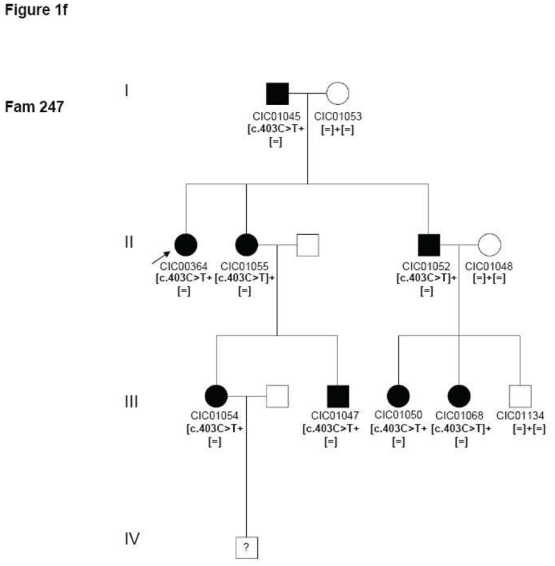
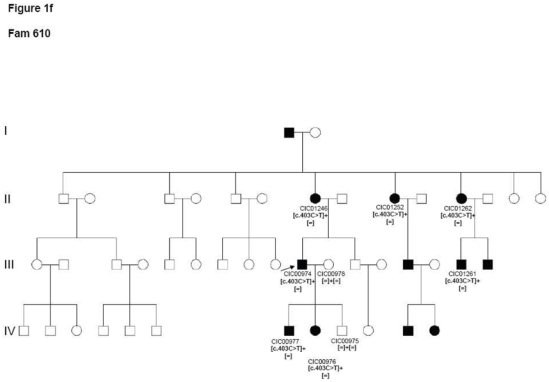
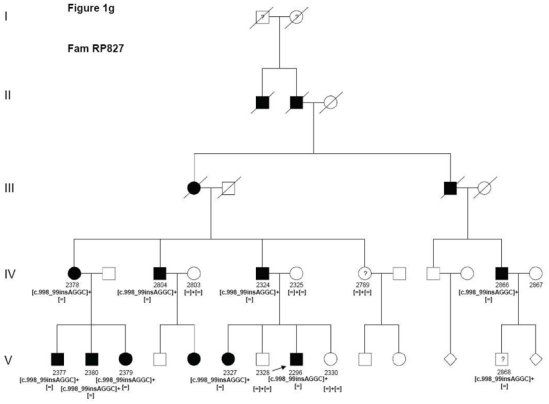
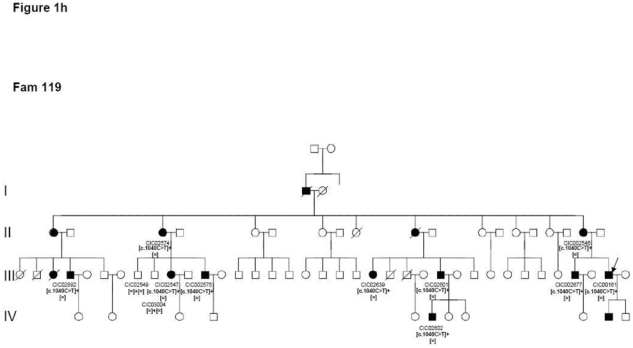
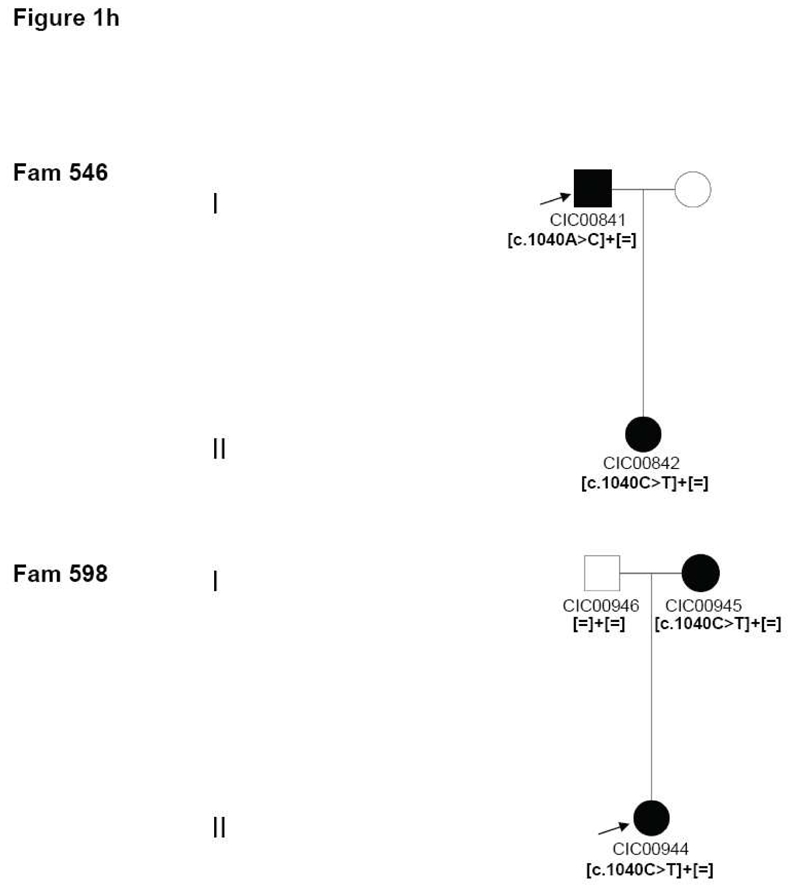
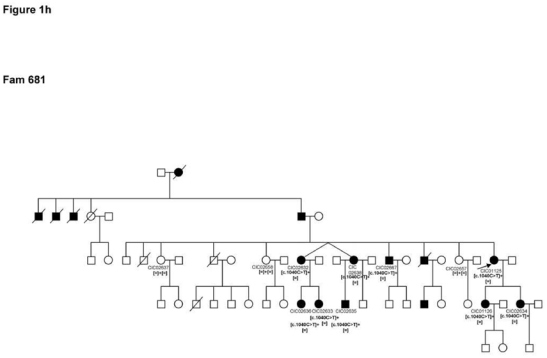
Pedigrees of adRP patients with RHO mutations and co-segregation in available family members. Filled symbols represent affected and unfilled unaffected persons. Squares indicate males, circles females. Arrows reflect the index patients.
