

Abstract
It is becoming increasingly recognized that G protein-coupled receptors physically interact. These interactions may provide a mechanism for allosteric modulation of receptor function. In this study, we examined this possibility by using an established model system of a receptor heteromer consisting of μ and δ opioid receptors. We examined the effect of a number of μ receptor ligands on the binding equilibrium and association and dissociation kinetics of a radiolabeled δ receptor agonist, [3H]deltorphin II. We also examined the effect of δ receptor ligands on the binding equilibrium and association and dissociation kinetics of a radiolabeled μ receptor agonist, [3H][d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin ([3H]DAMGO). We show that μ receptor ligands are capable of allosterically enhancing δ receptor radioligand binding and vice versa. Thus, there is strong positive cooperativity between the two receptor units with remarkable consequences for ligand pharmacology. We find that the data can be simulated by adapting an allosteric receptor model previously developed for small molecules, suggesting that the ligand-occupied protomers function as allosteric modulators of the partner receptor's activity.
Introduction
G protein-coupled receptors (GPCRs) comprise one of the largest gene families in the mammalian genome that respond to a wide range of stimuli, including biogenic amines, amino acids, peptides, lipids, nucleosides, and large polypeptides. GPCRs are involved in a variety of biological processes, including neurotransmission, metabolism, and cellular differentiation, among others, and are therefore important targets for drug development (Rozenfeld et al., 2006; Kenakin and Miller, 2010). Many therapeutic agents target the orthosteric site of GPCRs (the site to which the endogenous ligand binds). These drugs either activate (agonists) or block (antagonists) receptor function. More recently, efforts have been made toward the identification of drugs that do not directly bind to the orthostheric site but are able to efficiently modulate GPCR function (Soudijn et al., 2004; Ma et al., 2009; Duvoisin et al., 2010). The advantage of this approach is the development of drugs that have fewer side effects and are better able to distinguish between GPCR subtypes.
A number of studies have shown that GPCRs can form dimers or oligomers (for the sake of simplicity, throughout the text, we will refer to oligomers as dimers; complexes consisting of two or more identical monomers, also known as protomers, as homomers; and complexes of two different protomers as heteromers). The existence of GPCR homomers and heteromers has been shown to occur in heterologous cells, in cell lines endogenously expressing receptors, in primary cell cultures, and in a few cases in intact tissues (for reviews, see Rios et al., 2001; Prinster et al., 2005; Rozenfeld and Devi, 2010b). In some cases, GPCR heteromerization has been shown to be essential for the formation of a functional receptor; the best known examples are GABAB and some taste and odorant receptors (White et al., 1998; Nelson et al., 2002; Neuhaus et al., 2005). In other cases, studies show that GPCR heteromerization leads to the modulation of the pharmacological, signaling, and trafficking properties of individual protomers (Rios et al., 2001; Prinster et al., 2005; Milligan, 2009).
We have shown previously that heteromerization of μ opioid receptor (μOR) with δ opioid receptor (δOR) leads to the modulation of receptor binding and signaling properties (Gomes et al., 2000, 2004). We found that although coexpression of δOR alone did not affect μOR activity, occupancy of δOR by a selective δOR agonist or antagonist significantly enhanced the potency and intrinsic activity of μOR agonists in cells or brain regions coexpressing both receptors (Gomes et al., 2000, 2004). For example, the selective δOR antagonist TIPPψ was capable of increasing the intrinsic activity of morphine or DAMGO as measured using guanosine 5′-O-(3-thio)triphosphate binding or extracellular signal-regulated kinases 1/2 phosphorylation assays (Gomes et al., 2000, 2004). This increase in intrinsic activity was also seen in SK-N-SH cells that endogenously express both μOR and δOR and in spinal cord membranes from wild-type animals but not from animals lacking δOR (Gomes et al., 2004).
In this study, we examined whether the reciprocal also occurred (i.e., whether μOR ligands could modulate the binding properties of δOR). We then examined whether one protomer could act as an allosteric modulator of the other protomer by examining the dissociation kinetics of radiolabeled bound agonist in the absence and presence of ligands to the heteromeric partner. To explore the occurrence of allosteric modulation, we examined whether simulations using the ternary complex mathematical model developed for small molecule modulators of GPCRs would generate data similar to those obtained experimentally with μOR-δOR heteromers. We find that both experimental saturation and enhancement curves can be simulated in this model by using the affinities of the ligands and by defining the binding cooperativity as the cooperative relationship between the orthosteric ligand A and the ligand-occupied receptor B. This suggests that a ligand-occupied protomer can be simulated in a ternary complex model as a “binding allosteric entity” of its heteromeric partner.
Materials and Methods
Materials.
[d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO) and Tyr-d-Ala-Phe-Glu-Val-Val-Gly (deltorphin II) were from Sigma/RBI (Natick, MA). d-Phe-Cys-Tyr-d-Trp-Orn-Thr-Pen-Thr-NH2 (CTOP) was from Peninsula Laboratories Inc. (San Carlos, CA). β-Endorphin, BNTX, met-enkephalin, endomorphin 1, endomorphin 2, naloxone, naloxonazine, and d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2 (CTAP) were from Tocris (Ballwin, MO). [3H]DAMGO and [3H]deltorphin II were from PerkinElmer Life and Analytical Sciences (Waltham, MA). Fentanyl, methadone, morphine, and etonitazene were from Dr. Eric J. Simon (New York University School of Medicine, New York, NY). Tyr-Ticψ(CH2NH)-Phe-Phe (TIPPψ) was from Dr. Peter Schiller (Institut de Reserches Cliniques de Montreal, Montreal, ON, Canada).
Cell Culture and Transfection.
Chinese hamster ovary (CHO) cells stably expressing Flag-μOR or Flag-δOR or coexpressing Flag-μOR and myc-δOR (in a ratio of 1:4, 1:6, or 1:40) were generated using Lipofectamine reagent (Invitrogen, Carlsbad, CA) and grown as described previously (Gomes et al., 2003). SK-N-SH cells that express endogenous μOR and δOR (2:1 ratio) were grown in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and penicillin-streptomycin (Invitrogen).
Membrane Preparation.
Membranes were prepared from SK-N-SH cells, CHO cells expressing either Flag-tagged μOR or myc-tagged δOR, or cells coexpressing μOR and δOR, as described previously (Gomes et al., 2003).
Saturation Binding Assays.
