

Abstract
Electron transfer may be concerted with proton transfer. It may also be concerted with the cleavage of a bond between heavy atoms. All three events may also be concerted. A model is presented to analyze the kinetics of these all-concerted reactions for homogeneous or electrochemical reduction or oxidation processes. It allows the estimation of the kinetic advantage that derives from the increase of the bond-breaking driving force resulting from the concerted proton transfer. Application of the model to the electrochemical reductive cleavage of the O–O bond of an organic peroxide in the presence of a proximal acid group illustrates the applicability of the model and provides an example demonstrating that electron transfer, heavy-atom bond breaking, and proton transfer may be all concerted. Such analyses are expected to be useful for the invention, analysis, and optimization of reactions involved in contemporary energy challenges as well as for the comprehension of major biochemical processes, a number of which involve electron and proton transfer together with cleavage of bonds between heavy atoms.
Keywords: electrochemistry, proton-coupled electron transfer, dissociative electron transfer
Bond cleavage or bond formation accompanies electron transfer in a considerable number of natural or artificial processes. As summarized in Fig. 1, reductive electron transfers generate basic species, in the Lewis or Brønsted sense, that are prone to lose a base or gain an acid, as for example, but not necessarily, a proton (1). Conversely, oxidative electron transfers generate acid species that may gain a base or lose an acid, e.g., a proton.
Fig. 1.

Association between electron transfer and bond cleavage or formation involving either heavy atom or proton transfer (as shown between parentheses in the latter case). e- represents an electron exchange with an electrode in the case of an electrochemical process or a molecular electron donor/acceptor for homogeneous reactions.
Altogether, over the four reactions depicted in Fig. 1, proton transfer coupled with electron transfer may be involved in two cases, whereas heavy-atom bond cleavage or formation has a wider scope. These reactions may occur homogeneously with a molecular electron donor, or acceptor, or may be triggered heterogeneously at an electrode. An advantage of the electrochemical approach is an easy determination of the rate-driving force relationships, which may be obtained from the current-potential responses once the contribution of reactant transport has been taken into account. Mechanism analysis and modeling of the kinetics of the rate-determining steps are closely similar in the homogeneous and electrochemical cases. Uncovering the reaction mechanism and the kinetic controlling parameters in one case may thus be readily transposed to the other.
In most cases, the radicals formed (see Fig. 1) are easier to reduce, or to oxidize, than the starting molecules, giving rise to multielectron processes as are most practical natural or synthetic electron transfer reactions [according to the classical “ECE-DISP” mechanism in the electrochemical case (1)]. Under those circumstances, even if more than one electron is globally exchanged, the rate control concerns the initial electron transfer in conjunction with the changes in bonding depicted in Fig. 1.
In this framework, uncovering the mechanisms and modeling the kinetic characteristics of these reactions has attracted and continues to attract a great deal of attention concerning both heavy-atoms bonds (2–4) and proton transfers (5–11). As pictured in Fig. 1, bond formation or cleavage may occur in a stepwise manner (electron transfer first, followed by bond change as represented in the upper pathways of Fig. 1, or vice versa as represented in the lower pathways of Fig. 1) or in a concerted manner, electron transfer and bond change occurring in a single elementary step (horizontal pathways in Fig. 1).
Whichever the mechanism, the electron transfer process benefits globally from an increase of driving force offered by the bond cleavage or the proton transfer. The advantage of concerted pathways is that they skip the intermediates involved in the stepwise pathways. They may thus be the preferred pathways in so far as these intermediates are high in energy and/or the kinetic price to pay for concertedness is not too high. These are the reasons that demonstrating the occurrence of concerted pathways and modeling their dynamics attract a particularly active current attention. As far as modeling is concerned, the case of heavy-atom bond breaking or formation on the one hand and of proton transfer on the other should be distinguished. Semiclassical treatments of the reaction kinetics can be developed in the first case, whereas quantum mechanical treatments are required for what concerns the proton displacement in the second case (12).
So far, electron transfers associated with cleavage or formation of bond between heavy atoms on the one hand and proton-coupled electron transfers on the other have been considered as two separate categories. We address, in the present contribution, reactions in which electron transfer is accompanied both by bond cleavage and proton transfer, which may also be first steps of multielectron processes. Contemporary energy challenges do involve such types of reactions, concerning particularly the cleavage of oxygen–oxygen and carbon–oxygen bonds (13).
