

Abstract
A gain-of-function mutant of the lymphoid phosphatase Lyp (PTPN22) has recently been implicated with type 1 diabetes and other autoimmune diseases, suggesting that small-molecule inhibitors of Lyp could be useful for the treatment of autoimmunity. Virtual ligand screening (VLS) was applied in the search for hit compounds. Two different docking algorithms, FlexX and ICM, were used to screen a library of ‘drug-like’ molecules against two different 3D structures, representing Lyp’s catalytic site in both the inactive ‘open’ and active ‘closed’ conformation. Top scoring compounds of each VLS run were tested for their inhibitory activity against recombinant Lyp. Interestingly, VLS with both active and inactive conformation yielded very potent hits with IC50 values in the sub and low micromolar range. Moreover, many of these hits showed high docking scores only with one conformation. This was for instance the case with several 2-benzamidobenzoic acid derivatives, which specifically docked to the inactive open form. Tryptophan fluorescence measurements further supported a binding mode, in which these compounds seem to stabilize the phosphatase in its inactive conformation.
Keywords: Autoimmunity, Drug Design, Inhibitors, Phosphatases, Virtual Screening
Introduction
Tyrosine phosphorylation[i] plays an extremely important role in many processes that are characteristic of higher eukaryotes, such as cell-to-cell communication and coordination of the behavior of cell populations within multicellular organisms.[ii] This rapidly reversible post-translational modification is catalyzed by protein tyrosine kinases (PTKs) and reversed by protein tyrosine phosphatases (PTPs). Thereby, PTPs often play very specific, non-redundant, and highly regulated roles. The human genome contains at least 107 PTP genes with 81 genes encoding for active protein phosphtases. While PTKs have been considered as potential drug targets for some twenty years and PTK inhibitors are already on the market (e.g. Gleevec, Iressa, Tarceva), PTPs only recently have been implicated in human diseases, including cancer and diabetes.[iii,iv,v] The lymphoid tyrosine phosphatase Lyp,[vi] which is encoded by the ptpn22 gene, has a critical negative regulatory role in T cell receptor signaling. Recently, a single-nucleotide polymorphism in Lyp was discovered to correlate strongly with the incidence of type 1 diabetes[vii] and other autoimmune diseases, such as rheumatoid arthritis,[viii] juvenile rheumatoid arthritis,[ix] systemic lupus erythematosus,[x] and Grave’s disease.[vii] Since the autoimmunity-predisposing allele is a gain-of-function mutant,[xi] a specific small-molecule inhibitor of Lyp could be beneficial in treating these diseases.
Based on the increasing number of available three-dimensional structures of PTPs in recent years, in silico methods have become more and more popular as hit/lead discovery tools for tyrosine phosphatases.[xii] Depending on the conformation of the WPD-loop, which contains the catalytically essential general acid/base aspartic acid, two types of PTP structures can be typically found: The inactive ‘open’ conformation refers to the WPD-loop in distant position in regards to the catalytic pocket. Substrate or ligand binding to the bottom of the catalytic pocket causes the loop to shift by ~8 Å, forming the active ‘closed’ conformation.[xiii] Usually, only structures in the closed conformation are considered as suitable receptors to in silico screen for inhibitors thought to target the active site. Here, we present how virtual ligand screening (VLS) with a structure that contains the WPD-loop in open conformation can also lead to unique and potent hits. Analysis of the docking poses for these compounds as well as tryptophan fluorescence measurements suggest a binding mode that is very specific and seems to stabilize Lyp in its inactive conformation.
Results and Discussion
In silico screening
To identify hit compounds for Lyp by VLS, two docking algorithms, FlexX[xiv] and ICM[xv], were employed to screen a library of 27,030 compounds. A high-resolution crystal structure of Lyp’s catalytic domain in open conformation (LypO, PDB code: 2P6X) was used, as well as a homology model of Lyp in closed conformation (LypC) since a crystal or NMR structure was not available for the closed form. The modelled LypC could be structurally aligned to LypO with an RMSD of 1.52 Å when WPD-loop atoms were omitted (Figure 1A). However, the surface topology around the catalytic pocket, especially towards the WPD-loop, differed dramatically between the two structures (Figure 1B/C), suggesting that VLS should yield distinct hits for each receptor conformation. Four VLS experiments (LypC with FlexX/ICM and LypO with FlexX/ICM) were done, and compounds were ranked according to their docking scores. For each VLS run, the 20 best-ranked compounds were then chosen for evaluation. Since nine compounds were among more than one top 20 sets, a total of 71 compounds were purchased. Clustering these hits by Tanimoto distance revealed 10 different compound classes with at least two members and 14 singletons at a distance of 0.4 (Supporting Information Table 1S). As suspected, some of the clusters were very specific to only one receptor conformation.
