

Summary
The adapter protein MecA targets the transcription factor ComK for degradation by the ClpC/ClpP proteolytic complex, thereby negatively regulating competence in Bacillus subtilis. Here we show that MecA also decreases the frequency of transitions to the sporulation pathway as well as the expression of eps, which encodes synthesis of the biofilm matrix exopolysaccharide. We present genetic and biophysical evidence that MecA down-regulates eps expression and spore formation by directly interacting with Spo0A. MecA does not target Spo0A for degradation, and apparently does not prevent the phosphorylation of Spo0A. We propose that it inhibits the transcriptional activity of Spo0A~P by direct binding. Thus, in its interaction with Spo0A, MecA differs from its role in the regulation of competence where it targets ComK for degradation. MecA acts as a general buffering protein for development, acting by two distinct mechanisms to regulate inappropriate transitions to energy-intensive pathways.
Keywords: MecA, sporulation, biofilms, bimodal expression, Spo0A
Introduction
Bacillus subtilis expresses a variety of global life-style responses to stress, including competence for transformation, sporulation and biofilm formation. Because the developmental pathways are energy-intensive and both competence and sporulation cause the cessation of growth, high frequencies of transition would be disadvantageous when nutrients are plentiful. Consequently, mechanisms to minimize such inappropriate transitions are expected to increase fitness. However, it is plausible that even in the absence of stress these transition rates would be nonzero, providing a clonal population with a sub-population of cells insured in advance against sudden adversity (Dubnau & Losick, 2006, Veening et al., 2008, Losick & Desplan, 2008). Thus, rare competent or sporulating cells are present even in rapidly growing cultures. These considerations imply that selective pressure has approximated an optimal rate of transition for each adaptive pathway under conditions of plenty. In contrast, when nutrients are scarce and B. subtilis exits from exponential growth, the transition rates increase and cells choose from among these developmental adaptations, each of which has the potential to guarantee survival of at least a portion of the population. These transition rate increases occur in an orderly and coordinated manner using pathway-specific mechanisms. In other words, the transition rates are not fixed, but are adjusted by regulatory mechanisms to reflect changing circumstances.
Competence refers to a gene expression state in which cells can take up and integrate environmental DNA. The expression of competence genes provides a well-studied example of bimodal gene expression in bacteria (Maamar et al., 2007, Suel et al., 2007, Leisner et al., 2007). These genes are expressed in a minor fraction of the cells in a clonal population and the transition rate adjustments described above are well illustrated by the formation of competent cells. The expression of competence genes requires the transcription factor ComK, which is also an auto-activator, acting positively and directly at its own promoter (van Sinderen et al., 1995). ComK is expressed bimodally because of two additional factors acting in concert with this positive auto-regulation; stochastic variability (noise) in the basal amount of ComK per cell and the existence of a nonlinear response to ComK that permits only those cells above a threshold to auto-activate PcomK. During exponential growth, MecA lowers the probability that such variation will result in competence by selectively targeting ComK molecules for degradation by a complex of the AAA+ protein ClpC and the serine protease ClpP (Turgay et al., 1998). As a result, the auto-regulatory loop is rarely activated, transition probability is low and very few competent cells are present in growing cultures. As cells exit from exponential growth, the anti-adaptor ComS is synthesized in response to a quorum-sensing mechanism that measures population density. ComS competitively releases ComK from binding to MecA (Prepiak & Dubnau, 2007), reducing its rate of degradation and permitting stochastic fluctuations in the basal level transcription of comK to be manifested as a marked increase in the probability of transitions to competence. Thus, MecA acts to dampen the likelihood of transitions to competence particularly during exponential growth. In several respects, sporulation and early biofilm gene expression follow the pattern described for competence. Both exhibit low but non-zero probabilities of transition in growing cells and increased transition rates as cultures enter stationary phase.
The progression of B. subtilis to a biofilm-producing state is largely governed by the transcriptional regulator SinR (Branda et al., 2006) (Fig. 1A). SinR directly represses the eps operon, which encodes synthesis of biofilm-associated extracellular polysaccharide, as well as the yqxM-sipW-tasA operon, which encodes a protein component of the extracellular matrix (Branda et al., 2004, Branda et al., 2006). The activity of SinR is regulated not by a change in its amount, but rather by the selective production of SinI, which binds directly to SinR, preventing it from inhibiting the transcription of its target promoters (Bai et al., 1993). When SinI overcomes the repression due to SinR, matrix can be produced and a biofilm community can develop (Kearns et al., 2005).
