

Abstract
Ligand engagement by integrins induces receptor clustering and formation of complexes at the integrin cytoplasmic face that controls cell signaling and cytoskeletal dynamics critical for adhesion-dependent processes. This study searches for a subset of integrin effectors that coordinates both tumor cell invasion and resistance to the chemotherapeutic drug cisplatin in oral carcinomas. Candidate integrin effectors were identified in a proteomics screen of proteins recruited to clustered integrin αβ1, αvβ or α6β receptors in oral carcinomas. Proteins with diverse functions including microtubule and actin binding proteins, and factors involved in trafficking, transcription and translation were identified in oral carcinoma integrin complexes. Knockdown of effectors in the oral carcinoma HN12 cells revealed that p130Cas, Dek, Src and talin were required for invasion through Matrigel. Disruption of talin or p130Cas by RNA interference increased resistance to cisplatin, whereas targeting Dek, Src or zyxin reduced HN12 resistance to cisplatin. Analysis of the spreading of HN12 cells on collagen I and laminin I revealed that a decrease in p130Cas or talin expression inhibited spreading on both matrices. Interestingly, a reduction in zyxin expression enhanced spreading on laminin I and inhibited spreading on collagen I. Reduction of Dek, Src, talin or zyxin expression reduced HN12 proliferation by 30%. Proliferation was not affected by a reduction in p130Cas expression. We conclude that p130Cas, Src and talin function in both oral carcinoma invasion and resistance to cisplatin.
Keywords: integrin, cytoplasmic effectors, oral carcinoma, tumor invasion, cell spreading, proliferation, cisplatin, chemoresistance, Matrigel
1. Introduction
Patients with stage III and IV oral carcinoma have poor survival rates that have not significantly changed over four decades due, in part, to frequent metastasis and resistance to both radiation and chemotherapy of these tumors [1]. The role of the tumor stroma in chemoresistance has been an area of intense study over the last decade. Tumor-stromal fibroblasts form a dense or rigid stroma that may function as a barrier to prevent the penetration of therapeutics into tumor tissue [2] [3]. Adhesion of breast tumor cell lines to matrix secreted by stromal fibroblasts is sufficient to confer tumor cell resistance to chemotherapeutics [4] [5]. In our laboratory, we found that adhesion to the matrix secreted by oral carcinoma is sufficient to induce chemoresistance (A. Berrier, submitted manuscript). Adhesions within the tumor matrix comprised of collagens, fibronectins and laminins mediated by engagement of integrin αβ1, αvβ and α6β receptors regulates invasion and chemoresistance in a variety of tumors [6] [7] [8]. Even though integrins can regulate tumor invasion and chemoresistance, it is currently not known whether effectors downstream of integrins coordinate invasive potential with resistance to cisplatin.
We used a proteomics approach to identify integrin effectors in oral carcinoma. Recruitment to integrin cytoplasmic complexes was induced by clustering integrin receptors at the cell surface with anti-integrin antibodies immobilized on magnetic beads. The integrin-associated complexes were biochemically isolated, and the resulting tryptic peptides were analyzed by mass spectrometry. We characterized roles for a subset of the identified integrin effectors in oral carcinoma invasion and resistance to cisplatin.
2. Materials and methods
2.1. Integrin receptor clustering
HN4 and HN12 cells were isolated from the primary tumor lesion and lymph node respectively of a patient with tongue cancer [9]. HN12 and HN4 cells were cultured in DMEM supplemented with fetal bovine serum, penicillin/streptomycin and hydrocortisone in 10% CO2 [10]. Cells were placed in suspension at a density of 2×106 cells/ml in DMEM supplemented with 1% bovine calf serum and penicillin/streptomycin. The cells were incubated with magnetic beads (4.5 μm diameter) conjugated with the following anti-integrin receptor antibodies: integrin α6, GoH3, Santa Cruz Biotechnology, integrin αv, L230, ATCC, and integrin β1, K20, Beckman Coulter. The antibodies were covalently coupled to Tosylactivated M-450 Dynabeads (Invitrogen, Dynal) according to the manufacturer’s instructions. The integrins were clustered by incubating the beads with cells at a 5:1 ratio with shaking for 60 minutes at 37°C.