Prevalence of different RHO mutations in France
Together our study on autosomal dominant RP patients from France showed that 16.5% revealed novel or known RHO mutations. Mutations locations revealed no specific hot spots since they involved all exons. However, three mutations occurred at least in two families indicating that the p.Asn15Ser, p.Arg135Trp and the p.Pro347Leu substitutions in RHO are frequent causes of RP in this population.
Phenotypic characteristics of patients with RHO mutation
Thirty affected subjects, between age 8 and 62, from the 13 families found with RHO mutation underwent complete clinical examination. Their phenotypic details are summarized in Table 2. The group of patients reported here shows 3 distinct phenotypes and resemble either class A or B mutants from the classification proposed by Cideciyan and co-workers 21:
Table 2.
Clinical features of affected members from familiies with ausosomal dominant retinitis pigmentosa (adRP) due to RHO mutations.
| Family and RHO mutation | Patient | Sex | Age of diagnos | Symptoms | Age at exam | BCVA OD/OS | Cataract | Fundus | OCT | VF | ERG ISCEV standards |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Family PB41 c.44A>G p.15Asn>Ser |
2752 III.12 |
F | Mild night blindness | 39 | 20/20 20/20 |
- | Bone spicules in Lower sector | Normal foveal lamination | Central scotoma (15°) Isoptre V4 60°N, 70°T |
50% of normal value for scotopic responses 60% of normal value for photopic responses No implicit time shift |
|
| 2808 II.2 |
F | Night blindness | 59 | 20/20 +2.50 (−1.25; 170°) 20/20 +2.00 (−0.50; 50°) |
- | Bone spicules in Lower sector | Normal foveal lamination | Isoptre V4 65°N, 70°T | 20% of normal value for scotopic responses 35% of normal value for photopic rresponses No implicit time shift |
||
|
Family PB42 c.44A>G p.15Asn>Ser |
2929 II.4 |
F | No night blindness | 60 | 20/25 +3.25 (−1.00; 144°) 20/25 +2.75 (−0.50; 10°) |
- | Bone spicules in Lower sector | Normal foveal lamination | Isoptre V4 80°N, 70°T | 30% of normal value for scotopic responses Photopic 30Hz ERG slightly reduced No implicit time shift |
|
| 2927 II.8 |
M | 30 | PVFI at 30 No night blindness |
52 | 20/20 +2.50 (−2.75; 20°) 20/20 +2.75 (−2.50; 170°) |
- | Bone spicules in lower sector Epiretinal membrane OD/OS |
Normal foveal lamination | Isoptre V4 80°N, 90°T | 30% of normal value for scotopic responses 80% of normal value for photopic responses No implicit time shift |
|
| 2938 III.12 |
M | Mild photophobia No night blindness |
28 | 20/20 −0.25 (−0.50; 15°) 20/20 −0.5 |
- | Bone spicules in Lower sector | Normal foveal lamination | Normal | Normal scotopic responses Photopic 30Hz ERG slightly reduced No implicit time shift |
||
|
Family 155 c.263T>C p.Leu88Pro novel |
CIC 00218 | M | 15 | Night blindness since childhood photophobia at age 59 followed by progressive loss of central vision | 62 | 20/63 20/80 |
IOL at age 48 | bone spicules 360° some areas of central atrophy | Foveal thinning | Isoptre V4 20° central ODS | Not detectable |
|
Family 96/172 c.392T>C p.Leu131Pro |
CIC 00123 | M | 10 | Night blindness since childhood | 27 | 20/32 +1.25(−1.25)85° 20/32 +1(−1)90° |
- | bone spicules 360° some areas of central atrophy, few white dots | Normal foveal lamination | Isoptre V4 OD 15° central OS<10° | Not detectable |
| CIC 00249 | F | 30 | Night blindness since childhood progressive decreased vision and photophobia | 56 | 20/400 +3.505(−1.75)90° 20/500 +3(−1.50)90° |
+ | bone spicules 360° some areas of central, atrophy, no ring on AF | Foveal thinning | Isoptre III4 20° central ODS | Not detectable | |
| CIC 00799 | F | Teens | Night blindness since childhood | 31 | 20/32 −1(−1)50° 20/32 −1(−1.25)135° |
- | Few peripheral RPE changes 360°, white dots, small perifoveal ring of hyperAF | Normal foveal lamination | Isoptre II4 110° horizontally and 90° vertically | No responses detectable in scotopic conditions, some residual flicker responses | |
| CIC 00500 | F | Teens | Night blindness since childhood | 43 | 20/40 +0.25(−0.75)120° 20/40 +0(−1)60° |
+ | bone spicules 360° some areas of central atrophy, white dots, small perifoveal ring of hyperAF | Normal foveal lamination | Isoptre III4 60° horizontally and 30° vertically | Not detectable | |
| CIC 00501 | M | 28 | Night blindness since childhood | 51 | 20/40 +0.75(−0.25)95 20/40 pl(−1.25)90° |