For whole-cell binding, CHO cells (stably expressing only μOR or δOR or coexpressing μOR and δOR) or SK-N-SH cells (endogenously expressing μOR and δOR) were incubated with different concentrations (0–10 nM) of [3H]deltorphin II in the absence or presence of 10 nM concentrations of various ligands (as shown in the figures), as described previously (Gomes et al., 2000, 2003). Nonspecific binding was determined in the presence of 1 μM deltorphin II and was less than 5% of the total binding. For membrane binding, SK-N-SH cell membranes (50 μg) were incubated with [3H]deltorphin II (10 nM) in the absence or presence of either 10 nM DAMGO or 10 nM CTOP or with [3H]DAMGO (10 nM) in the absence or presence of either 10 nM deltorphin II or 10 nM TIPPψ. Nonspecific binding was determined in the presence of 1 μM diprenorphine and was less than 5% of the total binding.
Effect of μOR Ligands on [3H]deltorphin II Binding.
These studies were carried out in whole cells. CHO cells stably coexpressing Flag-μOR and myc-δOR were incubated with 6 nM [3H]deltorphin II in the absence or presence of different concentrations (0–1 nM) of μOR ligands as described previously (Gomes et al., 2000, 2003). For experiments using low concentrations (3 pM) of [3H]deltorphin II in the absence or presence of low concentrations (3 fM, 3 pM, and 3 nM) of the μOR antagonist CTOP, scintillation vials were counted for 10 min instead of 1 min because of low count numbers.
Effect of δOR Ligands on [3H]DAMGO Binding.
These studies were carried out in whole cells. CHO cells stably coexpressing Flag-μOR and myc-δOR were incubated with 10 nM [3H]DAMGO in the absence or presence of different concentrations (0–1 nM) of δOR ligands as described previously (Gomes et al., 2000, 2003). For experiments using low concentrations (3 pM) of [3H]DAMGO in the absence or presence of low concentrations (3 fM, 3 pM, and 3 nM) of the δOR antagonist TIPPψ, scintillation vials were counted for 10 min instead of 1 min because of low count numbers.
Effect of Pertussis Toxin Treatment.
For experiments carried out in whole cells, CHO cells coexpressing μOR-δOR were treated overnight with 50 ng/ml pertussis toxin followed by treatment with [3H]DAMGO (10 nM) in the absence or presence of TIPPψ (10 nM) or [3H]deltorphin II (6 nM) in the absence or presence of CTOP (10 nM) as described above. For studies using membrane preparations, they were pretreated with 50 ng/ml pertussis toxin for 3 h and used for binding assays as described above.
Association Assays.
These studies were carried out in whole cells. SK-N-SH cells (3 × 105 cells/well) were plated into a 24-well plate precoated with poly-d-lysine. After 48 h, the media were removed, and cells were incubated with 10 nM [3H]DAMGO in the absence or presence of 10 nM TIPPψ in assay buffer (50 mM Tris-Cl, pH 7.4, containing 0.32 M sucrose) for different time periods (0–60 min) at 37°C. Cells were also incubated with 6 nM [3H]deltorphin II in the absence or presence of 10 nM DAMGO or fentanyl. In another set of plates, cells were incubated with 10 nM [3H]DAMGO in assay buffer for 1 h at 37°C. Cells were then incubated with TIPPψ (10 nM) for different time periods (0–60 min). At the end of each incubation period, the assay buffer was removed and cells were lysed with 100 μl of 1 N NaOH, followed by neutralization with 100 μl of 1 N HCl. The supernatant was collected and radioactivity measured in a liquid scintillation counter.
Dissociation Assays.
These studies were carried out in whole cells. SK-N-SH cells, plated as described above for association assays, were incubated with 10 nM [3H]DAMGO or 6 nM [3H]deltorphin II in assay buffer for 1 h at 37°C as described previously (Gomes et al., 2000, 2003). The assay buffer was then removed, the plates were kept on ice, and cells were incubated with either 1 μM DAMGO in the absence or presence of 0.1, 1, or 10 nM TIPPψ or with 1 μM deltorphin II in the absence or presence of 0.1, 1, or 10 nM fentanyl for indicated time intervals (0–120 min). At the end of the incubation period, the assay buffer was removed, and cells were lysed with 100 μl of 1 N NaOH, followed by neutralization with 100 μl of 1 N HCl. The supernatant was collected and radioactivity measured in a liquid scintillation counter. Acid wash experiments to remove surface bound radiolabel indicate that <3% of [3H]DAMGO or [3H]deltorphin II was sequestered/internalized when using 50 mM Tris-Cl, pH 7.4, containing 0.32 M sucrose to carry out these studies (I. Gomes and L. A. Devi, unpublished observations).
Pharmacological Modeling.
The allosteric two-state receptor model as developed by Hall (2000) was taken as the starting point for pharmacological modeling. In this model an allosteric modulator B binds to the inactive (R) and/or active (R*) state of the receptor R and influences the binding of orthosteric ligand A to R and/or R*. In our dimeric scenario, we kept the model as is but equated one ligand-occupied receptor to B (for instance, the ligand-occupied δOR) while referring to the other (for instance μOR) receptor as R/R* and the orthosteric ligand as A. We preferred this simplicity to a more complicated model in which we would have to include more parameters (for instance, the equilibrium association constant between the two receptor monomers in the absence of any ligand).
We implemented the model in MatLab, version 7.1 (The MathWorks, Inc., Natick, MA), and composed a graphic interface to facilitate both input (various parameter settings) and output (simulated curves). The radiolabeled agonists ([3H]DAMGO and [3H]deltorphin II) were assumed to bind to receptor subpopulations R*, R*B, and RB (but not R) to yield [ARB], [AR*], and [AR*B]. Thus, the proportion of radioligand bound receptors versus all receptors is
which can be restated as
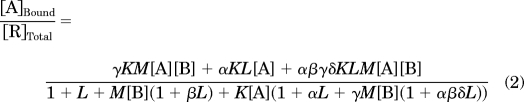 |
where [A] and [B] stand for the concentration of orthosteric ligand A and ligand-occupied receptor B. K and M are the association constant for ligand A and ligand-occupied receptor B binding to receptor R. α and β are the intrinsic efficacy of ligand A and ligand-occupied receptor B on receptor R, respectively. L is the isomerization constant between R and R*. γ is the binding cooperativity constant between orthosteric ligand A and ligand-occupied receptor B. δ is the activation cooperativity constant between ligand A and ligand-occupied receptor B. Eq. 2 was used for all the simulations.
Data Analysis.
One-site or two-site analysis of equilibrium binding parameters (Kd, Bmax, and pEC50 values) and kinetic rate constants (t1/2, koff values) were determined using Prism (ver. 4.0; GraphPad Software, San Diego, CA).
Results
Saturation Equilibrium Binding to μOR-δOR.