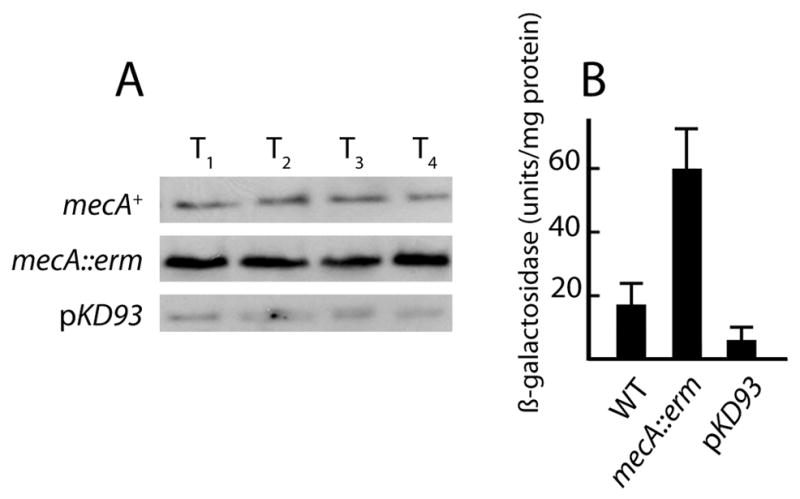
Fig. 1.

(A) Scheme showing the role of Spo0A~P as a master upstream regulator of both eps expression and transcription of sporulation genes. Spo0A~P activates a promoter in front of sinI (Shafikhani et al., 2002). The eps operon is directly repressed by SinR (Kearns et al., 2005). Generally, repression due to SinR is lifted when it is sequestered by the protein SinI (Bai et al., 1993). (B) eps-lacZ expression in mecA and pKD93 strains. Wild-type (BD4498, empty squares), mecA::erm (BD4538, filled squares) and pKD93 (BD4644, filled circles) strains were grown in LB and β-galactosidase activities were determined at the indicated times. The triangles show results for strains growing in MsGG medium; wild-type (BD4498, closed triangles) and pKD93 (BD4644, open triangles). (C) spoIIG-luc expression in mecA and pKD93 strains. Wild-type (PP533, black line), mecA (PP551, gray line) and pKD93 (PP565, dotted line) strains were grown in DSM in a plate reader and growth (OD at 600 nm) and light output was measured every 1.5 minutes. For both panels, “Time” is given as hours before and after the transition to stationary phase.
Because Spo0A~P is required not only for biofilm formation, but for competence (Hahn et al., 1995) and for the expression of sporulation genes (Molle et al., 2003) (Fig. 1A), this response regulator protein is the ultimate controller of all three developmental pathways. sinI is transcribed from two SigA-dependent promoters, one of which is activated by the binding of phosphorylated Spo0A (Gaur et al., 1988, Shafikhani et al., 2002). The master regulator Spo0A~P therefore lies upstream of sinI in biofilm development and activates eps by indirectly down-regulating SinR. AbrB, another regulator of biofilm formation (Hamon et al., 2004), represses sinI by binding directly to its promoter (Shafikhani et al., 2002) and may also regulate eps by direct binding (Murray et al., 2009), although the major regulation of eps transcription is certainly mediated by the SinI-SinR pathway. abrB transcription is down-regulated by Spo0A~P (Strauch et al., 1990) and AbrB activity is inhibited by the AbbA protein, which requires Spo0A~P for its production (Banse et al., 2008).
Spo0A activation is dependent on phosphorylation largely through a signal transduction cascade, known as the phosphorelay (Burbulys et al., 1991). Genes regulated by Spo0A exhibit a hierarchical response to the concentration of Spo0A~P; sinI and abrB have high affinity promoters whereas sporulation genes require higher levels of Spo0A~P for their activation (Fujita et al., 2005). Although like competence sinI is expressed bimodally, the mechanism by which only certain cells are selected for sinI expression is not clear but may be related to kinetic heterogeneity in the production of Spo0A~P because a strain that is spo0A-deficient expresses little sinI whereas the presence of a constitutively active Spo0A results in enhanced expression of sinI in all cells, suggesting that the availability of Spo0A~P is normally limiting for transitions to eps expression and biofilm formation (Chai et al., 2008).
We report here that inactivation of mecA causes an increased expression of eps and of sporulation genes, while over-expression of mecA has the reverse effect, decreasing eps and spore gene expression. Also, when mecA is inactivated, the fraction of cells expressing these genes increases, even during growth. A mutant form of Spo0A that does not require phosphorylation bypasses the inhibition of eps and spore gene transcription by MecA. We have found that MecA binds directly to Spo0A but does not target this protein for degradation as it does ComK. We propose that by preventing Spo0A from acting as a transcription factor, either by preventing its phosphorylation or by acting on Spo0A~P, MecA dampens transitions to biofilm development and spore formation, thus preventing inappropriate expression of these pathways and acting as a general buffer for developmental transitions in B. subtilis.
