

Abstract
Fidelity of chromosome segregation is ensured by a tension-dependent error correction system that prevents stabilization of incorrect chromosome–microtubule attachments. Unattached or incorrectly attached chromosomes also activate the spindle assembly checkpoint, thus delaying mitotic exit until all chromosomes are bioriented. The Aurora B kinase is widely recognized as a component of error correction. Conversely, its role in the checkpoint is controversial. Here, we report an analysis of the role of Aurora B in the spindle checkpoint under conditions believed to uncouple the effects of Aurora B inhibition on the checkpoint from those on error correction. Partial inhibition of several checkpoint and kinetochore components, including Mps1 and Ndc80, strongly synergizes with inhibition of Aurora B activity and dramatically affects the ability of cells to arrest in mitosis in the presence of spindle poisons. Thus, Aurora B might contribute to spindle checkpoint signalling independently of error correction. Our results support a model in which Aurora B is at the apex of a signalling pyramid whose sensory apparatus promotes the concomitant activation of error correction and checkpoint signalling pathways.
Keywords: centromere, hesperadin, kinetochore, reversine, tension
Introduction
The metaphase-to-anaphase transition is an irreversible transition of the cell cycle. Satisfaction of the spindle assembly checkpoint and subsequent activation of the ubiquitin ligase anaphase promoting complex/cyclosome (APC/C) ultimately lead to the destruction of cyclin B and securin, causing mitotic exit and sister chromatid separation (Musacchio and Salmon, 2007). To be accurate, chromosome segregation requires that all sister chromatid pairs are bioriented, which implies that the two sister chromatids of each chromosome are bound to opposite spindle poles.
Kinetochores link chromosomes to microtubules (Cheeseman and Desai, 2008; Santaguida and Musacchio, 2009). The so-called KMN network (from the initials of its Knl1, Mis12 and Ndc80 subcomplexes), a 10-subunit assembly, provides the microtubule-binding interface of kinetochores (Cheeseman et al, 2006; DeLuca et al, 2006). Kinetochores host an error correction mechanism that clears improper kinetochore–microtubule attachments. The observation that syntelic attachments (both kinetochores bound to the same pole) are intrinsically unstable, unless tension is artificially exercised on them, led to propose that tension is required to stabilize kinetochore–microtubule attachments (Nicklas and Koch, 1969).
Aurora B (Ipl1 in Saccharomyces cerevisiae), a serine/threonine (S/T) kinase, is a subunit of a chromosome passenger complex that is recruited to centromeres during mitosis (Carmena et al, 2009). Aurora B is a crucial component of a tension sensor at centromeres and kinetochores and its depletion or inhibition results in the accumulation of mal-attachments (Biggins and Murray, 2001; Tanaka et al, 2002).
Kinetochores also host the spindle assembly checkpoint, whose effector, the mitotic checkpoint complex (MCC), prevents APC/C activation until all chromosomes have bioriented (reviewed in Musacchio and Salmon, 2007). Checkpoint components include kinases, such as Bub1, BubR1, Mps1 and Prp4, as well as protein–protein interaction components, such as Mad1, Mad2 and the Rod–Zwilch–Zw10 complex (RZZ) (Musacchio and Salmon, 2007). Within the MCC, Mad2, Bub3 and BubR1 form a complex with Cdc20, an APC/C co-activator required for targeting crucial APC/C substrates at the metaphase–anaphase transition. When engaged in the MCC, Cdc20 is unable to target its substrates, so that entry into anaphase becomes inhibited (Musacchio and Salmon, 2007).
The exact relationship between tension-dependent error correction and checkpoint status is elusive (Khodjakov and Rieder, 2009; Nezi and Musacchio, 2009; Santaguida and Musacchio, 2009; Khodjakov and Pines, 2010; Lampson and Cheeseman, 2010; Maresca and Salmon, 2010). It has been proposed that the spindle checkpoint is exquisitely sensitive to microtubule attachment, regardless of whether the attachment is under tension (Khodjakov and Rieder, 2009; Khodjakov and Pines, 2010). Within this scheme, error correction and the checkpoint are distinct molecular entities, monitoring lack of tension or attachment, respectively, and the role of Aurora B in the checkpoint is indirect: error correction, which is Aurora B dependent, generates conditions, likely including unattached kinetochores, which in turn activate an Aurora B-independent checkpoint (Khodjakov and Rieder, 2009; Khodjakov and Pines, 2010) (model 1, Figure 1A). In an alternative view, the spindle checkpoint and error correction are viewed as co-regulated phenomena, both of which require Aurora B (Santaguida and Musacchio, 2009; Maresca and Salmon, 2010). According to this view, tensionless kinetochores (including unattached or incorrectly attached kinetochores) signal to the SAC in an Aurora B-dependent manner. Concomitantly, Aurora B activity is required to prevent premature stabilization of kinetochore–microtubule attachments, thus protecting from errors (model 2, Figure 1B).
Figure 1.

Schematic description of role of Aurora B in error correction and the spindle checkpoint. (A) Model 1: Aurora B is required for error correction. Correction generates unattached kinetochores, or incompletely attached kinetochores, re-activating the checkpoint. Aurora B does not contribute to checkpoint signalling from unattached kinetochores. Thus, in model 1 Aurora B contributes extrinsically to the checkpoint by generating, upon correction, a condition that supports the checkpoint, such as unattached kinetochores. SAC, spindle assembly checkpoint. (B) Model 2: In this scheme, Aurora B is additionally required for the checkpoint response from unattached kinetochores, that is it is an intrinsic component of the checkpoint. (C) A test for model 1. When Aurora B is inhibited, error correction is impaired, and therefore the checkpoint response from an incorrect attachment cannot fire. If a spindle poison is added to create an unattached kinetochore independently of error correction, the checkpoint response is normal even when Aurora B has been inhibited. (D) The same test is applied to model 2. In this case, unattached kinetochores cannot bypass inhibition of Aurora B, because Aurora B is also required for the checkpoint response from unattached kinetochores.
Regardless of which camp is chosen, it is generally agreed that microtubules are required for creating attachment and tension, so that in their absence (e.g. in the presence of microtubule-depolymerizing agents such as nocodazole), the checkpoint cannot be satisfied. This allowed the development of a test to assess models 1 and 2. The original implementation of this test was utilized to conclude that Ipl1/Aurora B is not involved in the checkpoint response from unattached kinetochores in S. cerevisiae (Biggins and Murray, 2001) (Figure 1C and D). In brief, the goal of the test is to combine inhibition of Aurora B with microtubule depolymerization by spindle poisons. In case of model 1, this predicts that the checkpoint should work normally under these conditions, because the function of Aurora B in the creation of unattached kinetochores during error correction is bypassed by microtubule depolymerization. Conversely, loss of potency of the checkpoint response when inhibiting Aurora B in the presence of unattached kinetochores would confirm an intrinsic role in the checkpoint independently of error correction (Figure 1C and D). Despite the availability of this assay, however, the controversy continued to flourish, largely because the precise conditions to neutralize the effects from impaired error correction on the checkpoint response when inhibiting Aurora B have not been standardized, and the results accumulated apparently in support of each of the two competing hypotheses (Biggins and Murray, 2001; Kallio et al, 2002; Ditchfield et al, 2003; Hauf et al, 2003; Petersen and Hagan, 2003; King et al, 2007; Vader et al, 2007; Vanoosthuyse and Hardwick, 2009).