To characterize the binding properties of μOR-δOR heteromers, we used heterologous cells (CHO) expressing recombinant receptors and native cells (SK-N-SH) expressing endogenous receptors. We had previously shown that low nonsignaling doses of δOR ligands [e.g., deltorphin II, TIPPψ, naltriben, BNTX, and N,N-diallyl-Tyr-Aib-Aib-Phe-Leu (ICI 174,864)] increased the binding of radiolabeled μOR agonists such as DAMGO or morphine in cells coexpressing μOR-δOR (Gomes et al., 2000, 2004). In the present study, we examined whether this effect was reciprocal [i.e., whether low doses of μOR ligands increased the binding of a radiolabeled δOR agonist, [3H]deltorphin II, in whole cells coexpressing μOR-δOR]. We found that in cells expressing only δOR, there are no significant changes in Kd and Bmax for [3H]deltorphin II binding in the absence or presence of low doses of three different μOR ligands (DAMGO, fentanyl, and morphine) (Table 1). However, in cells coexpressing both μOR and δOR, the addition of these μOR ligands enhanced radiolabeled δOR agonist binding (Table 1). We observe significant increases in agonist Bmax values in these two cell lines (Table 1), particularly in the presence of DAMGO (∼1.5- and ∼2-fold in CHO and SK-N-SH cells, respectively). This phenomenon (observed in whole cells) is also seen in membrane preparations from cells coexpressing μOR-δOR but not expressing solely μOR or δOR (Supplemental Fig. 1, A and B). This increase in radiolabeled agonist binding to δOR in the presence of μOR ligands (and vice versa) is referred to in the text as “heteromer-mediated” binding. Next, we examined whether heteromer-mediated binding is modulated by pertussis toxin pretreatment. We find that we detect lower levels of heteromer-mediated binding in whole cells or membranes from cells coexpressing μOR-δOR that were treated with pertussis toxin compared with control cells or membranes not treated with pertussis toxin (Supplemental Fig. 1, C and D). We also examined whether changes in the ratio of μOR to δOR affected heteromer-mediated binding. For this, we carried out binding studies with radiolabeled μOR agonist [3H]DAMGO in the absence or presence of the δOR antagonist TIPPψ and with radiolabeled δOR agonist [3H]deltorphin II in the absence or presence of the μOR antagonist CTOP in whole cells expressing μOR to δOR in ratios of 1:4, 1:6, and 1:40. We observe a decrease in heteromer-mediated binding as the ratio of μOR:δOR is increased from 1:4 to 1:40 (Supplemental Fig. 1E). Taken together, these results show that binding of a selective radiolabeled agonist to one protomer can be potentiated by a selective antagonist to the partner protomer, and this “heteromer-mediated binding” is affected by G-protein inactivation and the relative ratio of the two receptors.
TABLE 1.
Effects of μOR ligands on [3H]deltorphin II binding
CHO whole cells expressing δOR, CHO whole cells coexpressing μOR and δOR in a ratio of 1:4, and SK-N-SH whole cells endogenously expressing μOR and δOR in a ratio of 2:1 were incubated with [3H]deltorphin II (0–6 nM) in the absence or presence of 10 nM DAMGO, fentanyl, or morphine, and ligand binding was determined as described under Materials and Methods. Data represent mean ± S.E.M. (n = 3).
|
Kd |
Bmax |
|||||
|---|---|---|---|---|---|---|
| δ | μ-δ | SK-N-SH | δ | μ-δ | SK-N-SH | |
| nM | fmol/mg protein | |||||
| Control | 0.4 ± 0.1 | 1.3 ± 0.4 | 0.2 ± 0.1 | 39 ± 2 | 62 ± 4 | 93 ± 4 |
| + Fentanyl | 0.6 ± 0.2 | 1.0 ± 0.3 | 0.7 ± 0.2 | 42 ± 3 | 82 ± 5** | 173 ± 10** |
| + DAMGO | 0.6 ± 0.2 | 0.6 ± 0.2 | 0.5 ± 0.1 | 42 ± 3 | 93 ± 6** | 184 ± 10** |
| + Morphine | 0.4 ± 0.1 | 0.6 ± 0.2 | 1.4 ± 0.4** | 39 ± 2 | 78 ± 4** | 178 ± 13** |
P < 0.01 vs. control, Dunnett's test.
Enhancement of Radiolabeled Agonist Binding to δOR by μOR Ligands.
Next we characterized the “heteromer-mediated binding” by examining the ability of a panel of 12 μOR ligands to modulate agonist binding to δOR (Table 2). These ligands included both agonists (partial and full) and antagonists (neutral and inverse agonists) of the μOR. The agonists included endogenous peptides and synthetic peptidic or nonpeptidic compounds, whereas the antagonists were either of peptidic or nonpeptidic nature. As seen from Fig. 1, the μOR ligands increased agonist binding of [3H]deltorphin II to δOR receptors. The μOR ligands exhibited differences in their maximal capability to potentiate [3H]deltorphin II binding as well as in their potency. A comparison of the maximal enhancement (efficacy) and pEC50 (−log EC50) values shows the clinically relevant synthetic agonist fentanyl to be highly efficacious (96% compared with CTAP, which showed maximum enhancement and hence is taken as 100%), whereas both the antagonist naloxone and the endogenous ligand met-enkephalin were found to be least efficacious (27 and 17%, respectively) in increasing agonist binding to δOR (Fig. 1; Table 2). It is noteworthy that the EC50 values are all in the picomolar to femtomolar range, well below the binding affinities of these ligands to their cognate receptor (see pKi values in Table 2). Because the EC50 values are very low, we examined whether it was possible to detect specific binding using a very low concentration (3 pM) of [3H]deltorphin II in the absence or presence of ultralow and low concentrations of the μOR antagonist CTOP (3 fM, 3 pM, and 3 nM). We found that in the presence of increasing concentrations of CTOP, an increase in specific binding was observed (Supplemental Fig. 2B), whereas in the absence of the μOR antagonist CTOP, we observed no detectable specific binding with 3 pM [3H]deltorphin II. This enhancement of agonist binding by ultralow doses of the ligand to the partner receptor suggests allosteric modulation of ligand binding by heteromerization.
TABLE 2.