Association of bond cleavage (or formation) and proton transfer with electron transfer may result in various degrees of concertedness up to the point where the three events are all concerted. The ensuing competition between reaction pathways is depicted in Fig. 2. For the sake of simplicity, the representation is limited to the case of a reduction, accompanied by the cleavage of a heavy-atom bond and by proton transfer, showing the various possible three-step, two-step and totally concerted pathways. Transposition to other cases, involving oxidation and/or bond formation or different sequencing of the bond changes and proton transfer, will follow the same principles.
Fig. 2.

Stepwise and concerted pathways in reactions where electron transfer is coupled with heavy-atom bond cleavage (Y–X) and proton transfer to an accepting molecule (B). ED, electron donor.
Modeling the kinetics of the rate-determining steps in the stepwise pathways may rely on existing treatments concerning outer-sphere electron transfer (14, 15) in the three-step case and, in the two-step cases, concerted heavy-atom bond breaking (4, 16) and concerted proton transfer (8, 17–20). This is not the case for the all-concerted pathway. The first purpose of the present contribution was therefore to model the kinetics of reactions in which electron transfer is concerted with both cleavage of a heavy-atom bond and with proton transfer so as to derive the relationship between rate and driving force and express the parameters that govern the intrinsic reactivity for both homogeneous and electrochemical cases.
In a second part, we illustrate the application of the model with an experimental example carefully selected for its simplicity; namely, the electrochemical reductive cleavage of an O–O bond helped by the presence of a proximal carboxylic acid group (see molecule 1 in Fig. 3) as compared to the methyl ester derivative of the same molecule (see molecule 2 in Fig. 3).
Fig. 3.

Peroxide molecules selected to illustrate the application of the model for the kinetics of electron transfer concerted with both heavy-atom bond cleavage and proton transfer.
It is interesting to note in this connection that the catalase activity (which involves the reductive cleavage of the hydrogen peroxide O–O bond) of heme hydroperoxidase models is enhanced by the close vicinity of a hanging acid group (21). However, the mechanism does not seem to belong to the “all concerted” category, the role of the acid being rather to inhibit a homolytic cleavage pathway (22). It has also been recently reported that the presence of a proximal acid group favors the production of water over hydrogen peroxide in the reduction of dioxygen by cobalt corrole complexes (13, 23).
Results and Discussion
Modeling Reactions in Which Electron Transfer Is Concerted with both Cleavage of a Heavy-Atom Bond and Proton Transfer.
We may combine the approaches that have been followed previously to model dissociative electron transfer with no accompanying proton transfer (4, 16) on the one hand and the proton-coupled electron transfer with no heavy-atom bond breaking on the other. (8, 17–20). Fig. 4 provides an outline of the main principles to be used to model the reaction kinetics so as to derive the rate law and the accompanying governing parameters. As in the simple concerted proton-electron transfer (CPET) case, two successive applications of the Born–Oppenheimer approximation lead to defining the transition state in terms of a heavy-atom coordinate and the preexponential factor in terms of a proton displacement coordinate. Concerning the first of these applications, heavy-atom reorganization involves the solvent molecules, the vibrations of reactant bonds not being cleaved in the reaction, and, most importantly, the contribution of bond cleavage. Regarding the latter, it seems appropriate to use the same approximation for the potential energy curves as for concerted dissociative electron transfers with no accompanying proton transfers; i.e., a Morse curve for the reactants and a repulsive Morse curve for the products (equal to the repulsive part of the reactant Morse curve) as shown in Fig. 4 (4, 16). For dissociative electron transfers with no accompanying proton transfers, the model has indeed been validated by a number of experimental homogeneous and electrochemical examples including reactions involving the concerted cleavage of O–O bonds (2–4).
Fig. 4.

Potential energy curves for the reorganization of the heavy atoms of the system, restricted for the sake of simplicity to the Morse curves representing the contribution of bond breaking (for the other contributions, see Eq. 1) and for the proton displacement concerted with electron transfer (Upper Inset). ZPER and ZPE≠, zero-point energies in the initial and transition states, respectively. Other symbols are defined in the text.
