

Abstract
Electron transfer flavoprotein - ubiquinone oxidoreductase (ETF-QO) is an iron-sulfur flavoprotein that accepts electrons from electron-transfer flavoprotein (ETF) and reduces ubiquinone from the Q-pool. ETF-QO contains a single [4Fe-4S]2+,1+ cluster and one equivalent of FAD, which are diamagnetic in the isolated oxidized enzyme and can be reduced to paramagnetic forms by enzymatic donors or dithionite. Mutations were introduced by site-directed mutagenesis of amino acids in the vicinity of the iron-sulfur cluster of Rhodobacter sphaeroides ETF-QO. Y501 and T525 are equivalent to Y533 and T558 in the porcine ETF-QO. In the porcine protein, these residues are within hydrogen bonding distance of the Sγ of the cysteine ligands to the iron-sulfur cluster. Y501F, T525A, and Y501F/T525A substitutions were made to determine the effects on midpoint potential, activity, and EPR spectral properties of the cluster. The integrity of the mutated proteins was confirmed by optical spectra, EPR g-values, and spin-lattice relaxation rates, and the cluster to flavin point-dipole distance was determined by relaxation enhancement. Potentiometric titrations were monitored by changes in the CW EPR signals of the cluster and semiquinone. Single mutations decreased the mid-point potentials of the iron-sulfur cluster from +37 mV for wild type to −60 mV for Y501F and T525A and to −128 mV for Y501F/T525A. Lowering the midpoint potential resulted in a decrease in steady-state ubiquinone reductase activity and in ETF semiquinone disproportionation. The decrease in activity demonstrates that reduction of the iron-sulfur cluster is required for activity. There was no detectable effect of the mutations on the flavin midpoint potentials.
Electron transfer flavoprotein-ubiquinone oxidoreductase (ETF-QO) is a monotopic protein that is located in the inner mitochondrial membrane (1). ETF-QO is the electron acceptor for electron transfer flavoprotein (ETF) which catalyzes the oxidation of nine flavoprotein dehydrogenases and two N-methyl dehydrogenases (2, 3). Ubiquinone is the physiological electron acceptor for ETF-QO and transfers the electrons to cytochrome bc1 complex (Complex III). ETF-QO is essential for the oxidation of fatty acids and some amino acids (1, 3, 4). Inherited deficiencies of this protein result in a severe metabolic disease known as multiple acyl-CoA dehydrogenase deficiency or glutaric academia type II (5). Interest in the pathophysiology of this metabolic disease and the role of the enzyme in oxidative metabolism motivates investigation of the kinetic and thermodynamic properties of ETF-QO.
ETF-QO contains a [4Fe-4S]2+,1+ cluster and a single equivalent of flavin adenine dinucleotide (FAD). Both centers are diamagnetic in the oxidized as-isolated enzyme. In the wild type protein the redox midpoint potentials of the two centers are similar and it is only possible to poise the system in a state where both the [4Fe-4S]+ cluster and the flavin are partially in the paramagnetic forms (4). Enzymatic reduction of the wild type protein with octanoyl-CoA adds approximately two electrons to the system, distributed between the iron-sulfur cluster, the flavin semiquinone, and the flavin hydroquinone. Strong chemical reducing agents such as dithionite fully reduce the iron-sulfur cluster and reduce the flavin to hydroquinone. To elucidate the roles of the two redox active centers it would be helpful to selectively modify the redox potential of the iron-sulfur cluster. Some of the environmental factors that modulate redox potentials include solvent accessibility, hydrogen bonding to cysteine Sγ ligated to the cluster, backbone amide dipoles and local charge (6). Computational methods predicting the effects of protein environment on the redox potentials of iron-sulfur clusters are now complemented by direct determination of cluster potentials in site directed mutants (7–10). In particular, Denke et al. have shown alteration of the midpoint potential and catalytic activity of the Rieske iron-sulfur protein by eliminating hydrogen bonds to the Sγ sulfur atoms bound to the cluster (8).
Recently, the crystal structure of porcine ETF-QO (2.7 Å) was determined in the presence and absence of bound ubiquinone (UQ) (11). The structure shows that the three functional domains, i.e., the iron-sulfur cluster, FAD, and ubiquinone domains, are closely packed and share structural elements. The spatial relationship between the flavin and the cluster are shown in Figure 1. The distance of closest approach between the flavin (C8) and UQ (O2) is 9.9 Å, which is much shorter than the 18.8 Å distance between cluster (Fe3) and UQ (O2). A survey of intramolecular electron-transfer pathways in proteins concluded that the upper limit for direct transfer was 14 Å (12). Thus, the shorter distance between the flavin and UQ suggests that the flavin is responsible for the reduction of UQ and not the cluster. There is no structural information available on the docking sites of ETF to ETF-QO, but the relative proximity of the cluster to the enzyme surface (~8 Å), as opposed to the flavin (>14 Å), suggests that the cluster might accept electrons from ETF. The midpoint potentials for flavin semiquinone (+28 mV) and cluster (+47 mV) are similar and the Fe3 atom of the cluster is 11.5 Å from the C8 atom in the isoalloxazine ring, consistent with rapid equilibration between the two centers (1). Zhang et al. proposed (11) that the cluster may serve as a redox-poising or an electron storage site for the flavin as in NADH-UQ oxidoreductase (Complex I) (13). The structure reveals that the cluster is supported through hydrogen bonds between Sγ atoms of the four cysteines and the polypeptide chain. Amino acids H503, T558, and Y533 in porcine ETF-QO form weak hydrogen bonds to cysteine Sγ that could modulate the redox potential of the iron-sulfur cluster, Figure 1. These sites are of interest to investigate the impact that hydrogen bonding to the iron-sulfur cluster has on enzyme activity and electronic structure of the cluster. Porcine and human ETF-QO have 98% sequence identity, and human and R. sphaeroides ETF-QO have 67% sequence identity (Watmough, N. J., Frerman, F. E., Butt, J. N. (2007), unpublished results), which predicts closely related tertiary structures (14). The equivalents of porcine T558 and Y533 are Y501 and T525 in R. sphaeroides ETF-QO. The mutations Y501F, T525A, and Y501F/T525A were introduced to eliminate the hydrogen bonds to cysteine Sγ atoms of the [4Fe-4S] cluster and determine the impact of the hydrogen bonds on redox potentials and activity.
Figure 1.

Relative locations of the iron-sulfur cluster, the residues that were mutated, and the FAD based on the crystal structure of porcine ETF-QO (2GMH). The distance of closest approach between the cluster (Fe3) and the isoalloxazine ring (C8) of the FAD is 11.5 Å. Residues Y533 and T558 form hydrogen bonds to Sγ in C559 and C556, respectively.
2. Experimental
Cloning, site directed mutagenesis and overexpression
The ETF-QO gene was located in the R. sphaeroides genome, chromosome 1 (RSP1777f), by BLAST search using the human amino acid sequence as the query. R. sphaeroides DNA was isolated with the DNeasy kit (Qiagen) from cells grown on LB medium at 30°C. The gene was amplified using the long PCR system (Roche) according to the manufacturer’s instructions except that reactions contained 5% dimethylsulfoxide. No product was obtained when the dimethylsulfoxide concentration was below 2%. The primers were: forward primer, GACCGCCA/TATGACCGAGCAGACTCCC; reverse primer, GCGGTCAGA/AGCTTACATGTTGGGATAGTTCGG. The underlined sequences indicate the NdeI and Hind III recognition sites and the backslash indicates the cut site, respectively. Under these conditions a single product, ~1.7 kb, was generated. The product was purified by agarose gel electrophoresis and ligated into the pCR-TOPO cloning vector (Invitrogen). The gene was removed from the pCR-TOPO cloning vector by digestion with NdeI and Hind III and ligated into the pET21a expression vector, which had been digested with NdeI and Hind III, and purified by agarose gel electrophoresis. E. coli BL21 DE3 was transformed and recombinants were selected and screened for synthesis of a protein, Mr ≈ 60,000, after addition of IPTG, that crossreacted with antisera raised against porcine ETF-QO. The wild type protein and mutants were overexpressed in E. coli C43, a derivative of BL21 DE3 (15). Site-directed mutagenesis was carried out using the Stratagene Quickchange system according to the manufacturer’s instructions.