Results
MecA is a negative regulator of eps expression and of sporulation
This study initiated from two separate observations on mecA mutant strains growing on solid media. These observations were made using a laboratory strain IS75. The first such observation was that mecA colonies presented a rough-textured appearance. Because over-production of the biofilm-associated exopolysaccharide encoded by the eps operon yields similar appearing colonies, we constructed a mecA::erm eps::tet eps-lacZ strain (BD3980). This strain exhibited smoother colonies, like those of the wild-type, suggesting that the rough colony phenotype was indeed due to over-expression of eps. To further characterize the role of MecA in eps expression, we examined an eps-lacZ transcriptional fusion in mecA+ (BD4498) and mecA (BD4538) backgrounds and in a strain carrying a multi-copy plasmid (pKD93) that over-expresses mecA (BD4644) from a constitutive promoter on the vector (Kong et al., 1993). (Over-expression of MecA in this strain in documented below (Fig. 6)). This experiment was done in a complex medium (LB) because mecA strains do not grow in the medium commonly used to study biofilm formation (not shown). The mecA strain showed increased expression of eps-lacZ throughout growth. In the experiment shown in Fig. 1B eps-lacZ expression in the mecA strain is elevated about 4-fold throughout growth. In three additional experiments in which samples were taken at T1, the mecA strain showed an average increase over the wild-type strain of 5.7-fold. Expression of eps-lacZ was nearly eliminated in the pKD93 background (Fig. 1B). In MsGG medium, which is customarily used to study biofilm formation, the over-expression of MecA markedly decreased the increase in eps-lacZ expression that took place at T0. We conclude that MecA is a negative regulator of eps, confirming the inference from colony morphology.
Fig. 6.

Detection of Spo0A protein by immunoblotting in wild-type (BD2149), mecA::erm (BD2148) and pKD93 (PP493) strains. The cultures were grown in DSM and samples were taken at the times indicated on the growth curve (panel C) for immunoblotting with anti-Spo0A and anti-MecA antisera (panels A and B respectively. The three strains grew identically and a curve is shown only for the wild-type strain. Equal amounts of protein were loaded in each lane. (D) BD2148 and PP493 were grown in DSM to just after T1 and puromycin (200 mg/ml) was added to inhibit protein synthesis. Samples were taken for blotting with anti-Spo0A antiserum (both strains) and with anti-MecA antiserum (PP493 only) at the indicated times. The arrows show the position of the Spo0A (top two panels) and Mec (bottom panel) signals.
The second observation was that the over-expression of mecA from a multicopy plasmid resulted in translucent colonies, typical of strains with an early block in spore formation. This phenotype was confirmed by spore counts in sporulation medium (DSM). In a typical experiment, after 24 and 48 hours growth, a strain that carried pKD93 (PP493) exhibited sporulation frequencies of 1.5 × 10−5 and 4.9 × 10−5 respectively, while at these times, an isogenic strain which did not over-express mecA (BD630) achieved sporulation frequencies of 0.32 and 0.66. The appearance of the PP493 colonies suggested that the MecA-induced block in spore formation occurred at an early stage. To test this inference, the effect of MecA on early spore gene expression was determined by monitoring light output from a fusion of the spoIIG promoter to firefly luciferase. We have shown elsewhere that this luc reporter may be used in B. subtilis as a real-time reflection of the rate of transcription from a given promoter (N. Mirouze, P. Prepiak and D. Dubnau, submitted). For this experiment, luminometry and OD600 measurements were made in a temperature-controlled plate reader on cultures growing in the presence of luciferin. Fig. 1C shows that mecA over-expression prevented spoIIG transcription. Conversely, the inactivation of mecA resulted in a reproducible increase in spoIIG expression, manifested only after the culture had entered stationary phase. Expression of a spoIIE-lacZ fusion was also inhibited in the presence of pKD93, as shown below in Fig. 5. These results demonstrate that MecA limits the expression of early Spo0A-dependent sporulation genes.
Fig. 5.