Recently, however, it was shown that a rigorous assessment of whether Aurora B is implicated in checkpoint signalling through the test in Figure 1 requires that microtubules are completely removed, which is only true at very high concentrations of microtubule-depolymerizing drugs (Yang et al, 2009). By definition, the checkpoint cannot be satisfied at very high concentrations of microtubule-depolymerizing agents (as there are no microtubules left), providing a condition for assessing the role of Aurora B in the checkpoint independently from its effects on error correction. At suboptimal concentrations of spindle poisons, residual microtubules contribute to checkpoint satisfaction when the error correction function of Aurora B is inhibited, therefore accelerating mitotic exit (Yang et al, 2009).
The study concluded that previous positive evidence supporting an involvement of Aurora B in the checkpoint independently of error correction was biased by insufficient concentrations of microtubule-depolymerizing agents (Yang et al, 2009). In a previous characterization of the effects of hesperadin, potent small-molecule inhibitor of Aurora B, on checkpoint duration, an inhibitor concentration of 100 nM was typically used (Ditchfield et al, 2003; Hauf et al, 2003). At this concentration of hesperadin, there is a strong dependence of mitotic duration on nocodazole concentration, with cells living mitosis much more rapidly at low nocodazole concentrations than at high-nocodazole concentrations (Hauf et al, 2003; Yang et al, 2009).
An undemonstrated assumption in many studies with small-molecule inhibitors, including those with Aurora B, is that the enzymatic activity of the target is completely inhibited at the typical concentrations of inhibitors used, or anyway that residual activity is insufficient to sustain the normal function of the enzyme. Here, we decided to adopt the rigorous framework offered by the addition of high-nocodazole concentrations to re-evaluate the effects of Aurora B inhibitors on the spindle assembly checkpoint. Our results are consistent with a role of Aurora B in checkpoint signalling independently of error correction.
Results and discussion
Effects on mitotic arrest from inhibiting Aurora B in low or high nocodazole
It has been argued that the duration of the mitotic arrest in the presence of 100 nM hesperadin (Hauf et al, 2003) may depend on the concentration of nocodazole (Yang et al, 2009). We confirmed this result using a range of nocodazole concentrations (Figure 2A). At low nocodazole concentrations, HeLa cells concomitantly treated with 100 nM hesperadin left mitosis significantly more rapidly than at high-nocodazole concentrations. These results are consistent with the hypothesis that inhibition of error correction in the presence of residual microtubules reduces the duration of the mitotic arrest by satisfying the spindle checkpoint (Yang et al, 2009).
Figure 2.

Effects of hesperadin and reversine on the spindle checkpoint. (A) HeLa cells were treated as indicated in the scheme. STA, single thymidine arrest. Cells were monitored by time-lapse video microscopy as they transited through mitosis. The histogram reports the time in mitosis (measured as the time of a cell's rounding up) at the indicated nocodazole concentrations and at a fixed concentration of hesperadin of 100 nM. At the same hesperadin concentration, there were significant differences in the time spent in mitosis depending on nocodazole concentrations. Values represent mean and s.d. and were calculated from at least 50 cells for each condition. (B) Cells were filmed like in (A) in the presence of the indicated concentrations of inhibitors and nocodazole.
As expected, the increase in the duration of mitotic arrest saturates at high concentrations of nocodazole (Figure 2A). Based on these observations, and in line with Yang et al (2009), we opted to use nocodazole at 3.3 μM as a working concentration under which it can be safely assumed that checkpoint satisfaction is virtually impossible due to complete depolymerization of microtubules. At this concentration of nocodazole, tubulin appears completely diffuse (Yang et al, 2009; Santaguida et al, 2010). Furthermore, the duration of the mitotic arrest in the presence of 100 nM hesperadin increased very modestly at higher concentrations of nocodazole in comparison to the duration of 3.3 μM, suggesting that this concentration largely satisfies the requirement that microtubules are depolymerized (Figure 2A). We also reasoned that growing concentrations of nocodazole might increase the risk of unspecific interference with other cellular processes (however, we demonstrate in Supplementary Figure S4 that our conclusions remain valid even at higher concentrations of nocodazole or in the presence of a different microtubule-depolymerizing agent). Based on these considerations, we consider 3.3 μM nocodazole an appropriate concentration for testing the role of Aurora B on checkpoint signalling independently of error correction.
We therefore compared the duration of the checkpoint-dependent mitotic arrest in HeLa cells treated with 0.33 and 3.3 μM nocodazole at different concentrations of hesperadin. As a control for checkpoint override, we used reversine, a bona fide ATP-competitive inhibitor of the spindle checkpoint kinase Mps1 (Santaguida et al, 2010). At both concentrations of nocodazole, we observed a strong dose-dependent effect on the duration of the mitotic arrest (Supplementary Table SI; Figure 2B). At 100 nM hesperadin, the checkpoint response was significantly but not dramatically affected, with cells undergoing override at ∼700 min rather than >1100 min in control cells at high-nocodazole concentrations (Figure 2B; Supplementary Table SI) (at which point control cells usually undergo slippage, whereby they exit M-phase spontaneously despite the continued presence of checkpoint-activating offences (Brito and Rieder, 2006)). Yang et al (2009) observed even milder effects on checkpoint duration in the presence of 100 nM hesperadin at 3.2 μM nocodazole in RPE1 cells. At 0.5 and 1.0 μM hesperadin, however, we observed a very strong reduction in the duration of the checkpoint, both in low and high nocodazole. The reduction was inferior but close to that observed with 1 μM reversine (a concentration causing complete checkpoint abrogation, Supplementary Table SI and Figure 2B).
Altogether, these results support the contention that high doses of nocodazole are required to rigorously assess the role of Aurora B (Yang et al, 2009). Furthermore, the results provide an initial indication that Aurora B activity is required for the checkpoint response in the absence of microtubules.
Effects on kinetochore localization of checkpoint proteins from inhibiting Aurora B
Lack of kinetochore localization of Mad2 or Mad1 (the Mad2 kinetochore receptor, displaying identical kinetochore localization timing as Mad2) strongly correlates with checkpoint weakening or impairment. Because kinetochore localization of Mad2 is impaired at 0.3 μM nocodazole, but normal at 3.2 μM in the presence of 100 nM hesperadin (Yang et al, 2009), Rieder and colleagues concluded that Mad2 was absent from kinetochores upon inhibition of Aurora B because residual microtubules in low nocodazole were ‘stripping’ Mad2 from kinetochores (Yang et al, 2009), the normal route through which Mad2 is removed from kinetochores (Musacchio and Salmon, 2007).
We found that at 100 nM hesperadin, whether Mad1 is found at kinetochores depends on the protocol of drug exposure. When hesperadin was added after prolonged nocodazole arrest, Mad1 localization at 3.3 μM nocodazole, but not at 0.33 μM, was normal, as reported previously for Mad2 (Yang et al, 2009) (Figure 3A). However, if hesperadin was added together with nocodazole prior to entry into mitosis, Mad1 failed to localize to kinetochore at both low and high-nocodazole concentrations (Figure 3B). Based on these results, we surmise that there is probably a less stringent requirement for Aurora B activity to retain Mad1 and Mad2 at kinetochores than there is for their initial recruitment to kinetochores.
Figure 3.