Enhancement of agonist binding to δOR by low doses of μOR ligands
CHO whole cells coexpressing μOR and δOR in a ratio of 1:4 were incubated with [3H]deltorphin II (6 nM) in the absence or presence of different concentrations (0–1 nM) of different μOR ligands (endogenous ligands, agonists, and antagonists/inverse agonists, respectively), and ligand binding determined as described under Materials and Methods. Data represent mean ± S.E.M. (n = 3). The pEC50 and percentage stimulation values were derived from the curves in Fig. 1.
| Ligand | pEC50 | Max. Enhancement (CTAP = 100%) | pKi | Ligand Activity |
|---|---|---|---|---|
| % | ||||
| CTAP | 12.77 ± 0.15 | 100 ± 5 | 8.27a | Antagonist |
| DAMGO | 12.30 ± 0.23 | 97 ± 6 | 8.7b | Full agonist |
| Fentanyl | 12.87 ± 0.12 | 96 ± 4 | 9.4b | Full agonist |
| Methadone | 13.66 ± 0.28 | 88 ± 5 | 9.1b | Partial agonist |
| Etonitazene | 12.82 ± 0.20 | 87 ± 5 | 9.96c | Full agonist |
| Naloxonazine | 12.68 ± 0.22 | 76 ± 3 | 10.3b | Antagonist |
| Endomorphin-2 | 14.56 ± 0.24 | 72 ± 4 | 7.8d | Full agonist |
| β-Endorphin | 12.08 ± 0.42 | 67 ± 8 | 9.0b | Full agonist |
| Endomorphin-1 | 13.79 ± 0.34 | 66 ± 4 | 9.4b | Full agonist |
| Morphine | 13.39 ± 0.24 | 65 ± 7 | 7.9b | Partial agonist |
| Naloxone | 12.48 ± 0.47 | 27 ± 4 | 9.0b | Antagonist |
| Met-enkephalin | 12.72 ± 0.58 | 17 ± 4 | 9.2b | Full agonist |
Data from Onali and Olianas, 2004.
Data from Raynor et al., 1994.
Data from Zernig et al., 1995.
Data from Harrison et al., 1999.
Fig. 1.

Concentration-dependent enhancement by μOR ligands of [3H]deltorphin binding. CHO whole cells coexpressing μOR and δOR (in a ratio of 1:4) were incubated with [3H]deltorphin II (6 nM) in the absence or presence of different concentrations (0–1 nM) of either DAMGO, β-endorphin, naloxone, met-enkephalin, fentanyl, methadone, endomorphin 1, endomorphin 2, CTAP, etonitazene, naloxonazine, or morphine and ligand binding determined as described under Materials and Methods. Data represent mean ± S.E.M. (n = 3).
Enhancement of Radiolabeled Agonist Binding to μOR by δOR Ligands.
We also carried out concentration-effect curves for the enhancement of agonist binding to μOR by six different δOR ligands (Table 3). In this case, the modulatory activity of the δOR ligands was expressed as a percentage of the maximum enhancement observed for [3H]DAMGO binding with the δOR antagonist TIPPψ (and this was taken as 100%). We found that among the δOR ligands, the agonist deltorphin II and the antagonist TIPPψ were both highly potent (pEC50 values of 11.99 and 11.04, respectively) and efficacious (89 and 100%, respectively) in increasing [3H]DAMGO binding (Supplemental Fig. 2A; Table 3). Other δOR ligands had an intermediate efficacy (the two antagonists/inverse agonists naltriben and BNTX), whereas the two agonists, (+)-4-[(αR)-α-((2S,5R)-4-allyl-2,5-dimethyl-1piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide (SNC80) and [d-Pen2,d-Pen5]-enkephalin were least efficacious in that they were only marginally active, precluding the determination of a pEC50 value (Table 3). A comparison between pEC50 and pKi values (the “orthosteric” affinity of these ligands for the δOR) showed a 10- to 1000-fold difference between these two potencies/affinities (Table 3). We also examined whether we could detect measurable specific binding using a very low concentration (3 pM) of [3H]DAMGO in the absence or presence of very low concentrations of the δOR antagonist TIPPψ (3 fM, 3 pM, and 3 nM). We found that in the absence of the δOR antagonist TIPPψ, we observed no specific binding with 3 pM [3H]DAMGO; however, in the presence of increasing concentrations of TIPPψ, an increase in detectable specific binding was observed (Supplemental Fig. 2B). These results are consistent with a role for “allosterism” in heteromer-mediated binding.
TABLE 3.
Enhancement of agonist binding to μOR by low doses of δOR ligands
CHO whole cells coexpressing μOR and δOR in a ratio of 1:4 were incubated with [3H]DAMGO (10 nM) in the absence or presence of different concentrations (0–1 nM) of different δOR ligands (agonists and antagonists/inverse agonists), and ligand binding was determined as described under Materials and Methods. Data represent mean ± S.E.M. (n = 3). The pEC50 and percentage stimulation values were derived from the curves in Supplemental Fig. 2. pKi values are from Toll et al., 1998.
| Ligand | pEC50 | Max. Enhancement (TIPPψ = 100%) | pKi | Ligand Activity |
|---|---|---|---|---|
| % | ||||
| TIPPψ | 11.04 ± 0.16 | 100 ± 6 | 9.0 | Inverse agonist |
| Deltorphin II | 11.99 ± 0.13 | 89 ± 1 | 8.8 | Full agonist |
| Naltriben | 10.92 ± 0.10 | 78 ± 1 | 10.0 | Antagonist |
| BNTX | 10.88 ± 0.19 | 26 ± 2 | 8.4 | Antagonist |
| SNC80 | N.D. | 14 | 8.9 | Full agonist |
| DPDPE | N.D. | 2 | 8.8 | Full agonist |
Association Kinetics of Radioligand Agonist Binding in the Presence of μOR or δOR Ligands.
Next, we characterized the heteromer-mediated binding by examining the time course of μOR ligand-mediated enhancement of radiolabeled agonist binding to δOR. Whole cells coexpressing μOR and δOR were incubated for various time periods with 6 nM [3H]deltorphin II in the absence or presence of 10 nM DAMGO or fentanyl. We find that [3H]deltorphin II exhibits association rates (t1/2) of ∼9.5 ± 0.5 min in the absence of μOR ligands, ∼14.3 ± 2 min (p < 0.05, one-way ANOVA) in the presence of DAMGO, and ∼18.3 ± 2.1 min (p < 0.01, one-way ANOVA) in the presence of fentanyl (Fig. 2). We also examined the time course of δOR ligand-mediated enhancement of radiolabeled μOR agonist binding to μOR. Cells were incubated for various time periods with 10 nM [3H]DAMGO in the absence or presence of 10 nM TIPPψ. We find that [3H]DAMGO exhibits a t1/2 of ∼9.9 ± 1.8 min in the absence and ∼18.9 ± 2.7 min (p < 0.01) in the presence of the δOR ligand TIPPψ (Fig. 3A). These results emphasize that a δOR ligand can modulate the association kinetics of a μOR ligand and vice versa via a GPCR-GPCR allosteric modulation phenomenon as long as these ligands actually target different receptors within the heterodimeric entity.
