

Abstract
Coccoliths are micrometer scale shields made from 20 to 60 individual calcite (CaCO3) crystals that are produced by some species of algae. Currently, coccoliths serve as an important sink in the global carbon cycle, but decreasing ocean pH challenges their stability. Chalk deposits, the fossil remains of ancient algae, have remained remarkably unchanged by diagenesis, the process that converts sediment to rock. Even after 60 million years, the fossil coccolith crystals are still tiny (< 1 μm), compared with inorganically produced calcite, where one day old crystals can be 10 times larger, which raises the question if the biogenic nature of coccolith calcite gives it different properties than inorganic calcite? And if so, can these properties protect coccoliths in CO2 challenged oceans? Here we describe a new method for tracking dissolution of individual specimens, at picogram (10-12 g) resolution. The results show that the behavior of modern and fossil coccoliths is similar and both are more stable than inorganic calcite. Organic material associated with the biogenic calcite provides the explanation. However, ancient and modern coccoliths, that resist dissolution in Ca-free artificial seawater at pH > 8, all dissolve when pH is 7.8 or lower. Ocean pH is predicted to fall below 7.8 by the year 2100, in response to rising CO2 levels. Our results imply that at these conditions the advantages offered by the biogenic nature of calcite will disappear putting coccoliths on algae and in the calcareous bottom sediments at risk.
Keywords: marine carbon cycle, ocean buffer capacity, dissolution rate, atomic force microscopy, single particle dissolution
As atmospheric CO2 levels rise, pH in the oceans decreases, threatening marine organisms. Some species of planktonic algae (1), the coccolithophorids, help control calcite equilibria, contributing to the buffer capacity of the global ocean and sediments. Coccolithophorids also thrived in ancient seas, producing the vast beds of chalk that now serve as aquifers for drinking water and reservoirs for oil and gas. Particle size, and thus pore size, in chalk remain very small (Fig. 1), in spite of the natural tendency for surface free energy to drive Ostwald ripening. Ostwald ripening is the process where small crystals dissolve and larger ones grow (2), for example, grain coarsening in limestone (3). However, after 60 million years, the biogenic calcite crystals in chalk are still less than a micrometer whereas calcite crystals synthesised in the laboratory are a factor of at least 5 times larger, after only hours to days (Fig. 1B). Why have chalk particles remained so small? And does this resistance to recrystallization protect coccoliths in acidifying oceans? This work was designed (i) to explore the possibility that the answer lies in the biogenic nature of the calcite and (ii) to discover its implications for modern ocean acidification and the carbon cycle.
Fig. 1.

SEM images of (A) chalk extracted from more than 60 million year old sediments below the North Sea and (B) calcite formed inorganically under ambient conditions in the laboratory (about a day).
Algae form the individual crystals they need for making coccoliths on templates of complex sugars (4–7). Polysaccharides extracted from cultured algae inhibit calcite growth by blocking some crystal surface sites (8) and mature coccoliths are protected from dissolution by an organic coating (7). Previous work has also suggested the presence of organic material associated with coccoliths from chalk (9–11). Direct comparison of dissolution rates for biogenic and inorganic calcite would go far to elucidate the role of organic material, but it is not trivial to collect enough pure, clean biogenic calcite for bulk experiments. Cultured samples inevitably retain some proteins from the algae cells, which complicates determining a pure calcite dissolution rate. In ancient material, the proteins are assumed to have been eaten millions of years ago when the algae sank to the ocean floor, so proteins are not a problem, but chalk invariably also contains quartz, flint, clay, and fossils of other microorganisms. Although single coccoliths have been studied previously (7, 12, 13), dissolution rates were difficult to extract because mass and surface area could not be determined directly. To circumvent these problems, we developed a method that directly tracks mass change of single, individual specimens by monitoring the change in oscillation frequency of a cantilever to which they were glued. Periodic SEM images verified what was there, what dissolved, and provided estimates of surface area.