E. coli C43 was transformed with the plasmid and a starter culture was grown overnight to an OD600nm= 3 to 5 from glycerol stock solutions in a LB media. The next day 700 mL of LB media was inoculated with 1 mL of starter culture containing 100 μg/mL ampicillin, 2 μM riboflavin and 40 μM Fe3+ complexed with 8-hydroxyquinoline. The synthesis of the enzyme was induced at OD600nm= 0.6–1.0 by the addition of 0.5 mM IPTG to each flask and the cells were grown for ~16 h at 30°C until the OD600nm= 5. The cells were collected by centrifugation and washed twice with cold phosphate buffered saline. The cell pellets were homogenized in 4 times (w/v) of 20 mM Tris-HCl, 50 mM NaCl, 10% glycerol, and 0.1 mM DTT at pH 7.4 with protease inhibitors, and then cell lyses was achieved by two passages through a French pressure cell. The disrupted cells were centrifuged for 90 minutes at 100,000×g and the sediment was diluted to 20 mg protein/mL in the same buffer solution and extracted with 2.5:1 ratio of dodecyl β-D maltoside and protein. The concentrated stock detergent solution was made in the same buffer and was added to the resuspended membranes over 5–10 minutes. The membranes were extracted for 90 minutes at 4°C and the solution was centrifuged for 1 h at 100,000×g. The supernatant was loaded on a column (2.5×16 cm) of Q-Sepharose (fast flow) that was equilibrated with 20 mM Tris-HCl, 150 mM NaCl, 0.02% Triton, and 0.1 mM DTT at pH 7.4. The loaded sample was washed with 1.5 L 20 mM Tris-HCl, pH 7.4, containing 250 mM NaCl, 0.02% Triton X-100, 0.1 mM DTT, and the protein was eluted with a linear gradient of 500 mL of the wash buffer and 500 mL of 20mM Tris-HCl, pH 7.4, containing 500 mM NaCl, 0.02% Triton X-100, and 0.1% DTT. The protein usually was 90–95% pure at this point as judged by SDS-page gel. Further purification was done by chromatography on a column (1×8 cm) of Source 15Q using the same gradient scaled to the dimensions of the column. BioBeads SM-2 Absorbent (BioRad) were added to remove the Triton and the purified protein was dialyzed in 20 mM Hepes(K+) at pH 7.4. Ubiquinone was not present in the isolated protein. The concentrations of the enzymes were determined spectrophotometrically using ε430nm = 24.0 mM−1 cm−1 (Watmough, N. J., Frerman, F. E., Butt, J. N. (2007), unpublished results).
Enzyme Assays
ETF-QO quinone reductase activity was determined spectrophotometrically in a coupled reaction containing 20 mM Hepes(K+) buffer at pH 7.4, 2 μM medium chain acyl-CoA dehydrogenase (MCAD), 2 μM porcine ETF, 100 μM octanoyl CoA, 0.2 mM dodecamaltoside, and 55 μM Q1 (coenzyme Q1, Sigma Chemical Co.) at 25°C (16). The reaction was initiated by the addition of ETF-QO (10 nM in resulting solution) and ubiquinone reduction was monitored by the decrease of absorbance at 275nm, (Δε = 7.4 mM−1 cm−1).
Disproportionation of ETF1e- semiquinone catalyzed by ETF-QO was assayed spectrophotometrically under anaerobic conditions as described by Beckmann and Frerman (2). Reaction mixtures were prepared containing 10 mM Tris-HCl at pH 7.5 with 20 mM glucose and 10 μM ETF. The absorption spectrum of oxidized ETF was recorded, and then ETF was reduced quantitatively to the semiquinone with dithionite (sodium dithionite, >82%, Fluka) (17). Reactions were initiated by the addition of a stock solution of ETF-QO.
Analytical Methods
Iron was determined by the method of Beinert (18). Flavin was determined by measurement of the absorbance of free FAD released from the enzyme by addition of SDS 0.2% using ε450 = 11.3 mM−1 cm−1 (19).
Spectrophotometric Titrations
ETF-QOs were reduced either enzymatically using octanoyl-CoA as the electron donor or directly with dithionite (3, 4, 20). ETF-QO at about 30μM was enzymatically reduced in stoppered cuvettes containing 20 mM Tris-HCl at pH 7.4 containing 8 mM CHAPS, 2μM human MCAD, 2μM human ETF, and 20 mM β-D-glucose. The reaction mixtures were made anaerobic by 10 cycles of alternate evacuation and purging with argon. Residual oxygen was removed by addition of glucose oxidase (1 unit/mL) and catalase (24 units/mL) (4). ETF-QO was titrated with octanoyl-CoA and absorption spectra following each addition were measured after the optical spectrum had been stable for 10 minutes. Full reduction of ETF-QO by dithionite was performed under the same conditions, but without the other enzymes and octanoyl-CoA. Dithionite was prepared as a 5 mM solution in anaerobic 0.1 M sodium pyrophosphate at pH 8.0.
Potentiometric titrations
Mediated potentiometric titrations were carried out in an anaerobic vessel under continuous N2 (g) flow at 4 °C using dithionite as reductant (21). The solution potential was monitored using a Ag/AgCl platinum Orion 9678BNWP ORP electrode filled with 4M KCl and a Fisher Scientific Accumet Basic pH/mV meter model. The ORP electrode was calibrated using an Orion 967901 ORP standard. All values are converted to a Standard Hydrogen Electrode (SHE) scale by addition of 200 mV. The following mediator dyes were added to facilitate redox equilibrium (21): 2,3,5,6-tetramethyl-p-phenylendiamine (+260 mV), 2,6-dichlorophenol indophenol (+217 mV), phenozine methosulphate (+80 mV), methylene blue (+11 mV), pyocyanine (−34 mV), indigo carmine (−125 mV), and 9,10-anthraquinone 2,6-disulphonic acid (−185 mV). The mixture of mediators was added to ~35–50 μM enzyme at a final concentration of 25 μM of each dye in 20mM Hepes, 20% glycerol (v/v) at pH 7.4. Aliquots of a dithionite stock solution were added to poise the potential. The sample was continuously stirred during the titration. Equilibrium was judged to have occurred when the potential changed less than 2 mV in 5 minutes. Samples of poised enzyme were transferred anaerobically to 4 mm OD quartz EPR tubes, and the solutions were immediately frozen in liquid nitrogen. The tubes were flame sealed and stored at −80 °C.
EPR spectroscopy
Continuous wave (CW) EPR spectra of the FAD semiquinone signal were collected at ~9.2–9.3 GHz on a Varian E109 spectrometer with a GaAsFET amplifier, a TE102 rectangular cavity, and a Varian liquid-nitrogen-cooled gas flow system. The spectra were recorded using the following operating conditions: 108 K, 1.0 G modulation amplitude at 100 kHz modulation, a microwave power of 5 μW, 128 ms time constant, and averaging ten 200 G scans. The semiquinone concentration was calculated by double integration of the EPR signal and compared to the double integral for the signal from a tempol standard (0.65 mM) recorded under the same conditions except for 0.5 G modulation amplitude. Quantitation of the semiquinone signal was performed at 108 K instead of at the lower temperatures that were used to quantitate the signal for the [4Fe-4S]+ because the relaxation rates for the semiquinone become so slow at lower temperatures that it is difficult to obtain spectra that are not power-saturated. In addition, at 108 K the relaxation rates for the [4Fe-4S]+ signal are so fast that it is not detected and overlap of signals is not a problem.