- | bone spicules 360° some areas of central atrophy, no ring on AF | Foveal thinning | Isoptre V4 20° | Not detectable | |
|
Family 247 c.403C>T p.Arg135Trp |
CIC 00364 | F | Night blindness since childhood | 52 | 20/40 +3.75(−0.25)35° 20/63 +5.75(−1)130° |
+ | bone spicules 360° some areas of central atrophy, no ring on AF | Foveal thinning | Isoptre III4 30° | Not detectable | |
|
Family 610 c.403C>T p.Arg135Trp |
CIC 00974 | M | 10 | Night blindness | 37 | 20/40 −8(−2.75)0° 20/40 −8(−2.25)175° |
- | Peripheral RPE/choroidal atrophy with bone spicules 360°, small perifoveal ring of hyperAF | Foveal thinning | Isoptre III4 170° horizontal x100° vertical | Not detectable |
| CIC 00976 | F | 8 | Night blindness | 8 | 20/63 −2(−3.25)0° 20/40 +0.75(−2.25)180° |
- | Nearly normal fundus, Perifoveal ring of hyperAF | Normal foveal lamination | Isoptre III4 140° horizontal; 100° vertical | Both scotopic and photopic amplitudes reduction* | |
| CIC 00977 | M | 10 | Night blindness | 11 | 20/20 +0.25(−2.75)5° 20/20 +0.25(−2.50)170° |
- | Few RPE changes with no bone spicules, Bilateral CME, Perifoveal ring of hyperAF | CME | Isoptre III4 140° horizontal X120° vertical | Scotopic responses 10% of normal Photopic responses 50% normal Both amplitude reduction and implicit time shift |
|
|
Family 475 c.620T>A p.Met207Lys |
CIC 00716 | M | 23 | None | 23 | 20/13 0(−1)160° 20/15 0(−1.50)15° |
- | Preserved macula besides some perifoveolar RPE clumps; Moderate salt and pepper appearance of retinal periphery | Normal foveal lamination | Normal | Scotopic response amplitudes 80% of normal, normal photopic responses, no implicit time shift |
| CIC 00715 | F | 38 | Decreased VA Some degree of night vision disturbances |
46 | 20/25 +1(−0.50)160° HM | - | Patchy chorioretinal atrophy with some RPE clumps in posterior pole and mid periphery No pale disc and no narrowing of blood vessels salt and pepper aspect in retinal periphery; no bone spicules |
OD normal foveal lamination OS foveal thinning |
OD normal OS normal peripheral isoptre |
65% of normal for scotopic response amplitudes and 90% for scotopic responses; No implicit time shift |
|
| CIC 00717 | F | 40 | Night blindness since age 40 Decreased VA |
58 | 20/200 +1.75(−0.50)20° 20/25 +2.25(−0.50)140° |
+ | Patchy chorioretinal atrophy with some RPE clumps in posterior pole and mid periphery No pale disc and no narrowing of blood vessels, salt and pepper aspect in retinal periphery; no bone spicules |
OD foveal thinning OS normal foveal lamination |
Normal peripheral isoptre | Not performed | |
| CIC 02599 | F | 26 | None | 26 | 20/15 ODS with no correction | - | Normal aspect of posterior poles besides some perifoveolar RPE clumps and one small area of atrophy; Moderate salt and pepper appearance of retinal periphery | Normal foveal lamination | normal | Not performed | |
|
Family RP827 c.998_999insAGGC p. Ser333GlyfsX22 |
2296 V.8 |
M | 13 | Night blindness at early childhood PVFI at 13 Intense photophobia | 34 | 20/40 +5.50 (−0.50; 165°) 20/30 +5.00 (−0.50; 170°) |
+ | Bones spicules 360° CME |
CME | 15° | Not detectable |
| 2327 V.6 |
F | 11 | Night blindness at 11 PVFI at 20 Photophobia at 25 |
38 | 20/100 +5.00 20/400 +6.00 (−0.75; 60°) |
+ | Bones spicules 360° Foveal photoreceptor loss |
Foveal thinning | 15° | Not detectable except for residual 30 Hz flicker ERG | |
| 2379 V.3 |
F | childhoo d | Night blindness since early childhood Photophobia at 5 |
45 | 20/30 (−2.00; 90°) 20/40 (−1.25; 105°) |
+ | Bones spicules 360° small Perifoveal ring of hyperAF | Foveal thinning | 20° | Not detectable | |
| 2324 IV.5 |
M | 50 | Night blindness at early childhood PVFI at 25 Photophobia | 60 | 20/400 +1.50 (−1.00; 95°) 20/200 +2.00 (−1.00; 85°) |
IOL | Bones spicules 360° | Foveal thinning | 10° | Not detectable except for residual 30 Hz flicker ERG | |
|
Family 394 c.1031A>C p.Gln344Pro, novel |
CIC 00590 | F | 32 | Night blindness since age 4 PVFI since age 19 |
53 | 20/125 −1.50(−1)35° 20/200 −1.25(−1)150° |
IOL at 48 | bone spicules 360°, no ring on AF, perifoveal atrophy | Foveal thinning | 20° | Not detectable |
| CIC 00592 | H | 13 | Moderate night blindness | 13 | 20/25 +1.5(−1.75)170° 20/32 +2(−2.75)175° |
- | peripheral RPE changes 360° with white dots, Perifoveal ring of hyperAF | Normal foveal lamination | normal | Scotopic responses 10% of normal Photopic responses 80% normal Both amplitude reduction and implicit time shift |