The forward irreversible reaction is a third-order reaction expressed in the homogeneous case as
| [1] |
Introducing the activation free energy, ΔG≠, and the third-order preexponential factor, 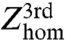 , the rate constant,
, the rate constant,  , is obtained from
, is obtained from
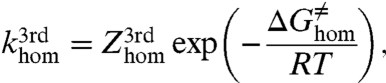 |
[2] |
with
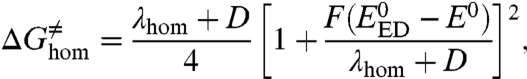 |
[3] |
where E0 is the standard potential relative to the all-concerted reaction and 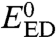 is the standard potential of the electron donor. The reorganization energy, λhom + D, comprises the homolytic dissociation energy of the Y–X bond, D, and a term, λhom, that includes the energy for the solvent reorganization and internal reorganization in the electron donor and in Y–X molecule, besides the cleavage of the bond. As in the case of simple CPET reactions, the preexponential factor combines the formation of the precursor complex, the degree of adiabaticity of electron transfer, and the effect of proton tunneling at the transition state. The third-order character of such reactions does not prevent their occurrence, as shown, for example, by the homogeneous oxidation of phenol by RuIII(bpy)3 with hydrogen phosphate as the proton acceptor (24). In systems where the proton donor is attached to the structure that bears the cleavable heavy-atom bond, as in the illustrating example discussed below (Fig. 3), Eq. 1 is replaced by a second-order reaction-rate law, leading to
is the standard potential of the electron donor. The reorganization energy, λhom + D, comprises the homolytic dissociation energy of the Y–X bond, D, and a term, λhom, that includes the energy for the solvent reorganization and internal reorganization in the electron donor and in Y–X molecule, besides the cleavage of the bond. As in the case of simple CPET reactions, the preexponential factor combines the formation of the precursor complex, the degree of adiabaticity of electron transfer, and the effect of proton tunneling at the transition state. The third-order character of such reactions does not prevent their occurrence, as shown, for example, by the homogeneous oxidation of phenol by RuIII(bpy)3 with hydrogen phosphate as the proton acceptor (24). In systems where the proton donor is attached to the structure that bears the cleavable heavy-atom bond, as in the illustrating example discussed below (Fig. 3), Eq. 1 is replaced by a second-order reaction-rate law, leading to
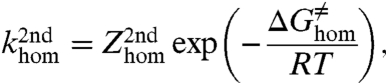 |
with the same expression of the free energy of activation as given by Eq. 3.
In the electrochemical case, a first and rather crude approach consists in considering that among all the electronic states of the electrons in the electrode, only those located at the Fermi level contribute to the reaction. Then, the rate law of the irreversible all-concerted reaction is
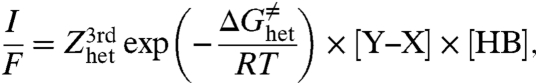 |
[4] |
with
 |
[5] |
where [Y–X] and [HB] are the concentrations of the indicated species at the electrode surface, I is the current density, and E is the electrode potential. The reorganization energy, λhet, includes the energy for solvent reorganization and internal reorganization in the Y–X molecule, besides the cleavage of the bond.
In the case where the proton donor is attached to the structure that bears the cleavable heavy-atom bond, as in the illustrating example discussed below (Fig. 3) Eq. 4 is replaced by a second-order reaction-rate law:
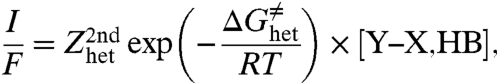 |
[6] |
with the same expression as in Eq. 5 for the activation free energy.
If all electronic states are taken into account, by analogy to the case of outer-sphere electron transfers, the rate law can be written as (25–27)
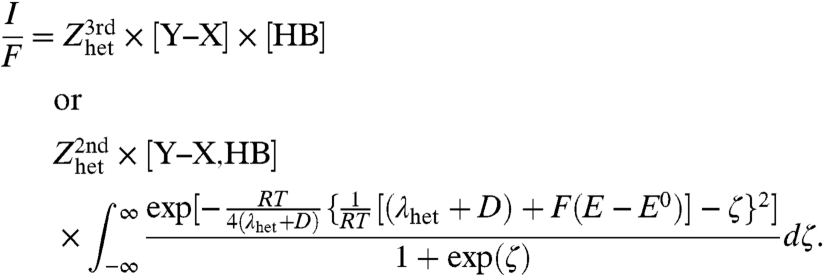 |
[7] |
As far as assignment of the all-concerted pathway is concerned, an interesting question is whether or not the appearance of a hydrogen/deuterium (H/D) kinetic isotope effect may be used as diagnostic criterion. At first sight, the answer is expected to be positive by reference to CPET reactions with no heavy-atom bond breaking where small but significant (of the order of 2) H/D kinetic isotope effects are commonly observed (8–10, 28). There is, however, an important difference between these reactions and the present ones, namely the large irreversibility caused by the breaking of the heavy-atom bond, as pictured in Fig. 4, which results in a larger reorganization energy due to the contribution of D. To overcome this intrinsic barrier, a large driving force is required. It follows that the transition state closely resembles the initial state. One consequence is that, in the heavy-atom transition state, the •Y X- moiety is much less basic than in the products’ geometry because the Y–X bond is not completely broken. This appears in Fig. 4 when one compares the proton energy profiles of the •Y X-⋯HB electronic states in the Upper Inset of Fig 4 and in the Right Lower Inset of the same figure. The result is that the intersection of the proton energy profiles of reactants and product electronic states in the transition state is likely to be close to the zero-point energy level. This indicates that the overlap of proton vibronic states is large and therefore insensitive to isotope substitution. If the second consequence of the close resemblance between transition state and initial state (namely, ZPE≠ ≈ ZPER) is taken into account, then the H/D kinetic isotope effect is predicted to be negligible in spite of the concerted character of the reaction. In other words, the absence of H/D kinetic isotope effect is related to the cleavage of the heavy-atom bond lagging behind proton transfer. Indeed, at the transition state, when electron and proton are transferred, the distance between two fragments is small, much smaller than in the final state of the system.