The CW EPR spectra of the [4Fe-4S]+ were recorded at 9.35–9.5 GHz on a locally constructed spectrometer (22) with a GaAsFET amplifier, Bruker split-ring resonator and Oxford CF 935 cryostat. The operating conditions were: 10 to 40 K, 5.0 G modulation amplitude at 100 kHz modulation frequency, and microwave powers of 5 and 10 μW. The [4Fe-4S]+ concentration was calculated by double-integration of spectra recorded at 15 and 18 K and compared to double integrals of signals for a standard sample of CuEDTA at the same temperatures. The operating conditions for the standard were: 5.0 G modulation amplitude at 100 kHz modulation frequency, and microwave powers between 5 and 10μW. Spectra were recorded with sweep widths between 850 and 1000 G and averaging of 3–10 scans. Resonator background spectra for 20 mM Tris-HCl at pH 7.4 containing 20% glycerol were recorded under identical conditions and subtracted from spectra of the iron-sulfur cluster.
Electron spin echo (ESE) experiments were performed at 8 to 70 K on a Bruker E580 with a split-ring resonator and Oxford CF 935 cryostat. Inversion recovery was used to measure the electron spin-lattice relaxation rates for the [4Fe-4S]+ at g ~1.92 between 8 and 16 K. The recovery curves for [4Fe-4S]+ were analyzed by fitting with a sum of two exponentials. The short component was attributed to spectral diffusion processes and the long component was assigned as the T1 relaxation time. Although the impact of rapid spectra diffusion processes on recovery processes can be minimized by lengthening the pump pulse in saturation recovery experiments, the processes often contribute a fast component to inversion recovery curves at low temperatures (23). Inversion recovery curves for the semiquinone signals were recorded at 24–70 K. As discussed below, in an enzymatically reduced sample of Y501F/T525A essentially all of the iron-sulfur is in the diamagnetic +2 oxidation state. Thus, the relaxation rates for semiquinone in this sample are values in the absence of relaxation enhancement. These values were used to analyze the relaxation enhancement and determine inter-spin distances for the other mutants. The enhancement of the relaxation rates for the semiquinone was analyzed using the program MENOSR (24) and the methodology that previously was applied to spin-labeled metmyoglobin (25).
EPR Simulations
CW EPR spectra of dithionite reduced [4Fe-4S]+ at 15 K were simulated using the program MONMER (26) to determine g-values and first-derivative peak-to-peak linewidths. The fits to the low temperature spectra for the mutants were better for a sum of linewidths than for a single linewidth. The linewidths of the [4Fe-4S]+ signal are temperature dependent above about 25 K and at higher temperatures the spin-lattice relaxation rates for the [4Fe-4S]+ were calculated from the temperature-dependent contributions to the CW linewidths. The CW spectra from 25 K to 40 K were simulated using the locally written program SATMON (27) in which the lineshape is a Gaussian distribution of Lorentzian spin packets characterized by T2. In the temperature range where linewidths are temperature-dependent, it was assumed that T1 = T2 for [4Fe-4S]+. A detailed account of the calculations can be found elsewhere (23). The temperature dependence of T1 for [4Fe-4S]+ was fit with the function (23)
Where T is the temperature in Kelvin, ARam is the coefficient for the contribution from the Raman process, θD is the Debye temperature, J8 is the transport integral, , Aorb is the coefficient for the contribution from the Orbach process, and Δorb is the energy separation between the ground and excited state for the Orbach process. Relaxation enhancement calculations were performed using the program MENOSR (24). Input parameters include the g-values for both paramagnetic centers and the relaxation rates in the absence of spin-spin interaction. Adjustable parameters in the calculation are the inter-spin distance and the fraction of semiquinone interacting with a paramagnetic [4Fe-4S]+.
Midpoint Potential Calculations
Peak-to-peak signal amplitudes, rather than double integration of the EPR signals, were used to calculate [ox]/[red] for the cluster in the titration curves, because of the overlap of the signals for [4Fe-4S]+ and semiquinone. The [4Fe-4S]+ the signal at gz value was selected for the calculations because it is relatively strong and does not overlap with the resonator background signal. The relative concentrations at each point in the titration were calculated based on the assumption that the limiting value at low potential corresponded to 100% of the protein. The midpoint potential of the [4Fe-4S]2+,1+ cluster was calculated by nonlinear least square fitting to the plot of [ox]/[red] versus E (mV) using the Nernst equation, where E is the measured equilibrium at each point and Em is the midpoint potential for an n=1 reduced iron sulfur cluster. The midpoint potentials for the first (Em1) and second (Em2) electron transfers to the FAD were determined using the Nernst equations as previously described (28). The error bars in the experimental midpoint potential values are from the sum of squares of the best fits and the uncertainty of the ORP electrode poise.
3. Results and Discussion
Characterization of the mutants
ETF-QO contains a [4Fe-4S]2+,1+ cluster and a single equivalent of FAD. Both are diamagnetic in the oxidized as-isolated enzyme. The optical spectra (320 to 650 nm) for the three mutants Y501F, T525A, and Y501F/T525A are very similar to that for the wild type protein (Figure 2). The 4:1 iron:flavin ratios (Table 1) and the optical spectra indicate that the mutations did not disrupt the protein structure. The lineshapes for the CW spectra of the semiquinone at 108 K in the three mutants are indistinguishable from that of wild type protein. CW EPR spectra at 15 K for dithionite-reduced samples of mutant and wild-type ETF-QO are shown in Figure 3. At this temperature and at the microwave powers optimized to record the spectrum of the [4Fe-4S]+, the overlapping contribution from semiquinone is severely power-saturated so its contribution was edited out of the spectra of the mutants. The vertical lines mark the g-values for the cluster in the wild type enzyme. The mutations caused changes in g-values that are larger for T525A than for Y501F and are larger along gy and gx than along gz (Table 2). The changes in g-values are small, relative to the range of g-values that are observed for [4Fe-4S]+ clusters (29, 30), which supports the conclusion that the mutations caused only minor changes in protein structure. Interpretation of the g-value changes will require molecular orbital calculations. For Y501F and T525A, it is proposed that a single hydrogen bond is deleted and that two hydrogen bonds are deleted in Y501F/T525A. For all of the mutants, the linewidths are substantially broader than for the wild type protein, which is attributed to g-strain (31). The loss of hydrogen bonds to the cysteine Sγ cluster ligands may make the structure less well-defined. The temperature dependence of the spin lattice relaxation rate, 1/T1, at the gy turning point in the spectrum of [4Fe-4S]+ was measured for the 3 mutants and the wild type protein (Figure 4). Analysis of the temperature dependence of the relaxation in terms of contributions from Raman and Orbach process (32) showed that the energy of the low-lying excited state ΔE = 205 ± 5 K (140 cm−1) is unchanged by the mutations. Each of these observations supports the conclusion that the mutations did not cause major structural changes.
Figure 2.
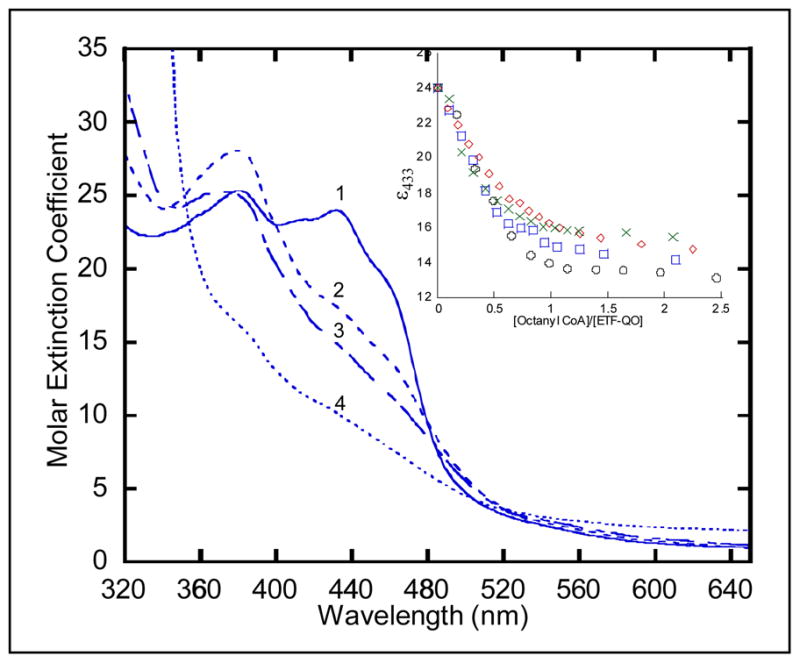
Anaerobic reductive titration of Y501F ETF-QO. The solution contained ~30 μM R. sphaeroides Y501F ETF-QO, 2 μM human ETF, and 2 μM MCAD in 20 mM Hepes(K+) at pH 7.4. The oxidized sample (spectrum 1) was titrated with octanyl-CoA. Spectra were recorded every 5–10 minutes until less than 0.001 a.u. change per minute in absorbance was observed. The inset shows the Δε433 nm vs. mole ratio of titrant for (○) native, (
 ) Y501F, (
) Y501F, (
 ) T525A, and (
) T525A, and (
 ) Y501F/T525A. Spectra 2 and 3 correspond to mole ratios of 0.5:1 and 1:1, respectively, and spectrum 4 is the dithionite reduced sample of Y501F ETF-QO.