Bypass of pKD93 inhibition by sad67. Strains carrying pKD93 and Pspac-sad67 (BD4628) (A) or pKD93 and Pspac-spo0A (BD4692) (B and C) were grown in LB until 1 hour before T0. The cultures were split and IPTG (1 mM) was added to half of each culture. Growth was continued for two hours and samples were collected for Western blotting with anti-SinI antiserum. The two lanes in panel C are identical to those in B except that they have been enhanced to more clearly show the absence of induction. The vertical line is an indication that panel A is from a different gel than panels B and C. For both Pspac strains, induction of Spo0A and the Sad67 protein was verified by stripping the gels and probing with Spo0A antiserum (not shown). Panel D shows bypass of spoIIE-lacZ expression in the presence of pKD93 in cultures growing in DSM. The solid and empty symbols show results from cultures incubated with and without IPTG, respectively. The squares show results for a Pspac-sad67 spoIIE-lacZ strain (PP485). Only one line is shown for the empty and filled squares. The circles show spoIIE-lacZ expression for a spoIIE-lacZ strain carrying pKD93 (PP487). The ellipses show expression in a Pspac-sad67 spoIIE-lacZ pKD93 strain. Time zero is defined as the time when IPTG (1 mM) was added (at T0).
MecA biases the fraction of cells entering the biofilm and sporulation pathways
Prior studies had shown that eps is expressed in only a few percent of the cells in a given population (Chai et al., 2008). To determine whether mecA affected the proportion of cells expressing eps, we examined mutant strains by fluorescence microscopy, using a fusion of the eps promoter to cyan fluorescent protein (eps-cfp). These strains, eps-cfp (BDBD4621), eps-cfp mecA::erm (BD4642) and eps-cfp pKD93 (BD4643), were grown in LB to T1 and samples were prepared from each for fluorescence microscopy. In the wild-type background (Fig. 2A), 3.9% of the cells expressed eps-cfp, confirming the published results (Chai et al., 2008). This was increased in the mecA strain (Fig. 2B) to 35.6% and in the pKD93 strain (Fig. 2C) we were unable to find any cells expressing eps, among approximately 5,000 cells examined. These results show that MecA acts to limit the number of eps-expressing cells, but is not the only factor responsible for this limitation, because even in its absence expression did not occur in all the cells. The 9-fold increase in the fraction of expressing cells in the mecA strain is approximately consistent with the 5.7-fold increase in eps-lacZ noted above.
Fig. 2.

Heterogeneous expression of eps-cfp and spoIIE-gfp in mecA and pKD93 strains. Strains were grown in LB and sampled at T1. Panels A, B and C present typical fields from the wild-type (BD4621), mecA::erm (BD4642) and pKD93 (BD4643) strains respectively. Cell bodies were stained with propidium iodide and pseudocolored red. CFP fluorescence was pseudocolored cyan and overlayed on the propidium iodide channel. For panels D and E, a strain expressing spoIIE-gfp was grown in DSM to T-1. Images showing GFP fluorescence were overlayed on DIC images. Panels D and E show images from strains with the wild-type (PP480) and mecA::erm (PP479) backgrounds respectively.
A similar experiment was carried out with a fusion of the spoIIE promoter to GFP using cultures growing in DSM until T-1, well before the normal onset of sporulation. In the wild-type strain, occasional fluorescent cells were observed. For example, in the field shown in Fig. 2D, which was selected to include a fluorescent cell, one such cell is visible among about 274 cells. The frequency of such cells is below 0.1%. In the mecA null strain field (Fig. 2E), 9 fluorescent cells are evident among 389 cells. In a strain over-expressing mecA from a multicopy plasmid, no fluorescent cells were detected among several thousand examined (not shown). Corresponding results were obtained by measuring the frequency of spores among cells during logarithmic growth in DSM. In the wild-type strain, the measured frequency of heat resistant spores was 1.4 × 10−7. In the mecA strain the frequency was 5 × 10−3, while no spores were detected in the pKD93 strain. As with competence and eps expression, it appears that MecA limits rare transitions to sporulation in growing cultures.
To explore the reasons for these two seemingly unrelated MecA-associated phenotypes, affecting eps and early spore gene expression, we carried out a series of epistasis experiments employing mutations in regulatory genes known to affect biofilm and spore development.
MecA regulates eps expression largely through the SinI/SinR pathway
To investigate where mecA acts in the eps regulatory pathway, we sought to determine if the effect of mecA over-expression on eps-lacZ expression could be bypassed by inactivation of sinR, a direct repressor of eps (Kearns et al., 2005). As expected, the sinR::cat strain (BD4544) showed elevated eps expression, approximately 28-fold higher than the amount seen in the wild-type strain (BD4498) (Fig. 3A). Although the pKD93 strain (BD4644) was greatly reduced in the expression of eps, the pKD93 sinR::cat strain (BD4549) expressed about 40% as much eps-lacZ as the sinR::cat strain, demonstrating substantial bypass of the mecA over-expression effect by inactivation of sinR and suggesting that MecA exerts much of its effect on eps expression upstream of sinR.