Figure 1.

A) Alignment of crystal structure of Lyp with WPD-loop in open conformation (2P6X.pdb, green) and homology model of Lyp with WPD-loop in closed conformation (blue-white). B/C) Active site surface representation of Lyp crystal structure with WPD-loop in open conformation (B) and Lyp homology model with WPD-loop in closed conformation (C). Structures were aligned, and pictures represent the exact same view. The color code of the MOLCAD surfaces represents the normalized electrostatic potential (red: most positive, purple: most negative). The white arrows indicate the catalytic pocket with P-loop.
Evaluation of screening hits
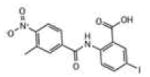
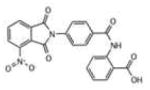
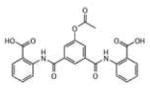
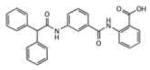
To evaluate the inhibitory activity of the 71 hits, a 96-well plate phosphatase assay was applied. At 40 μM compound concentration, inhibitory activity was determined as percentage inhibition compared to a dimethylsulfoxide (DMSO) control (Supporting Information Table 1S). The overall performance in generating active compounds was very similar among the 4 VLS runs. This result was completely unexpected for the two runs using LypO, because this structure does not resemble the active receptor conformation. Interestingly, there was only little overlap of high docking scores for each hit among the different VLS conditions. Moreover, VLS with Lyp in open conformation yielded several potent inhibitors that completely failed to dock into the closed conformation. For instance, the 2-benzamidobenzoic acid (2-BBA) derivatives, which all ranked among the top 21 hits (inhibition >90%), exhibited high docking scores only with LypO. Their IC50 values for Lyp ranged from 0.947 to 22.9 mM (Table 1).
Table 1.
Docking scores and inhibitory activity of 2-benzamidobenzoic acids sorted by their IC50 values against recombinantLyp.
| # | Structure | IC50, μM | LypC FlexX[a] | LypC ICM[b] | LypO FlexX[c] | LypO ICM[d] |
|---|---|---|---|---|---|---|
| 8 |
|
0.947 | −29.56 | −15.94 | −32.04 | −37.51 |
| 7 |
|
0.966 | −30.16 | −10.69 | −32.55 | −29.21 |
| 6 |
|
1.69 | −28.37 | −17.14 | −28.42 | −41.73 |
| 14 |
|
4.85 | −27.95 | −12.75 | −45.68 | −33.68 |
| 12 |
|
7.75 | −25.09 | −7.09 | −37.23 | −23.90 |
| 11 |
|
11.0 | −22.71 | −5.82 | −29.77 | −39.93 |
| 21 |
|
22.9 | −18.40 | −9.81 | −30.96 | −43.33 |
FlexX docking score with LypC
ICM docking score with LypC
FlexX docking score with LypO
ICM docking score with LypO.
When we evaluated the binding mode of these compounds, all 2-BBAs, except 7, occupied surface areas in LypO that are not accessible with the WPD-loop in closed conformation (Figure 2). The docking poses suggested that each molecule binds to both the phosphate-binding loop (P-loop) at the bottom of the catalytic pocket as well as the distant WPD-loop – a binding mode that may stabilize the WPD-loop in the open conformation. Only 7 occupied a position within the active site of LypO that resembled the one found with LypC with the benzoic acid moiety not able to bind to the P-loop because of steric hindrance of the iodine substituent. Instead, the nitrophenyl group was found to bind into the catalytic pocket, which was confirmed by testing 2-benzamido-5-iodobenzoic acid that lacks the nitro group and had a 157 fold less inhibitory activity against Lyp. For all other 2-BBAs, the benzamidobenzoic acid group was found to form a complex network of hydrogen-bonding interactions with residues within the P-loop, including the invariant Arg-233, as shown for compound 12 (Figure 3). In addition, 12 was also found to specifically interact with amino acid residues within the WPD-loop region. In particular, one of the two phenyl groups seemed to fit perfectly into a hydrophobic pocket, which is formed by parts of the WPD-loop and the β3-strand in LypO. These hydrophobic interactions of 12 with the WPD-loop seem to contribute a great deal of binding energy and therefore should be important for inhibitory activity. Since 12 did not dock into the closed receptor conformation with reasonable scores, the observed docking pose may well reflect the actual binding mode, in which an active site inhibitor stabilizes the WPD-loop in open conformation. In fact, during the preparation of this manuscript Zhong-Yin Zhang’s group reported a co-crystal structure of Lyp and an active site inhibitor, in which Lyp was also found in an open conformation.[xvi]
Figure 2.