Proton-Assisted Reductive Cleavage of an O–O Bond as an Illustrative Example of an Electron Transfer Concerted with both Heavy-Atom Bond Cleavage and Proton Transfer.
We now discuss the electrochemistry of compound 1 (Fig. 3) as an example of the coupling between electron transfer, heavy-atom bond breaking, and proton transfer from a proximal carboxylic acid group attached to the structure that bears the O–O bond.
The electrochemical reduction of aliphatic peroxides and most aliphatic endoperoxides, studied in an aprotic solvent, such as dimethylformamide (DMF), consists in a dissociative electron transfer in which the O–O bond is cleaved concertedly with electron transfer, thus generating a broken anion radical, with the charge and unpaired electron located on one and the other oxygen atom (29–32) as sketched in Fig. 3. The broken anion radical is then further reduced at a potential more positive than the reduction potential of the starting peroxide with eventual protonation of the alcoholate, resulting in a total two-electrons per mole stoichiometry, which is nevertheless governed by the kinetics of the first concerted bond-breaking electron transfer. In any electrochemical nondestructive techniques, e.g., cyclic voltammetry (27, 33) , the current-potential response is indicative of this first one-electron uptake, or removal, even though the overall stoichiometry is two-electrons per molecule.
Compound 1 (Fig. 3) was selected so as to test the idea that the presence of a proximal acid group could greatly facilitate the reductive cleavage of the O–O bond by protonation of the alcoholate. The total gain in driving force consequently expected should approximately correspond to the difference of pKa between the alcohol that is formed upon reduction and the proximal acid; i.e., 19 pH units [from the pKa of tert-butanol, 32.4, and the pKa of acetic acid, 13.3, in DMF (34)], equivalent to -1.11 eV in terms of free energy.
The effect of this large increase of the reaction driving force on the electrochemistry of compound 1 was tested by cyclic voltammetry in DMF (Fig. 5). Comparison with the cyclic voltammetric response of the corresponding methyl ester, compound 2, in which such proton transfer may not occur, is shown in Fig. 5 so as to demonstrate the role played by the presence of the proximal acid group and the ensuing increase of the driving force.
Fig. 5.

Thick gray lines: cyclic voltammetry of 2 mM compound 1 (Left) and compound 2 (Right) in DMF + 0.1 M n-Bu4NBF4 at 0.2 V/s, at a glassy carbon disk electrode at 22 °C. Black thin lines: simulation (see text) with E0(1) = -0.37, E0(2) = -1.48 V vs. SCE, (λhet + D)(1) = 2.6, (λhet + D)(2) = 2.07 eV, 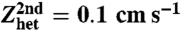 ; diffusion coefficients of compounds 1 and 2, 7 × 10-6 cm2 s-1.
; diffusion coefficients of compounds 1 and 2, 7 × 10-6 cm2 s-1.
Besides their total irreversibility, the most striking feature of the cyclic voltammograms in Fig. 5 is the very large gap, of more than 700 mV (Table 1), separating the peak potentials of compounds 1 and 2, attesting the very significant effect of the presence of the proximal acid group.
Table 1.
Cyclic voltammetric peak potentials at 0.2 V/s
*In the presence or absence of 1% CH3OH.