Fig. 2.

Association kinetics of δOR radioligand agonist binding. SK-N-SH whole cells endogenously expressing μOR and δOR (2:1 ratio) were incubated with 6 nM [3H]deltorphin II in the absence or presence of 10 nM DAMGO or fentanyl for different time periods (0–60 min) at 37°C and ligand binding determined as described under Materials and Methods. Data represent mean ± S.E.M. (n = 3).
Fig. 3.

Association kinetics of μOR radioligand agonist binding. A, SK-N-SH whole cells were incubated with 10 nM [3H]DAMGO in the absence or presence of 10 nM TIPPψ for different time periods (0–30 min) at 37°C and ligand binding determined as described under Materials and Methods. B, SK-N-SH whole cells were incubated with 10 nM [3H]DAMGO for 1 h at 37°C. Cells were then incubated for different time periods (0–30 min) with TIPPψ (10 nM) and ligand binding determined as described under Materials and Methods. Data represent mean ± S.E.M. (n = 3).
We then examined the time course of the effect of the δOR ligand TIPPψ on [3H]DAMGO binding (Fig. 3B). Whole cells coexpressing μOR-δOR were allowed to equilibrate with [3H]DAMGO for 1 h at 37°C; this was followed by the addition of TIPPψ (10 nM) at time = 0, and the specific binding of [3H]DAMGO to μOR was determined over a time up to 30 min. We found that the addition of TIPPψ led to a rapid increase in [3H]DAMGO binding to μOR-δOR with a t1/2 ∼ 2 ± 0.7 min (Fig. 3B). These results support the hypothesis that the presence of a δOR ligand actually allows the μOR ligand to access a new population of μOR of high affinity for μOR ligand. The time lapse for such a phenomenon to reach its plateau is fast (∼2 min), which suggests that interconversion events of existing conformations of μOR from low affinity to high affinity for μOR or that subcellular conformation/interaction switches of prebounded partner proteins such as G-proteins (see Supplemental Fig. 1) are involved, rather than a recruitment of physically nonaccessible μOR populations.
Dissociation Kinetics of Radioligand Agonist Binding to δOR in the Absence and Presence of a μOR Ligand and Vice Versa.
To further analyze the potential allosteric nature of the findings obtained in the equilibrium binding and association kinetic studies, we examined the dissociation kinetics of radiolabeled ligand binding to cognate receptors in the absence or presence of the ligand to the partner receptor. In initial experiments, we carried out dissociation kinetics at 37°C and observed that more than 70% of bound radiolabeled ligand dissociated within 3 min of incubation with excess of unlabeled ligand (I. Gomes and L. A. Devi, unpublished observations). To slow down the dissociation process, the dissociation kinetic experiments were carried out at 4°C after equilibration with the radiolabeled ligand at 37°C. The change in temperature did not induce significant changes in the initial level of bound radioactivity (Supplemental Fig. 3A). Under these conditions, a semi-log plot of the dissociation of [3H]deltorphin II with time after the addition of 1 μM unlabeled deltorphin II exhibited an apparent koff value of 0.17 ± 0.012 min−1 and a half-life of 4.2 ± 0.63 min (Fig. 4A). We find that the rate of [3H]deltorphin II dissociation (induced by 1 μM unlabeled deltorphin II) is significantly retarded in a concentration-dependent manner in the presence of either a μOR agonist such as fentanyl (apparent koff from 0.17 to 0.04 min−1)(Fig. 4A) or antagonists such as CTOP (Supplemental Fig. 3B). A reciprocal study carried out with [3H]DAMGO as the radioligand for μOR shows that the rate of [3H]DAMGO dissociation (induced by 1 μM unlabeled DAMGO) was also slowed down in the presence of TIPPψ (apparent koff from 0.13 to 0.04 min−1)(Fig. 4B). To see whether these effects were specific to μOR-δOR heteromers, we also examined the effect of (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo-[1,2,3-d,e]-1,4-benzoxazin-6-yl]-1-naphthalenyl-methanone (WIN55212-2) (a CB1 cannabinoid receptor agonist) and clonidine (α2A adrenergic receptor agonist) on the dissociation kinetics of [3H]DAMGO. These ligands were chosen because previous studies showed that CB1 and α2A receptors can form heteromers with μOR and that they exhibit antagonistic interactions in signaling studies (Jordan et al., 2003; Rios et al., 2006; Vilardaga et al., 2008). Consistent with this, we find that CB1 and α2A receptor agonists did not slow down (but increased) the rate of [3H]DAMGO dissociation (Supplemental Fig. 3C). Taken together, these results with retardation of the dissociation kinetics by selective ligands suggest a strong positive allosteric modulation of ligand binding within the heteromeric complex of μOR and δOR.
Fig. 4.

Dissociation kinetics of [3H]deltorphin II (A) or [3H]DAMGO (B) binding. A, SK-N-SH whole cells endogenously expressing μOR and δOR were incubated with 6 nM [3H]deltorphin II for 1 h at 37°C. The supernatant was removed, the plates were kept on ice, and cells were incubated with unlabeled 1 μM deltorphin II in the absence or presence of either 0.1, 1, or 10 nM of fentanyl for different time intervals (0–120 min) and ligand binding was determined as described under Materials and Methods. B, SK-N-SH whole cells were incubated with 10 nM [3H]DAMGO for 1 h at 37°C. The supernatant was removed, the plates were kept on ice, and cells were incubated with unlabeled 1 μM DAMGO in the absence or presence of either 0.1, 1, or 10 nM TIPPψ for different time intervals (0–120 min) and ligand binding determined as described under Materials and Methods. Data represent mean ± S.E.M. (n = 3). *, p < 0.05; **, p < 0.01, Dunnett's test.
Principles of Allosteric Modulation Applied to the μOR-δOR Heteromeric Complex.
Our observations that δOR ligands cause a retardation in the dissociation kinetics of radiolabeled μOR agonists and vice versa prompted us to examine a pharmacological model of allosteric modulation. The rationale was the strong parallel between our findings and similar observations with respect to receptor-ligand kinetics influenced by allosteric small molecules. A schematic of how one protomer in a receptor dimer, occupied by a ligand, can modulate the binding properties and thus the function of the other protomer is shown in Fig. 5, A and B. This is quite similar to the ability of some small molecule modulators to change the receptor conformation and modulate the binding of the orthosteric ligand (e.g., the hormone or the neurotransmitter for that receptor). In this view, retardation of apparent dissociation kinetics is equivalent to positive cooperativity between the two protomers, slowing down the global dissociation of the orthosteric ligand for one of the two protomer populations, a so-called allosteric enhancement.