|
|
Family 119 c.1040C>T p.Pro347Leu |
CIC 00161 | H | 11 | Night blindness | 42 | 20/32 plano(−1)90° 20/25 plano(−0.50)80° |
- | Bone spicules 360°, Perifoveal ring of hyperAF | Normal foveal lamination | 20° | Not detectable |
|
Family 546 c.1040C>T p.Pro347Leu |
CIC 00841 | H | Teens | Night blindness since early childhood, PVFI at 25, recent photophobia | 42 | 20/40 0(−1)115° 20/63 −1(−0.25)15° |
+ | bone spicules 360° White dots Bilateral CME, small perifoveal ring of hyperAF |
CME | Isoptre III4 20° |
Not detectable |
|
Family 598 c.1040C>T p.Pro347Leu |
CIC 00944 | F | 10 | Night blindness | 14 | 20/20 +2(−1.25)5° 20/20+1.5(−05)10° |
- | Some peripheral RPE changes over 360°, CME, Perifoveal ring of hyperAF | CME | normal | Scotopic responses 10% of normal Photopic responses 80% normal Both amplitude reduction and implicit time shift |
| CIC 00945 | F | 9 | Night blindness | 43 | 20/32 +3.5(−1.25)10° 20/32 +3.75(−0.75)5° |
- | bone spicules 360°, no ring on AF | Foveal thinning | 20° | Not detectable | |
|
Family 681 c.1040C>T p.Pro347Leu |
CIC 01125 | F | 49 | Night blindness | 56 | 20/400 +1.50(−0.75)20° 20/200 +3.25(−0.75)10° |
OD IOL OS + |
Few bone spicules 360° some areas of central atrophy, incomplete perifoveal ring of hyperAF | Foveal thinning | Isoptre III4 40° |
Not detectable |
| CIC 01126 | F | 29 | Night blindness | 36 | 20/20 20/20 |
- | Few bone spicules 360° Perifoveal ring of hyperAF |
Normal foveal lamination | Isoptre III4: 150° horizontally X 60° vertically | No detectable scotopic responses Some residual flicker responses |
PVFI = Peripheral Visual Field Impairment, VF = Visual Field, IOL = Intraocular lens, BCVA: Best Corrected Visual Acuity, OD/OS: Right eye/Left eye, RPE: Retinal pigment epithelium, HM: Hand Motion; CME: Cystoid Macular Edema;
ERG performed with skin electrodes which precluded us to have a precise quantification of abnormalities, AF: autofluoresce., OCT: Optical Coherence Tomography.
a generalized rod-cone dysfunction observed in patients carrying mutations (p.Leu88Pro, p.Leu131Pro, p.Arg135Trp, p.Ser334GlyfsX20, p.Gln344Pro, p.Pro347Leu), which resemble the class A mutants.
a sector RP associated with the p. Asn15Ser mutation and
a restricted chorioretinal dystrophy predominant at the posterior pole associated with the p.Met207Lys substitution. Due to the more restricted phenotype, we classified the two latter mutations as class B mutations.
In generalized forms, symptoms are classical for RP with no obvious phenotype/genotype differences and are dominated by night blindness, from early childhood, progressive peripheral visual field constriction and late photophobia. Age at time of diagnosis varies from 8 to 49 with a majority within the teenage years, earlier than restricted diseases. Central vision ranges from 20/20 to 20/400. It decreases with age, after peripheral visual field impairment, and is usually relatively conserved up to the 5th decade. However, in 8/21 patients, atrophic changes within the macula occur after the mid-twenties and compromised further central vision. Some degree of cataract or intraocular lens is present as early as 34 in 11/21 patients. Fundus examination, shows in most patients classical RPE changes in the periphery with intraretinal pigment migrations, sign of photoreceptor cell death, increasing with age. White dots are present in 5 patients who are 43 or younger associated with three genotypes are in our series: p.Leu131Pro, p.Gln344Pro and p.Pro347Leu. OCT findings are summarized in Table 2. There was no correlation between OCT abormalities and genotype. Cystoid Macular Edema (CME) is present in 4/30 patients in association with 4 different genotypes. A perifoveal ring of hyper-autofluorescence is present 13/18 patients for whom fundus autofluorescence imaging has been performed. Absence of this ring is associated with irregular loss of autofluorescence within the macula in relation with atrophic changes (Figure 3). ERG responses are usually undetectable for both scotopic and photopic recordings after 30 or show only residual photopic Flicker responses. When ERGs are detectable, in younger patients, they usually show more decreased amplitudes for scotopic than photopic responses with implicit time shift, consistent with generalized rod-cone dysfunction.