Fig. 3.

Effects of sinR, abrB and clpC inactivation on eps-lacZ expression in wild-type (BD4498) and pKD93 (BD4643) backgrounds. Strains carrying the indicated mutations, were grown in LB and samples taken for β-galactosidase determination at T1. (A) The following strains, all carrying eps-lacZ, were used for this experiment: BD4498 (wild-type), BD4644 (pKD93), BD4544 (sinR::cat), BD4549 (sinR::cat pKD93), BD4623 (abrB::cat), BD4615 (sinR::kan abrB::cat), BD5113 (abrB::cat pKD93), BD4624 (abrB::cat sinR::kan pKD93). (B) The following strains carrying eps-lacZ were used for this experiment: wild-type (BD4498), sinR::cat (BD4544), abrB::cat (BD4623), spo0A::kan (BD4928), spo0A::kan sinR::cat (BD4929), spo0A::kan abrB::cat eps-lacZ (BD4930), spo0A::kan abrB::cat sinR::kan (BD4931), spo0A::kan mecA::erm (BD5589), mecA::erm (BD4538). (C) The following strains were used for this experiment: wild-type (BD4498), pKD93 (BD4644), clpC::tet (BD4580), clpC::tet pKD93 (BD5114). The presence of the pKD93 plasmid indicates that mecA is over-expressed. The numbers in parentheses refer to the number of independent measurements for each strain and the whiskers show standard deviations.
We next determined whether MecA affected the expression of sinI, which encodes an antagonist of SinR (Bai et al., 1993). Western blots, using antiserum raised against SinI, were performed on extracts of the wild-type, mecA and pKD93 strains. Fig. 4A shows that there is less SinI in the pKD93 strain, and more in the mecA strain than in the wild-type. These data led us to suspect that MecA decreases eps expression at least in part by depressing the level of SinI. Fig. 4B shows that similar effects were observed with a sinI-lacZ translational fusion reporter strain, showing that MecA down-regulates the expression of sinI-lacZ.
Fig. 4.
The amount of SinI protein and the expression of sinI-lacZ in mecA and pKD93 strains. (A) Immunoblot using anti-SinI antiserum on extracts of wild-type (BD4498), mecA::erm (BD4538) and pKD93 (BD4644) strains grown in LB to the indicated times. Equal amounts of total protein were loaded on each lane. (B) β-galactosidase activities produced from sinI-lacZ in wild-type (BD4555), mecA::erm (BD4568) and pKD93 (BD4557) strains grown in LB to T1. The data for panels A and B were derived from three independent measurements. The presence of the pKD93 plasmid indicates that mecA is over-expressed. The whiskers show standard deviations.
AbrB, like SinR, is a negative regulator of eps (Hamon et al., 2004, Chu et al., 2008, Winkelman et al., 2009) and has recently been reported to bind directly to the promoter of the eps operon (Murray et al., 2009). AbrB also appears to repress sinI (Shafikhani & Leighton, 2004). As expected from the published results, the abrB mutation raised expression of eps-lacZ in the mecA+ background about 3.7-fold, a far less dramatic effect than seen with the sinR knockout (Fig. 3A). To test whether the MecA effect on eps could be bypassed by inactivation of abrB as it was by the loss of sinR function, an eps-lacZ abrB pKD93 strain (BD5113) was tested (Fig. 3A). Unlike inactivation of sinR, the abrB mutation was not able to raise the low level of eps-lacZ expression due to MecA overproduction. However, the inactivation of both abrB and sinR (BD4624) bypassed the pKD93 phenotype to a level nearly twice that achieved by inactivation of sinR alone in the pKD93 background. Interestingly, the level of expression in the pKD93 sinR abrB strain was about half the level in the sinR abrB strain, suggesting that MecA can limit eps transcription by another pathway, not involving SinR or AbrB.
Taken together, these data suggest that MecA exerts its negative effect on eps most importantly by potentiating the activity of the major repressor SinR. We also conclude from the data in Fig. 4 that the MecA effect on SinR activity is likely due to a negative effect on the production rather than the activity of SinI.
Spo0A is a potential target for the regulation of eps by MecA
Because both abrB and sinI are controlled by the transcription factor Spo0A~P, a simple hypothesis is that the MecA effect is exerted through this response regulator. This would also explain the ability of MecA to limit sporulation, which has an absolute requirement for Spo0A~P.