†In the presence of 1% CD3OD.
‡In volts vs. SCE.
It is also interesting, albeit less important to note that the peak height of compound 2 is twice that of compound 1. This is easily explained by the occurrence of a “father–son” reaction (1), in the case of compound 1, in which half of compound 1 is used to neutralize, by means of its acid functionality, the negative charge produced by the reduction of the other half (SI Appendix).
Examining the various mechanistic possibilities summarized in Fig. 2, it appears that the two outer-sphere electron transfer routes are ruled out by the general concerted, dissociative character of electron transfer to aliphatic peroxides as already mentioned. Among the two remaining possibilities, the two-step pathway consists in a first irreversible concerted electron transfer and bond-breaking step followed by a downhill protonation step. In such a situation, the kinetics of the reaction does not respond to the increase of driving force offered by the follow-up protonation but is simply driven by the thermodynamics of the first step. The peak potential of compound 1 should in this case be similar to that of compound 2, where protonation is not involved. The observed difference of 700 mV between the two peak potentials at 0.2 V/s therefore rules out the occurrence of this two-step mechanism.
We are therefore left with the all-concerted pathway, the kinetics of which is expected to respond to the driving force increase resulting from protonation. The theoretical model discussed above may then be applied (Y = X = O) in its electrochemical version for second-order process leading to Eq. 5 and lower Eq. 7.
Another interesting observation is the lack of H/D kinetic isotope effect (Table 1), which may look surprising for an all-concerted pathway but is in fact expected for the reasons discussed in the preceding section. This observation also indicates that the degree of adiabaticity of the whole reaction is not a reflection of proton tunneling but possibly of the degree of adiabaticity of electron transfer itself. Concerted electron transfer—bond-breaking reactions of peroxides, with no associated proton transfer, have been observed to be characterized by a large degree of nonadiabaticity, leading to small values of the preexponential factor (30). It follows that the preexponential factor is expected to be similar for the concerted bond cleavage–proton–electron transfer that compound 1 undergoes and for the concerted bond cleavage–electron transfer taking place with compound 2. The main difference between the reduction of compound 1 and the reduction of compound 2 therefore concerns the driving force of the reaction. The cyclic voltammetric responses in Fig. 5 may thus be simulated according to Eqs. 5 and 7 (35) as shown in Fig. 5. For the standard potential of the concerted bond cleavage–electron transfer undergone by compound 2, we took the same value as previously determined for di-tert-butyl peroxide (30) E0(2) = -1.48 V vs. SCE. The standard potential for compound 1 is then obtained by adding to E0(2) the previously estimated increase in driving force deriving from proton transfer, thus resulting in E0(1) = -0.37 V vs. SCE. Simulation of the responses of compounds 1 and 2 (Fig. 5) with the same value of the preexponential factor resulted in the following values:  , (λhet + D)(1) = 2.6 eV, (λhet + D)(2) = 2.07 eV. The small value of the preexponential factor is, for the reasons given earlier, a reflection of the nonadiabaticity of electron transfer, a characteristic already noted with the reductive cleavage of other peroxides with no accompanying proton transfer (30).
, (λhet + D)(1) = 2.6 eV, (λhet + D)(2) = 2.07 eV. The small value of the preexponential factor is, for the reasons given earlier, a reflection of the nonadiabaticity of electron transfer, a characteristic already noted with the reductive cleavage of other peroxides with no accompanying proton transfer (30).
It is noteworthy that not only the location and height of the cyclic voltammetric responses of both compounds are correctly reproduced by the simulation but also their shape.
What can be the reasons that the parameter λhet + D is smaller for compound 2 than for compound 1? The reorganization energy, λhet, is mainly concerned with solvent reorganization that we may estimate as λ0 ≈ 1 eV, for both compounds 1 and 2, from  (where a is the radius of the sphere equivalent to the volume where the negative charge is located) (28), noting that the excess of solvent reorganization energy due to the concerted proton transfer is negligible as compared to the solvent reorganization energy resulting from electron transfer. As concerns heavy-atom intramolecular reorganization that may result from the concerted proton transfer, the quantum chemical calculations described later have shown that geometrical changes and therefore intramolecular reorganization, occurring upon electron transfer besides bond breaking, may be considered as negligible. The bond dissociation energies may therefore be estimated as D(1) = 1.6 eV, D(2) = 1.07 eV.