Figure 3.
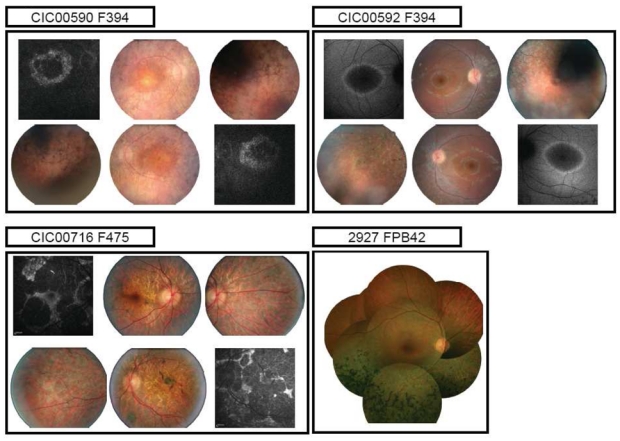
Fundus and autofluorescence pictures of 3 index patients with distinct adRP phenotypes (diffuse, sector RP and restricted chorioretinal atrophy)
Sector RP was seen in 2 families (PB41 and PB42) carrying the same p.Asn15Ser change. Five patients, from age 28 to 60, underwent full ophthalmic examination. Night blindness is an inconstant sign in these subjects who all retain a normal central vision with inferior peripheral field defect correlated with fundus abnormalities. ERG responses show decreased scotopic responses with additional photopic abnormalities in some patients. There is however no implicit time shift consistent with a restricted rod-cone dysfunction.
One additional family, F475 with a novel p.Met207Arg, shows also restricted chorioretinal degeneration. Phenotype-genotype correlations are described in more details elsewhere (Audo et al., 2010, in press). Briefly, onset of symptoms appears in the fourth decade in this family with moderate night blindness and asymmetric visual loss. Affected family members show patchy areas of chorioretinal atrophy within the posterior pole (Figure 3) with decreased ERG response amplitudes for both scotopic and photopic responses and no implicit time shift consistent with restricted disease.
DISCUSSION
The current study reports mutation spectrum on the rhodopsin gene in a cohort of patients from 2 major French centres and further outlines phenotypic variability associated with rhodopsin mutation showing both, generalized or sectorial retinal degeneration. To the best of our knowledge to date only two studies on RHO mutations in a French cohort were published: One describing the prevalence of RHO mutations in Southern France and the other reported on the identification of 5 new mutations with no information on prevalence and ethnic origin 13, 26.
The overall prevalence of RHO mutations in our cohort is 16.5%. This is consistent with previous reports on European cohorts including Spain (20%)10, Germany (16%)11, Italy (16%)12 and Southern France (10%).13 This is higher than reports from China (2–7%) 14, 15, 27, Japan (0–6%) 16 India (0 – 2%) 17 and South-Africa (7%) 18. Studies from the UK and Norway revealed higher numbers with 30–50% 8, 9, 28. However, the studied cohorts were small (12–20 families) and thus these results must be validated in larger cohorts. In the US population RHO mutations were shown to account for up to 30% of adRP 3–7. The prevalence of the p.Pro23His mutation in the US has been reported as high as 12% of adRP due to a founder effect from a common British ancestor 29. This mutation has never been found in European cohorts of adRP 30, including the current report, nor in Asian cohorts 31, 32, which would account for differences in the overall RHO mutation prevalence between the American population and reports from other populations.
Three novel changes were identified in the current study: p.Leu88Pro, p.Met207Lys and Gln344Pro.
The p.Leu88Pro substitution leads to a severe generalized rod-cone dystrophy phenotype in the patients. Disease-causing mutations have already been reported for the surrounding residues (namely p.Val87Asp and p.Gly89Asp) and misfolding has been hypothesised as a pathogenic mechanism 4, 33. The Leucine in 88 is located within the alpha helix of the second transmembrane domain of rhodopsin. The residue at this position is not invariant among Metazoan organism (Figure 2b), but shows always hydrophobic characteristics, necessary for the maintenance of this alpha helix. The substitution of the leucine by a proline would induce a kink in the helix and destabilize the protein through rhodopsin misfolding. This would classify the p.Leu88Pro within class II after Mendes and colleagues 19.
The novel p.Met207Arg substitution was associated with unusual chorioretinal atrophy. Mutation consequences are discussed elsewhere (Audo et al., 2010, in press) and would suggest a change in sterical constraints within the retinal binding pocket.
The c.1031A>C change in exon 5 leading to a p.Gln344Pro substitution was associated with a severe generalized rod-cone dystrophy. Gln at this position is evolutionary highly conserved (Figure 2b). It is located in the C-term external loop and it is unlikely that mutations in this residue would induce a misfolding. This would classify our novel change p.Gln344Pro in class I after Mendes and colleagues 19. Previously a c.1030C>T change leading to a p.Gln344Stop was associated with normal phototransduction function but with mislocalization 34. Furthermore, Tai and co-workers identified the direct interaction between a dynein light-chain subunit and the C-terminus of rhodopsin, which is important for the correct protein transport of post-Golgi rhodopsin-containing vesicles along the microtubules up to the outer segment 35. Different C-terminal mutations were unable to interact with this domain and thus led to a trafficking defect. A similar mechanism can be advocated for the novel reported change p.Gln344Pro.