Specimens were attached to cantilevers (initial resonant frequency, f0 ∼ 330 kHz; spring constant, k ∼ 40 N m) with approximately 100 pg of glue. Specimen mass was much less than cantilever mass, so change during dissolution, Δm, could be derived from the resonance frequency difference (14, 15):
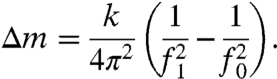 |
[1] |
We used pure, optical quality Iceland spar as the inorganic calcite control for comparison with pure lab grown calcite, commercial calcite powder, cultured samples of Coccolithus pelagicus, and intact fossil coccoliths from chalk. Each tip, with its specimen, was submerged in 2 mL of solution for several minutes, rinsed with filtered, calcite-saturated ultrapure deionized (DI) water and dried. Oscillation frequency was measured in air. Each submerge-rinse-dry-measure sequence began with a fresh aliquot of solution and provided one datum on the plots of Fig. 2 and Fig. 3.
Fig. 2.

Dissolving calcite. (A) Mass loss of pure, synthetic calcite crystals in pure DI water; (B) SEM of the single particle (mass ∼4.4 ng) glued to a cantilever; (C) glue residue after dissolution; (D) mass loss of one Iceland spar calcite cleavage fragment exposed to ASW in tests where Ca concentration was slightly above saturation and where the solution was Ca-free and finally pure DI water; (E) calcite mass approximately 420 pg; and (F) glue residue after dissolution was complete.
Fig. 3.

Dissolving coccoliths. (A) Mass loss from one cultured coccolith exposed to Ca-free ASW (Ωcalcite estimated in each solution aliquot at the end of the dissolution period, i.e., representing each data point, < 10-8) and then to pure water (estimated Ωcalcite in each aliquot < 10-12); (B) a coccolith from C. pelagicus imaged at t = 6,000 s; (C) after complete dissolution, an organic relic beautifully reflects coccolith structure and morphology; (D) mass loss from a 60 million year old coccolith (Arkhangelskiella sp. cf. cymibiformis); (E) initial SEM image, then exposure to Ca-free ASW, pH 8.4 (t > 1,500 s) removed loose particles (arrows), presumed to be nonbiogenic calcite, but the coccolith remained; (F) the same coccolith after 3,200 s, just before exposure to pure water; and (G) complete dissolution left an unstructured relic.
Results and Discussion
Fig. 2A shows an example of mass loss with time for a single crystal extracted from pure, synthetic calcite powder (Fig. 2B) in DI water. The results for all of the experiments are listed in Table 1. Specimen mass was derived from the difference between the initial cantilever-glue-specimen and the cantilever-glue at the end of the experiment (Fig. 2C). Measurement of mass loss is extremely precise, with uncertainty in the femtogram (10-15 g) range. It was these differences in the rate of mass loss for the various samples that we used to make the conclusions for this work. However, to be able to compare our rates for single particles with those of previously published, macroscopic experiments, we have also derived the dissolution rate by normalizing the rate of mass loss with surface area.
Table 1.
Initial specimen mass, rate of mass loss, estimated surface area, and dissolution rate
| Pure water, pH 5.6 | Ca-free ASW, pH 8.2–8.4 | Ca-free ASW, pH 7.8 | ||||||
| Mass (pg)* | Surface area (μm2) | Rate of mass loss ± σ (fg/s)† | Dissolution rate (μmol/m2s) | Rate of mass loss ± σ (fg/s) | Dissolution rate (μmol/m2s) | Rate of mass loss ± σ (fg/s) | Dissolution rate (μmol/m2s) | |
| Pure synthetic calcite | 4,440 | 845 | 648 ± 4 | 11.5 | 105 ± 4 | 1.4 | ||
| Inorganic Iceland spar calcite | 420 | 100 | 103 ± 2 | 10.0 | 57 ± 2 | 5.2 | ||
| Commercial calcite powder | 13,700 | 2,450 | 650 ± 2 | 2.7 | ||||
| C. pelagicus cultured | 408 | 325 | 268 ± 14 | 9.0 | 0 ± 2 | 0 | - | |
| 390 | 300 | 246 ± 22 | 7.6 | 0 ± 8 | 0 | - | ||
| 531 | - | - | - | - | - | 38 ± 6 | ||
| Ancient coccoliths | 830 | 180 | 377 ± 16 | 20.6 | 0 ± 6 | 0 | - | |
| 200 | 130 | 243 ± 37 | 18.7 | 0 ± 8 | 0 | - | ||
| 127 | 160 | 68 ± 6 | 4.2 | 0 ± 0.7 | 0 | - | ||
| 500 | 126 | - | - | - | 82 ± 14 | 6.5 | ||
| 440 | 150 | - | - | - | 72 ± 6 | 4.8 | ||
*pg, picogram (10-12 g)
†fg, femtogram (10-15 g)
Surface Area Estimates and Dissolution Rate.