) Y501F/T525A. Spectra 2 and 3 correspond to mole ratios of 0.5:1 and 1:1, respectively, and spectrum 4 is the dithionite reduced sample of Y501F ETF-QO.
Table 1.
Activity assays, iron/flavin ratios, midpoint potentials, and enzymatic titration values of native and mutant ETF-QO samples.
| Sample | Iron/− Flavin Ratio | [4Fe4S]+1,+2 Em (mV) | Q/•Q− Em1 (mV) | •Q−/QH2 Em2 (mV) | Reductive Titration Δε430 | Quinone Reductase Activity (s Activity (s−1) | Disproportionation Activity (s Activity (s−1) |
|---|---|---|---|---|---|---|---|
| wild type | 4.0:1 | +37 ± 7 | +38 ± 12 | −62 ± 16 | 10453 | 24.2 (100%) | 8.3 (100%) |
| Y501F | 4.0:1 | −64 ± 5 | +39 ± | −56 ± 8 | 9616 | 8.9 (37%) | 4.1 (51%) |
| T525A | 4.1:1 | −58 ± 6 | +56 ± | −54 ± 7 | 9328 | 8.5 (35%) | 5.1 (62%) |
| Y501F/T525A | 3.8:1 | −128 ± 4 | +50 ± 5 | −55 ± 4 | 8433 | 1.8 (8%) | 0.66 (8%) |
Figure 3.

X-band (9.35 to 9.5 GHz) CW EPR spectra at 15 K of ETF-QO reduced with excess dithionite, recorded with 0.005 mW power. The saturated semiquinone signal was deleted from the spectra of the mutants to aid in comparison. The anaerobic samples contained 50 mM Tris HCl at pH 7.4 and 20% glycerol. The vertical lines are the g-values for native R. sphaeroides ETF-QO.
Table 2.
g-values and linewidths (G) for [4Fe-4S]+ at 15 K and Semiquinone at 105 K
| Sample | [4Fe4S]+ | Linewidthc | [4Fe4S]+ | Linewidthc | [4Fe4S]+ | Linewidthc | SQb |
|---|---|---|---|---|---|---|---|
| gza | (G) | gya | (G) | gxa | (G) | g | |
| Wild Type | 2.089 | 22 | 1.934 | 15 | 1.875 | 29 | 2.0036 |
| Y501F | 2.088 | 28 | 1.933 | 30 | 1.873 | 37 | 2.0036 |
| T525A | 2.097 | 35 | 1.913 | 30 | 1.857 | 48 | 2.0036 |
| Y501F/T525A | 2.095 | 33 | 1.921 | 35 | 1.868 | 51 | 2.0036 |
The average uncertainty of the iron-sulfur cluster g-values was ~± 0.003.
The average uncertainty of the semiquinone g-values was ~± 0.0005.
Peak-to-peak first derivative linewidths were simulated using MONMER. Average uncertainty of the linewidth was ± 2 G for gz and gx and ± 10 G for gy.
Figure 4.
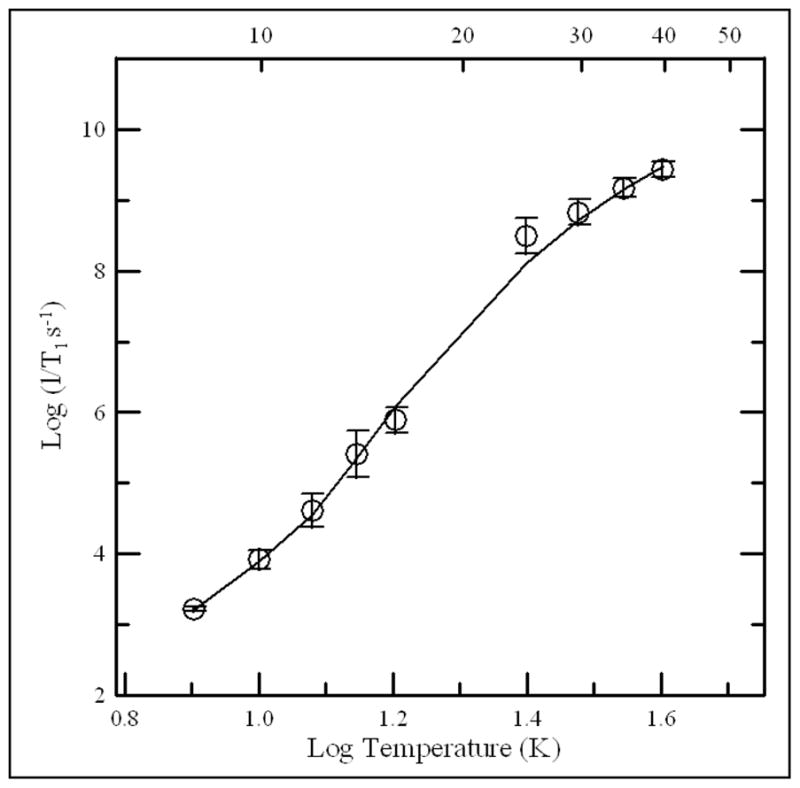
Temperature dependence of spin-lattice relaxation rates is shown for dithionite reduced [4Fe-4S]+ in R. sphaeroides ETF-QO. The circles are the average values for the native and mutant samples and the error bars reflect the standard deviations for the four proteins. The fit line is the sum of contributions from the Raman and Orbach processes. The Debye temperature was fixed at 100 K and the energy of the low-lying excited state was ΔE = 205 (140 cm−1).
Potentiometric Titration
The EPR signals associated with the FAD semiquinone and [4Fe-4S]+ were used to monitor the redox titrations (Figures 5 and 6). The calculated midpoint potentials are shown in Table 1. The Em for [4Fe-4S]+1,+2 in the wild type enzyme (+37 ± 11 mV) is within experimental uncertainty of Em for the mammalian enzyme (+47 ± 10 mV) (28). The Y501F and T525A single mutations lower Em to −64 ± 9 mV and −58 ± 10 mV, respectively. The Y501F/T525A double mutation lowers Em to −128 ± 9 mV. These decreases in the midpoint potentials are consistent with prior observations that hydrogen bonds to cysteine Sγ of cluster ligands tune the redox behavior of the cluster (8, 33–36). For example, the deletion of residues that are hydrogen bonded to cysteine Sγ lowered the midpoint potentials in R. sphaeroides and Saccharomyces cerevisiae Rieske protein, which is attributed to a decrease in charge density around the sulfur atoms (8, 33).
Figure 5.

Potentiometric titration curves are shown of the semiquinone for R. sphaeroides (○) native, (
) Y501F, (
) T525A, and (
) Y501F/T525A ETF-QOs. The semiquinone spectra were recorded at 105 K to avoid saturations of the EPR signal and the midpoint potentials were calculated from Nernst equations described by Paulsen (41).
Figure 6.