Fig. 5.

A and B, scheme of proposed allosteric modulation of a heterodimeric receptor complex. A, standard model of allosteric modulation. Cooperativity (arrow) between the binding of an orthosteric ligand (OL) and a small molecule allosteric modulator (AM). According to this concept, allosteric modulation would take place on one protein building block only. B, dimeric allosteric modulation. One protomer occupied by its OL (indicated by the rectangle and arrow) may function as an AM and influence the binding of the OL to the interacting partner protomer. In this concept, allosteric modulation involves the two receptor partners. The allosteric modulation in this situation may be larger than in A, hence the bigger font size for AM. C to F, simulated binding curves for Table 1 and Fig. 1. The control curve in Fig. 5d was simulated in the absence of ligand-occupied receptor B using the same parameter values of L, K, and α. C and D, the experimental (C) and simulated (D) saturation binding curves for [3H]deltorphin II on SK-N-SH cells in the absence and presence of DAMGO or morphine. The parameter values used were L = 0.01, K = 6.3 × 108, M = 1013, α = 100, β = 1, δ = 1, [B] = 10−8, γ values are given in the figure. E and F, the experimental (E) and simulated (F) enhancement curves for [3H]deltorphin II binding to CHO whole cells coexpressing μ and δ opioid receptors in the presence of either fentanyl or naloxone. The parameter values used were L = 0.01, K = 6.3 × 108; M = 1013, α = 100; β = 1; δ = 1; [A] = 6 × 10−9; γ values are given in the figure.
In the present study, we applied the equations used for allosteric modulation by small drug-like molecules (Hall, 2000), assuming that the modulator in this case is a ligand-bound receptor partner rather than a small drug-like molecule. The model has two explicit assumptions: 1) one receptor protomer exists in an active as well as an inactive state and 2) to this both the orthosteric ligand and the other protomer bind. Because we used radiolabeled agonists, we assumed the concentration of the complex between the orthosteric agonist and the inactive receptor state to be negligible. We found that we are able to accurately simulate allosteric modulation because the experimental and simulated curves closely match (Fig. 5, C–F). The orthosteric affinities of the ligands (Tables 2 and 3) were sufficient to simulate both saturation (Fig. 5, C and D) and enhancement curves (Fig. 5, E and F), the latter with low picomolar EC50 values. This required only changes in the values for γ, the parameter defining the binding cooperativity between orthosteric ligand A and ligand-occupied receptor B. Taken together, our data indicate that the experimental findings are simulated in a straightforward manner within the framework of an allosteric receptor model (originally designed for the interaction of small molecules with a receptor monomer) in which the ligand-occupied second protomer behaves as the allosteric ligand. These results suggest that the principles of allosteric modulation are applicable to partners in heteromeric GPCRs.
Discussion
Previous studies have shown that GPCRs form heteromeric complexes leading to the modulation of the properties of individual protomers (Rios et al., 2001; Prinster et al., 2005; Milligan 2009). These studies have suggested heteromerization as a potential mechanism for allosteric modulation of receptor activity and function. This could be at the level of individual protomers in the heteromeric complex. To explore this, we used an established model system of a receptor heteromer, the complex between μOR and δOR. In equilibrium binding experiments, using a radiolabeled probe for one protomer and a low ligand concentration targeted to the partner protomer in the μOR-δOR heteromer, we found an enhancement of radioligand binding, the consequence of an increase in the ratio R*/R rather than an increase in affinity (Kd values). This excludes a competitive interaction at the orthosteric binding site of either of the two receptor types, which would be suggested by a decrease in affinity without changes in receptor density, and displacement rather than enhancement of radioligand binding. It is noteworthy that we observed no relationship between the affinities of μOR or δOR ligands for their orthosteric site on μOR or δOR (pKi values) and their potency in enhancing ligand binding to δOR or μOR (pEC50 values), respectively (Tables 2 and 3). This suggests that although the binding of a ligand to the modulating-protomer partner is the initial trigger for the protomer-protomer interactions, it is not necessary that the ligand remain in its binding site to sustain the change. Thus, cross-protomer allosteric modulation could represent a “pseudo-irreversible state” commonly seen in enzymatic substrate-product reactions rather than the “fast equilibrium” characteristic of the steady-state kinetics of ligand-receptor interactions.
These observations are probe-specific, because the increase in receptor density was observed with some radiolabeled ligands, such as DAMGO, morphine as μOR probes, or deltorphin II as a δOR probe (Gomes et al., 2000, 2004; Table 1), and not with others, such as radiolabeled diprenorphine, naloxone, or [d-Pen2,d-Pen5]-enkephalin (data not shown). Thus, this phenomenon could be not only probe-specific but also agonist-specific. The observations are robust when μOR/δOR are expressed at a ratio of 1:4 and are practically absent at a ratio of 1:40 (Supplemental Fig. 1). This is consistent with the idea that allosteric interactions between μOR and δOR protomers would be influenced by the relative levels of expression of μOR and δOR in a cell. In this context, we recently found that μOR-δOR heteromer abundance can be up-regulated by long-term morphine administration, pharmacological chaperones, and endogenous chaperones, such as receptor transport protein 4. This leads to changes in ligand binding and signaling properties of the heteromer (Rozenfeld and Devi, 2007, 2010a; Décaillot et al., 2008; Gupta et al., 2010).
Allosteric interactions could occur at the level of μOR and δOR protomers, where one ligand-occupied protomer functions as an allosteric enhancer of the other protomer. We find that the allosteric receptor model developed for small molecule modulators (Hall, 2000) could simulate the saturation and enhancement curves observed with μOR-δOR heteromers by using the ligands' orthosteric affinities and changing the values for the parameter defining the binding cooperativity between orthosteric ligand A and ligand-occupied receptor B. However, other factors could contribute to the observed enhancement in binding seen in cells coexpressing μOR-δOR. That positive binding cooperativity is observed at 4°C (albeit to a lesser extent than at 37°C) and in membrane preparations (Supplemental Fig. 1, A and B) suggests that downstream events after receptor activation are not required for this phenomenon to occur. However, this does not rule out the participation of proteins that are preassociated with the heteromer, such as G-proteins. If the positive binding cooperativity with μOR-δOR heteromers involves heteromer-associated Gαi proteins, then this would be lost after pretreatment with pertussis toxin, which uncouples the G-protein from the receptor (Chabre et al., 2009). We observe positive cooperativity in μOR-δOR heteromer binding after pertussis toxin treatment, although to a much smaller extent than in the absence of pertussis toxin (Supplemental Fig. 1, C and D), suggesting the involvement of Gαi proteins in this phenomenon and that the binding cooperativity observed in the presence of pertussis toxin could be due largely to direct protomer interactions.