Dissolution rates depend on surface area as well as dislocation density, the number of edges, crystal orientation, and the nucleation density of etch pits (16). Most of these variables are difficult to estimate, so therefore contribute to uncertainty in normalizing dissolution rates. For these experiments, we determined the specimen-solution contact area using dimensions derived from SEM images, which introduces several more sources of uncertainty: Images transform 3-D objects to 2-D, so the angle of view distorts dimensions; pictures were only taken from one side, so material behind the field of view was neglected; and the surface area that was protected by glue, although small, was difficult to estimate. For the cultured coccoliths, we derived surface area by analogy with an elliptical plate. In addition to the actual specimen that we dissolved, we also used average measurements for the two ellipse axes and thickness from SEM and atomic force microscopy (AFM) images taken perpendicular to the flat dimension from other specimens of the same species and batch. For the fossil coccoliths, dimensions were taken in the same way from other samples of the same species in the source chalk. For the calcite specimens, we derived surface area by two methods from SEM images and from their mass and a simple model.
To test for consistency between estimates from SEM images, we used the synthetic calcite and assumed spherical geometry for the rounded rhombohedral crystals, such as is shown in Fig. 2A. The dissolution rate would then be linked essentially to one parameter: the radius. This trick removes the need for determining the area because the mass of the sphere gives the surface area through the radius. The mass loss for a sphere with a uniform surface would have the following relationship with time:
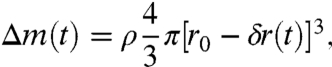 |
[2] |
where Δm(t) represents the residual mass after time, t; ρ, the density of calcite (2.71 g/cm3); and [r0 - δr(t)], the radius at time, t, after dissolution of an incremental part of the radius, δr. Dissolution rate per unit area is assumed to be constant, so the rate of decrease in the radius is constant:
| [3] |
where R represents the thickness of the layer lost per unit time. By inserting Eq. 3 into Eq. 2 the dissolution rate can be found by extracting the third order fitting coefficient, K3,
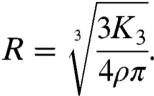 |
[4] |
Fitting the mass loss with time curve with a third order polynomial gives an average rate of mass loss. For the curve shown in Fig. 2A for pure calcite, mass loss is 4.3 Å/s. For comparison, the thickness of a calcite step that is one molecular layer high on the rhombohedral face is 3 Å. Converting to conventional units for bulk dissolution experiments gives a dissolution rate of 11.5 μmol/m2s. From the SEM images, we estimated the surface area to be about 8 × 10-8 m2. Assuming that the initial changes in surface area are small, a linear fit to the early data from the same curve gives a dissolution rate of 8.1 μmol/m2s. The closeness of the dissolution rates derived from the polynomial and the tangential fits provides confidence that the surface area assumptions are not too crude.
As a specimen dissolves, its surface area gradually decreases, so for all measurements we used only data from the first part of the curves where dissolution began for determining the rate of mass loss. In all cases, we used the surface area estimated from images. Although repeat surface area estimates gave results that were within a few percent, the true level of error is nearly impossible to estimate. Therefore, we have not reported uncertainty for surface area, nor for the dissolution rates derived using them. In fact, the research questions of this study were designed to provide yes or no answers, not requiring a definition of uncertainty. Conclusions are based on the rates of mass loss, which are very precise. We compare dissolution rates only at the order of magnitude level.
Calcite Dissolution.