(where a is the radius of the sphere equivalent to the volume where the negative charge is located) (28), noting that the excess of solvent reorganization energy due to the concerted proton transfer is negligible as compared to the solvent reorganization energy resulting from electron transfer. As concerns heavy-atom intramolecular reorganization that may result from the concerted proton transfer, the quantum chemical calculations described later have shown that geometrical changes and therefore intramolecular reorganization, occurring upon electron transfer besides bond breaking, may be considered as negligible. The bond dissociation energies may therefore be estimated as D(1) = 1.6 eV, D(2) = 1.07 eV.
It is noteworthy that the first of these values is closely similar to the bond dissociation energy reported for di-tert-butyl peroxide [1.60 ≤ D ≤ 1.77 eV (29)], where no interaction between the fragments resulting from cleavage is expected. Interpretation of the fact that the bond dissociation energy for compound 2 is significantly smaller rests on the existence of such interactions in the case of compound 2 in the framework of a somewhat more refined model of concerted bond-breaking electron transfer reaction than the raw version that we have used so far. This more sophisticated model takes into account the weak interaction, DP, which may exist between the two fragments resulting from bond breaking (Fig. 6), in the framework of what has been called “sticky dissociative electron transfer” (27, 36).
Fig. 6.

Schematic potential energy curves for the “sticky dissociative electron transfer” model (full line) as compared to the noninteraction classical model (dotted line for the product state).
Eq. 5 is then replaced by
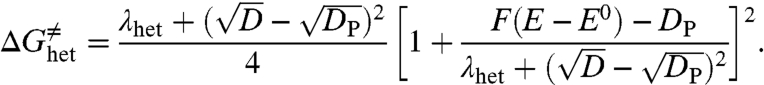 |
[8] |
Evidence for the existence of such interactions in the gas phase for compound 2 but not for compound 1 is indeed provided by quantum mechanical calculations that led to the structures displayed in Fig. 7 (the calculations were carried out on a slightly simplified structure where the cyclohexyl ring has been replaced by two hydrogens; see Materials and Methods). In line with the quite different O–O distances in compounds 1 and 2, no significant interaction was found in 1red, whereas a 0.2 eV interaction energy was found for 2red (SI Appendix). Most of this interaction is wiped out in a polar solvent such as DMF, but a residual interaction energy of DP = 0.05 eV suffices to explain the observed behavior in the framework of the “sticky dissociative electron transfer” as results from the application of Eq. 8.
Fig. 7.

Structure of compounds 1 and 2 at the oxidized and reduced states from quantum chemical calculations (see Materials and Methods).
Concluding Remarks
A model has been devised to analyze the kinetics of electron transfer reactions that are concerted with both heavy-atom bond breaking and proton transfer for homogeneous and electrochemical reduction or oxidation processes. The activation-driving force relationship is quadratic and involves as main reorganization parameter the sum of solvent and internal reorganization energies on the one hand and the homolytic bond dissociation energy on the other. Application of this relationship allows one to estimate the kinetic advantage that derives from the increase in the driving force of the reaction resulting from the concerted proton transfer. Because heavy-atom bond breaking is an irreversible process, the transition state closely resembles the initial state. The intersection of the proton energy profiles of reactants and product electronic states in the transition state (see the Inset of Fig. 4) is consequently close to the zero-point energy level. Therefore, the overlap of both proton vibronic states is large and consequently insensitive to isotope substitution, making the H/D kinetic isotope effect negligible is most cases in spite of the proton transfer being concerted with electron transfer. Another consequence is that the preexponential factor reflects, besides the formation of the precursor complex, the degree of adiabaticity of the electron transfer with little contribution arising from proton tunneling.
The electrochemical cleavage of the O–O bond of an organic peroxide in the presence of a proximal acid group has illustrated the applicability of the model demonstrating that electron transfer, heavy-atom bond breaking, and proton transfer may be all concerted.
These results are expected to be useful for the invention, analysis, and optimization of reactions involved in contemporary energy challenges as well as for the comprehension of major biochemical processes, a number of which involve electron and proton transfer together with cleavage of bonds between heavy atoms.
Materials and Methods
The electrochemical kinetics was obtained from cyclic voltammetric experiments on a glassy carbon electrode carefully polished before each run. Synthesis and characterization of compounds 1 and 2 were performed according to ref. 37. More details on the methods and materials used, including chemicals, cyclic voltammetry, father–son reactions, and quantum chemical calculations, are given in the SI Appendix.
Supplementary Material
Acknowledgments.