Effects of hesperadin and reversine on the spindle checkpoint. (A) Kinetochore localization of the checkpoint protein Mad1 in HeLa cells. The scheme of the experiment is shown (numbers above arrows indicate hours). Cycling cells were treated with 0.3 or 3.3 μM nocodazole for 12 h. MG132 was first added and then hesperadin. After 90 min, cells were fixed and processed for indirect immunofluorescence. CREST is a kinetochore marker. MG132 was added to retain cells in mitosis in the presence of hesperadin. Bar=5 μm (B) Kinetochore localization of Mad1 was evaluated as in (A), but hesperadin and nocodazole were added before cells entered mitosis. DTA, double thymidine arrest. (C) Immunoprecipitation of Cdc20 and analysis of interacting proteins at the indicated concentration of reversine or hesperadin. HeLa cells were arrested in nocodazole for 12 h. After shake-off, MG132 was added, and after 30′ the indicated inhibitors were added for further 90 min in the continued presence of MG132. Cells were then harvested and processed for immunoprecipitation. (D, E) The phosphorylation state of BubR1 and Bub1 was evaluated in mitotic cells treated with nocodazole and the indicated concentration of hesperadin or reversine. Cells were treated as discussed in (C). (F) Concentrations of inhibitors required for the indicated percent inhibitions of Aurora B and Mps1 were calculated as explained in Materials and methods.
Outer kinetochore organization is not grossly affected by Aurora B inhibitors
Results so far are consistent with the hypothesis that Aurora B is implicated in checkpoint signalling regardless of its proven function in error correction. Further confirming this idea, hesperadin, like reversine, promoted MCC dissociation in a dose-dependent manner (Figure 3C). Mitotic phosphorylation of BubR1 and Bub1 was also dramatically reduced (Figure 3D and E), indicating that both Aurora B and Mps1 contribute to their mitotic phosphorylation.
In the experiments in Figure 3A and B, reduced or absent Mad1 localization in high nocodazole is unlikely to be caused by Mad1 ‘stripping’, as the latter requires microtubules (Hoffman et al, 2001). As an alternative explanation, we asked if the inability of Mad1 to reach the kinetochore was due to a defect in kinetochore assembly when Aurora B is inhibited. Indeed, it has been proposed that Aurora B contributes to outer kinetochore assembly (Emanuele et al, 2008). Other studies, however, confute the idea that Aurora B is important for kinetochore assembly (Liu et al, 2010; Santaguida et al, 2010; Welburn et al, 2010). In agreement with the latter studies, we found that the levels of kinetochore-localized Ndc80 and Knl1, two components of the so-called KMN network that are crucially implicated in the recruitment of the checkpoint proteins, appear to localize to kinetochores essentially normally even at the high concentrations of hesperadin that prevent Mad1 localization in high nocodazole (Supplementary Figure S1). Kinetochore localization of Ndc80 relies on core kinetochore components, including CENP-I and the Mis12 complex (Hori et al, 2003; Kline et al, 2006; Liu et al, 2006; McAinsh et al, 2006). That kinetochore localization of Ndc80 is largely unaffected indicates that the core structure of the kinetochore is preserved in the presence of Aurora B inhibitors. In summary, although we cannot rule out that the localization of additional kinetochore components, not considered in our analysis, is affected when Aurora B is inhibited, we suspect that reduced localization of checkpoint components is unlikely to be caused by an overt defect in the assembly of the kinetochore. Further evidence in support of this contention is discussed in the context of Figure 6.
What is the ‘right’ concentration of an Aurora B inhibitor?
Results so far indicate that hesperadin has negative consequences on the checkpoint even when microtubules have been completely depolymerized to exclude effects from inhibiting error correction. Thus, our results challenge the contention that Aurora B influences the checkpoint exclusively through error correction (Yang et al, 2009). We note that this contention was based on the undemonstrated assumption that 100 nM hesperadin is sufficient to completely abrogate Aurora B activity, but our results on the duration of the mitotic arrest at different doses of hesperadin (Supplementary Table SI; Figure 2B) suggest that this might not be the case. This issue is further addressed in experiments presented in Figures 4, 5 and 6.
Figure 4.

Specificity of effects of Aurora B inhibitors on the spindle checkpoint. (A) The indicated human cell lines were released from a single thymidine arrest and treated with nocodazole (3.3 μM) and the indicated inhibitors. Cells were analysed by time-lapse video microscopy. This protocol, illustrated schematically in Figure 2A, was also used for (C, D). Time spent in mitosis was evaluated based on mitotic rounding up of cells. Values represent mean and s.d. and were calculated from at least 50 cells for each condition. (B) Immunoprecipitation of Cdc20 and analysis of interacting proteins at the indicated concentration of reversine, hesperadin or their combination. Cells were treated as indicated. (C, D) The same experiment as in (A) was carried out with different inhibitors or inhibitor combinations in HeLa cells. (E) Asynchronously growing HeLa cells were treated with 3.3 μM nocodazole for 6 h. Mitotic cells were collected by shake-off, re-plated in the presence of the indicated inhibitors and filmed by time-lapse video microscopy. (F) The experiment was conducted as described in (E), but cells were kept in 3.3 μM nocodazole for 12 h before shake-off, re-plating and filming. Under these conditions, cells treated with hesperadin alone did not leave mitosis earlier than control cells. Cells treated with hesperadin and reversine, however, did.
Figure 5.

Combination of Mps1 and Aurora B inhibition has synergistic effects on checkpoint. (A) HeLa cells were released from a single thymidine arrest (STA) and treated with 3.3 μM nocodazole and the indicated inhibitors. Time spent in mitosis was evaluated based on mitotic rounding up of cells by time-lapse video microscopy. Values represent the mean (s.d. are collected in Supplementary Table SI) and were calculated from at least 50 cells for each condition. (B) Kinetochore localization of the spindle checkpoint proteins Mad1 and Zwilch in HeLa cells was evaluated at 3.3 μM nocodazole, upon addition of the indicated inhibitors and of MG132 (to suppress mitotic exit in the presence of the inhibitors). Cells were fixed and processed for indirect immunofluorescence. Bar=5 μm. (C) Quantification of Mad1 and Zwilch localization from the experiment in (B). Values are calculated from at least 90 cells from three independent experiments. (D) Immunoprecipitation of Cdc20 and analysis of interacting proteins at the indicated concentration of reversine, hesperadin or their combination. (E) Loewe additivity analysis of the experiment in (A). For the three indicated hesperadin:reversine ratios, the fractional inhibition was plotted against the inhibitor concentration. Fractional inhibition was calculated and the curves for reversine (red), hesperadin (green) and their combination (blue) were fitted with a Hill function (see Materials and methods for details). Based on the additivity formula (Chou, 1991): 1=C1/C1x+C2/C2x (see Materials and methods), we plotted the hypothetical additivity curves for different inhibitors combination ratios (1:1, 2:1, 1:2). We also fitted the experimental combination curves for the same ratios and compared them with the hypothetical additivity curves. If the experimental combination curve lies on the left of the additivity curve, synergy is present. Combining hesperadin and reversine produces synergy when the two drugs are used in the three different combination ratios (see also Supplementary Figure S5). To represent the combination data quantitatively, we also plotted the theoretical combination curves for the three ratios 1:1, 2:1, 1:2 (Chou, 1991; Chou, 2006). On the x axis of these plots we have the fractional inhibition and on the y axis the combination index. We show that the CI is <1 for different effect levels and for the three combination ratios (see Supplementary Figure S5). Strong synergy is observed (CI<0.3) for a large range of fractional inhibition. As an example, in order to produce an effect of about 85%, we should use about 560 nM reversine or >1000 nM hesperadin; but, if we use them in 1:1 combination, about 55 nM reversine+55 nM hesperadin are sufficient. In this case the CI is about 0.13.