The Iceland spar calcite was optically pure and had less than 1% impurities, mainly Mg2+, Mn2+, Sr2+, and Fe2+ (17). We selected a fresh cleavage fragment with about the same mass as the cultured coccoliths. In ultrapure DI water, where pH was 5.6, dissolution was 10 μmol/m2s (Fig. 2D and Table 1), which was close to that of the pure lab grown calcite (8–11.5 μmol/m2s).
The dissolution rate for commercial calcite powder is slow in comparison (2.7 μmol/m2s) (Table 1 and Fig. S1). This slow rate can easily be explained. Although reagent grade commercial calcite powder contains less than 1% inorganic impurities, there is a considerable concentration of organic compounds. During the manufacturing process, simple organic additives such as citric acid or sugar are added to inhibit crystal growth, thus enhancing nucleation and ensuring a fine-grained, fluffy powder preferred by customers. This organic material is observable on the crystal surfaces (18) and although commercial calcite powder is inorganically produced, the added organic compounds inhibit dissolution, making it behave much more like biogenic calcite. However, we determined the dissolution rate of this material so we could confirm that our single specimen results are comparable with data for commercial powder from bulk experiments reported in the literature [close to 5 μmol/m2s at pH 8 (16)]. Dissolution rates for commercial powders are expected to vary because each supplier uses a different, often secret, recipe for developing their product.
The standard recipe for artificial seawater (ASW) (19) produces a solution that is supersaturated with respect to calcite. For a few experiments, we produced a solution with pH = 8.2 and 75% of the CaCl2, to make it only slightly supersaturated, so no dissolution was expected (Fig. 2D). In ASW at the same pH, but initially free of Ca (Ωcalcite = 0), the Iceland spar dissolved, but at a rate slower than at pH 5.6, also as expected.
Coccolith Dissolution.
We chose C. pelagicus because for a modern species its diameter is relatively large (∼12 μm), similar to those found in chalk. In DI water at equilibrium with air (pH 5.6, Ωcalcite = 0), specimens dissolved readily, with rates similar to the pure, inorganic Iceland spar (Fig. 3A and Table 1). Ancient coccoliths, when exposed to DI water, such as the specimen of Arkhangelskiella sp. cf. cymibiformis (20) shown in Fig. 3D, behaved in the same way. For most fossil specimens, the rate of mass loss was in the same range as for cultured material (200–400 fg/s) (Table 1 and Fig. S2). In low pH solutions, far from equilibrium, their biogenic nature offers no hindrance for dissolution. The dissolution rate estimates for ancient coccoliths are high (18–20 μmol/m2s), a factor of 2 higher than that of pure inorganic calcite, which we attribute to large uncertainty in estimating surface area. Fossil coccolith morphology is extremely complex so an error in surface area by a factor of 2 would not be unrealistic.
In Ca-free ASW at pH 8.2, modern and ancient coccoliths behave the same, and unexpectedly, they are stable, in spite of solution undersaturation with respect to calcite. In the initial Ca-free ASW, Ωcalcite was zero and by the time the sample was extracted from each of the aliquots, we estimate that Ωcalcite was about 10-8, far from equilibrium. The noise in the data (Fig. 3A, t < 6,000 s; and Fig. 3D, 1,500 < t < 3,200 s) during the ASW experiments results from adventitious particles (∼10–20 pg) that attach and detach. SEM images taken initially (Fig. 3F) and after exposure to Ca-free ASW (Fig. 3 B and E) confirmed that the coccoliths were intact. Some fossil coccoliths are accompanied by small particles (such as those indicated by the arrows, Fig. 3F). These dissolve in Ca-free ASW (Fig. 3D, t < 1,500 s), their solubility and morphology are reminiscent of nonbiogenic calcite and account for minor mass loss during the early stages of fossil coccolith dissolution. Because the SEM image from the end of the Ca-free ASW experiments proves that the coccolith itself remains intact, one can conclude that there is an important difference between the stability of the biogenic coccoliths and the inorganically formed calcite. SEM images (Fig. 1A) reveal that although euhedral crystals of calcite exist in chalk, and are probably nonbiogenic, coccolith fragments are abundant, which suggests that whatever inhibits coccolith dissolution in Ca-free ASW also plays a dominant role in keeping chalk particles small.