We thank Prof. K.A. Woerpel (New York University, New York) for helpful advice on the synthesis of compounds 1 and 2. Partial financial support from the Agence Nationale de la Recherche (Programme blanc PROTOCOLE) is gratefully acknowledged.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1104952108/-/DCSupplemental.
References
- 1.Savéant J-M. Elements of Molecular and Biomolecular Electrochemistry. New York: Wiley-Interscience; 2006. Chap 2. [Google Scholar]
- 2.Houmam A. Electron transfer initiated reactions: Bond formation and bond dissociation. Chem Rev. 2008;108:2180–2237. doi: 10.1021/cr068070x. [DOI] [PubMed] [Google Scholar]
- 3.Costentin C, Robert M, Savéant J-M. Electron transfer and bond breaking: Recent advances. Chem Phys. 2006;324:40–56. [Google Scholar]
- 4.Savéant J-M. Elements of Molecular and Biomolecular Electro-chemistry. New York: Wiley-Interscience; 2006. Chap 3. [Google Scholar]
- 5.Huynh MHV, Meyer TJ. Proton-coupled electron transfer. Chem Rev. 2007;107:5004–5064. doi: 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reece SY, Nocera DG. Proton-coupled electron transfer in biology: Results from synergistic studies in natural and model systems. Annu Rev Biochem. 2009;78:673–699. doi: 10.1146/annurev.biochem.78.080207.092132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dempsey JL, Winkler JR, Gray HB. Proton-coupled electron flow in protein redox machines. Chem Rev. 2010;110:7024–7039. doi: 10.1021/cr100182b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costentin C, Robert M, Savéant J-M. Concerted proton-electron transfers: Electrochemical and related approaches. Acc Chem Res. 2010;43:1019–1029. doi: 10.1021/ar9002812. [DOI] [PubMed] [Google Scholar]
- 9.Costentin C, Robert M, Savéant J-M. Update 1 of: Electrochemical approach to the mechanistic study of proton-coupled electron transfer. Chem Rev. 2010;110:PR1–PR40. doi: 10.1021/cr100038y. [DOI] [PubMed] [Google Scholar]
- 10.Rhile IJ, et al. Concerted proton-electron transfer in the oxidation of hydrogen-bonded phenols. J Am Chem Soc. 2006;128:6075–6088. doi: 10.1021/ja054167+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Markle TF, Rhile IJ, DiPasquale AG, Mayer JM. Probing concerted proton-electron transfer in phenol-imidazoles. Proc Natl Acad Sci USA. 2008;105:8185–8190. doi: 10.1073/pnas.0708967105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borgis D, Hynes JT. Dynamical theory of proton tunneling transfer rates in solution: General formulation. J Chem Phys. 1993;170:315–346. [Google Scholar]
- 13.Nocera DG. Chemistry of personalized solar energy. Inorg Chem. 2009;48:10001–10017. doi: 10.1021/ic901328v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marcus RA. On the theory of electron-transfer reactions: VI. Unified treatment for homogeneous and electrode reactions. J Chem Phys. 1965;43:679–701. [Google Scholar]
- 15.Hush NS. Electron transfer in retrospect and prospect 1: Adiabatic electrode processes. J Electroanal Chem. 1999;460:5–29. [Google Scholar]
- 16.Savéant JM. A simple-model for the kinetics of dissociative electron-transfer in polar-solvents. Application to the homogeneous and heterogeneous reduction of alkyl-halides. J Am Chem Soc. 1987;109:6788–6795. [Google Scholar]
- 17.Hammes-Schiffer S, Stuchebrukhov AA. Theory of coupled electron and proton transfers. Chem Rev. 2010;110:6939–6960. doi: 10.1021/cr1001436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venkataraman C, Soudackov AV, Hammes-Schiffer S. Theoretical formulation of nonadiabatic electrochemical proton-coupled electron transfer at metal-solution interfaces. J Phys Chem C. 2008;112:12386–12397. [Google Scholar]
- 19.Costentin C, Robert M, Savéant J-M. Electrochemical concerted proton and electron transfers. Potential-dependent rate constant, reorganization factors, proton tunneling and isotope effects. J Electroanal Chem. 2006;588:197–206. [Google Scholar]