Figure 6.

Synergistic effects from inhibiting Aurora B and other checkpoint and kinetochore proteins. (A) HeLa cells were depleted of Nuf2 (a subunit of the Ndc80 complex), Mps1 or Aurora B by RNAi. Depletion of Nuf2 also destabilizes the Ndc80 subunit of the complex (Meraldi et al, 2004). (B, C) HeLa cells were released from a double thymidine arrest and treated with 3.3 μM nocodazole and the indicated inhibitors in combination with the indicated RNAi-based depletions. Cells were analysed by time-lapse video microscopy. Time spent in mitosis was evaluated based on mitotic rounding up of cells. Values represent mean and s.d. and were calculated from at least 50 cells for each condition.
On the other hand, using hesperadin (and reversine) at relatively high concentrations, up to 1 μM, raises significant concerns about the specificity of its effects. To address such concerns, we determined that hesperadin is inactive against a set of checkpoint and mitotic kinases (Supplementary Figure S2; the specificity of reversine has been discussed (Santaguida et al, 2010)). Furthermore, in discussing this objection, it should be noted that checkpoint signals from a single unattached kinetochore are sufficient to maintain a mitotic arrest (Rieder et al, 1995), strongly suggesting that the checkpoint network is designed to achieve amplification. The exact topology of the checkpoint network is unknown, so that the way in which signal amplification is achieved remains unclear. But we argue that because of the amplification properties of the network, it may be necessary to achieve very significant inhibition of its activity before a penetrant checkpoint phenotype is observed when chromosomes are unattached. Indeed, small residual amounts (2–5%) of the checkpoint kinase Bub1 are compatible with a checkpoint response in nocodazole, whereas its full depletion causes checkpoint failure (Meraldi and Sorger, 2005; Perera et al, 2007). Similarly, while depletion of the subunits of the Ndc80 complex causes a checkpoint defect, small residual amounts are compatible with strong mitotic arrest in nocodazole (Martin-Lluesma et al, 2002; DeLuca et al, 2003; McCleland et al, 2003; Meraldi et al, 2004).
To provide a quantitative framework to these ideas, we predicted the inhibitory effects of hesperadin or reversine on Aurora B or Mps1 after measuring their catalytic parameters. With 2 mM ATP, a concentration approximating the ATP concentration in cells (Traut, 1994), we predict that 0.18–1.9 μM reversine or 0.13–1.4 μM hesperadin might be respectively required to achieve inhibition of Mps1 or Aurora B activity from 90 to 99% (Figure 3F; Supplementary Figure S3). Factors such as limited inhibitor permeability, inhibitor modification and competition from other active sites, likely further decrease the active inhibitor concentration in cells. Thus, that doses of hesperadin or reversine as high as 1–2 μM are required for checkpoint override is expected and unsurprising.
Mps1 and Aurora B inhibitors synergize
To build a stronger case for a direct role of Aurora B in the checkpoint, we asked if such a role could be exacerbated under conditions of partial inhibition of other checkpoint components. For this, we initially tested the effects from combining Aurora B and Mps1 inhibitors on the checkpoint response. At 200 nM, hesperadin or reversine each mildly but significantly affected the timing of the checkpoint response to high nocodazole (Figure 4A; Supplementary Table SI). When the two inhibitors were combined, each at 100 nM, a dramatic checkpoint defect was exposed (Figure 4A). Similar results were obtained in the osteosarcoma U2OS cell line and the non-transformed immortalized epithelial cell line hTERT-RPE1 (Figure 4A). The same effect was also observed at 10 μM nocodazole or 15 μM colchicine, ruling out an off-target effect of the spindle poison on the checkpoint, or an effect of residual microtubules on checkpoint satisfaction (Supplementary Figure S4). The combination of 100 nM hesperadin and 100 nM reversine caused a dramatic decrease in the levels of MCC, indicative of its disassembly and of checkpoint override (Figure 4B).
Hesperadin has relatively modest selectivity for Aurora B (Hauf et al, 2003), leaving open the possibility that its effects on the checkpoint are due to inhibition of other Aurora family members, Aurora A and Aurora C. To address this concern, we carried out inhibitor combination experiments with additional Aurora inhibitors, including ZM447439, reported to have 20-fold Aurora B/Aurora A selectivity and five-fold Aurora B/Aurora C selectivity (Ditchfield et al, 2003; Girdler et al, 2006), and AZD1152, reported to have >1000-fold Aurora B/Aurora A selectivity and >17-fold Aurora B/Aurora C selectivity (Mortlock et al, 2007). As an alternative Mps1 inhibitor, we used Mps1-IN-1 (Kwiatkowski et al, 2010). In all cases, we observed very dramatic effects from the combined inhibition of Aurora B and Mps1 (Figure 4C and D).
The localization experiments in Figure 3A and B suggest the possibility that the effects of Aurora B inhibitors on the checkpoint response might depend on whether the Aurora B inhibitors are added prior to entry into mitosis or after entry into mitosis. Specifically, these results suggest the possibility that Aurora B is required to initiate the checkpoint response, but not to maintain it. To test this idea, we collected mitotic cells by shake-off 6 h after the addition of nocodazole and added hesperadin, reversine or their combination. The results in Figure 4E demonstrate that under these conditions, inhibitor-treated cells exited mitosis prematurely, indicating that Aurora B is not only required for instating checkpoint signalling, but also for maintaining it. When cells were harvested after a 12-h mitotic arrest, we noted that the ability of Aurora B and Mps1 inhibitors, or their combination, to drive mitotic exit was comparatively reduced, although not abrogated (Figure 4F). It is difficult to explain these observations, but we speculate that they might be related to defined physiological changes in cells facing a prolonged arrest with high concentrations of spindle poisons, and possibly finalized to prevent re-entry in the cell cycle.
Formal analysis using Loewe's additivity hypothesis
The experiments above suggest the possibility that combining Aurora B and Mps1 inhibitors has a more than additive adverse effect on the checkpoint. To explore this systematically, we analysed the effects from combining hesperadin and reversine at different ratios in high nocodazole (Figure 5A). As little as 10 nM hesperadin reduced the duration of the checkpoint arrest to one third at 100 nM reversine (from 754 to 256 min), whereas 25 nM hesperadin caused dramatic checkpoint failure (Figure 5A; Supplementary Table SI). In isolation, 100 nM reversine or 25 nM hesperadin had negligible effects on the localization of Mad1 or Zwilch to kinetochores in high nocodazole, whereas their combination evicted them from kinetochores (Figure 5B and C) and caused significant MCC disassembly (Figure 5D). Being caused by very low hesperadin concentrations, these dramatic effects are likely due to specific Aurora B inhibition.
We adopted the Loewe additivity hypothesis (Loewe, 1953) and the Chou and Talalay method (Chou and Talalay, 1981, 1984; Chou, 1991) to investigate the effect of hesperadin and reversine combinations on the timing of mitotic exit from 3.3 μM nocodazole-induced arrest. At several relative ratios (three of which are shown in Figure 5E), the effects on the checkpoint from combining the two inhibitors denoted a very small combination index (CI), indicative of very strong synergy between the inhibitors (CI<1 indicates synergy and CI=0.3 indicates strong synergy) (Figure 5E; Supplementary Figure S5).