2.2. Isolation of integrin complexes and proteomics analysis
Approximately 1×107 cells bound to beads were lysed in CSK buffer (0.5% Triton X-100, 3 mM MgCl2, 300 mM NaCl, 300 mM sucrose, 10 mM Pipes, pH 6.8 with protease inhibitor cocktail (Roche), 1 mM sodium vanadate, 5 mM NaF and 1 mM PMSF) and bath sonicated for 20 seconds to reduce sample viscosity. The beads were washed 5x with CSK buffer. Bound protein complexes were eluted in boiling 2% SDS, 62.5 mM Tris, pH 6.8. The proteins in the bead supernate were concentrated with 12% TCA then washed 3x with cold acetone and air dried. Samples were digested with trypsin, and analysis of the tryptic peptides and comparisons between the samples was performed using contrast 2.0 software [11] [12]. The proteomics protocols and data are available in the Supplementary materials section.
2.3. Calcium phosphate transfection
HN12 cells were placed in suspension in antibiotic-free DMEM supplemented with serum at a density of 1×105 cells/ml. Cells were transfected using calcium phosphate precipitation (Invitrogen). For each target gene, three pairs of stealth siRNA duplexes were pooled (50 nM total, Invitrogen) and transfected. Four days later, cells were harvested for assays and Western blot analysis.
2.4. Invasion assay
Transfected HN12 cells were replated in transwell chambers with 8 μm pores. The top of the filter was coated with matrigel (1:20 dilution) and the filter bottom was coated with collagen I (0.5 mg/ml). Cells were seeded in the top chambers in serum-free medium. The bottom chambers contained either complete or serum-free medium [13]. The HN12 cells were allowed to invade for 16 hours at 37°C. Invading cells were stained with cytotracker orange and fixed (PBS 4% formaldehyde and 5% sucrose). At least 10 digital images of filter bottoms from three different wells were acquired with an inverted microscope (Olympus IX81) using a 20x objective, with a digital Homamatsu electron multiplier CCD camera. Cell numbers were counted by a “blinded” investigator and statistical analysis of the data was performed using the student’s t-test.
2.5. Cell spreading assay
Transfected HN12 cells were resuspended in serum-free medium containing 0.1% BSA. Multiwell 24-well plates were coated with collagen I or laminin I at 10 μg/mL and blocked with 0.1% BSA in PBS [14]. 5×104 cells were allowed to spread for 60 minutes at 37°C. Adherent cells were fixed (5% sucrose 4% formaldehyde in PBS) and phase contrast digital images were acquired using an inverted microscope (Olympus IX81). Cells with a cytoplasm encircling the nucleus were scored as spread [14]. Statistical analysis of the data was performed with the student’s t-test.
2.6. MTT proliferation assay
MTT (3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide) assays were performed according to the manufacturer’s instructions (ATCC). Purple tetrazolium was quantified by absorbance at 570 nm using a multiwell plate reader (Biotek, Synergy2), subtracting background determined in the absence of cells. Statistical analysis of the data was performed with the student’s t-test.
2.7. HN12 matrix
HN12 cells were cultured on collagen I (20 μg/mL) coated tissue culture dishes for at least 12 days in complete DMEM supplemented with ascorbic acid (50 μg/mL). The matrices were denuded of cells as described [15] [16].
2.8. Cisplatin resistance
HN12 cells were placed in suspension in serum-free medium and 4×104 cells were added to each well containing HN12 matrix. Cisplatin (1 μM and 30 μM, EMD chemicals) was resuspended in vehicle DMSO (final concentration <0.1%, Sigma) and both reagents were incubated with matrix adherent cells for 48 hours. Subsequently, the MTT assay measured cell proliferation, normalized relative to DMSO-treated cultures. The proliferation IC50 for cisplatin in HN12 cells adherent to collagen I is 30 μM and serum levels of cisplatin in patients is approximately 10 μM.
2.9. Western blotting
Whole cell lysates were prepared by addition of 5X SDS sample buffer and syringe passaging of the lysates to shear genomic DNA. Lysates containing equivalent levels of actin were tested for steady-state protein levels of the siRNA target. Primary antibodies included mouse anti-p130Cas, BD Biosciences, mouse anti-talin, Sigma, rabbit anti-zyxin, Sigma, mouse anti-Dek, BD Biosciences, mouse anti-Src, Upstate Biotechnology and secondary antibodies were HRP conjugated (GE Healthcare). The blots were incubated with chemiluminescence ECL reagent (Amersham ECL plus, GE Healthcare) and then exposed to Amersham hyperfilm (GE Healthcare) and developed using a Konica Minolta automatic film developer (Model SRX-101A). The autoradiograms were scanned and images imported into Adobe Photoshop to quantitate band intensities.