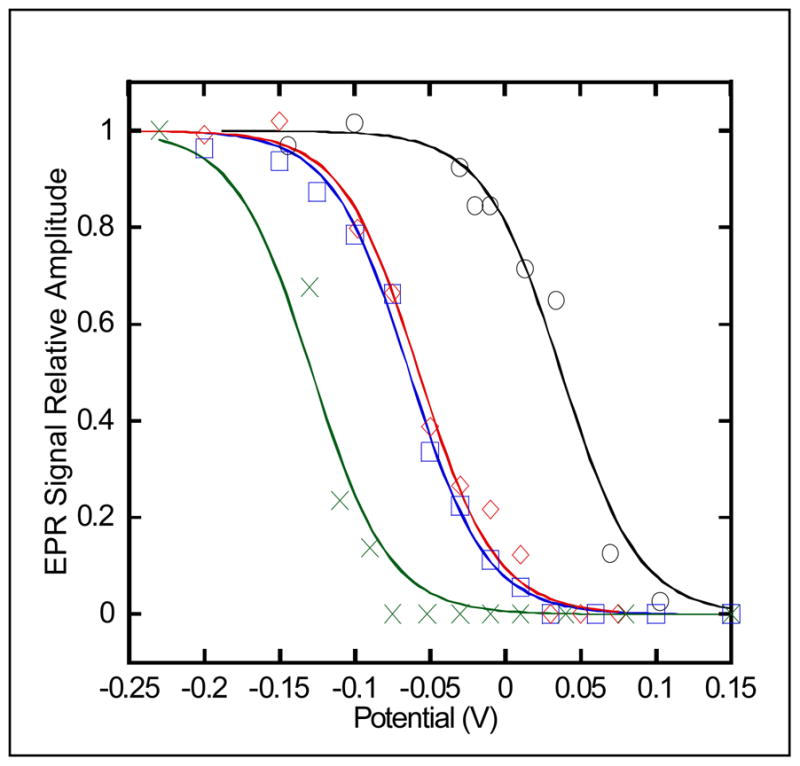
Potentiometric titration curves are shown of the iron-sulfur cluster for R. sphaeroides (○) native, (
) Y501F, (
) T525A, and (
) Y501F/T525A ETF-QOs. The oxidation-reduction midpoint potentials of the cluster were determined by the increase in the X-band EPR signal at 15 K ([4Fe-4S]+) and fit using a single Nernst curve of n=1.
Figure 5 shows the FAD potentiometric titration curves for wild type, Y501F, T525A, and Y501F/T525A mutants of R. sphaeroides ETF-QO. The average midpoint potentials for quinone/anionic semiquinone (Q/SQ) and anionic semiquinone/quinol (SQ/QH2) are E1m = 45 ± 15 mV and E2m = −57 ± 10 mV, respectively, independent of mutation. The previously reported midpoint potentials for porcine ETF-QO flavin were E1m = +28 and E2m = −6 ± 15 mV, respectively (28). Thus, the quinone/anionic semiquinone midpoint potential for R. sphaeroides is similar to mammalian ETF-QO, but the anionic semiquinone/quinol midpoint potentials are lower by ~50 mV. A possible explanation for this difference is the nature to the hydrogen bond donor to N1-C2O of the flavin. Threonine is the hydrogen bond donor in virtually all other known ETF-QOs, but this threonine is substituted by asparagine at position 338 in R. sphaeroides. An asparagine at this site would result in a weaker hydrogen donor and would lower the redox potential of the flavin. The maximum FAD semiquinone EPR signal was at about −15 ± 5 mV for wild type enzyme and mutations, which is about 25 mV lower than calculated based on the midpoint potentials for porcine ETF-QO (28). The larger difference between the two potentials for R. sphaeroides than for porcine ETF-QO predicts a larger maximum semiquinone signal (28). The potentiometric titration results indicate that changes in redox potential of the iron-sulfur cluster do not impact the redox potentials of the flavin. Therefore, the cluster does not seem to have a regional redox-poising effect (modulation effect) on the flavin.
The number of electrons added to each of the redox active centers as a function of the solution potential is shown in Figure 7. For the wild type protein, at the potential that corresponds to the maximum intensity of the semiquinone signal, approximately two electrons have been added to the protein, but for the single mutants only about 1.2 to 1.3 electrons have been added. The decrease in the midpoint potential for the [4Fe-4S]2+,1+ cluster means that less of the cluster is reduced under these conditions. For the double mutant the shift in the midpoint potential for the cluster is so large that almost none is reduced at this solution potential. Absorption Spectra and Reductive Titration: Changes in the optical spectra of ETF-QO have been used previously to monitor the extent of its reduction, which can be achieved either enzymatically using octanoyl-CoA as the electron donor or chemically with dithionite (1, 2, 28). The absorption spectra for oxidized Y501F and for samples reduced with 0.5:1, 1:1 octanoyl-CoA:ETF-QO, and excess dithionate are shown in figure 2. Each mole of octanoyl CoA can transfer two-electrons to the protein. The peaks in spectrum of the oxidized enzyme at about 430 and 380 nm are from the FAD (37). The semiquinone has a maximum absorbance at about 380 nm, so the absorbance at this wavelength increases as semiquinone is formed. However, the semiquinone absorbs much less strongly at 430 nm than FAD so the absorbance at this wavelength decreases as semiquinone is formed. The maximum absorbance for the hydroquinone is at shorter wavelength and it absorbs much less strongly at either 380 or 436 nm than either quinone or semiquinone. The absorption from the iron-sulfur cluster contributes throughout this region but does not have distinctive features (38) and is weaker for [4Fe-4S]+ than for [4Fe-4S]2+.
Figure 7.
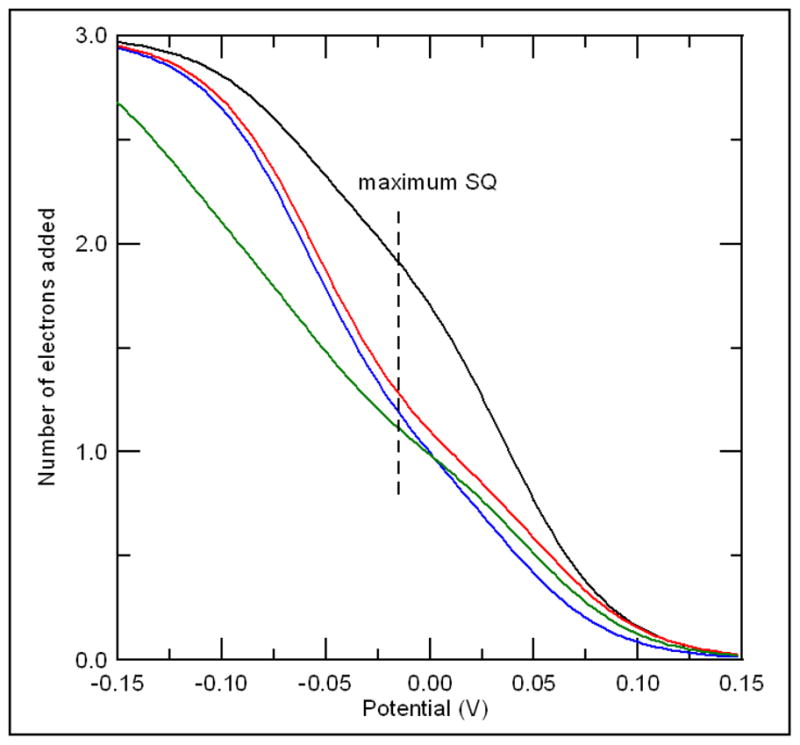
Number of electrons added to the protein as a function of potential for native (__), Y501F (
 ), T525A (
), T525A (
 ), and Y501F/T525A (
), and Y501F/T525A (
 ) calculated from the mid-point potentials listed in Table 1, using the Nernst equation.
) calculated from the mid-point potentials listed in Table 1, using the Nernst equation.