Synergy between partial outer kinetochore defects and Aurora B inhibition
We performed additional experiments by exploiting partial or complete depletions of checkpoint proteins through RNAi (Figure 6A). RNAi-based depletion of MPS1 caused complete checkpoint override (Figure 6B), but we found it difficult to modulate the levels of Mps1 with sufficient robustness to achieve partial depletion (not shown). Partial depletion of Aurora B by RNAi (Figure 6B), on the other hand, was compatible with a long-term arrest in high nocodazole, but not when reversine was used at 250 nM. Also in this case there was a very strong synergistic effect (Figure 6B; Supplementary Table SI).
The KMN network complex is implicated in the recruitment of all checkpoint proteins (with the exclusion of Aurora B) (Santaguida and Musacchio, 2009). Within the KMN network, the Ndc80 complex has been implicated in a pathway of recruitment of the RZZ complex, Mad1 and Mad2 (Martin-Lluesma et al, 2002). Consistently, RNAi-based depletion of Ndc80 and Nuf2, two components of the Ndc80 complex, results in complete inhibition of the checkpoint response (Meraldi et al, 2004). On the other hand, suboptimal depletions result in a strong mitotic arrest even in the absence of spindle poisons, possibly because a residual checkpoint response is mounted in the presence of residual Ndc80 complex (Martin-Lluesma et al, 2002; DeLuca et al, 2003). We confirmed that partial RNAi-based depletion of Nuf2 was compatible with prolonged checkpoint arrest in high nocodazole (Figure 6C). Addition of hesperadin at 100 nM caused a dramatic effect on the duration of the checkpoint response, with cells being completely unable to arrest in mitosis in high nocodazole (Figure 6C).
Thus, even defects in kinetochore assembly can sensitize cells to Aurora B inhibition and cause a checkpoint defect in high nocodazole. These results further argue against the possibility, discussed above, that Aurora B inhibitors can, by themselves, dramatically affect kinetochore assembly.
Conclusions
The work presented here lends credit to the hypothesis that Aurora B has a role in the spindle checkpoint independently of error correction. This hypothesis has been formulated several times in the past (Kallio et al, 2002; Ditchfield et al, 2003; Hauf et al, 2003; Petersen and Hagan, 2003; King et al, 2007; Vader et al, 2007; Vanoosthuyse and Hardwick, 2009), and regularly challenged on the ground that it is difficult to identify conditions in which the well-established influence on error correction from inhibiting Aurora B can be reliably excluded from the analysis of the role of Aurora B in the checkpoint (for recent discussions, see Khodjakov and Rieder, 2009; Nezi and Musacchio, 2009; Santaguida and Musacchio, 2009; Khodjakov and Pines, 2010; Lampson and Cheeseman, 2010; Maresca and Salmon, 2010).
Our results question the contention that Aurora B is exclusively an extrinsic checkpoint component whose influence on the checkpoint is merely a consequence of its participation in error correction. If error correction and the spindle checkpoint were molecularly distinct, then the inhibition of error correction should only influence the ability to correct improper attachments, but not the checkpoint response, as predicted for the test of model 1 (Figure 1C). We show instead, under conditions in which inhibition of error correction is not expected to have detrimental effects on the intensity of the checkpoint response, due to the presence of very high concentrations of microtubule depolymerizers, that the spindle checkpoint response is very severely impaired when Aurora kinase activity becomes inhibited, as expected for model 2 (Figure 1D).
Several previous studies have suggested the possibility that Aurora kinase activity is completely (Biggins et al, 1999; Yang et al, 2009) or partially (Ditchfield et al, 2003; Hauf et al, 2003) dispensable for the checkpoint response to unattached kinetochores. We suspect that the observations on which these conclusions were based might have been caused by residual kinase activity of mutant proteins or incomplete inhibition with small-molecule inhibitors. For instance, our results strongly argue that 100 nM hesperadin achieves significant but incomplete inhibition of Aurora B activity, in line with the partial inhibition of the spindle checkpoint observed in HeLa cells under these conditions (Hauf et al, 2003).
Previously, partial Aurora B and Bub1 inhibition has been shown to have synergistic effects on checkpoint inhibition (Meraldi and Sorger, 2005; Morrow et al, 2005). The results were interpreted as reflecting the existence of two distinct arms of the checkpoint response, a tension-dependent arm relying on Aurora B and an attachment sensitive arm relying on Bub1. We show that Aurora B and Mps1 strongly synergize in the checkpoint. Because complete inhibition of Aurora B, Bub1 or Mps1 in isolation leads to a checkpoint defect in high nocodazole (with high doses of hesperadin in the case of Aurora B, with high doses of reversine or RNAi in the case of Mps1, with RNAi in the case of Bub1), these kinases appear to operate within a single pathway, and synergy results from inhibiting this pathway concomitantly at distinct nodes. Understanding the topology of the checkpoint network that explains these results is the challenge for future studies.
By showing that Aurora B might contribute to checkpoint signalling independently of error correction, we provide the basis for a better molecular understanding of the checkpoint sensory apparatus. The ability of Aurora B to phosphorylate substrates in the kinetochore depends on their distance from Aurora B, which changes during the course of attachment (Liu et al, 2009). In this respect, Aurora B might contribute to the ability of kinetochores of monitoring intrakinetochore stretch, an increase of ∼35 nm in the distance between inner and outer kinetochore components when tension is built (Maresca and Salmon, 2009; Uchida et al, 2009). The exact molecular changes imposed by intrakinetochore stretch, and the way Aurora B might measure them, remain a matter for conjecture (Liu et al, 2009; Maresca and Salmon, 2009, 2010; Santaguida and Musacchio, 2009). We note that the physical distances associated with intrakinetochore stretch, ∼35 nm, resemble the molecular scale of the proteins involved (e.g. the Ndc80 complex has a long axis of 55–60 nm). This suggests that the pool of Aurora B responsible for the checkpoint response might reside very closely to its substrates. The very high local concentration of checkpoint-relevant kinase-substrate pairs at unattached kinetochores might explain why very little residual Aurora kinase activity is compatible with the checkpoint response.
Materials and methods
Cell culture and synchronization
HeLa cells and U2OS cells were grown in Dulbecco's modified Eagle medium (Euroclone) supplemented with 10% fetal bovine serum (Hyclone) and 2 mM L-glutamine. hTERT-RPE1 cells were grown in Minimal Essential Medium: HAM's F12K Medium 1:1 supplemented with 10% fetal bovine serum (Hyclone), 15 mM HEPES, 0.5 mM sodium pyruvate. Nocodazole (at concentrations indicated in each figure or figure legend), thymidine (2 mM) and Colchicine (15 μM) were obtained from Sigma-Aldrich. MG132 (Calbiochem) was used at 10 μM throughout.
RNAi
Previously described siRNA duplexes were used to repress Aurora B (Ditchfield et al, 2003), Mps1 (Jelluma et al, 2008) and Nuf2 (Meraldi et al, 2004). siRNA duplexes were purchased from Dharmacon Research and transfected using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions.