The stability of the biogenic calcite in Ca-free seawater at pH 8.2 contrasts dramatically with the dissolution of pure, inorganic calcite at the same pH. The SEM image, Fig. 3C, suggests why. After dissolution, a relic remains. This insoluble material is an exquisite reflection of the original coccolith morphology. Reproducibility experiments proved that all cultured coccoliths left such a coating, but some collapsed after dissolution. The mass of the relic material could not be determined precisely because it is less than the noise level in the experiment (10–20 pg). For consideration, the mass of a 30-nm thick organic coating covering an area of about 300 μm2 would be approximately 10 pg. Such a protective layer is consistent with evidence of organic coatings on cultured coccoliths presented by Henriksen et al. (7). The material remaining after dissolution of fossil coccoliths lacked form and was difficult to differentiate from the glue on the cantilever, but comparing Fig. 3G with Fig. 2 C and F, Fig. S3, and Fig. S4 provides a hint that formless, insoluble residue also remains after dissolution of ancient coccoliths.
Although cultured and ancient specimens were stable in Ca-free ASW at pH 8.2 or higher, both types dissolved completely at pH 7.8 in spite of organic coating (Fig. S4 and S5). Rates of mass loss and dissolution at the lower pH (Table 1) were in the same range as pure calcite dissolving in solutions at pH > 8. In light of ocean acidification from increasing CO2 emissions (13, 21–23), this result is important. Although some algae species benefit from increased CO2 (24), our results supplement those of Riebesell et al. (25), that more acidic oceans will put coccolith stability at risk. Models predict that ocean pH will decrease below 7.8 sometime between year 2060 and 2100 (24), but some areas of surface ocean, where seawater concentrations are locally different, are likely to dip sooner to levels promoting coccolith dissolution. Another aspect is the stability of coccoliths on the seafloor. Under normal conditions, algae flourish at the ocean surface, where seawater is oversaturated with respect to calcite. When they die, they sink. Below the lysocline, also called the carbonate compensation depth (CCD) (3,000–4,500 m below sea level), they dissolve (26). If overall pH of ocean water decreases, the lysocline will rise in the water column, increasing the volume of water where coccoliths are unstable (27, 28). At pH < 8.2, where calcite is undersaturated, for every CO2 molecule that contributes to increased ocean acidity, one carbonate ion will be released from calcite to seawater,
| [5] |
consuming buffer capacity also in the calcareous mud that forms bottom sediments.
Implications.
In spite of being different species, and separated in age by more than 60 Ma, ancient and modern coccoliths behave remarkably similarly. Biogenic calcite is more robust than inorganic calcite; inorganic calcite dissolves in Ca-free ASW at pH 8.2, whereas biogenic calcite does not. However, in the same solutions, biogenic calcite dissolves when pH decreases from > 8.0 to 7.8, which has several implications. The organic material that protects coccoliths from dissolution at conditions slightly undersaturated with respect to calcite contributes to inhibiting the recrystallization that is necessary for diagenesis. This organic material has kept chalk particles very small, even in comparison with inorganic calcite crystals only a day old. In modern oceans, coccoliths are an important sink in the carbon cycle. In scenarios where oceans are acidified, either by the natural equilibration of CO2 from air or by anthropogenic attempts at carbon sequestration in deep ocean waters, our results imply that if pH drops to 7.8 or lower in volumes of the ocean where calcite is undersaturated, the protection offered by the biogenic nature of calcite will disappear, putting coccoliths on algae and in the calcareous bottom sediments at risk.
Materials and Methods
For the ancient material, we selected coccoliths from very lightly crushed core plugs. The chalk came from a known oil-bearing formation in the North Sea basin, but for the location where this particular core was taken there was geological and analytical evidence that oil had never come into contact with the sample. The specimens that we used were taken from inside the core to avoid contamination from air, drilling fluid, and fingers. The fossil coccoliths were 100–800 pg, so we chose a large modern species for comparison. We cultured C. pelagicus, PLY no. 182G, from the Plymouth Algal Culture Collection, in F/2 medium with artificial seawater, at 18 °C under constant illumination (29). For harvesting, we centrifuged the coccolithophore culture, removed the supernatant, and rinsed the pellet with Iceland spar equilibrated water to prevent dissolution of the coccoliths. The solid material was then spread onto a glass slide. Single coccoliths were located using an optical microscope and mounted onto the cantilever.