To further explore the involvement of Spo0A and hence the plausibility of our hypothesis that MecA acts on eps expression via Spo0A, we carried out additional epistasis experiments. Fig. 3B shows that as expected, eps expression was totally dependent on spo0A (BD4928) and that inactivation of mecA does not bypass the dependence of eps expression on spo0A; a double spo0A mecA mutant (BD5589) fails to express eps-lacZ. We then determined whether inactivation of abrB and sinR, singly or together, could bypass the depressing effect of spo0A inactivation on the expression of eps as they did for over-expression of MecA. Inactivation of either sinR (BD4929) or abrB (4930) bypassed the spo0A requirement to a level somewhat in excess of the wild-type level, but much below the level achieved by the sinR mutant, suggesting that without Spo0A neither repressor was present at a concentration sufficient to limit eps transcription. Indeed, inactivation of both repressors (BD4931) restored eps expression in the spo0A background to approximately the level exhibited by the sinR mutant. It is important to recognize that in the spo0A knockout mutant, AbrB accumulates and exerts a depressing influence on eps expression in excess of that in the wild-type situation. Clearly, SinR is the major repressor of eps transcription (Kearns et al., 2005).
Fig. 3A demonstrated that inactivation of both sinR and abrB partially bypassed the negative effect of MecA on eps expression. Fig. 3B demonstrates further that eliminating repression by both SinR and AbrB is sufficient to restore substantial eps expression when Spo0A is absent. In total, these results enhance the plausibility of the hypothesis that MecA may be regulating eps by interfering with Spo0A.
However, the experiments presented so far do not determine whether MecA limits the amount of Spo0A protein in the cell, the phosphorylation of Spo0A or the activity of Spo0A~P as a transcription factor.
The MecA inhibition of spoIIE and sinI expression is bypassed by the sad67 mutation
To further investigate the role of MecA, we utilized the sad67 mutation that makes this Spo0A factor independent of phosphorylation for its activity and removes 19 amino acid residues from the receiver domain of this transcription factor (Ireton et al., 1993). For the first experiment we used a construct in which the Sad67 protein was expressed from the IPTG-inducible Pspac promoter in a strain that over-expressed mecA (BD4628) and we determined the amount of SinI in the cell by Western blotting. Fig. 5A shows that two hours after induction, bypass of MecA overproduction by Sad67 took place. In contrast, induction of the wild-type spo0A under control of the Pspac promoter (BD4692) did not bypass MecA overproduction (Fig. 5B, C).
A similar experiment was carried out to test the ability of the Sad67 protein to bypass the over-production of MecA with spoIIE-lacZ as the reporter (Fig. 5D). As expected, MecA overproduction completely prevents the expression of spoIIE-lacZ in a strain carrying pKD93 (PP487), as it did for spoIIG-lacZ (Fig. 1C). When the Pspac-sad67 construct was induced by the addition of IPTG in a strain carrying the mecA over-expressing plasmid (PP488), spoIIE-lacZ was strongly expressed, overcoming the inhibition by MecA (Fig. 5D). Even the leaky expression of sad67 from the uninduced Pspac promoter was sufficient to achieve a delayed, intermediate level of spoIIE-lacZ expression. The induced expression in the presence of pKD93 was about two-thirds of that achieved in an isogenic strain lacking pKD93 (PP485), which was unaffected by the addition of IPTG. The sad67 bypass of the ability of pKD93 to inhibit expression of both spoIIE and sinI is consistent with the hypothesis that MecA interferes with the phosphorylation of Spo0A, although it can also be explained by a failure of this mutant protein to interact with MecA (see below).
ClpC participates in the regulation of eps
MecA controls the cellular levels of the competence transcription factor ComK by regulated proteolysis, acting as an adaptor to target ComK for degradation by the ClpC/ClpP protease (Turgay et al., 1997). Because the known functions of MecA and ClpC are so intimately connected, it appeared possible that ClpC would also be involved in the regulation of eps expression. Specifically, MecA may target Spo0A for degradation by ClpC/ClpP. The sad67 bypass experiments might then be explained as a failure of MecA to target the Sad67 protein, which lacks 19 residues from its receiver domain. Fig. 3C shows that the inactivation of clpC indeed causes increased expression of eps (in BD4580), indicating that ClpC does play a negative role in the regulation of eps. However, the level of eps-lacZ expression in the clpC::tet strain is nearly 4-fold lower than that reached in a mecA loss-of-function mutant (compare Figs. 3C with 3B and 1B).