3. Results
3.1. The selective recruitment of proteins to integrin αβ1, αVβ or α6β complexes in oral carcinoma
We hypothesized that specific integrin effectors critical for invasion and chemoresistance are recruited to integrin αvβ, α6β and αβ1 receptors in oral carcinoma [17] [18] [19]. To test this notion, we first determined whether clustering integrin receptors with αv, α6 or β1 subunits in HN12 cells resulted in the specific recruitment of distinct effectors to each receptor. HN12 cells are an oral squamous cell carcinoma (OSCC) metastatic line that retains their invasive phenotype in vitro [10]. Integrins were clustered at the cell surface of HN12 cells for 1 hour with magnetic beads covalently coupled to anti-integrin αvβ, α6β or αβ1 antibodies. The proteins recruited to the integrin receptors were biochemically extracted and the sequences of tryptic peptides were identified in an unbiased manner. Elucidation of peptides that are unique to each integrin complex was performed with contrast 2.0 software [11] [12]. Peptides identified in a particular integrin complex are listed according to the receptor complex in Table 1. These results suggest that oral carcinoma proteins are selectively recruited to particular integrin receptors. A functionally diverse set of proteins were isolated in the complexes including microtubule and actin binding proteins, and factors involved in trafficking, transcription and translation. Each complex contained proteins that are not currently defined as integrin effectors and two examples from each complex are huntingtin-associated protein 1, twinkle, fragile X mental retardation syndrome rp2, opioid growth factor receptor, Dek oncogene and quaking. A well known component of integrin-based cell adhesion complexes such as talin was identified in each of the receptor complexes. P130Cas was isolated in α6β and αβ1 complexes. Dek, Src and zyxin were observed in only the α6β complex. The complete proteomics data set is provided in supplementary materials.
Table 1.
HN12 proteins recruited to particular integrin receptors. Integrin αvβ, α6β or αβ1 receptors were each clustered on the surface of HN12 cells. Tryptic peptides in the integrin complexes were identified and contrast analysis performed to reveal peptides present in only one of the integrin receptor complexes. The protein and corresponding GenBank accession number is shown. The complete data set for HN12 and HN4 cells is available as supplementary material.
| Integrin αv complexes |
| aconitase 1 (gi 55663279) |
| arp6 (gi 11968057) |
| dedicator of cytokinesis 7, 9 (gi 12698087, gi 24308029) |
| huntingtin-associated protein 1 (gi 10241694) |
| janus kinase 1 (gi 102469034) |
| kinesin family member 15, 2C (gi 9910266, gi 14250610) |
| laminin, gamma 2 (gi 55663092) |
| map1b protein (gi 66267565) |
| nestin (gi 38176300) |
| rabconnectin-3 beta isoform 1 (gi 73747877) |
| tensin (gi 66529407) |
| twinkle (gi 39725942) |
| wiz protein (gi 71051630) |
| Integrin β1 complexes |
| calmodulin regulated spectrin- ap1 (gi 40538730) |
| cdc2-like 1 isoform 6 (gi 16332366) |
| fgfr1/bcr chimaeric fusion protein (gi 16444914) |
| fragile X mental retardation syndrome rp2 (gi 4758410) |
| kinesin light chain 2 (gi 12383062) |
| microtubule-actin crosslinking factor 1 (gi 55959649) |
| mitogen- and sapk-2 (gi 3411161) |
| opioid growth factor receptor (gi 33286446) |
| ptpase, non-receptor type 13 (gi 18375646) |
| pumilio homolog 2 (gi 13491168) |
| p120 catenin (gi 3152827) |
| rack7 isoform 1 (gi 86143624) |
| retinoblastoma binding protein 2 (gi 435778) |
| suppression of tumorigenicity 5 (gi 13509322) |
| thyroid receptor-interacting protein 6 (gi 91208423) |
| ttf1 protein (gi 62022496) |
| Integrin α6 complexes |
| activating transcription factor 2 (gi 22538422) |
| arfaptin 2 (gi 6912602) |
| beta tubulin 1, class VI (gi 13562114) |
| calponin 2,3 (gi 4758018, gi 4502923) |
| coronin, actin binding protein, 2A (gi 16554585) |
| dek oncogene (gi 4503249) |
| Filamin-binding LIM protein-1 (gi 27462701) |
| galectin-8 (gi 6625728) |
| importin 4 (gi 62460637) |
| mapkk2 (gi 13489054) |
| mixed lineage kinase ZAK (gi 7542537) |
| n-cadherin (gi 1335229) |
| por1 (gi 1292866) |
| quaking (gi 56566044) |
| rap1A (gi 54696200) |
| rho gef1 (gi 30583681) |
| slingshot 2 (gi 37674210) |
| stat 3 (gi 21618340) |
| spindlin (gi 112293285) |