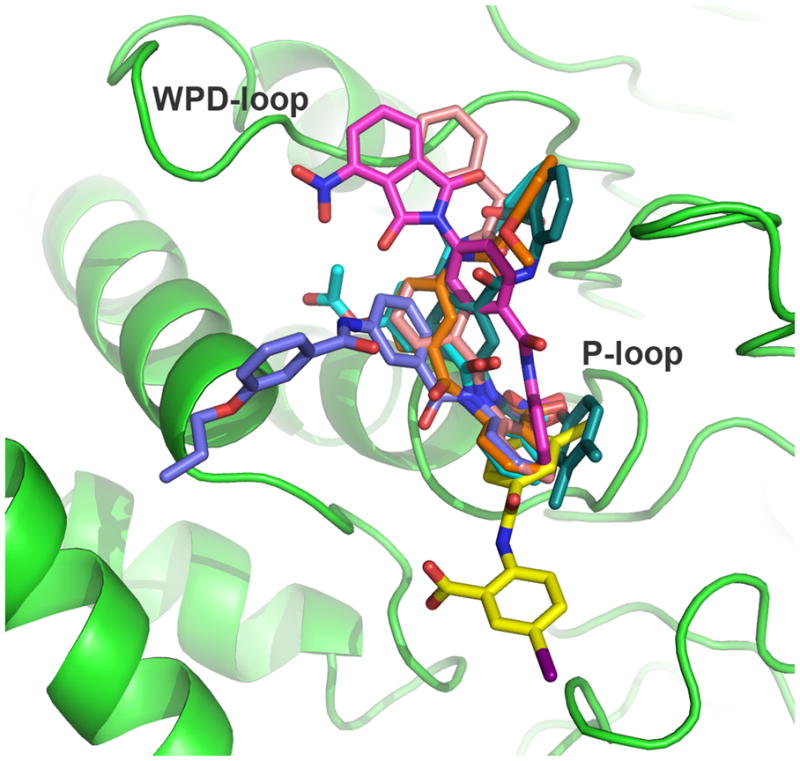
Ribbon diagram of Lyp in the inactive open conformation with FlexX docking solutions of compounds 6 (magenta), 7 (yellow), 8 (blue), 11 (green), 12 (pink), 14 (cyan), and 21 (orange). Compounds are shown in stick representation.
Figure 3.

A) Ribbon diagram of compound 12 docked to Lyp in open conformation using FlexX. Amino acid residues of the catalytic pocket and the WPD-loop as well as compound 12 are shown in stick representation. The P-loop is colored in light blue; the WPD-loop is colored in yellow. Dashed yellow lines indicate hydrogen-bonding interactions between 12 and Lyp. B) Surface diagram of same docking solution as in (A). White arrow indicates a hydrophobic pocket that is formed by parts of the WPD-loop and the β3-strand.
Tryptophan fluorescence measurements
To give further evidence that the 2-BBAs may stabilize the inactive conformation, we used fluorescence spectroscopy to measure the tryptophan emission of recombinant Lyp. Although there are a total of six tryptophan residues within the catalytic domain, none of them is near the active site except Trp-193 of the WPD-loop. At 100, 25, 6.25, or 1.56 μM, all 2-BBAs quenched the tryptophan emission concentration dependent to various extends compared to a control, with 7 having the highest quenching effect (Figure 4). In addition, a red shift of the emission maximum was observed in the presence of compounds, compared to DMSO alone. This shift was between 3 and 7 nm for all compounds except for 7, which caused a significant higher shift of 15 nm. Since a substantial conformational change of the WPD-loop will cause a more profound effect on the fluorescence emission of Trp-193, this data supports the notion that all 2-BBAs but 7 stabilize Lyp in an open conformation.
Figure 4.