Immunofluorescence microscopy and antibodies for immunofluorescence
Immunofluorescence microscopy was carried out on cells fixed using PFA 4% in PBS, permeabilized using Triton X-100 0.1% in PBS, then treated with BSA 4% in PBS as blocking agent and incubated with the proper antibodies diluted in BSA 4% in PBS. Incubation with primary and secondary antibodies was performed as described previously (Taylor et al, 2001). Antibodies against Mad1, BubR1, Bub1, Cenp-C and Zwilch have been described (Taylor et al, 2001; De Antoni et al, 2005; Trazzi et al, 2009; Civril et al, 2010). Additional antibodies for immunofluorescence were anti-Centromeric antibodies (working dilution 1:50, Antibodies Inc.) and mouse anti-HEC1 (human NDC80) (working dilution 1:1000, Genetex Clone 9G3.23). Cy3- and Cy5-labelled and Alexa-488-labelled secondary antibodies for immunofluorescence were from Jackson Immunoresearch and Invitrogen, respectively. DNA was stained with 4′,6-diamidino-2-phenylindole. The coverslips were mounted using Mowiol mounting media. Cells were imaged using a Leica TCS SP2 confocal microscope equipped with a × 63 NA 1.4 objective lens using the LCS 3D software (Leica). Images were imported in Adobe Photoshop CS3 (Adobe Systems Inc.) and levels were adjusted.
Antibodies for immunoblotting
The following antibodies were used for immunoblotting: rabbit anti-Aurora B (working dilution 1:1000, Abcam); rabbit anti-Bub1 (working dilution 1:2000, Abcam); mouse anti-BubR1 (working dilution 1:1000, Transduction Laboratories, BD); mouse anti-Mps1 (working dilution 1:2000, Upstate); rabbit anti-pH3 Ser10 (working dilution 1:1000, Abcam); rabbit anti-Cdc20 (working dilution 1:500, Santa Cruz); mouse anti-Hec1 (human Ndc80) (working dilution 1:1000, Genetex Clone 9G3.23); mouse anti-Bub3 (working dilution 1:2000, Transduction lab, BD); mouse anti-Mad2 (clone AS55-A12) was produced at the IFOM-IEO campus monoclonal antibody facility (De Antoni et al, 2005).
Immunoprecipitations
HeLa cells were harvested by trypsinization and lysed in lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1% glycerol, 1% Triton X-100, 5 mM EGTA, 1.5 mM MgCl2, 10 mM Na3VO4, 50 mM NaF, 10 mM Na4-pyrophosphate, protease inhibitor cocktail (Roche)) for 20 min on ice and then sonicated. Cell lysates were centrifuged for 45 min at 13 000 r.p.m. at 4°C. Equivalent amounts of soluble protein lysates were incubated with mouse anti-Cdc20 (2 μg antibody/mg lysate—Santa Cruz) for 12 h at 4°C followed by incubation with protein G Sepharose beads (Pharmacia) at 4°C for 2 h. The beads were washed three times in lysis buffer and proteins were eluted in SDS sample buffer.
Video microscopy
Live cell imaging was performed using an IX70 inverted microscope (Nikon) equipped with an incubation chamber (Solent Scientific) maintained at 37°C in an atmosphere of 5% CO2. Movies were acquired using a × 20 magnification objective controlled by ScanR software (Olympus).
In vitro kinase assays
In vitro kinase assays were performed and analysed as previously described (Santaguida et al, 2010).
ADP luminescent assay
Kinetic analyses of Aurora B45−344:INCENP835−903 (Santaguida et al, 2010) and Mps11−857 (full-length human MPS1, Invitrogen) were performed using a luminometric kinase assay varying the concentration of ATP using the ADP-Glo reagents (Promega). In all, 5 nM Aurora B kinase was assayed in a 10-μl reaction containing 25 mM Tris (pH 7.6), 10 mM MgCl2, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, varying concentrations of ATP and 5 μM histone H3 (Roche) and followed for 15 min. In all, 50 nM Mps1 kinase was assayed in a 10-μl reaction containing 12.5 mM Tris (pH 7.6), 10 mM MgCl2, 1 mM EGTA, 0.01% Triton X-100, varying concentrations of ATP and 6 μM MAD1:MAD2 complex as substrate and followed for 30 min. The overall reaction rate was determined as the slope of the linearly increasing phase of the reaction. Each data point was collected in duplicate and kinetic parameters were obtained using GraphPad Prism v3.0 software.
Additivity analysis
To define fractional inhibition, we considered 70 min spent as a mitotically rounded-up cell as corresponding to a 100% drug effect (no delay in mitotic exit) and about 1100 min as a 0% effect (maximum duration of mitosis, corresponding to no drug addition). The effect is therefore intended as the percent reduction of time required for mitotic exit. So, if a drug (or a combination of drugs) produces a mitotic exit time equal to x minutes, we say that the effect produced is ((1100–70)–(x–70))/(1100–70)=(1030–(x–70))/1030. In order to apply Chou and Talalay method (Chou and Talalay, 1981, 1984; Chou, 1991), we first fitted dose–effect curves for single inhibitors with Hill functions of the form E=Cn/(kn+Cn); here E is the percent effect deriving from a drug concentration equal to C of a single drug and k and n are coefficients to be fitted. From the Chou model (Chou, 2006) we have that, if Cx1 and Cx2 are the doses of drugs 1 and 2 (respectively) that produce an effect equal to x when used alone and if C1 and C2 are the doses of the same drugs in combination that give rise to that effect, the combination is additive if the quantity C1/C1x+C2/C2x is equal to one. This means that the total dose of the two drugs in combination simply is equal to equi-effective doses of the two drugs used alone; in other words, no total dose sparing advantages derive from using the drugs together. The quantity C1/C1x+C2/C2x is called the ‘CI’ and is a way of comparing the effect of a drug combination with the effects of single inhibitors. A CI value which is <1 indicates a synergistic effect deriving from the combination and for a certain effect level; on the contrary, CI>1 indicates antagonism. A value CI<0.3 is usually considered as an indicator of a strong synergistic effect.
Determination of the Kd for hesperadin, ZM447439 and reversine
To determine the degree of kinase activity inhibition (defined as the percent reduction of the initial maximal velocity) at different inhibitor concentrations, reported in Figure 3F, we carried out a simulation of the dose–response curves for the three kinase-inhibitor pairs shown in Figure 3F. For this, we generated a system of ordinary differential equations that describes both the phosphorylation reaction and the effect of the inhibitor. The equations are based on two simplifications. In the first we assume that most of the enzyme is bound to the substrate, which is justified by the fact that in the experiment we use (enzyme)=5 nM and (substrate)=5 μM. As a consequence of this assumption, we neglect the free enzyme. The second simplification is the standard quasi-steady state approximation for the enzyme–substrate and enzyme–inhibitor complexes. Given these two assumptions, we introduce the following variables: I, inhibitor; A, ATP; P, phosphorylated substrate (which equals ADP); and E, enzyme (which is always bound to the substrate). Moreover, we keep track of the following complexes: E–A, formed by the substrate-bound enzyme and ATP, and E–I, formed by the substrate-bound enzyme and the inhibitor. We adopt the following reaction scheme
(1) E+A↔E–A (kon, koff)
(2) E–A → E+P (kcat)
(3) E+I↔E–I (kon_i, koff_i)
which can be translated in the system of algebraic-differential equations:
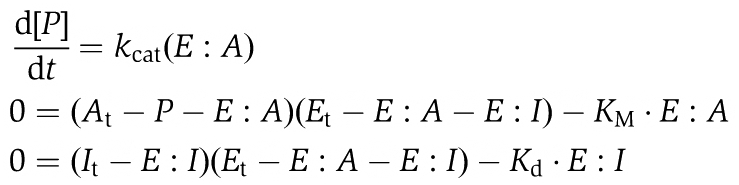 |
where KM=(koff+kcat)/kon and Kd=koff_i/kon_i. KM and kcat were measured for the three enzymes considered: for Aurora B KM=16.5 μM and kcat=62 per min; for Mps1 KM=4.9 μM and kcat=3.2 per min (Supplementary Figure S3A–D). The dose–response curves were calculated by letting the reaction proceed in the presence of initial concentrations of enzyme (Et)=5 nM and ATP (At)=50 μM (where the subscript t stands for ‘total’) for 1 h, the same duration of the reported experimental inhibition curves (Santaguida et al, 2010). Different points of the dose–response curves were calculated by increasing the amount of total inhibitor (It). The amount of phosphorylated substrate (i.e. ADP produced) was plotted against the total amount of inhibitor. The only unknown parameter in the system of equations is the Kd of the inhibitor for the enzyme's active site. To get an estimate for Kd we imposed to the simulation that at 60 min, the initial substrate has been halved in the presence of a concentration of inhibitor equal to the experimentally calculated IC50. All other points shown in the curves in Supplementary Figure S3E–G were not fitted but simulated based on this Kd. All numerical simulations were carried out with XPP-AUT, a free software program developed by Professor Bard Ermentrout (Department of Mathematics, University of Pittsburgh).