| trim41 protein (gi 48257198) |
| zyxin (gi 4508047) |
3.2. Requirement for integrin effectors in oral carcinoma invasion through matrigel
Preliminary studies demonstrated that HN12 invasion through Matrigel is disrupted after treatment with integrin β1 function-blocking antibodies (unpublished observations, A Berrier). Integrin linkage to the cytoskeleton is critical for cell traction during integrin-dependent mesenchymal motility [20]. Of the proteins identified in the proteomics screen p130Cas, talin, Src and zyxin were selected for further analysis based upon their function in dynamic regulation of the actin cytoskeleton [21] [22]. Dek nuclear activities regulate chemoresistance in melanomas [23], however the function of Dek and these other effectors in oral carcinoma invasion and chemoresistance is currently not known. We first examined whether these selected integrin effectors are required for HN12 cell invasion through Matrigel. Suppression of protein expression for each of the candidate genes in HN12 cells was achieved by transfection with a pool of three pairs of siRNA duplexes. Disruption of steady-state protein levels by approximately 80% was routinely observed. Transfected HN12 cells were seeded in transwell chambers coated with Matrigel. Invading cells were stained with cytotracker orange and cell numbers counted. The HN12 cells failed to invade through Matrigel if the bottom chamber lacked the chemoattractants provided by serum (BSA, Figure 1A). Quantitation of the number of invading HN12 cells with serum in the bottom chamber (Figure 1B) revealed that supression of either talin or p130Cas inhibited HN12 tumor cell invasion to less than 5% (p130Cas p=0.002, talin p<0.005) of control cells. Supression of Dek and Src reduced invasion to levels that are 49.7% and 61.8% of wild type (Dek p=0.03, Src p=0.018). Suppression of zyxin failed to significantly reduce HN12 cell invasion. Thus, p130Cas, Dek, Src and talin each regulate oral carcinoma invasion.
Fig 1.
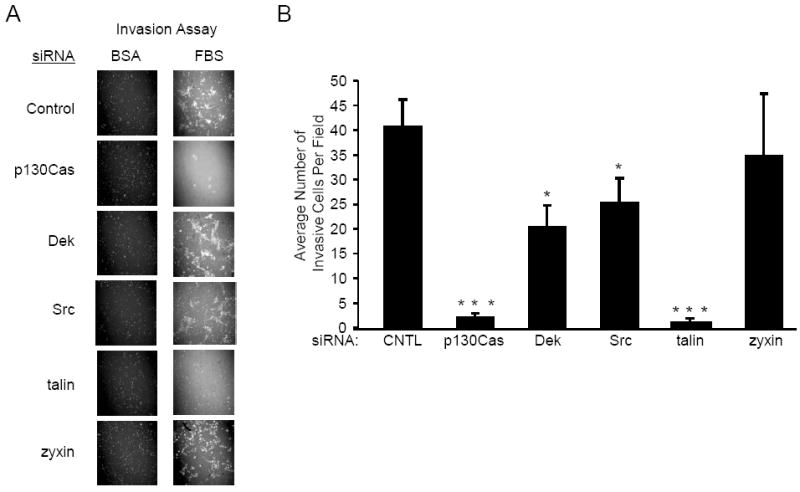
Disruption of p130Cas, Dek, Src or talin inhibits HN12 invasion. HN12 cells were transfected with control, p130Cas, Dek, Src, talin or zyxin siRNA. Transfected cells were seeded into transwell chambers coated with matrigel. A. The bottom chamber contained BSA (left) or FBS (right). Shown are invading cells stained with cytotracker orange. B. Quantitation of invasion by fluorescence imaging. The average number of invading cells is shown in the bar graph. The error bars indicate standard error. This experiment was performed in triplicate in 3 separate trials. Student’s t-test p values are indicated, * p < 0.05, ** p < 0.01, *** p < 0.005.
3.3 Requirement for integrin effectors in oral carcinoma spreading on collagen I and laminin I
The inhibition of HN12 invasion associated with disruption of integrin effectors could have resulted from loss of cell-matrix adhesion. To evaluate cell-matrix adhesion, the spreading of cells on laminin I and collagen I was examined since spreading on these matrix proteins is dependent on integrin signaling [24]. The role of the selected effectors in HN12 cell spreading on these matrices has not been reported previously. HN12 cells with knockdown of the selected effectors were replated on collagen I or laminin I and allowed to spread for 1 hour. Phase contrast images of the adherent cells were acquired with an inverted microscope (Figure 2A). Analysis of cell morphology revealed that a higher percentage of cells spread if the HN12 cells were adherent to collagen I in comparison to laminin I (control cells on collagen 94.5% spread, on laminin 56% spread, p=0.02) (Figure 2B, C). The knockdown of talin reduced the percentage of spread cells by 40% (p=0.0001) on collagen and 64% (p=0.0005) on laminin. Reduction of p130Cas expression inhibited spreading by 5% (p=0.001) on collagen and 26% (p=0.018) on laminin. Interestingly, knockdown of zyxin inhibited spreading on collagen by 9% (p=0.007) and enhanced spreading on laminin by 36% (p=0.0067). In contrast, a reduction in Dek or Src expression did not significantly change the percentages of spread cells on either matrix. P130Cas, talin, and zyxin each regulate HN12 spreading on collagen I and laminin I. Thus, among these integrin effectors, disruption of invasion does not correlate with modulation of cell spreading.