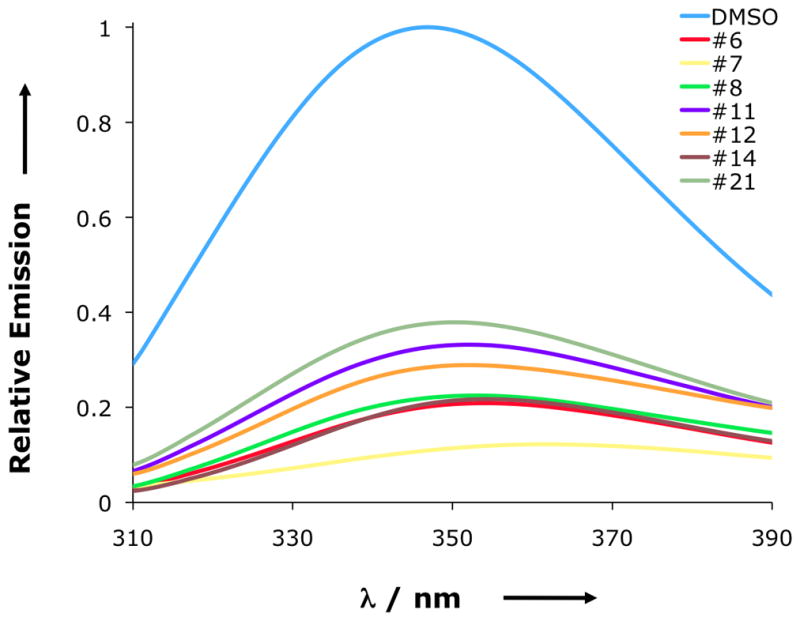
Normalized tryptophan fluorescence emission of Lyp catalytic domain (1 μM) in the presence of 2% DMSO or 100 μM inhibitors with an excitation wavelength of 285 nm.
Conclusion
We have shown how in silico screening with a tyrosine phosphatase structure in its inactive ‘open’ conformation can yield very potent inhibitors. These compounds still bind to amino acid residues inside the catalytic pocket with functional groups that mimic the phosphotyrosine group of the natural substrate. However, substantial portions of the molecules seem to interact with a surface area outside the catalytic pocket that would normally be occupied by the WPD-loop in closed conformation. This observation strongly suggests a binding mode different from natural substrates or other known PTP inhibitors. Notably, these interactions seem to be very strong and specific in nature, and most likely stabilize the open conformation of the WPD-loop. This principle may apply more broadly to PTPs and also other enzymes that undergo substantial conformational cycling during catalysis. Our best compounds exhibit a pool of inhibitors that will be further tested for selectivity and cell-based activity, and may serve as starting point for developing Lyp inhibitors that potentially could be used to treat autoimmune diseases.
Experimental Section
Compound library
To generate a virtual library of ‘drug-like’ compounds, the 50,000 molecules containing DIVERSet™ library from ChemBridge (ChemBridge, Inc.) was reduced by compounds that didn’t meet certain criteria, which were calculated using the ICM software package (Molsoft, L.L.C.). First, compounds with so-called ‘bad groups’ were removed from the set, which contained in drugs unwanted functional groups.[xvii] Second, compounds with a calculated ‘drug-likeness’ score lower than −0.5 were removed. Third, compounds with cLogP values lower than −0.5 and greater than 5 were removed. And fourth, compounds with PSA values greater than 140 were removed from the set, resulting in a library of 27,030 compounds with molecular weights lower than 600. The library was saved as SD file.
LypC homology modeling
The SWISS-MODEL comparative protein modeling server[xviii] (http://swissmodel.expasy.org/) was used to generate a homology model for Lyp in closed conformation (LypC). A high-resolution structure of the close homolog PTP1B (sequence identity 38.6%) in closed conformation (PDB code 1PTY) was used as a specific template structure. SwissModel Automatic Modelling Mode[xviii] was used to generate the model of LypC, which comprised amino acids 44–288. The model structure was assessed by three different methods, Anolea, Gromos, and Verify3D, which all suggested a reliable model prediction for most parts of the structure, including the active site.