Determination of the predicted concentrations of inhibitors in vivo
To predict the amount of inhibitors required for inhibiting Aurora B and Mps1 in vivo, we assumed a concentration of ATP in cells of 2 mM (Traut, 1994) and cellular concentrations of each kinase of 1 nM. Moreover, we assumed, as done for the measurements in vitro, that the substrates of the enzymes are more abundant than the enzymes (i.e. no free enzymes). We then used the differential-algebraic equations described above to calculate the initial rate of the reaction in the presence of different doses of inhibitors, using the kinetic parameters measured in vitro. We took the initial rate of the reaction without inhibitors as 100%, and we identified the concentration of inhibitors that would reduce it to 50, 10, 5 and 1%.
Supplementary Material
Acknowledgments
We thank Stephen S Taylor, Tarun Kapoor and the members of the Musacchio laboratory for many helpful discussions, and Nathanael Gray for providing Mps1-IN-1. Work in the Musacchio laboratory is generously funded by the Association for International Cancer Research (AICR), the Telethon Foundation, the Seventh Framework Program European Research Council grant KINCON and the Integrated Project MitoSys (grant agreement 241548), the Italian Association for Cancer Research (AIRC), the Fondo di Investimento per la Ricerca di Base (FIRB), the Cariplo Foundation and the Human Frontier Science Program. SS is a graduate student of the European School of Molecular Medicine and is supported by a fellowship from the Italian Foundation for Cancer Research (FIRC).
Footnotes
The authors declare that they have no conflict of interest.
References
- Biggins S, Murray AW (2001) The budding yeast protein kinase Ipl1/Aurora allows the absence of tension to activate the spindle checkpoint. Genes Dev 15: 3118–3129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins S, Severin FF, Bhalla N, Sassoon I, Hyman AA, Murray AW (1999) The conserved protein kinase Ipl1 regulates microtubule binding to kinetochores in budding yeast. Genes Dev 13: 532–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito DA, Rieder CL (2006) Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol 16: 1194–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmena M, Ruchaud S, Earnshaw WC (2009) Making the Auroras glow: regulation of Aurora A and B kinase function by interacting proteins. Curr Opin Cell Biol 21: 796–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM, Chappie JS, Wilson-Kubalek EM, Desai A (2006) The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell 127: 983–997 [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Desai A (2008) Molecular architecture of the kinetochore-microtubule interface. Nat Rev Mol Cell Biol 9: 33–46 [DOI] [PubMed] [Google Scholar]
- Chou TC (1991) The median-effect principle and the combination index for quantitation of synergism and antagonism. In Synergism and Antagonism in Chemotherapy, Chou TC, Rideout DC (eds) pp 61–102. San Diego: Academic Press [Google Scholar]
- Chou TC (2006) Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58: 621–681 [DOI] [PubMed] [Google Scholar]
- Chou TC, Talalay P (1981) Generalized equations for the analysis of inhibitions of Michaelis-Menten and higher-order kinetic systems with two or more mutually exclusive and nonexclusive inhibitors. Eur J Biochem 115: 207–216 [DOI] [PubMed] [Google Scholar]
- Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22: 27–55 [DOI] [PubMed] [Google Scholar]
- Civril F, Wehenkel A, Giorgi FM, Santaguida S, Di Fonzo A, Grigorean G, Ciccarelli FD, Musacchio A (2010) Structural analysis of the RZZ complex reveals common ancestry with multisubunit vesicle tethering machinery. Structure 18: 616–626 [DOI] [PubMed] [Google Scholar]
- De Antoni A, Pearson CG, Cimini D, Canman JC, Sala V, Nezi L, Mapelli M, Sironi L, Faretta M, Salmon ED, Musacchio A (2005) The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr Biol 15: 214–225 [DOI] [PubMed] [Google Scholar]
- DeLuca JG, Gall WE, Ciferri C, Cimini D, Musacchio A, Salmon ED (2006) Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell 127: 969–982 [DOI] [PubMed] [Google Scholar]
- DeLuca JG, Howell BJ, Canman JC, Hickey JM, Fang G, Salmon ED (2003) Nuf2 and Hec1 are required for retention of the checkpoint proteins Mad1 and Mad2 to kinetochores. Curr Biol 13: 2103–2109 [DOI] [PubMed] [Google Scholar]
- Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, Taylor SS (2003) Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol 161: 267–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuele MJ, Lan W, Jwa M, Miller SA, Chan CS, Stukenberg PT (2008) Aurora B kinase and protein phosphatase 1 have opposing roles in modulating kinetochore assembly. J Cell Biol 181: 241–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girdler F, Gascoigne KE, Eyers PA, Hartmuth S, Crafter C, Foote KM, Keen NJ, Taylor SS (2006) Validating Aurora B as an anti-cancer drug target. J Cell Sci 119 (Pt 17): 3664–3675 [DOI] [PubMed] [Google Scholar]
- Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters JM (2003) The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol 161: 281–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DB, Pearson CG, Yen TJ, Howell BJ, Salmon ED (2001) Microtubule-dependent changes in assembly of microtubule motor proteins and mitotic spindle checkpoint proteins at PtK1 kinetochores. Mol Biol Cell 12: 1995–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T, Haraguchi T, Hiraoka Y, Kimura H, Fukagawa T (2003) Dynamic behavior of Nuf2-Hec1 complex that localizes to the centrosome and centromere and is essential for mitotic progression in vertebrate cells. J Cell Sci 116 (Pt 16): 3347–3362 [DOI] [PubMed] [Google Scholar]
- Jelluma N, Brenkman AB, van den Broek NJ, Cruijsen CW, van Osch MH, Lens SM, Medema RH, Kops GJ (2008) Mps1 phosphorylates borealin to control Aurora B activity and chromosome alignment. Cell 132: 233–246 [DOI] [PubMed] [Google Scholar]
- Kallio MJ, McCleland ML, Stukenberg PT, Gorbsky GJ (2002) Inhibition of aurora B kinase blocks chromosome segregation, overrides the spindle checkpoint, and perturbs microtubule dynamics in mitosis. Curr Biol 12: 900–905 [DOI] [PubMed] [Google Scholar]