Fig 2.
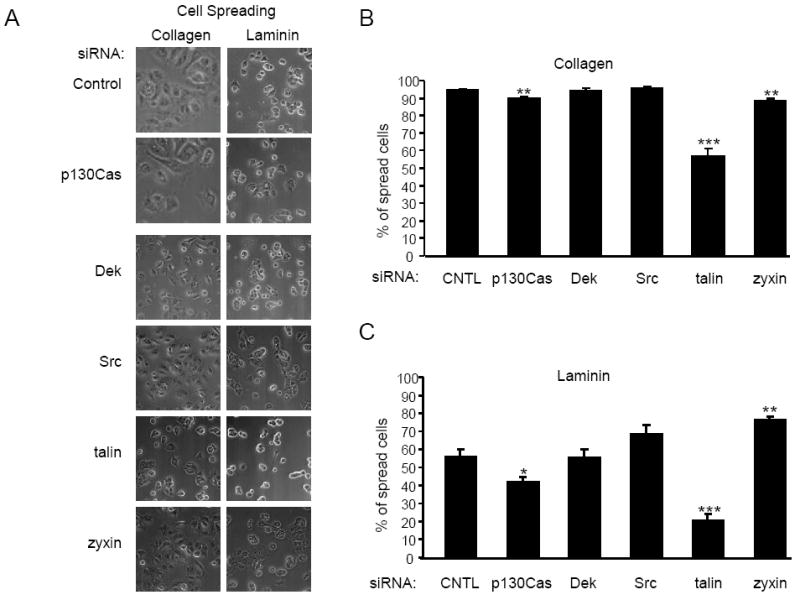
Knockdown of talin and p130Cas inhibits HN12 spreading on collagen and laminin. HN12 cells transfected with control, p130Cas, Dek, Src, talin or zyxin siRNA were seeded on glass coverslips coated with collagen I or laminin I at 10 μg/ml. A. Phase contrast images of the cells adherent to collagen (left) or laminin (right). The percentage of cells that spread on collagen (B) or laminin (C) is shown in the bar graphs. The error bars indicate standard error. The experiment was performed 3 times. Student’s t-test p values are indicated, * p < 0.05, ** p < 0.01, *** p < 0.005.
3.4. Role of integrin effectors in cisplatin resistance
Cisplatin is a commonly used chemotherapeutic for oral and tongue cancers, in spite of the fact that nearly 80% of these patients have cisplatin resistant tumors or will develop after treatment recurrences that are cisplatin resistant [25]. HN12 cells are resistant to cisplatin at 10 μM [26] and recently our laboratory observed that adhesion to HN12 matrix increases HN12 resistance to cisplatin at 30 μM (manuscript submitted, A. Berrier). Integrin function is required for resistance to chemotherapeutics in breast cancer lines adherent to stromal matrix [5]. Our studies focused on whether the selected integrin effectors regulate HN12 chemoresistance induced by adhesion to HN12 matrix. We tested whether the knockdown of the selected effectors could alter HN12 resistance to cisplatin. Cells with targeted disruption of integrin effectors were seeded on HN12 matrix and adherent cells were treated with vehicle DMSO or cisplatin (1 and 30 μM) and proliferation was measured by MTT assay after 48 hours in serum-free medium (Figure 3A). For these cisplatin-resistant cells on HN12 matrix, transfection with control siRNA and treatment with cisplatin at 1 μM unexpectedly but reproducibly increased proliferation 2.96-fold (p=0.01) relative to vehicle DMSO-treated cells. Proliferation was 1.32-fold (p=0.07) higher following treatment with 30 μM cisplatin in comparison to vehicle DMSO-treated cells. The targeted reduction of Dek, Src and zyxin showed similar levels of proliferation for the DMSO and 30 μM cisplatin-treated cells suggesting a slight increase in chemosensitivity (comparison of 30 μM cisplatin, control vs Src p=0.03, vs zyxin p=0.027). In contrast, targeted reduction of p130Cas increased proliferation 1.76-fold in 30 μM cisplatin compared to the DMSO controls (comparison of 30 μM cisplatin, control vs p130Cas p=0.033). Of interest, targeting talin increased HN12 proliferation 4.64- and 2.28- fold after treatment with 1 μM and 30 μM cisplatin respectively (comparison of control vs talin, 1 μM cisplatin p=0.02, 30 μM cisplatin p=0.01). Therefore, reductions in p130Cas and especially talin expression increased chemoresistance.