VLS with FlexX
FlexX algorithm was applied as part of the SYBYL software package (version 7.2, Tripos, Inc.), and calculations were run on a Linux workstation. The homology model of the closed conformation and the crystal structure of the open conformation (PDB code 2P6X) were prepared with the structure preparation tool as implemented in SYBYL. AMBER charges were assigned, orientations of side chain amides were corrected, and hydrogens were added and their positions optimized by energy minimization using AMBER7 FF99 force field. The receptor site was defined as an 8 Å radius within the catalytic cysteine 227. 3D coordinates of the compounds were generated using CONCORD as implemented in SYBYL and saved as MOL2 files. For docking, acids were deprotonated whereas amines were protonated to account for their charged state at pH 6. The 30 lowest energy poses for each compound and their corresponding Total-Scores (FlexX scores) were calculated.
VLS with ICM
ICM docking algorithm was applied as part of the ICM software package (version 3.4, Molsoft, L.L.C.), and calculations were run on a PowerPC G5 workstation. The protein structures were converted into ICM objects, charges were assigned, orientations of side chain amides were corrected, and hydrogens were added and their positions optimized by energy minimization using MMFF force field. The receptor site was defined as an 8 Å radius within the catalytic cysteine 227. 3D coordinates of the compounds were generated and charges were assigned using the implemented compound preparation tool.
Recombinant Lyp
Lyp catalytic domain (residues 2–309) was cloned into a pGEX-4T-1 vector, and the plasmid was transformed into Escherichia coli BL21 DE3 competent cells (Stratagene). For expression, an overnight-culture was diluted in LB/kanamycin medium and grown at 37°C to a cell density of A600 = 0.6. Isopropyl-1-thio-d-galactopyranoside at 0.4 mM was used to induce protein overexpression for 4 hours at room temperature. Harvested cells were resuspended in 10 ml Extraction Solution #1 (50 mM Tris-HCl pH 8.0, 1 mM EDTA, 5 mM β-mercaptoethanol, 0.005% NaN3, 0.08 mg/ml lysozyme, 1 mM phenylmethylsulfonyl fluoride) per gram of cells and stirred at 4°C for 30 minutes. Then, 1 ml of 10x Extraction Solution # 2 (1.5 M NaCl, 100 mM CaCl2, 100 mM MgSO4, 20 μg/ml DNase, 50 μg ovomucoid) per 10 ml cell suspension was added. After centrifugation at 17,000 rpm, the clear supernatant was loaded onto an affinity column with 5 ml of Glutathione Sepharose 4B beads (GE Healthcare), which were pre-equilibrated with wash buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 5 mM β-mercaptoethanol). GST-Lyp was eluted using 10 mM of glutathione in wash buffer. The active fraction was collected and further purified on a Superdex S-200 column that was pre-equilibrated with 20 mM Tris-HCl pH 8.0/1 mM DTT. The purified GST-Lyp was homogeneous as shown from SDS-PAGE with a single band.
Lyp enzymatic assay
Lyp-catalyzed hydrolysis of 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP, Invitrogen Inc.) in the presence of compounds was assayed at 30°C in a 60 μl 96-well format reaction system in 20 mM Bis-Tris, pH 6.0 assay buffer having an ionic strength of 20 mM (adjusted with NaCl) and containing 1 mM dithiotreitol, 1% PEG 8000, and 5% DMSO. At 40 μM compound concentration, the initial rate at 48 μM DiFMUP concentration (equal to the corresponding Km value) was determined using a FLx800 micro plate reader (Bio-Tek Instruments, Inc.), an excitation wave length of 360 nm and measuring the emission of the fluorescent reaction product 6,8-difluoro-7-hydroxy-4-methylcoumarin (DiFMU) at 460 nm. The nonenzymatic hydrolysis of the substrate was corrected by measuring the control without addition of enzyme.
IC50 measurements
With the same buffer system, substrate, and reader as above, the initial rate at 48 μM DiFMUP was determined at various concentrations of the compound, ranging from 1 mM to 1 nM. The nonenzymatic hydrolysis of the substrate was corrected by measuring the control without addition of enzyme. The IC50 value was determined by plotting the relative activity versus inhibitor concentration and fitting to Equation 1 using the software GraphPad Prism® (GraphPad Software, Inc.).
| (Eq. 1) |
In this case, Vi is the reaction velocity when the inhibitor concentration is [I], V0 is the reaction velocity with no inhibitor, and IC50 = Ki + Ki[S]/Km.