The over-expression of MecA down-regulates comK transcription even when clpC is inactivated, because MecA binding is sufficient to prevent ComK from interacting with the comK promoter even when degradation of ComK by ClpC/ClpP cannot occur (Kong & Dubnau, 1994). In the present case, ClpC inactivation prevented mecA over-expression from down-regulating eps transcription to the level of the pKD93 clpC+ strain (Fig. 3C). This dramatic difference and the lesser effect of clpC compared to mecA inactivation hint that ClpC and MecA may act differently in regulating the expression of eps and comK.
MecA does not target Spo0A for degradation, inhibit the phosphorelay or dephosphorylate Spo0A~P
Despite the difference just noted, it seemed possible that MecA was targeting Spo0A for degradation. As noted above, if MecA did this by direct binding, the Sad67 bypass might be due to a failure of MecA to bind to this mutant protein. More generally, if MecA were involved in the degradation of Spo0A, we would expect more of this protein to be present in a mecA-deficient strain and less in the pKD93 strain. We therefore compared the amounts of Spo0A and MecA by Western blotting during growth in DSM of mecA+(BD2149), mecA (BD2148) and pKD93 (PP493) strains (Fig. 6). Panel B shows that the pKD93 strain accumulates excess MecA compared to the wild-type strain as expected and that the amounts of MecA accumulated in each of these two strains did not vary markedly as the cultures entered stationary phase. Panel A shows that very little Spo0A was detectable just before T0. Importantly, in sample 1, which was taken just before T0 and in sample 2, taken just after T0, the amounts of Spo0A in the three strains were similar. Thereafter (sample 3, 4 and 5), Spo0A did accumulate to a lesser extent in the pKD93 strain than in the wild-type. In the mecA strain, excess Spo0A was detected in sample 5. We have done a similar experiment in LB and again found no noticeable differences in the amounts of Spo0A among these three strains at T-1 and at 30 minutes after T0 (not shown). We interpret these results as follows. When a culture growing in DSM enters stationary phase, Spo0A becomes phosphorylated, thereby enhancing transcription from the SigH-dependent spo0A promoter (Strauch et al., 1992, Fujita & Sadaie, 1998). We suggest that MecA limits the phosphorylation or the activity of Spo0A~P, and as a result, different amounts of Spo0A protein are present in the mutant and wild-type strains only after T0. Note that the absence of MecA has an effect on the amount of Spo0A only in sample 5, between T2 and T3. This is consistent with the results in Fig. 1C, which show an increased transcription of spoIIG in a mecA strain only after about T1.
As another test of the hypothesis that MecA may target Spo0A for degradation or otherwise decrease its stability, we added puromycin (200 μg/ml) just after T1 to ΔmecA (BD2149) and pKD93 (PP493) strains and collected samples at intervals for Western blotting to detect decay of the Spo0A signal. During 40 minutes incubation in the presence of puromycin, little decay was discernable in either strain (Fig. 6D). The PP493 samples were also probed using anti-MecA antiserum, and decay was readily detected, with an estimated half-life between 10 and 15 minutes, showing that the puromycin was working. This experiment demonstrates that Spo0A is quite stable under these conditions, even in a MecA over-expressing strain, making it quite unlikely that the latter protein modulates the decay of Spo0A.
Finally, to further pursue the issue of Spo0A degradation, we used an in vitro degradation assay to test whether or not MecA directly targets Spo0A for degradation by ClpC/ClpP. Fig. 7B shows that in the presence of MecA, ClpC, ClpP and ATP, ComK is targeted for degradation as expected (Turgay et al., 1997). Also as expected, when ComK was omitted, MecA itself was degraded (Fig. 7A) but only in the presence of ATP. These controls demonstrate that the protein preparations were active, behaving as expected. In contrast, no degradation of either His-tagged (Fig. 7A) or untagged versions of Spo0A (Fig. 7C) was observed. These preparations of Spo0A were active in binding to radiolabeled sinI and abrB promoter fragments (not shown). Taken together, our in vivo (Fig. 6) and in vitro (Fig. 7) results suggest strongly that MecA does not exert its negative effect by targeting Spo0A for degradation, nor does it otherwise affect the total amount of Spo0A protein in the cell.
Fig. 7.