Fig 3.
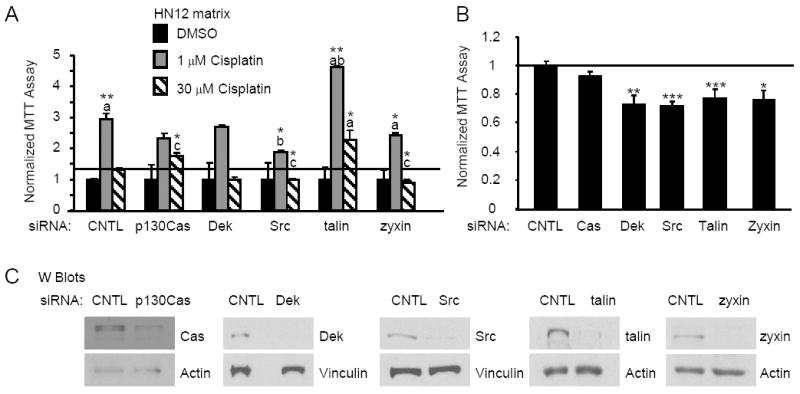
Knockdown of p130Cas, Src, talin and zyxin alters HN12 resistance to cisplatin. HN12 cells transfected with control, p130Cas, Dek, Src, talin or zyxin siRNA were seeded onto HN12 matrix and treated for 48 hours with cisplatin or vehicle DMSO as indicated. A. The average proliferation measured by MTT for HN12 cells is shown in the bar graph. The data are normalized to the DMSO-treated transfectants. The error bars indicate standard error. This experiment was performed in triplicate in two separate trials. Statistical comparisons a=control/DMSO, b=control/1 μM cisplatin, c=control/30 μM cisplatin. B. Transfectants were seeded onto HN12 matrix in serum and proliferation measured by MTT and normalized to control siRNA. This experiment was performed in triplicate in 3 separate trials. The error bars indicate standard error. C. Western blots of siRNA-transfectants. Lysates with equivalent levels of actin were analyzed for the protein level of the knockdown. Student’s t-test p values are indicated, * p < 0.05, ** p < 0.01, *** p < 0.005.
Since integrin signaling regulates tumor cell proliferation, we hypothesized that the selected integrin effectors might alter HN12 proliferation in the absence of cisplatin [27]. To address this concern, HN12 cells with knockdowns of each of the effectors were replated on HN12 matrix in serum and proliferation measured 48 hours later (Figure 3B). Control and p130Cas siRNA-treated cells displayed similar levels of proliferation. Dek, Src, talin and zyxin siRNA-treated cells showed 32%, 34%, 28% and 29% respective reductions in proliferation (comparison to control, Dek p=0.009, Src p=0.0019, talin p=0.0037, zyxin p=0.016). Representative Western blots are shown confirming the reduction in steady-state protein levels in the siRNA-treated HN12 cells (Figure 3C). Approximately 80% reduction in target gene expression was routinely observed following treatment with siRNA duplexes. Hence, Dek, Src, talin and zyxin similarly affect the proliferation of oral carcinoma adherent to HN12 matrix yet these effectors have different roles in chemoresistance. A summary of the integrin effector phenotypes in oral carcinoma is provided in Table 2.
Table 2.
Summary of integrin effector siRNA phenotypes in oral carcinoma.
| Spreading on | |||||
|---|---|---|---|---|---|
| siRNA | Invasion | laminin | collagen | Cisplatin resistance | Proliferation |
| p130Cas | ↓↓↓ | ↓ | ↓ | ↑ | — |
| Dek | ↓↓ | — | — | ↓ | ↓ |
| Src | ↓↓ | — | — | ↓ | ↓ |
| talin | ↓↓↓ | ↓↓ | ↓↓ | ↑ | ↓ |
| zyxin | — | ↑ | ↓ | ↓ | ↓ |
4. Discussion
Integrin mediated adhesions to extracellular matrix within the tumor microenvironment regulate both tumor invasion and resistance to chemotherapeutics [6] [7] [8]. Whether integrin-associated proteins coordinate invasive potential with chemoresistance is poorly understood. Proteomics analysis of clustered integrin αvβ, α6β or αβ1 receptors was performed to identify candidate integrin effectors in oral carcinomas. One outcome of the proteomics analysis of integrin complexes was the demonstration that oral carcinoma proteins are selectively recruited to particular integrin receptors. Of the oral carcinoma integrin effectors studied, the targeted reduction of talin had the most potent impact on adhesion-dependent processes including invasion, spreading and chemoresistance. These studies demonstrate that p130Cas, Src and talin regulate pathways important for both invasion and chemoresistance in response to adhesions within the tumor matrix.