For the pure calcite control, we used a chip of optical quality Iceland spar calcite (Ward’s Scientific), that was about the same mass as the cultured coccoliths. We also precipitated pure calcite crystals by mixing solutions of CaCl2 and Na2CO3. Literature values for the calcite dissolution rate have most often been determined on commercial calcite powder, which is free of trace element contamination, but which contains organic growth inhibitors, such as citric acid or sugar, added during manufacture to promote abundant nucleation and high surface area. The growth inhibitors undoubtedly affect dissolution rate, but we also experimented with this material so that we could compare with literature values for bulk calcite dissolution rates.
For all solutions, we used ultrapure DI water (MilliQ). Its resistivity was 18.2 MΩcm. Glass- and plasticware were washed and rinsed with DI water to minimize contamination by adventitious carbon. For preparing the artificial seawater, we used reagent grade salts and followed the recipe of Kester et al. (19). ASW is supersatured with calcite, risking precipitation during the experiments, so we used 75% of the recommended CaCl2 to make a slightly supersaturated solution for a couple of experiments. For most of them, made with Ca-free ASW, we simply omitted the CaCl2.
We designed the experiments to examine mass loss at conditions very far from equilibrium. For calcite, the saturation index, SIcalcite or Ωcalcite, is defined as
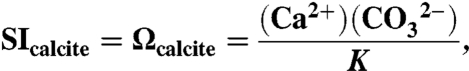 |
where I represents the activity of species I and K represents the thermodynamic equilibrium constant at standard temperature, pressure, and infinite dilution. In the initial aliquots of DI and Ca-free ASW, Ωcalcite was essentially zero. If a full coccolith, 500 pg mass, dissolved in a single 2 mL aliquot of DI, Ωcalcite would be ∼10-10.5. If it dissolved in Ca-free ASW, Ωcalcite would be ∼10-6, but dissolution was carried out during 10 to 100 steps, each in a separate, fresh aliquot, so by the end of each dissolution step, Ωcalcite in the aliquot would be one or two orders of magnitude lower, very far from equilibrium.
Mass Measurements.
To measure the mass and mass loss of individual specimens, we measured changes in the oscillation frequency of an AFM cantilever with the specimen attached. We used Olympus AC160TS cantilevers and the micromanipulators of an Asylum MFP3D AFM on a stand-alone base. The spring constant for each cantilever was derived from its thermal spectrum (30).
To attach the specimen on the cantilever, we dipped its end into a tiny spot of glue (Dana Lim Universal Epoxy 335), so that about 100 pg adhered, then we fished a likely particle from the thin layer of material spread on a clean, glass microscope slide. We used an optical microscope to find the particles, but the resolution was not good enough to see their quality. Specimens were chosen mostly by their accessibility to the cantilever. After the glue had cured for a day, the resonant frequency of the particle-cantilever assembly was recorded in air, using the standard AC-mode tuning routine of the software (Asylum MFP3D in Igor Pro 6.02). Initial specimen mass was then determined using Eq. 1. Some samples were examined using SEM to inspect for quality. From these samples, we could estimate surface area. Preparing the samples was tricky. In a couple of cases, glue covered the specimen, but it was observable on SEM images and data from dissolution experiments on these specimens clearly showed no mass loss in Ca-free ASW or in DI water.
All experiments were made at room temperature, 23 °C. All solutions were at equilibrium with atmospheric CO2 and pH was measured just before the experiment began. The pH electrode was calibrated just prior to each experiment using standard buffers at pH 7 and 10. For each dissolution, 2 mL of the chosen solution was filtered through a 0.22 μm acetate filter into an open AFM liquid cell, so the solutions were at equilibrium with CO2 in air at all times. The cantilever-specimen assembly was subsequently submerged for a time that varied from 2–10 min, depending on the experiment, then it was rinsed with filtered, calcite-saturated DI water, and finally dried by removing the remaining droplet by touching it with the corner of lint- and oil-free absorbent paper. The resonant frequency was recorded again. The sequence was repeated, using a fresh aliquot of solution each time and each sequence resulted in one datum for mass loss with time. The difference between the final and original cantilever mass provided the mass of the glue. The amount of calcite dissolved in each aliquot, and pH change, were negligible. The system was always very far from equilibrium, i.e., Ωcalcite approaching zero. Mass loss rates were derived from a linear fit to the relevant parts of the data. The results recorded in Table 1 have uncertainty ± 1σ.