The 10 other families identified with RHO mutation showed already described changes. The p.Asn15Ser mutation was identified in two different families from a similar region of France and thus represents probably a founder effect. Asn15 represent one of the important N-glycosylation sites of RHO. Thus the underlying pathogenic mechanism of the p.Asn15Ser was proposed to be trafficking defect 36.
The p.Leu131Pro mutation was identified in a large family from Martinique with typical diffuse rod-cone dystrophy, type A from Cideciyan and co-workers 21. This amino-acid substitution is assumed to lead to misfolding 37. Since this exchange has also been previously reported in another study from France 26, it may represent a major mutation in the affected French population.
The p.Arg135Trp was found in two unrelated families and was associated with typical severe diffuse rod-cone dystrophy, type A from Cideciyan and co-workers 21 as previously reported 38 39. Of notes, none of the examined patients in these 2 families demonstrated the white dots previously described in association with this genotype 39, 40. An explanation would be that the examined patients were either too young or too old to exhibit this distinct feature since, Oh and co-worker have reported the transient nature of these white dots appearing in the second decades of life then fading to leave place to RPE atrophy and bone spicules. It is also noteworthy that these white dots, which are located at the level of the RPE, are not specific of the p.Arg135Trp mutation since it was also seen in association with other RHO mutations in our series and may represent a non-specific sign of photoreceptor degeneration (see table 2 on clinical data).
The p.Pro347Leu mutation was the most prevalent, found in 4 families which would represent 5% of our adRP families. This mutation has also been reported in other populations 8, 10, 29, 32, 41. Although the 4 families studied herein were unrelated and from different geographical origin, a founder effect cannot be excluded. Haplotype analysis was not performed for this study. However, the gene location is a known hotspot through a higher probability of C>T transition due to a CpG sequence 3 and 6 disease causing amino-acid substitutions have reported at this location (see http://www.retina-international.org/sci-news/rhomut.htm). Again, it was suggested that for these substitutions a trafficking defect represent the pathogenic mechanism. Patients carrying the p.Pro347Leu mutation have a comparable phenotype as patients carrying the p.Gln344Pro and p.Ser334GlnfsX20 changes, all being located at the C-terminus, with early onset-night blindness, and generalized severe rod-cone dystrophy with loss of central vision in the 5th decades. The severity of the disease associated with C-terminal changes within the cytoplasmic domain is well documented in the literature 42–44 showing a worse prognosis compared in particular to the p.Pro23His mutation located in the N-terminal intradiscal/extracellular portion of the protein 43, 44. Our cohort in whom genotype-phenotype correlation was performed is still too small to judge the severity associated with a specific mutation but recurrent follow-up will further address this question.
One additional criterion that will need to be further precisely evaluated is the course of macular involvement: perifoveal and foveal atrophy is not uncommon in our series (see table 2 with clinical details) as well as cystoid macular edema which was present in 4/31 patients with no genotype-specificity. These macular changes are responsible for decreased central vision and their prevention should be the major target of future therapeutic interventions.
Further longitudinal studies will precise the course of the disease for each genotype and will help identifying suitable markers and therapeutic windows for photoreceptor rescue, gene replacement or cell based therapies.
Acknowledgments
The authors are grateful to patients and family members described in this study, to Thierry Léveillard, Dominique Santiard-Baron, Christine Chaumeil and clinical staff for their help in DNA collection and clinical staff from the Centre National de Référence Maladies Rares in Montpellier, Béatrice Bocquet and Delphine Coustes-Chazalette for their help with DNA collection, Gabor Mátyás, Institute of Medical Genetics in Zurich for providing the purification and sequencing protocol used herein. The project was financially supported by Foundation Fighting Blindness (I.A.), ANR (SS.B), Foundation Voir et Entendre and BQR, Université Pierre et Marie Curie6 (C.Z), PHRC national adRP (C H).
Footnotes
Data regarding family 475 have been presented as a poster at the ARVO 2009 meeting and are also published elsewhere (Audo et al. 2010, in press).