Known mechanisms of resistance to cisplatin include an increase in cisplatin efflux, a decrease in cisplatin uptake, an increase in DNA repair and disruption of apoptosis [28]. Inhibition of apoptosis is a likely scenario for how p130Cas and talin deficiencies induce chemoresistance since p130Cas controls apoptosis and talin regulates prostate cancer anoikis [29] [30]. It is possible that oral carcinomas with reductions in p130Cas/talin may fail to trigger apoptosis after exposure to cisplatin potentially through a disruption of the activation of death-associated caspases. Alternatively, the reduction of p130Cas or talin expression may enhance resistance to cisplatin as a result of aberrant regulation of focal adhesion signals that trigger proliferation.
Future studies will examine the molecular mechanism(s) that connect p130Cas/talin to invasion and chemoresistance. Understanding integrin pathways that are utilized for invasion and chemoresistance may provide novel insights into development of chemotherapeutics for oral carcinomas.
Supplementary Material
Acknowledgments
We thank the Katrina Visiting Faculty Program sponsored by the NCMHD/NIDCR/NIH, the NIDCR intramural research program and LSUHSC-NO School of Dentistry for support, and the Yates Laboratory for the proteomics. The expertise provided by Hynda Kleinman and Deborah Philp in invasion assays and Becky Worthylake in microscopy are greatly appreciated. We also thank the Department of Oral and Craniofacial Biology for their core facilities.
Abbreviations
- OSCC
oral squamous cell carcinoma
- siRNA
small inhibitory ribonucleic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11:9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 2.Yoshida Y, Kurokawa T, Nishikawa Y, Orisa M, Kleinman HK, Kotsuji F. Laminin-1-derived scrambled peptide AG73T disaggregates laminin-1-induced ovarian cancer cell spheroids and improves the efficacy of cisplatin. Int J Oncol. 2008;32:673–681. [PubMed] [Google Scholar]
- 3.Hassid Y, Eyal E, Margalit R, Furman-Haran E, Degani H. Non-invasive imaging of barriers to drug delivery in tumors. Microvasc Res. 2008;76:94–103. doi: 10.1016/j.mvr.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Puigvert JC, Huveneers S, Fredriksson L, op het Veld M, van de Water B, Danen EH. Cross-talk between integrins and oncogenes modulates chemosensitivity. Mol Pharmacol. 2009;75:947–955. doi: 10.1124/mol.108.051649. [DOI] [PubMed] [Google Scholar]
- 5.Serebriiskii I, Castello-Cros R, Lamb A, Golemis EA, Cukierman E. Fibroblast-derived 3D matrix differentially regulates the growth and drug-responsiveness of human cancer cells. Matrix Biol. 2008;27:573–585. doi: 10.1016/j.matbio.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rathinam R, Alahari SK. Important role of integrins in the cancer biology. Cancer Metastasis Rev. 2010;29:223–237. doi: 10.1007/s10555-010-9211-x. [DOI] [PubMed] [Google Scholar]
- 7.Morin PJ. Drug resistance and the microenvironment: nature and nurture. Drug Resist Updat. 2003;6:169–172. doi: 10.1016/s1368-7646(03)00059-1. [DOI] [PubMed] [Google Scholar]
- 8.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cardinali M, Pietraszkiewicz H, Ensley JF, Robbins KC. Tyrosine phosphorylation as a marker for aberrantly regulated growth-promoting pathways in cell lines derived from head and neck malignancies. Int J Cancer. 1995;61:98–103. doi: 10.1002/ijc.2910610117. [DOI] [PubMed] [Google Scholar]
- 10.Yeudall WA, Miyazaki H, Ensley JF, Cardinali M, Gutkind JS, Patel V. Uncoupling of epidermal growth factor-dependent proliferation and invasion in a model of squamous carcinoma progression. Oral Oncol. 2005;41:698–708. doi: 10.1016/j.oraloncology.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR., 3rd Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol. 1999;17:676–682. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 12.Bern M, Goldberg D, McDonald WH, Yates JR., 3rd Automatic quality assessment of peptide tandem mass spectra. Bioinformatics. 2004;20(Suppl 1):i49–54. doi: 10.1093/bioinformatics/bth947. [DOI] [PubMed] [Google Scholar]