- 20.Costentin C, Robert M, Savéant J-M. Reorganization energies and pre-exponential factors in the one-electron electrochemical and homogeneous oxidation of phenols coupled with an intramolecular amine-driven proton transfer. Phys Chem Chem Phys. 2006;12:13061–13069. doi: 10.1039/c0cp00017e. [DOI] [PubMed] [Google Scholar]
- 21.Chng LL, Chang CJ, Nocera DG. Catalytic O-O activation chemistry mediated by iron hangman porphyrins with a wide range of proton-donating abilities. Org Lett. 2003;5:2421–2424. doi: 10.1021/ol034581j. [DOI] [PubMed] [Google Scholar]
- 22.Soper JD, Kryatov SV, Rybak-Akimova EV, Nocera DG. Proton-directed redox control of O-O bond activation by heme hydroperoxidase models. J Am Chem Soc. 2007;129:5069–5075. doi: 10.1021/ja0683032. [DOI] [PubMed] [Google Scholar]
- 23.Dogutan DK, et al. Hangman corroles: Efficient synthesis and oxygen reaction chemistry. J Am Chem Soc. 2011;133:131–140. doi: 10.1021/ja108904s. [DOI] [PubMed] [Google Scholar]
- 24.Bonin J, Costentin C, Louault C, Robert M, Savéant J-M. Water (in water) as an intrinsically efficient proton acceptor in concerted proton electron transfers. J Am Chem Soc. 2011;133:6668–6674. doi: 10.1021/ja110935c. [DOI] [PubMed] [Google Scholar]
- 25.Levich VG. Present state of the theory of oxidation-reduction in solution (bulk and electrode reactions) In: Delahay P, Tobias CW, editors. Advances in Electrochemistry and Electrochemical Engineering. New York: Wiley; 1955. pp. 250–371. [Google Scholar]
- 26.Gosavi S, Marcus RA. Nonadiabatic electron transfer at metal surfaces. J Phys Chem B. 2000;104:2067–2072. [Google Scholar]
- 27.Savéant J-M. Elements of Molecular and Biomolecular Electrochemistry. New York: Wiley-Interscience; 2006. Chap 1. [Google Scholar]
- 28.Costentin C, Robert M, Savéant J-M. Concerted proton-electron transfers in the oxidation of phenols. Phys Chem Chem Phys. 2010;12:11179–11190. doi: 10.1039/c0cp00063a. [DOI] [PubMed] [Google Scholar]
- 29.Antonello S, Musumeci M, Wayner DDM, Maran F. Electroreduction of dialkyl peroxides. Activation-driving force relationships and bond dissociation free energies. J Am Chem Soc. 1997;119:9541–9549. [Google Scholar]
- 30.Donkers RL, Maran F, Wayner DDM, Workentin MS. Kinetics of the reduction of dialkyl peroxides. New insights into the dynamics of dissociative electron transfer. J Am Chem Soc. 1999;121:7239–7248. [Google Scholar]
- 31.Donkers RL, Workentin MS. Kinetics of dissociative electron transfer to ascaridole and dihydroascaridole—Model bicyclic endoperoxides of biological relevance. Chem Eur J. 2001;7:4012–4020. doi: 10.1002/1521-3765(20010917)7:18<4012::aid-chem4012>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 32.Najjar F, et al. Electrochemical reduction of G3-factor endoperoxide and its methyl ether: Evidence for a competition between concerted and stepwise dissociative electron transfer. Chem Eur J. 2007;13:1174–1179. doi: 10.1002/chem.200600445. [DOI] [PubMed] [Google Scholar]
- 33.Bard AJ, Faulkner LR. Eletrochemical Methods. 2nd Ed. Hoboken, NJ: John Wiley and Sons; 2001. Chap 6. [Google Scholar]
- 34.Andrieux CP, Gamby J, Hapiot P, Savéant J-M. Evidence for inverted region behavior in proton transfer to carbanions. J Am Chem Soc. 2003;125:10119–10124. doi: 10.1021/ja035268f. [DOI] [PubMed] [Google Scholar]
- 35.Andrieux CP, Gamby J, Hapiot P, Savéant J-M. Evidence for inverted region behavior in proton transfer to carbanions. J Am Chem Soc. 2003;125:10119–10124. doi: 10.1021/ja035268f. [DOI] [PubMed] [Google Scholar]
- 36.Rudolph M. Digital simulations on unequally spaced grids: Part 2. Using the box method by discretisation on a transformed equally spaced grid. J Electroanal Chem. 2003;543:23–39. [Google Scholar]
- 37.Ramirez A, Woerpel KA. Synthesis of 1,2-dioxolanes by annulation reactions of peroxycarbenium ions with alkenes. Org Lett. 2005;7:4617–4620. doi: 10.1021/ol051703u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.