References
- 1.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 2.Dryja TP, McEvoy JA, McGee TL, Berson EL. Novel rhodopsin mutations Gly114Val and Gln184Pro in dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2000;41:3124–3127. [PubMed] [Google Scholar]
- 3.Dryja TP, Hahn LB, Cowley GS, McGee TL, Berson EL. Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991;88:9370–9374. doi: 10.1073/pnas.88.20.9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sung CH, Davenport CM, Hennessey JC, et al. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991;88:6481–6485. doi: 10.1073/pnas.88.15.6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Macke JP, Davenport CM, Jacobson SG, et al. Identification of novel rhodopsin mutations responsible for retinitis pigmentosa: implications for the structure and function of rhodopsin. Am J Hum Genet. 1993;53:80–89. [PMC free article] [PubMed] [Google Scholar]
- 6.Sohocki MM, Daiger SP, Bowne SJ, et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat. 2001;17:42–51. doi: 10.1002/1098-1004(2001)17:1<42::AID-HUMU5>3.0.CO;2-K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sullivan LS, Bowne SJ, Birch DG, et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest Ophthalmol Vis Sci. 2006;47:3052–3064. doi: 10.1167/iovs.05-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inglehearn CF, Keen TJ, Bashir R, et al. A completed screen for mutations of the rhodopsin gene in a panel of patients with autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1992;1:41–45. doi: 10.1093/hmg/1.1.41. [DOI] [PubMed] [Google Scholar]
- 9.Grondahl J, Riise R, Heiberg A, Leren T, Christoffersen T, Bragadottir R. Autosomal dominant retinitis pigmentosa in Norway: a 20-year clinical follow-up study with molecular genetic analysis. Two novel rhodopsin mutations: 1003delG and I179F. Acta Ophthalmol Scand. 2007;85:287–297. doi: 10.1111/j.1600-0420.2006.00820.x. [DOI] [PubMed] [Google Scholar]
- 10.Milla E, Maseras M, Martinez-Gimeno M, et al. Genetic and molecular characterization of 148 patients with autosomal dominant retinitis pigmentosa (ADRP) Arch Soc Esp Oftalmol. 2002;77:481–484. [PubMed] [Google Scholar]
- 11.Bunge S, Wedemann H, David D, et al. Molecular analysis and genetic mapping of the rhodopsin gene in families with autosomal dominant retinitis pigmentosa. Genomics. 1993;17:230–233. doi: 10.1006/geno.1993.1309. [DOI] [PubMed] [Google Scholar]
- 12.Ziviello C, Simonelli F, Testa F, et al. Molecular genetics of autosomal dominant retinitis pigmentosa (ADRP): a comprehensive study of 43 Italian families. J Med Genet. 2005;42:e47. doi: 10.1136/jmg.2005.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bareil C, Hamel C, Pallares-Ruiz N, Arnaud B, Demaille J, Claustres M. Molecular analysis of the rhodopsin gene in southern France: identification of the first duplication responsible for retinitis pigmentosa, c. 998999ins4. Ophthalmic Genet. 1999;20:173–182. doi: 10.1076/opge.20.3.173.2282. [DOI] [PubMed] [Google Scholar]
- 14.Zhao K, Xiong S, Wang L, Wang L, Cui Y, Wang Q. Novel rhodopsin mutation in a Chinese family with autosomal dominant retinitis pigmentosa. Ophthalmic Genet. 2001;22:155–162. doi: 10.1076/opge.22.3.155.2225. [DOI] [PubMed] [Google Scholar]
- 15.Zhang XL, Liu M, Meng XH, et al. A complete screen for mutations of the rhodopsin gene in a panel of Chinese patients with autosomal dominant retinitis pigmentosa. Chin Med Sci J. 2005;20:30–34. [PubMed] [Google Scholar]
- 16.Ando Y, Ohmori M, Ohtake H, et al. Mutation screening and haplotype analysis of the rhodopsin gene locus in Japanese patients with retinitis pigmentosa. Mol Vis. 2007;13:1038–1044. [PMC free article] [PubMed] [Google Scholar]
- 17.Gandra M, Anandula V, Authiappan V, et al. Retinitis pigmentosa: mutation analysis of RHO, PRPF31, RP1, and IMPDH1 genes in patients from India. Mol Vis. 2008;14:1105–1113. [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts L, Ramesar R, Greenberg J. Low frequency of rhodopsin mutations in South African patients with autosomal dominant retinitis pigmentosa. Clin Genet. 2000;58:77–78. doi: 10.1034/j.1399-0004.2000.580114.x. [DOI] [PubMed] [Google Scholar]
- 19.Mendes HF, van der Spuy J, Chapple JP, Cheetham ME. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. 2005;11:177–185. doi: 10.1016/j.molmed.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 20.Heckenlively JR, Rodriguez JA, Daiger SP. Autosomal dominant sectoral retinitis pigmentosa. Two families with transversion mutation in codon 23 of rhodopsin. Arch Ophthalmol. 1991;109:84–91. doi: 10.1001/archopht.1991.01080010086038. [DOI] [PubMed] [Google Scholar]
- 21.Cideciyan AV, Hood DC, Huang Y, et al. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc Natl Acad Sci U S A. 1998;95:7103–7108. doi: 10.1073/pnas.95.12.7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M. ISCEV Standard for full-field clinical electroretinography (2008 update) Doc Ophthalmol. 2009;118:69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 23.Hood DC, Bach M, Brigell M, et al. ISCEV guidelines for clinical multifocal electroretinography (2007 edition) Doc Ophthalmol. 2008;116:1–11. doi: 10.1007/s10633-007-9089-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neidhardt J, Barthelmes D, Farahmand F, Fleischhauer JC, Berger W. Different amino acid substitutions at the same position in rhodopsin lead to distinct phenotypes. Invest Ophthalmol Vis Sci. 2006;47:1630–1635. doi: 10.1167/iovs.05-1317. [DOI] [PubMed] [Google Scholar]