The inset in Figure 2 shows the change in ε433 as a function of added octanoyl-CoA for wild type and mutant ETF-QOs. The initial decrease in absorbance is faster for Y501F/T525A than for other samples because in this case all of the reducing equivalents are added to the FAD, for which there is a larger change in absorbance, and not into the cluster. For the other samples some of the reducing equivalents are going into the cluster, for which the change in absorbance is smaller. The limiting values ofε433 are higher for Y501F/T525A than for the other mutants because the octanoyl CoA is not a strong enough reductant to reduce the cluster in this mutant (Table 1). The midpoint potential for the octanoyl-octenoyl-CoA couple is only −40 mV (39) which does not provide sufficient thermodynamic driving force to fully reduce the iron-sulfur clusters in the mutants. The changes in ε433 are semi-quantitative indicators of the extent of reduction of the sample but are not as informative as the EPR spectra and would not be sufficient to determine the midpoint potentials.
A linear change in the absorbance at 404 nm was used previously as an empirical parameter to monitor the change from oxidized ETF-QO to fully reduced species based on a Δε404nm = 4.35 ± 0.15 mM−1cm−1 per reducing equivalent (2, 28). This Δε404nm is the net effect of changes in both the FAD and iron-sulfur absorbance upon reduction. Mutant enzymes have a larger increase at absorbance 380 nm than wild type and native porcine ETF-QO (20) see figure 2. The increase in amplitude at 380 nm for the mutant samples is more pronounced because the underlying contribution from the [4Fe-4S]2+ has not decreased as much as in the wild type R. sphaeroides, porcine or human proteins. The ability to distinguish changes in concentrations of individual species by EPR provides the information required to interpret the overlapping optical spectra.
Concentrations of [4Fe-4S]+
The potentiometric titration curves were based on relative concentrations. To determine the absolute concentrations, integrated EPR signal intensities were compared with integrals for standard samples. The resulting values for [4Fe-4S]+ are shown in Table 3. Values were obtained from the point on the titration curve where the semiquinone signal is maximum (Figure 5) and for sample reduced with excess dithionite. There is significant uncertainty in these absolute concentrations because the calculation depends on the accuracy with which the total protein concentration in the EPR sample is known. For wild type ETF-QO, T525A, and Y501F the concentration of [4Fe-4S]+ in the presence of excess dithionite accounts for essentially all of the protein in the sample. The smaller limiting concentration for Y501F/T525A, even in the presence of excess dithionite, is consistent with the more negative midpoint potential for this mutant. At the potentials for which maximum semiquinone signal is observed (−15 mV), the concentration of [4Fe-4S]+ in the wild type protein is almost as large as in the presence of excess dithionite, which is consistent with midpoint potentials. However, the decreases in midpoint potential for the mutants make the cluster more difficult to reduce, and therefore the concentrations of [4Fe-4S]+ at −15 mV are much smaller (Table 3 and Figure 7). Previous experiments have determined concentrations of [4Fe-4S]+ by CW EPR for porcine samples reduced with octanoyl CoA or dithionite to be ~75% and ~80–90%, respectively (20, 28), which is in good agreement with the results for the wild type R. sphaeroides protein.
Table 3.
Populations of paramagnetic species in wild type and mutant ETF-QOs samples.
| Sample | Redox State | % SQa | % [4Fe-4S]+ b |
|---|---|---|---|
| Wild Type | SQ max | 53 ± 7 | 86 |
| Dithionite | 14 | 90 | |
| Y501F | SQ Max | 52 ± 4 | 20 |
| Dithionite | 11 | 76 | |
| T525A | SQ Max | 44 ± 3 | 20 |
| Dithionite | 13 | 98 | |
| Y501F/T525A | SQ max | 44 ± 3 | 0 |
| Dithionite | 4 | 55 |
Determined using double integration of the semiquinone CW spectra relative to tempol standard. The percentages are relative to the total protein concentration. Values are accurate to ±5%.
Determined using double integration of the [4Fe-4S]+ cluster CW spectra relative to CuEDTA standard. The percentages are an average from duplicate measurements at 15 and 18 K and are relative to the total protein concentration. Values accurate to ±10%.
Concentrations of semiquinone
Based on the difference between the two midpoint potentials for the flavin reduction the concentration of semiquinone at the potential where the signal is maximized is calculated to be about 80% for all of the samples. The experimental values were in the range of 44 to 53%. The discrepancy between these values and 80% is greater than the uncertainties that are expected in the EPR quantitation. Some of the error may also arise from uncertainties in the midpoint potentials. However, the key observation is that the results are similar for all of the samples, which reinforces the conclusion that changes in the midpoint potentials for the iron-sulfur cluster do not impact the redox behavior of the FAD. In the samples with excess dithionite the concentrations of semiquinone are small for all of the samples.
Iron-semiquinone inter-spin distances
Relaxation enhancement is a powerful method to determine the point dipole distance between a rapidly relaxing transition metal center, such as [4Fe-4S]+ and a more slowly relaxing center such as the FAD semiquinone (40). It was used to determine a distance of 18.5 Å between the center of the iron sulfur cluster and the weighted average position of the spin densities of FAD in mammalian and R. sphaeroides ETF-QO (Fielding, G. R. Eaton, and S. S. Eaton, unpublished results.) In the wild-type samples most of the iron-sulfur cluster is in the paramagnetic [4Fe-4S]+ form. In the mutant samples a smaller fraction of the cluster is paramagnetic so the relaxation enhancement is smaller (Figure 8). The midpoint potential for Y501F/T525A is sufficiently low that the semiquinone signal at −15 mV could be used as the reference signal in the absence of relaxation enhancement. The relaxation rates for this semiquinone between 35 and 70 K were found to be indistinguishable from the values for the semiquinone in ETF.
Figure 8.
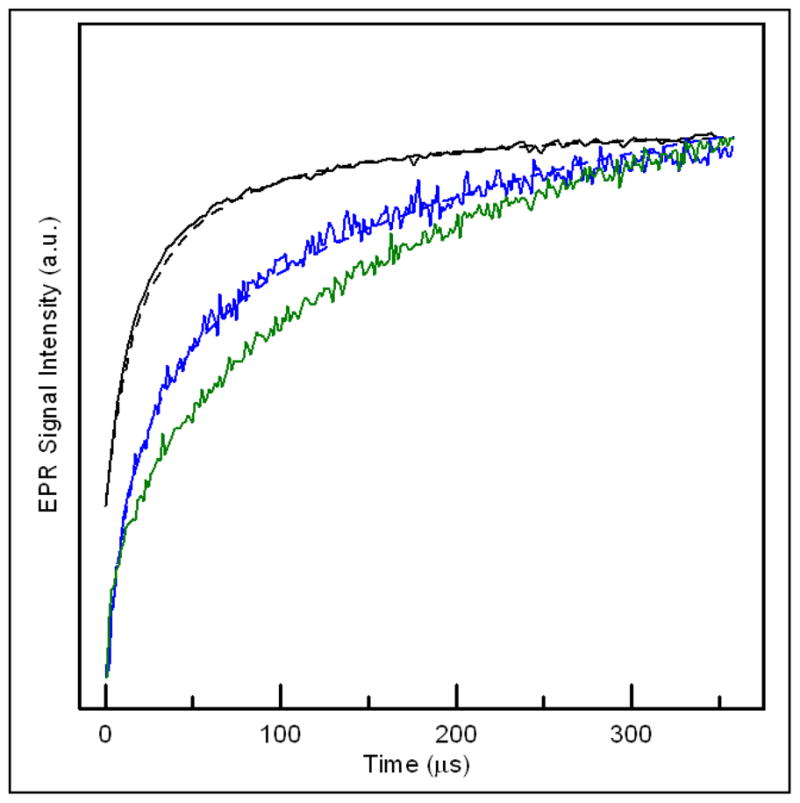
Inversion recovery signals for semiquinone in the enzymatically reduced native (top), Y501F (middle), and Y501F/T525A (bottom) samples of R. sphaeroides ETF-QO at 50 K. The curves are the sums of contributions from semiquinone with neighboring diamagnetic [4Fe-4S]2+ and paramagnetic [4Fe-4S]+. Differences in the inversion recovery curves result from the different populations of paramagnetic [4Fe-4S]+ present: native (~80%), Y501F (25%), and Y501F/T525A (0%). The dashed lines are simulated curves calculated with MENOSR for an average inter-spin distance of ~18.4 ± 1Å.