In most cases, we inspected the sample and cantilever with SEM before, during, and after the dissolution experiment. We did not coat the samples with a conducting film to improve imaging. To test if the electron beam had modified sample dissolution behavior, some dissolution experiments where performed without inspection by SEM. There was no detectable difference.
Measuring single specimen dissolution rates is extremely time consuming and very tricky. In total, we made about 50 experiments. In some cases, the sequence ended when the specimen flew off the cantilever during a measurement. In other cases, foreign particles attached to the cantilever or tip, destroying the mass/time relationship. When the purpose was to prove only the reproducibility of mass loss with time, SEM images were not taken, so surface area and dissolution rate could not be estimated.
Supplementary Material
Acknowledgments.
We sincerely thank F. Engstrøm, J.W. Nielsen, and L. Skovbjerg for discussion, L. Skovbjerg and J. Nissenbaum for Fig. 1, D. Belova and K.K. Sand for samples and Z. Balogh for SEM assistance. Comments from B. Watson and three anonymous reviewers helped sharpen the text. T.H. is grateful to the Danish National Natural Sciences Research Council (FNU) for the Skou Fellowship. The Nano-Chalk Venture is funded by the Danish National Advanced Technology Foundation, Maersk Oil and Gas A/S, and the University of Copenhagen.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1009447108/-/DCSupplemental.
References
- 1.Young JR, Didymus JM, Bown PR, Prins B, Mann S. Crystal assembly and phylogenetic evolution in heterococcoliths. Nature. 1992;356:516–518. [Google Scholar]
- 2.Madras G, McCoy BJ. Ostwald ripening with size-dependent rates: Similarity and power-law solutions. J Chem Phys. 2002;117:8042–8049. [Google Scholar]
- 3.Morse JW, Casey WH. Ostwald processes and mineral paragensis in sediments. Am J Sci. 1988;288:537–560. [Google Scholar]
- 4.Henriksen K, Stipp SLS, Young JR, Bown PR. Tailoring calcite: Nanoscale AFM of coccolith biocrystals. Am Mineral. 2003;88:2040–2044. [Google Scholar]
- 5.Henriksen K, Stipp SLS, Young JR, Marsh ME. Biological control on calcite crystallization: AFM investigation of coccolith polysaccharide function. Am Mineral. 2004;89:1709–1716. [Google Scholar]
- 6.Pouget EM, et al. The initial stages of template-controlled CaCO3 formation revealed by cryo-TEM. Science. 2009;323:1455–1458. doi: 10.1126/science.1169434. [DOI] [PubMed] [Google Scholar]
- 7.Henriksen K, Young JR, Bown PR, Stipp SLS. Coccolith biomineralization studied with atomic force microscopy. Palaeontology. 2004;47:725–743. [Google Scholar]
- 8.Henriksen K, Stipp SLS. Controlling biomineralization: The effect of solution composition on coccolith polysaccharide functionality. Cryst Growth Des. 2009;9:2088–2097. [Google Scholar]
- 9.Burki PM, Glasser LSD, Smith DN. Surface-coatings on ancient coccoliths. Nature. 1982;297:145–147. [Google Scholar]
- 10.Hamano M, Honjo S. Qualitative analysis of the organic matter preserved in Oligocene coccoliths. J Geol. 1969;75:607–614. [Google Scholar]
- 11.Suess E. Interaction of organic compounds with calcium carbonate—1. Association phenomena and geochemical implications. Geochim Cosmochim Acta. 1970;34:157–168. [Google Scholar]
- 12.Takahashi-Shimase K, Nakashima S. Shape changes of calcareous nannofossils upon aqueous dissolution as revealed by atomic force microscope measurements. Geophys Res Lett. 2004;31:14313. [Google Scholar]