Fluorescence emission measurements
Tryptophan emission spectra were recorded at room temperature on a MOS-250 spectrophotometer (BioLogic, Claix, France) at a scan speed of 60 nm/min and an emission bandwidth of 10 nm, using a 250 μl quartz cuvette. The concentration of recombinant Lyp catalytic domain was 1 μM in 20 mM Bis-Tris pH 6.0, containing 5 mM dithiothreitol (BioVectra) and 1% polyehtylenglycol (PEG 8000, Sigma). DMSO or compound in DMSO were added and mixed to a final concentrations of 100, 25, 6.25, and 1.56 μM and 2% DMSO. After 1 min incubation time, the fluorescence emission was measured at an excitation wavelength of 285 nm.
Acknowledgments
This work was supported by grants from the U.S. National Institutes of Health (AI053585 to TM and CA132121 to LT) and the Oxnard Foundation.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemmedchem.org or from the author.
References
- 1.Hunter T, Sefton BM. Proc Natl Acad Sci U S A. 1980;77:1311–1315. doi: 10.1073/pnas.77.3.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 3.Bialy L, Waldmann H. Angew Chem. 2005;117:3880–3906. [Google Scholar]; Angew Chem Int Ed. 2005;44:3814–3839. doi: 10.1002/anie.200461517. [DOI] [PubMed] [Google Scholar]
- 4.Tautz L, Pellecchia M, Mustelin T. Expert Opin Ther Targets. 2006;10:157–177. doi: 10.1517/14728222.10.1.157. [DOI] [PubMed] [Google Scholar]
- 5.Vang T, Miletic AV, Arimura Y, Tautz L, Rickert RC, Mustelin T. Annu Rev Immunol. 2008;26:29–55. doi: 10.1146/annurev.immunol.26.021607.090418. [DOI] [PubMed] [Google Scholar]
- 6.Cohen S, Dadi H, Shaoul E, Sharfe N, Roifman CM. Blood. 1999;93:2013–2024. [PubMed] [Google Scholar]
- 7.Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, MacMurray J, Meloni GF, Lucarelli P, Pellecchia M, Eisenbarth GS, Comings D, Mustelin T. Nat Genet. 2004;36:337–338. doi: 10.1038/ng1323. [DOI] [PubMed] [Google Scholar]
- 8.Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM, Conn MT, Chang M, Chang SY, Saiki RK, Catanese JJ, Leong DU, Garcia VE, McAllister LB, Jeffery DA, Lee AT, Batliwalla F, Remmers E, Criswell LA, Seldin MF, Kastner DL, Amos CI, Sninsky JJ, Gregersen PK. Am J Hum Genet. 2004;75:330–337. doi: 10.1086/422827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ladner MB, Bottini N, Valdes AM, Noble JA. Hum Immunol. 2005;66:60–64. doi: 10.1016/j.humimm.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 10.Kyogoku C, Langefeld CD, Ortmann WA, Lee A, Selby S, Carlton VE, Chang M, Ramos P, Baechler EC, Batliwalla FM, Novitzke J, Williams AH, Gillett C, Rodine P, Graham RR, Ardlie KG, Gaffney PM, Moser KL, Petri M, Begovich AB, Gregersen PK, Behrens TW. Am J Hum Genet. 2004;75:504–507. doi: 10.1086/423790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, Nika K, Tautz L, Tasken K, Cucca F, Mustelin T, Bottini N. Nat Genet. 2005;37:1317–1319. doi: 10.1038/ng1673. [DOI] [PubMed] [Google Scholar]
- 12.Tautz L, Mustelin T. Methods. 2007;42:250–260. doi: 10.1016/j.ymeth.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 13.Zhang ZY. Annu Rev Pharmacol Toxicol. 2002;42:209–234. doi: 10.1146/annurev.pharmtox.42.083001.144616. [DOI] [PubMed] [Google Scholar]
- 14.Rarey M, Kramer B, Lengauer T, Klebe G. J Mol Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 15.Totrov M, Abagyan R. Proteins. 1997;Suppl 1:215–220. doi: 10.1002/(sici)1097-0134(1997)1+<215::aid-prot29>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 16.Yu X, Sun JP, He Y, Guo X, Liu S, Zhou B, Hudmon A, Zhang ZY. Proc Natl Acad Sci U S A. 2007;104:19767–19772. doi: 10.1073/pnas.0706233104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hann M, Hudson B, Lewell X, Lifely R, Miller L, Ramsden N. J Chem Inf Comput Sci. 1999;39:897–902. doi: 10.1021/ci990423o. [DOI] [PubMed] [Google Scholar]
- 18.Arnold K, Bordoli L, Kopp J, Schwede T. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]