- Khodjakov A, Pines J (2010) Centromere tension: a divisive issue. Nat Cell Biol 12: 919–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodjakov A, Rieder CL (2009) The nature of cell-cycle checkpoints: facts and fallacies. J Biol 8: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King EM, Rachidi N, Morrice N, Hardwick KG, Stark MJ (2007) Ipl1p-dependent phosphorylation of Mad3p is required for the spindle checkpoint response to lack of tension at kinetochores. Genes Dev 21: 1163–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline SL, Cheeseman IM, Hori T, Fukagawa T, Desai A (2006) The human Mis12 complex is required for kinetochore assembly and proper chromosome segregation. J Cell Biol 173: 9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Jelluma N, Filippakopoulos P, Soundararajan M, Manak MS, Kwon M, Choi HG, Sim T, Deveraux QL, Rottmann S, Pellman D, Shah JV, Kops GJ, Knapp S, Gray NS (2010) Small-molecule kinase inhibitors provide insight into Mps1 cell cycle function. Nat Chem Biol 6: 359–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson MA, Cheeseman IM (2010) Sensing centromere tension: Aurora B and the regulation of kinetochore function. Trends Cell Biol (advance online publication 22 November 2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Vader G, Vromans M, Lampson M, Lens S (2009) Sensing chromosome bi-orientation by spatial separation of Aurora B kinase from kinetochore substrates. Science 323: 1350–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Vleugel M, Backer CB, Hori T, Fukagawa T, Cheeseman IM, Lampson MA (2010) Regulated targeting of protein phosphatase 1 to the outer kinetochore by KNL1 opposes Aurora B kinase. J Cell Biol 188: 809–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ST, Rattner JB, Jablonski SA, Yen TJ (2006) Mapping the assembly pathways that specify formation of the trilaminar kinetochore plates in human cells. J Cell Biol 175: 41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe S (1953) The problem of synergism and antagonism of combined drugs. Arzneimittelforschung 3: 285–290 [PubMed] [Google Scholar]
- Maresca TJ, Salmon ED (2009) Intrakinetochore stretch is associated with changes in kinetochore phosphorylation and spindle assembly checkpoint activity. J Cell Biol 184: 373–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresca TJ, Salmon ED (2010) Welcome to a new kind of tension: translating kinetochore mechanics into a wait-anaphase signal. J Cell Sci 123 (Pt 6): 825–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Lluesma S, Stucke VM, Nigg EA (2002) Role of Hec1 in spindle checkpoint signaling and kinetochore recruitment of Mad1/Mad2. Science 297: 2267–2270 [DOI] [PubMed] [Google Scholar]
- McAinsh AD, Meraldi P, Draviam VM, Toso A, Sorger PK (2006) The human kinetochore proteins Nnf1R and Mcm21R are required for accurate chromosome segregation. EMBO J 25: 4033–4049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleland ML, Gardner RD, Kallio MJ, Daum JR, Gorbsky GJ, Burke DJ, Stukenberg PT (2003) The highly conserved Ndc80 complex is required for kinetochore assembly, chromosome congression, and spindle checkpoint activity. Genes Dev 17: 101–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Draviam VM, Sorger PK (2004) Timing and checkpoints in the regulation of mitotic progression. Dev Cell 7: 45–60 [DOI] [PubMed] [Google Scholar]
- Meraldi P, Sorger PK (2005) A dual role for Bub1 in the spindle checkpoint and chromosome congression. EMBO J 24: 1621–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow CJ, Tighe A, Johnson VL, Scott MI, Ditchfield C, Taylor SS (2005) Bub1 and aurora B cooperate to maintain BubR1-mediated inhibition of APC/CCdc20. J Cell Sci 118 (Pt 16): 3639–3652 [DOI] [PubMed] [Google Scholar]
- Mortlock AA, Foote KM, Heron NM, Jung FH, Pasquet G, Lohmann JJ, Warin N, Renaud F, De Savi C, Roberts NJ, Johnson T, Dousson CB, Hill GB, Perkins D, Hatter G, Wilkinson RW, Wedge SR, Heaton SP, Odedra R, Keen NJ et al. (2007) Discovery, synthesis, and in vivo activity of a new class of pyrazoloquinazolines as selective inhibitors of aurora B kinase. J Med Chem 50: 2213–2224 [DOI] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED (2007) The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 8: 379–393 [DOI] [PubMed] [Google Scholar]
- Nezi L, Musacchio A (2009) Sister chromatid tension and the spindle assembly checkpoint. Curr Opin Cell Biol 21: 785–795 [DOI] [PubMed] [Google Scholar]
- Nicklas RB, Koch CA (1969) Chromosome micromanipulation. 3. Spindle fiber tension and the reorientation of mal-oriented chromosomes. J Cell Biol 43: 40–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera D, Tilston V, Hopwood JA, Barchi M, Boot-Handford RP, Taylor SS (2007) Bub1 maintains centromeric cohesion by activation of the spindle checkpoint. Dev Cell 13: 566–579 [DOI] [PubMed] [Google Scholar]
- Petersen J, Hagan IM (2003) S. pombe aurora kinase/survivin is required for chromosome condensation and the spindle checkpoint attachment response. Curr Biol 13: 590–597 [DOI] [PubMed] [Google Scholar]
- Rieder CL, Cole RW, Khodjakov A, Sluder G (1995) The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol 130: 941–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Musacchio A (2009) The life and miracles of kinetochores. EMBO J 28: 2511–2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Tighe A, D’Alise AM, Taylor SS, Musacchio A (2010) Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol 190: 73–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka TU, Rachidi N, Janke C, Pereira G, Gálová M, Schiebel E, Stark MJ, Nasmyth K (2002) Evidence that the Ipl1-Sli15 (Aurora kinase-INCENP) complex promotes chromosome bi-orientation by altering kinetochore-spindle pole connections. Cell 108: 317–329 [DOI] [PubMed] [Google Scholar]
- Taylor SS, Hussein D, Wang Y, Elderkin S, Morrow CJ (2001) Kinetochore localisation and phosphorylation of the mitotic checkpoint components Bub1 and BubR1 are differentially regulated by spindle events in human cells. J Cell Sci 114 (Pt 24): 4385–4395 [DOI] [PubMed] [Google Scholar]
- Traut TW (1994) Physiological concentrations of purines and pyrimidines. Mol Cell Biochem 140: 1–22 [DOI] [PubMed] [Google Scholar]
- Trazzi S, Perini G, Bernardoni R, Zoli M, Reese JC, Musacchio A, Della Valle G (2009) The C-terminal domain of CENP-C displays multiple and critical functions for mammalian centromere formation. PLoS One 4: e5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida KS, Takagaki K, Kumada K, Hirayama Y, Noda T, Hirota T (2009) Kinetochore stretching inactivates the spindle assembly checkpoint. J Cell Biol 184: 383–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vader G, Cruijsen CW, van Harn T, Vromans MJ, Medema RH, Lens SM (2007) The chromosomal passenger complex controls spindle checkpoint function independent from its role in correcting microtubule kinetochore interactions. Mol Biol Cell 18: 4553–4564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoosthuyse V, Hardwick KG (2009) A novel protein phosphatase 1-dependent spindle checkpoint silencing mechanism. Curr Biol 19: 1176–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welburn JPI, Vleugel M, Liu D, Yates JR III, Lampson MA, Fukagawa T, Cheeseman IM (2010) Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol Cell 38: 383–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Kenny AE, Brito DA, Rieder CL (2009) Cells satisfy the mitotic checkpoint in Taxol, and do so faster in concentrations that stabilize syntelic attachments. J Cell Biol 186: 675–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.