- 26.Souied E, Gerber S, Rozet JM, et al. Five novel missense mutations of the rhodopsin gene in autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1994;3:1433–1434. doi: 10.1093/hmg/3.8.1433. [DOI] [PubMed] [Google Scholar]
- 27.Chan WM, Yeung KY, Pang CP, et al. Rhodopsin mutations in Chinese patients with retinitis pigmentosa. Br J Ophthalmol. 2001;85:1046–1048. doi: 10.1136/bjo.85.9.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inglehearn CF, Tarttelin EE, Plant C, et al. A linkage survey of 20 dominant retinitis pigmentosa families: frequencies of the nine known loci and evidence for further heterogeneity. J Med Genet. 1998;35:1–5. doi: 10.1136/jmg.35.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dryja TP, McGee TL, Hahn LB, et al. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N Engl J Med. 1990;323:1302–1307. doi: 10.1056/NEJM199011083231903. [DOI] [PubMed] [Google Scholar]
- 30.Farrar GJ, Kenna P, Redmond R, et al. Autosomal dominant retinitis pigmentosa: absence of the rhodopsin proline----histidine substitution (codon 23) in pedigrees from Europe. Am J Hum Genet. 1990;47:941–945. [PMC free article] [PubMed] [Google Scholar]
- 31.Nakazawa M, Kikawa-Araki E, Shiono T, Tamai M. Analysis of rhodopsin gene in patients with retinitis pigmentosa using allele-specific polymerase chain reaction. Jpn J Ophthalmol. 1991;35:386–393. [PubMed] [Google Scholar]
- 32.Fujiki K, Hotta Y, Hayakawa M, et al. Point mutations of rhodopsin gene found in Japanese families with autosomal dominant retinitis pigmentosa (ADRP) Jpn J Hum Genet. 1992;37:125–132. doi: 10.1007/BF01899733. [DOI] [PubMed] [Google Scholar]
- 33.Sung CH, Schneider BG, Agarwal N, Papermaster DS, Nathans J. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991;88:8840–8844. doi: 10.1073/pnas.88.19.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sung CH, Makino C, Baylor D, Nathans J. A rhodopsin gene mutation responsible for autosomal dominant retinitis pigmentosa results in a protein that is defective in localization to the photoreceptor outer segment. J Neurosci. 1994;14:5818–5833. doi: 10.1523/JNEUROSCI.14-10-05818.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tai AW, Chuang JZ, Bode C, Wolfrum U, Sung CH. Rhodopsin’s carboxy-terminal cytoplasmic tail acts as a membrane receptor for cytoplasmic dynein by binding to the dynein light chain Tctex-1. Cell. 1999;97:877–887. doi: 10.1016/s0092-8674(00)80800-4. [DOI] [PubMed] [Google Scholar]
- 36.Kranich H, Bartkowski S, Denton MJ, et al. Autosomal dominant ‘sector’ retinitis pigmentosa due to a point mutation predicting an Asn-15-Ser substitution of rhodopsin. Hum Mol Genet. 1993;2:813–814. doi: 10.1093/hmg/2.6.813. [DOI] [PubMed] [Google Scholar]
- 37.Fuchs S, Kranich H, Denton MJ, et al. Three novel rhodopsin mutations (C110F, L131P, A164V) in patients with autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1994;3:1203. doi: 10.1093/hmg/3.7.1203. [DOI] [PubMed] [Google Scholar]
- 38.Pannarale MR, Grammatico B, Iannaccone A, et al. Autosomal-dominant retinitis pigmentosa associated with an Arg-135-Trp point mutation of the rhodopsin gene. Clinical features and longitudinal observations. Ophthalmology. 1996;103:1443–1452. doi: 10.1016/s0161-6420(96)30485-5. [DOI] [PubMed] [Google Scholar]
- 39.Oh KT, Oh DM, Weleber RG, et al. Genotype-phenotype correlation in a family with Arg135Leu rhodopsin retinitis pigmentosa. Br J Ophthalmol. 2004;88:1533–1537. doi: 10.1136/bjo.2004.043653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Souied E, Soubrane G, Benlian P, et al. Retinitis punctata albescens associated with the Arg135Trp mutation in the rhodopsin gene. Am J Ophthalmol. 1996;121:19–25. doi: 10.1016/s0002-9394(14)70530-6. [DOI] [PubMed] [Google Scholar]
- 41.Greenberg J, Franz T, Goliath R, Ramesar R. A photoreceptor gene mutation in an indigenous black African family with retinitis pigmentosa identified using a rapid screening approach for common rhodopsin mutations. S Afr Med J. 1999;89:877–878. [PubMed] [Google Scholar]
- 42.Berson EL, Rosner B, Sandberg MA, Weigel-DiFranco C, Dryja TP. Ocular findings in patients with autosomal dominant retinitis pigmentosa and rhodopsin, proline-347-leucine. Am J Ophthalmol. 1991;111:614–623. doi: 10.1016/s0002-9394(14)73708-0. [DOI] [PubMed] [Google Scholar]
- 43.Sandberg MA, Weigel-DiFranco C, Dryja TP, Berson EL. Clinical expression correlates with location of rhodopsin mutation in dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1995;36:1934–1942. [PubMed] [Google Scholar]
- 44.Oh KT, Longmuir R, Oh DM, et al. Comparison of the clinical expression of retinitis pigmentosa associated with rhodopsin mutations at codon 347 and codon 23. Am J Ophthalmol. 2003;136:306–313. doi: 10.1016/s0002-9394(03)00206-x. [DOI] [PubMed] [Google Scholar]