The inversion recovery curves for the semiquinone signal in samples prepared by octanoyl-CoA reduction were modeled using the program MENOSR (24). The populations of the semiquinone and [4Fe-4S]+ are similar to what is present at the potential that gives the maximum semiquinone signal. The contributions from semiquinone with neighboring diamagnetic [4Fe-4S]2+ and therefore no relaxation enhancement and with neighboring paramagnetic [4Fe-4S]+ that causes relaxation enhancement are superimposed in the inversion recovery curves. The impact of the cluster on the semiquinone is sufficiently large that T1 signals are readily distinguishable from interacting and non-interacting forms. For Y501F the distance determined by relaxation enhancement was in good agreement with the 18.5 Å calculated for wild type protein and the percent of the cluster in the paramagnetic form was 26 ± 3%, which is consistent with the population calculated from the midpoint potentials. The similarity in distances determined by relaxation enhancement for wild type and mutant protein is further evidence that the mutation did not disrupt the protein structure.
Activity and Disproportionation Assays
Quinone reductase activity (Table 3) is the rate at which electrons are shuttled through the enzyme. The specific activity, expressed as apparent substrate turnover, was 24.2 s−1 for the native enzyme. The turnover number decreased to ~8.7 s−1 for Y501F and T525A and 1.8 s−1 for Y501F/T525A. ETF-QO also catalyzes the disproportionation of ETF1e−, where ETF semiquinone serves as both an electron acceptor and donor. Rate constants were measured for the disproportionation of human ETF1e− by R. sphaeroides ETF-QOs (Table 1). The turnover number of 8.3 s−1 for wild-type ETF-QO is considerably lower with this heterolog coupled system than the 81.4 s−1 for the human ETF and ETF-QO homolog system (3). The use of ETF concentrations much less than that required for saturation (Km = 29.4, kcat = 25.6 s−1, Simkovic and Frerman, unpublished results) also contributed to the low turnover number. The turnover numbers for the mutant enzymes Y501F, T525A, and Y501F/T525A decreased to 4.1 s−1, 5.1 s−1, and 0.66 s−1, respectively. The similarities in activities and disproportionation turnover numbers for Y501F and T525A suggest that the decreases are due to the change in mid-point potential and not to a specific role of a particular hydrogen bond.
Electron Transfer Mechanism
Based on the crystal structure of porcine ETF-QO Zhang proposed that reduction of UQ by ETF-QO proceeds via the flavin, not the cluster (11). The cluster to UQ distance (~18.8 Å) is longer than the 14 Å distance required for efficient electron transfer between two redox centers (12). The distance between C6 of the flavin and O3 of UQ is 8.5 Å, which is shorter than the cluster to UQ distance. The point of entry is still ambiguous since either redox center may accept electrons from ETF, although the ETF-QO cluster is located closer to the surface (~8 Å) than the flavin (>14 Å). The decrease in both ubiquinone reductase and disproportionation activities when the midpoint potential of the [4Fe-4S]+ is decreased demonstrates that reduction of the cluster is required for activity. Denke described the role of the iron-sulfur cluster in ubiquinol reduction by the Rieske protein as the rate-limiting partial reaction (8). The role of the iron-sulfur cluster in ETF-QO may be similar.
4. Conclusion
Replacement of amino acids Y501 and T525 that are proposed to hydrogen bond to the cysteine Sγ ligands of the [4Fe-4S]+ cluster of R. sphaeroides ETF-QO by amino acids that do not form hydrogen bonds decreased the mid-point potentials by about 100 mV for a single mutation and about 165 mV for a double mutant. There was no accompanying change in the midpoint potentials for the flavin. The integrity of the mutated proteins was demonstrated by optical and EPR spectroscopy. In the mutants the quinone reductase activity and rates of disproportionation of ETF were dramatically decreased, which demonstrates that reduction of the iron-sulfur cluster is required for activity. The two mutations decreased the mid-point potentials by about the same amount and had similar impacts on activity, which demonstrates the correlation between redox potential of the [4Fe-4S]+ cluster and activity.
Acknowledgments
We are grateful to Dr. Jung-Ja Kim (Medical College of Wisconsin) for pre-publication information from the X-ray structure of porcine ETF-QO that informed our selection of Y501 and T525 in R. sphaeroides ETF-QO as hydrogen-bonded residues.
Abbreviations
- CW
continuous wave
- DTT
dithiothreitol
- EDTA
ethylenediamine tetraacetic acid
- EPR
electron paramagnetic resonance
- ETF
electron transfer flavoprotein
- ETF-QO
electron transfer flavoprotein ubiquinone oxidoreductase
- FAD
flavin adenine dinucleotide
- GaAsFET
gallium arsenide field effect transistor
- MCAD
medium chain acyl-CoA dehydrogenase
- ORP
oxidation reduction potential
- Q1
coenzyme Q1
- SDS
sodium dodecyl sulfate
- UQ
ubiquinone
Footnotes
This work was supported by the National Institutes of Health NIBIB EB002807 (GRE and SSE), The Children’s Hospital Research Foundation, Denver, CO (FEF), and by the BBRSC Underwood Fund (NW).
References
- 1.Ruzicka FJ, Beinert H. A new iron-sulfur protein of the respiratory chain: a component of the fatty acid oxidation pathway. J Biol Chem. 1977;252:8440–8445. [PubMed] [Google Scholar]
- 2.Beckmann JD, Frerman FE. Electron transfer flavoprotein-ubiquinone oxidoreductase from pig liver: purification and molecular, redox and catalytic properties. Biochemistry. 1985;24:3913–3921. doi: 10.1021/bi00336a016. [DOI] [PubMed] [Google Scholar]
- 3.Simkovic M, Degala GD, Eaton SS, Frerman FE. Expression of human electron transfer flavoprotein-ubiquinone oxidoreductase from a baculovirus vector: kinetic and spectral characterization of the human protein. Biochem J. 2002;364:659–667. doi: 10.1042/BJ20020042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beckmann JD, Frerman FE. Reaction of electron transfer flavoprotein with electron transfer flavoprotein-ubiquinone oxidoreductase. Biochemistry. 1985;24:3922–3925. doi: 10.1021/bi00336a017. [DOI] [PubMed] [Google Scholar]
- 5.Goodman SI, Binard RJ, Woontner MR, Frerman FE. Glutaric acidemia type II: gene structure and mutations of the electron transfer flavoprotein ubiquinone oxidoreductase (ETF-QO) gene. Mol Genet Metab. 2002;77:86–90. doi: 10.1016/s1096-7192(02)00138-5. [DOI] [PubMed] [Google Scholar]
- 6.Jang SB, Seefeldt LC, Peters JW. Modulating the midpoint potential of the [4Fe-4S] cluster of the nitrogenase Fe protein. Biochemistry. 2000;39:641–648. doi: 10.1021/bi991694v. [DOI] [PubMed] [Google Scholar]
- 7.Chen K, Bonagura CA, Tilley GJ, McEvoy JP, Jung Y-S, Armstrong FA, Stout CD, Burgess BK. Crystal structures of ferredoxin variants exhibiting large changes in [Fe-S] reduction potential. Nature Structural Biology. 2002;9:188–192. doi: 10.1038/nsb751. [DOI] [PubMed] [Google Scholar]
- 8.Denke E, Merbitz-Zahradnik T, Hatzfeld OM, Snyder CH, Link TA, Trumpower BL. Alteration of the midpoint potential and catalytic activity of the Rieske iron-sulfur protein by changes of amino acids forming hydrogen bonds to the iron-sulfur cluster. J Biol Chem. 1998;273:9085–9093. doi: 10.1074/jbc.273.15.9085. [DOI] [PubMed] [Google Scholar]