In a recent study by Han et al. (2009) a functional complementation assay was used to show that the minimal signaling unit for D2 dopamine receptors comprises two GPCRs and one heterotrimeric G protein. This signaling unit was shown to be activated by agonist binding to a single protomer, and binding of the second protomer by an inverse agonist enhanced signaling, whereas binding by an agonist blunted signaling. In prior studies, we found that binding of inverse agonists/antagonists to one receptor protomer enhances binding and signaling to the other protomer (Gomes et al., 2000, 2004). Additionally, we found that binding of an agonist to one protomer promotes the binding and signaling of the agonist to the second protomer (Gomes et al., 2000, 2004); this is in contrast to findings with D2 dopamine receptors, suggesting differences between receptor systems.
An intriguing finding of the present studies is that the pEC50 values are high, often corresponding to subpicomolar concentrations, compared with the “orthosteric” affinities (pKi values). This is particularly evident for the enhancing activity on the δOR with a difference of more than 5 log units for morphine (pEC50 − pKi), and an average 3- to 4-log unit difference for all other compounds. In this case, morphine, especially at low concentrations, does not so much activate the μOR as indirectly increase the activity of the δOR by enhancing the binding of δOR agonists. This raises the question of whether these high pEC50 values are physiologically relevant. In this context, studies have shown that ultralow doses (0.01–0.06 ng) of the δOR antagonist naltrindole augment the analgesic effects of spinally administered morphine to rats and inhibit the development of tolerance to morphine (Abul-Husn et al., 2007; McNaull et al., 2007). These results suggest that coadministration of morphine with ultralow doses of a δOR receptor antagonist could be clinically used to increase the analgesic efficacy of morphine by administering lower doses of morphine to obtain the same degree of analgesia but without the side effects associated with long-term morphine administration.
Allosteric enhancement of agonist binding and function by small molecules has been observed for several receptor systems and been comprehensibly described in many reviews (Christopoulos and Kenakin, 2002; Soudijn et al., 2004; May et al., 2007). Remarkably, concentration-effect relationships for small-molecule allosteric enhancers in these studies are reminiscent of the enhancement curves we describe for the μOR-δOR heteromer. This prompted us to examine the dissociation kinetics of the heteromer, because a small molecule allosteric enhancer is best identified by its retardation of the dissociation kinetics of the (orthosteric) ligand-receptor complex. We found a decrease in the apparent dissociation rate constants for the δOR ligand (as well as for the μOR ligand) when cotreated with the ligand for the partner receptor. This suggests a positive cooperativity between μOR and δOR protomers in the heteromeric complex. This is specific for μOR-δOR heteromers, because it is not observed with μOR-CB1 cannabinoid or with μOR-α2A adrenergic heteromers (Supplemental Fig. 3C). It is noteworthy that the semi-log plots of the dissociation curves for [3H]deltorphin II and [3H]DAMGO are curvilinear, suggestive of multiple sites/conformations with differing koff, which could correspond to receptor homomer and heteromer populations.
In conclusion, the allosteric modulation of GPCRs by dimerization/heteromerization serves as a novel concept for the design of synergistic ligand cocktails that target heteromeric receptor entities and differentially influence their activity. The identification of μOR-δOR heteromer selective small-molecule allosteric enhancers would not only facilitate studies to probe the nature of the interaction in detail but would also help in the development of drugs targeting μOR-δOR heteromers. The “double”' pharmacology (pEC50 and pKi values) of the opioid ligands described in this study poses both opportunities and challenges. An opportunity would be to design a partial agonist or positive allosteric modulator of the δOR that could maximally potentiate μOR agonist activity, allowing selective and potent analgesia in the spinal cord. Such a compound would not “hit” μOR homomers, thereby yielding much improved and needed tissue selectivity. The challenges in targeting GPCR heteromers are in defining the pairs of heteromers that “matter.” It would not suffice to determine the selectivity profile of a compound for a range of receptor homomers, which is the current practice in high-throughput screening. In addition, one would need a careful assessment of a receptor's natural heteromer partners and a subsequent study of their mutual allosteric modulation. Toward this end, generation of heteromer-selective antibodies such as those recently described for μOR-δOR heteromers (Gupta et al., 2010) are critical, and such antibodies could serve as much-needed tools that would facilitate examination of the distribution and regulation of heteromers in normal function and/or in pathologic conditions.
Supplementary Material
Acknowledgments
We thank R. Rozenfeld and I. Bushlin for critical reading of the manuscript and members of the Devi lab for helpful discussions.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
This work was supported by National Institutes of Health National Institute on Drug Abuse [Grants DA08863, DA019251] (to L.A.D.); and by the Dutch Top Institute Pharma project [Grant D1-105] (to A.P.IJ.)
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.070847.
- GPCR
- G protein-coupled receptor
- OR
- opioid receptor
- TIPPψ
- Tyr-Ticψ(CH2NH)-Phe-Phe
- DAMGO
- [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin
- deltorphin II
- Tyr-d-Ala-Phe-Glu-Val-Val-Gly
- CTOP
- d-Phe-Cys-Tyr-d-Trp-Orn-Thr-Pen-Thr-NH2
- BNTX
- 7-benzylidenenaltrexone maleate
- CTAP
- d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2
- CHO
- Chinese hamster ovary
- ICI 174,864
- N,N-diallyl-Tyr-Aib-Aib-Phe-Leu
- SNC80
- (+)-4-[(αR)-α-((2S,5R)-4-allyl-2,5-dimethyl-1piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide
- ANOVA
- analysis of variance
- WIN55212-2
- (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl) pyrrolo-[1,2,3-d,e]-1,4-benzoxazin-6-yl]-1-naphthalenyl-methanone.
Authorship Contributions
Participated in research design: IJzerman, Maillet, and Devi.
Conducted experiments: Gomes and Ye.
Performed data analysis: Gomes, IJzerman, and Maillet.
Wrote or contributed to the writing of the manuscript: Gomes, IJzerman, Maillet, and Devi.
Other: IJzerman and Devi acquired funding for the research.