- 13.Kleinman HK, Jacob K. Invasion assays. Curr Protoc Cell Biol. 2001;Chapter 12(Unit 12):12. doi: 10.1002/0471143030.cb1202s00. [DOI] [PubMed] [Google Scholar]
- 14.Berrier AL, LaFlamme SE. Cell-spreading assays. Methods Mol Biol. 2005;294:55–68. doi: 10.1385/1-59259-860-9:055. [DOI] [PubMed] [Google Scholar]
- 15.Vlodavsky I. Preparation of extracellular matrices produced by cultured corneal endothelial and PF-HR9 endodermal cells. Curr Protoc Cell Biol. 1999;1:1–14. doi: 10.1002/0471143030.cb1004s01. [DOI] [PubMed] [Google Scholar]
- 16.Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708–1712. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- 17.Ramos DM, But M, Regezi J, Schmidt BL, Atakilit A, Dang D, Ellis D, Jordan R, Li X. Expression of integrin beta 6 enhances invasive behavior in oral squamous cell carcinoma. Matrix Biol. 2002;21:297–307. doi: 10.1016/s0945-053x(02)00002-1. [DOI] [PubMed] [Google Scholar]
- 18.Zutter MM. Integrin-mediated adhesion: tipping the balance between chemosensitivity and chemoresistance. Adv Exp Med Biol. 2007;608:87–100. doi: 10.1007/978-0-387-74039-3_6. [DOI] [PubMed] [Google Scholar]
- 19.Cabodi S, del Pilar Camacho-Leal M, Di Stefano P, Defilippi P. Integrin signalling adaptors: not only figurants in the cancer story. Nat Rev Cancer. 2010;10:858–870. doi: 10.1038/nrc2967. [DOI] [PubMed] [Google Scholar]
- 20.Wolf K, Friedl P. Molecular mechanisms of cancer cell invasion and plasticity. Br J Dermatol. 2006;154(Suppl 1):11–15. doi: 10.1111/j.1365-2133.2006.07231.x. [DOI] [PubMed] [Google Scholar]
- 21.Hall A. The cytoskeleton and cancer. Cancer Metastasis Rev. 2009;28:5–14. doi: 10.1007/s10555-008-9166-3. [DOI] [PubMed] [Google Scholar]
- 22.Hervy M, Hoffman L, Beckerle MC. From the membrane to the nucleus and back again: bifunctional focal adhesion proteins. Curr Opin Cell Biol. 2006;18:524–532. doi: 10.1016/j.ceb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Riveiro-Falkenbach E, Soengas MS. Control of tumorigenesis and chemoresistance by the DEK oncogene. Clin Cancer Res. 2010;16:2932–2938. doi: 10.1158/1078-0432.CCR-09-2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berrier AL, Mastrangelo AM, Downward J, Ginsberg M, LaFlamme SE. Activated R-ras, Rac1, PI 3-kinase and PKCepsilon can each restore cell spreading inhibited by isolated integrin beta1 cytoplasmic domains. J Cell Biol. 2000;151:1549–1560. doi: 10.1083/jcb.151.7.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Specenier PM, Vermorken JB. Neoadjuvant chemotherapy in head and neck cancer: should it be revisited? Cancer Lett. 2007;256:166–177. doi: 10.1016/j.canlet.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 26.Kim HE, Krug MA, Han I, Ensley J, Yoo GH, Forman JD, Kim HR. Neutron radiation enhances cisplatin cytotoxicity independently of apoptosis in human head and neck carcinoma cells. Clin Cancer Res. 2000;6:4142–4147. [PubMed] [Google Scholar]
- 27.Stupack DG, Cheresh DA. Get a ligand, get a life: integrins, signaling and cell survival. J Cell Sci. 2002;115:3729–3738. doi: 10.1242/jcs.00071. [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez VM, Fuertes MA, Alonso C, Perez JM. Is cisplatin-induced cell death always produced by apoptosis? Mol Pharmacol. 2001;59:657–663. doi: 10.1124/mol.59.4.657. [DOI] [PubMed] [Google Scholar]
- 29.Kook S, Shim SR, Choi SJ, Ahnn J, Kim JI, Eom SH, Jung YK, Paik SG, Song WK. Caspase-mediated cleavage of p130cas in etoposide-induced apoptotic Rat-1 cells. Mol Biol Cell. 2000;11:929–939. doi: 10.1091/mbc.11.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakamoto S, McCann RO, Dhir R, Kyprianou N. Talin1 promotes tumor invasion and metastasis via focal adhesion signaling and anoikis resistance. Cancer Res. 2010;70:1885–1895. doi: 10.1158/0008-5472.CAN-09-2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.