- 9.Stephens PJ, Jollie DR, Warshel A. Protein control of redox potentials of iron-sulfur proteins. Chem Rev. 1996;96:2491–2513. doi: 10.1021/cr950045w. [DOI] [PubMed] [Google Scholar]
- 10.Langen R, Jensen GM, Jacob U, Stephens PJ, Warshel A. Protein control of iron-sulfur clusters redox potentials. J Biol Chem. 1992;267:25625–25627. [PubMed] [Google Scholar]
- 11.Zhang J, Frerman FE, Kim JJ. Structure of electron transfer flavoprotein ubiquinone oxidoreductase and electron transfer to the mitochondrial ubuiquinone pool. Proc Nat Acad Sci US. 2006;103:16212–16217. doi: 10.1073/pnas.0604567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principals of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 13.Sazanov LA, Hinchliffe P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science. 2006;311:1430–1436. doi: 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
- 14.Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5:823–826. doi: 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miroux B, Walker JE. Overproduction of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 16.Ramsay RR, Steenkamp DJ, Husain M. Reactions of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase. Biochem J. 1987;241:883–892. doi: 10.1042/bj2410883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Husain M, Steenkamp DJ. Electron transfer flavoprotein from pig liver mitochondria. A simple purification and reevaluation of some of the molecular properties. Biochem J. 1983;209:541–545. doi: 10.1042/bj2090541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beinert H. Micro methods for the quantitative determination of iron and copper in biological material. Meth Enzymol. 1978;54:435–445. doi: 10.1016/s0076-6879(78)54027-5. [DOI] [PubMed] [Google Scholar]
- 19.Siegel LM. Quantitative determination of noncovalently bound flavinx: types and methods of analysis. Meth Enzymol. 1978;53:419–429. doi: 10.1016/s0076-6879(78)53046-2. [DOI] [PubMed] [Google Scholar]
- 20.Johnson MK, Morningstar JE, Oliver M, Frerman FE. Electron paramagnetic resonance and magnetic circular dichroism studies of electron-transfer flavoprotein-ubiquinone oxidoreductase from pig liver. FEBS Letters. 1987;226:129–133. doi: 10.1016/0014-5793(87)80565-3. [DOI] [PubMed] [Google Scholar]
- 21.Dutton PL. Redox potentiomery: determination of mid-point potentials of oxidation reduction components of biological electron transfer systems. Meth Enzymol. 1978;54:411–435. doi: 10.1016/s0076-6879(78)54026-3. [DOI] [PubMed] [Google Scholar]
- 22.Quine RW, Eaton GR, Eaton SS. Pulsed EPR spectrometer. Rev Sci Instrum. 1987;58:1709–23. [Google Scholar]
- 23.Eaton SS, Eaton GR. Relaxation times of organic radicals and transition metal ions. Biol Magn Reson. 2000;19:29–154. [Google Scholar]
- 24.Rakowsky MH, More KM, Kulikov AV, Eaton GR, Eaton SS. Time-Domain Electron Paramagnetic Resonance as a Probe of Electron-Electron Spin-Spin Interaction in Spin-Labeled Low-Spin Iron Porphyrins. J Amer Chem Soc. 1995;117:2049–57. [Google Scholar]
- 25.Zhou Y, Bowler BE, Lynch K, Eaton SS, Eaton GR. Interspin distances in spin-labeled metmyoglobin variants determined by saturation recovery EPR. Biophys J. 2000;79:1039–1052. doi: 10.1016/S0006-3495(00)76358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toy AD, Chaston SHH, Pilbrow JR, Smith TD. Electron spin resonance study of the copper(II) chelates of certain monothio-β-diketones and diethyldithiocarbamate. Inorg Chem. 1971;10:2219–2225. [Google Scholar]
- 27.Rakowsky MH, Zecevic A, Eaton GR, Eaton SS. Determination of high-spin iron(III)-nitroxyl distances in spin-labeled porphyrins by time-domain EPR. J Magn Reson. 1998;131:97–110. doi: 10.1006/jmre.1997.1338. [DOI] [PubMed] [Google Scholar]
- 28.Paulsen KE, Orville AM, Frerman FE, Lipscomb JD, Stankovich MT. Redox properties of electron-transfer flavoprotein ubiquinone oxidoreductase as determined by EPR-spectroelectrochemistry. Biochemistry. 1992;31:11755–11761. doi: 10.1021/bi00162a012. [DOI] [PubMed] [Google Scholar]
- 29.Beinert H, Sands RH. In: Foundations of Modern EPR. Eaton GR, Eaton SS, Salikov KM, editors. World Scientific; Singapore: 1996. pp. 379–409. [Google Scholar]
- 30.McDevitt CA, Hanson GR, Noble CJ, Cheesman MR, McEwan AG. Charactization of the redox centers in dimethyl sulfide dehydrogenase from rhodovulum sulfidophilum. Biochemistry. 2002;41:15234–15244. doi: 10.1021/bi026221u. [DOI] [PubMed] [Google Scholar]
- 31.Dunham WR, Sands RH. g-Strain, ENDOR, and structure of active centers of two-iron ferredoxins. Biochem Biophys Res Comm. 2003;312:255–261. doi: 10.1016/j.bbrc.2003.09.228. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Y, Bowler BE, Eaton GR, Eaton SS. Electron Spin Lattice Relaxation Rates for S =1/2 Molecular Species in Glassy Matrices or Magnetically Dilute Solids at Temperatures between 10 and 300 K. J Magn Reson. 1999;139:165–174. doi: 10.1006/jmre.1999.1763. [DOI] [PubMed] [Google Scholar]
- 33.Kolling DJ. Atomic resolution structures of Rieske iron-sulfu protein: role of hydrogen bonds in tuning the redox potential of iron-sulfur clusters. Structure. 2007;15:29–38. doi: 10.1016/j.str.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iwagami SG, Creagh AL, Haynes CA, Borsari M, Felli IC, Piccioli M, Eltis LD. The role of a conserved tyrosine residue in high-potential iron sulfur proteins. Protein Sci. 2007;4:2562–2572. doi: 10.1002/pro.5560041213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giastas P, Pinotsis N, Efthymiou G, Wilmanns M, Kyritsis P, Moulis JM, Mavridis IM. The structure of the 2[4Fe-4S] ferredoxin from Pseudomonas aeruginosa at 1.32-A resolution: comparison with other high-resolution structures of ferredoxins and contributing structural features to reduction potential values. J Biol Inorg Chem. 2006;11:445–458. doi: 10.1007/s00775-006-0094-9. [DOI] [PubMed] [Google Scholar]
- 36.Dey A, Roche CL, Walters MA, Hodgson KO, Hedman B, Solomon EI. Sulfur K-edge XAS and DFT calculations on [Fe4S4]2+ clusters: effects of H-bonding and structural distortion on covalency and spin topology. Inorg Chem. 2005;44:8349–8354. doi: 10.1021/ic050981m. [DOI] [PubMed] [Google Scholar]
- 37.Gorelick RL, Schopfer LM, Ballou DP, Massey V, Thorpe C. Interflavin oxidation-reduction reactions between pig kidney general acyl-CoA dehydrogenase and electron transferring-flavoprotein. Biochemistry. 1985;24:6830–6839. doi: 10.1021/bi00345a015. [DOI] [PubMed] [Google Scholar]
- 38.Yano T, Yagi T, Sled VD, Ohnishi T. Expressions and characterization of the 66-kilodalton (NQO3) iron-sulfur subunit of the protein translocating NADH-quinone oxidoreductase of Paracoccus denitrificans. J Biol Chem. 1995;270:18624–18270. doi: 10.1074/jbc.270.31.18264. [DOI] [PubMed] [Google Scholar]
- 39.Lenn ND, Stankovich MT, Liu H. Regulation of the redox potential of general acyl-CoA dehydrogenase by substrate binding. Biochemistry. 1990;29:3709–3755. doi: 10.1021/bi00467a017. [DOI] [PubMed] [Google Scholar]
- 40.Kulikov AV, Likhtenshtein GI. The use of spin relaxation phenomena in the investigation of the structure of model and biological systems by the method of spin labels. Adv Mol Relax and Interact Proc. 1977;10:47–69. [Google Scholar]
- 41.Paulsen KE, Stankovich MT, Orville AM. Electron Paramagnetic Resonance Spectrochemical Titration. Methods Enzymol. 1993;227:396–411. doi: 10.1016/0076-6879(93)27016-a. [DOI] [PubMed] [Google Scholar]