References
- Abul-Husn NS, Sutak M, Milne B, Jhamandas K. (2007) Augmentation of spinal morphine analgesia and inhibition of tolerance by low doses of mu- and delta-opioid receptor antagonists. Br J Pharmacol 151:877–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabre M, Deterre P, Antonny B. (2009) The apparent cooperativity of some GPCRs does not necessarily imply dimerization. Trends Pharmacol Sci 30:182–187 [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. (2002) G protein-coupled receptor allosterism and complexing. Pharmacol Rev 54:323–374 [DOI] [PubMed] [Google Scholar]
- Décaillot FM, Rozenfeld R, Gupta A, Devi LA. (2008) Cell surface targeting of mu-delta opioid receptor heterodimers by RTP4. Proc Natl Acad Sci USA 105:16045–16050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvoisin RM, Pfankuch T, Wilson JM, Grabell J, Chhajlani V, Brown DG, Johnson E, Raber J. (2010) Acute pharmacological modulation of mGluR8 reduces measures of anxiety. Behav Brain Res 212:168–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Jordan BA, Gupta A, Trapaidze N, Nagy V, Devi LA. (2000) Heterodimerization of μ and δ opioid receptors: a role in opiate synergy. J Neurosci 20:RC110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Filipovska J, Devi LA. (2003) Opioid receptor oligomerization. Detection and functional characterization of interacting receptors. Methods Mol Med 84:157–183 [DOI] [PubMed] [Google Scholar]
- Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA. (2004) A role for heterodimerization of μ and δ opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci USA 101:5135–5139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Mulder J, Gomes I, Rozenfeld R, Bushlin I, Ong E, Lim M, Maillet E, Junek M, Cahill CM, et al. (2010) Increased abundance of opioid receptor heteromers after chronic morphine administration. Sci Signal 3:ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DA. (2000) Modeling the functional effects of allosteric modulators at pharmacological receptors: an extension of the two-state model of receptor activation. Mol Pharmacol 58:1412–1423 [DOI] [PubMed] [Google Scholar]
- Han Y, Moreira IS, Urizar E, Weinstein H, Javitch JA. (2009) Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat Chem Biol 5:688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison C, McNulty S, Smart D, Rowbotham DJ, Grandy DK, Devi LA, Lambert DG. (1999) The effects of endomorphin-1 and endomorphin-2 in CHO cells expressing recombinant mu-opioid receptors and SH-SY5Y cells. Br J Pharmacol 128:472–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan BA, Gomes I, Rios C, Filipovska J, Devi LA. (2003) Functional interactions between μ opioid and α2A-adrenergic receptors. Mol Pharmacol 64:1317–1324 [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. (2010) Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev 62:265–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Seager MA, Seager M, Wittmann M, Jacobson M, Bickel D, Burno M, Jones K, Graufelds VK, Xu G, et al. (2009) Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc Natl Acad Sci USA 106:15950–15955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A. (2007) Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol 47:1–51 [DOI] [PubMed] [Google Scholar]
- McNaull B, Trang T, Sutak M, Jhamandas K. (2007) Inhibition of tolerance to spinal morphine antinociception by low doses of opioid receptor antagonists. Eur. J Pharmacol 560:132–141 [DOI] [PubMed] [Google Scholar]
- Milligan G. (2009) G protein-coupled receptor hetero-dimerization: contribution to pharmacology and function. Br J Pharmacol 158:5–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson G, Chandrashekar J, Hoon MA, Feng L, Zhao G, Ryba NJ, Zuker CS. (2002) An amino-acid taste receptor. Nature 416:199–202 [DOI] [PubMed] [Google Scholar]
- Neuhaus EM, Gisselmann G, Zhang W, Dooley R, Störtkuhl K, Hatt H. (2005) Odorant receptor heterodimerization in the olfactory system of Drosophila melanogaster. Nat Neurosci 8:15–17 [DOI] [PubMed] [Google Scholar]
- Onali P, Olianas MC. (2004) G protein activation and cyclic AMP modulation by naloxone benzoylhydrazone in distinct layers of rat olfactory bulb. Br J Pharmacol 143:638–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinster SC, Hague C, Hall RA. (2005) Heterodimerization of G protein-coupled receptors: specificity and functional significance. Pharmacol Rev 57:289–298 [DOI] [PubMed] [Google Scholar]
- Raynor K, Kong H, Chen Y, Yasuda K, Yu L, Bell GI, Reisine T. (1994) Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol Pharmacol 45:330–334 [PubMed] [Google Scholar]
- Rios C, Gomes I, Devi LA. (2006) μ Opioid and CB1 cannabinoid receptor interactions: reciprocal inhibition of receptor signaling and neuritogenesis. Br J Pharmacol 148:387–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios CD, Jordan BA, Gomes I, Devi LA. (2001) G-protein-coupled receptor dimerization: modulation of receptor function. Pharmacol Ther 92:71–87 [DOI] [PubMed] [Google Scholar]
- Rozenfeld R, Décaillot F, IJzerman AP, Devi LA. (2006) Heterodimers of G protein-coupled receptors as novel and distinct drug targets. Drug Discov Today Ther Strateg 3:437–443 [Google Scholar]
- Rozenfeld R, Devi LA. (2007) Receptor heterodimerization leads to a switch in signaling: beta-arrestin2-mediated ERK activation by mu-delta opioid receptor heterodimers. FASEB J 21:2455–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenfeld R, Devi LA. (2010a) Exploring a role for heteromerization in GPCR signalling specificity. Biochem J 433:11–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenfeld R, Devi LA. (2010b) Receptor heteromerization and drug discovery. Trends Pharmacol Sci 31:124–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudijn W, Van Wijngaarden I, IJzerman AP. (2004) Allosteric modulation of G protein-coupled receptors: perspectives and recent developments. Drug Discov Today 9:752–758 [DOI] [PubMed] [Google Scholar]
- Toll L, Berzetei-Gurske IP, Polgar WE, Brandt SR, Adapa ID, Rodriguez L, Schwartz RW, Haggart D, O'Brien A, White A, et al. (1998) Standard binding and functional assays related to medications development division testing for potential cocaine and opiate narcotic treatment medications. NIDA Res Monogr 178:440–466 [PubMed] [Google Scholar]
- Vilardaga JP, Nikolaev VO, Lorenz K, Ferrandon S, Zhuang Z, Lohse MJ. (2008) Conformational cross-talk between alpha2A-adrenergic and mu-opioid receptors controls cell signaling. Nat Chem Biol 4:126–131 [DOI] [PubMed] [Google Scholar]
- White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, Barnes AA, Emson P, Foord SM, Marshall FH. (1998) Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature 396:679–682 [DOI] [PubMed] [Google Scholar]
- Zernig G, Issaevitch T, Broadbear JH, Burke TF, Lewis JW, Brine GA, Woods JH. (1995) Receptor reserve and affinity of mu opioid agonists in mouse antinociception: correlation with receptor binding. Life Sci 57:2113–2125 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.