- 13.Beaufort L, Probert I, Buchet N. Effects of acidification and primary production on coccolith weight: Implications for carbonate transfer from the surface to the deep ocean. Geochem Geophysics Geosystems. 2007;8:1–18. [Google Scholar]
- 14.Berger R, Gerber C, Lang HP, Gimzewski JK. Micromechanics: A toolbox for femtoscale science: Towards a laboratory on a tip. Microelectron Eng. 1997;35:373–379. [Google Scholar]
- 15.Igaki J, et al. Mechanical characteristics and applications of diamond-like carbon cantilevers fabricated by focused-ion-beam chemical vapor deposition. J Vac Sci Technol B Microelectron Nanometer Struct Process Meas Phenom. 2006;24:2911–2914. [Google Scholar]
- 16.Arvidson RS, Ertan IE, Amonette JE, Luttge A. Variation in calcite dissolution rates: A fundamental problem? Geochim Cosmochim Acta. 2003;67:1623–1634. [Google Scholar]
- 17.Harstad AO, Stipp SLS. Calcite dissolution: Effects of trace cations naturally present in Iceland spar calcites. Geochim Cosmochim Acta. 2007;71:56–70. [Google Scholar]
- 18.Stipp SL, Hochella MF. Structure and bonding enviroments at the calcite surface as observed with X-ray photoelectron-spectroscopy (XPS) and low energy diffraction (LEED) Geochim Cosmochim Acta. 1991;55:1723–1736. [Google Scholar]
- 19.Kester DR, Duedall IW, Connors DN, Pytkowic RM. Preparation of artificial seawater. Limnol Oceanogr. 1967;12:176–179. [Google Scholar]
- 20.Bramlette MN, Martini E. The great change in calcareous nannoplankton fossils between the Maestrichtian and Danian. Micropaleontology. 1964;10:291–322. [Google Scholar]
- 21.Feely RA, et al. Impact of anthropogenic CO2 on the CaCO3 system in the oceans. Science. 2004;305:362–366. doi: 10.1126/science.1097329. [DOI] [PubMed] [Google Scholar]
- 22.Sabine CL, et al. The oceanic sink for anthropogenic CO2. Science. 2004;305:367–371. doi: 10.1126/science.1097403. [DOI] [PubMed] [Google Scholar]
- 23.Key RM, et al. A global ocean carbon climatology: Results from global data analysis project (GLODAP) Global Biogeochem Cycles. 2004;18:4031. [Google Scholar]
- 24.Iglesias-Rodriguez MD, et al. Phytoplankton calcification in a high-CO2 world. Science. 2008;320:336–340. doi: 10.1126/science.1154122. [DOI] [PubMed] [Google Scholar]
- 25.Riebesell U, et al. Reduced calcification of marine plankton in response to increased atmospheric CO2. Nature. 2000;407:364–367. doi: 10.1038/35030078. [DOI] [PubMed] [Google Scholar]
- 26.Chen CTA, Feely RA, Gendron JF. Lysocline calcium carbonate compensation depth and calcareous sediments in the north Pacific Ocean. Pac Sci. 1988;42:237–252. [Google Scholar]
- 27.Orr JC, et al. Anthropogenic ocean acidification over the twenty-first century and its impact on calcifying organisms. Nature. 2005;437:681–686. doi: 10.1038/nature04095. [DOI] [PubMed] [Google Scholar]
- 28.Caldeira K, Wickett ME. Anthropogenic carbon and ocean pH. Nature. 2003;425:365. doi: 10.1038/425365a. [DOI] [PubMed] [Google Scholar]
- 29.Guillard RR, Ryther JH. Studies of marine planktonic diatoms. 1. Cyclotella nana Hustedt, and Detonula confervacea (cleve) Gran. Can J Microbiol. 1962;8:229–239. doi: 10.1139/m62-029. [DOI] [PubMed] [Google Scholar]
- 30.Hutter JL, Bechhoefer J. Calbration of atomic-force microscope tips. Rev Sci Instrum. 1993;64:1868–1873. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.