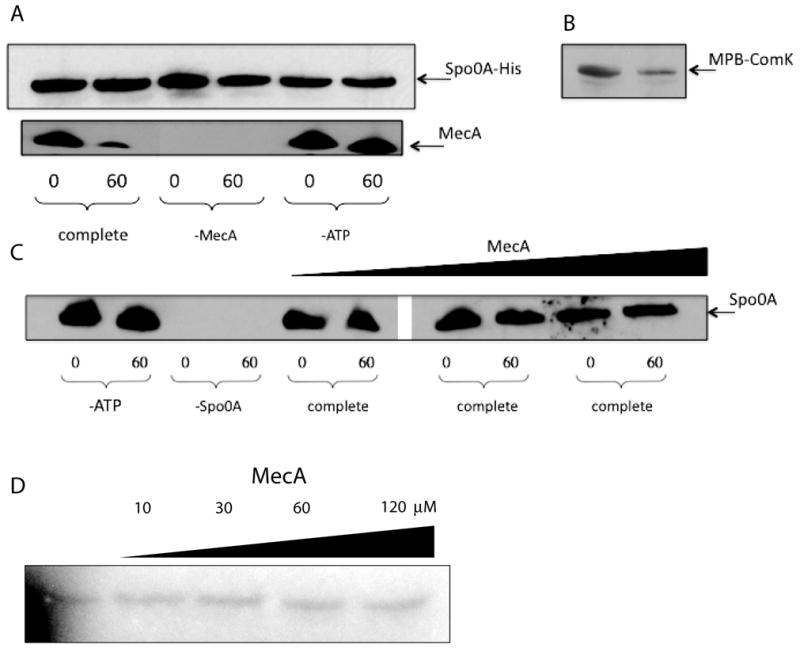
In vitro degradation assay. Spo0A-His, Spo0A, ComK-MBP and MecA were detected by immunoblotting with their cognate antisera. Samples were taken after zero and 60 minutes incubation. All incubation mixtures contained MecA, ClpC, ClpP, ATP and an ATP regenerating system unless otherwise indicated. (A) The incubation mixtures contained Spo0A-His. (B) The incubation mixture contained ComK-MBP, ClpC, ClpP, MecA and ATP. (C) Native Spo0A protein (0.3 μM) and ATP were included in these incubations, unless otherwise indicated and the concentration of MecA was varied in the 6 rightmost lanes (0.4 μM, 1.5 μM and 5 μM). Samples were run on the same gel, but an irrelevant portion was excised between lanes 6 and 7. (D) His-tagged KinA, Spo0F, Spo0B and untagged Spo0A (each at 0.2 μM) and various concentrations of MecA-His6 were incubated in the presence of 32P-γ-ATP as described in Experimental Procedures and following the published procedures (Fujita & Losick, 2003) and (Burbulys et al., 1991). The samples were autoradiographed after resolution by SDS-PAGE. Just before loading, bovine serum albumin was added to equalize the total protein loaded per lane. The gel was deliberately underexposed to ensure that the signals were within the sensitive range of film response. In control experiments, omission of the individual phosphorelay components prevented phosphorylation of Spo0A (not shown).
We next determined whether the addition of MecA would interfere with the phosphorylation of Spo0A in the presence of 32P-γ-ATP and the His-tagged phosphorelay components KinA, Spo0F and Spo0B. As shown in Fig. 7D, the addition of MecA-His6, even at concentrations up to 120 μM, did not decrease the yield of Spo0A~P. We conclude that MecA by itself neither interferes with the phosphorelay nor causes the dephosphorylation of Spo0A~P in the presence of the phosphorelay components.
MecA and Spo0A interact directly
The evidence presented so far suggests that MecA directly or indirectly interferes with the activity of Spo0A~P as a transcription factor. To test whether MecA binds directly to Spo0A or to the other phosphorelay proteins, we utilized surface plasmon resonance (SPR). MecA-His6 was immobilized to the surface of a CM5 chip using primary amine chemistry. Various concentrations of KinA, Spo0F, Spo0B or Spo0A were passed over the chip surface (Fig. 8A). With all of these proteins except Spo0A, the results were negative (not shown). This failure of MecA to interact with KinA, Spo0F or Spo0B is consistent with its failure to inhibit the phosphorelay reaction in vitro (Fig. 7D). In contrast, Spo0A exhibited a dose-dependent binding reaction with immobilized MecA within the low micromolar range of Spo0A concentrations. Similar results were obtained when anti-Spo0A antibodies that had been previously immobilized on the CM5 surface were used to capture Spo0A and MecA was then injected over the chip surface (not shown). When heat-denatured Spo0A was used, no appreciable binding was observed and the presence of bovine serum albumin injected together with the Spo0A did not inhibit the binding reaction, demonstrating the specificity of the interaction. As a final verification, non-His-tagged MecA prepared from an intein construct was used with similar results (not shown).
Fig. 8.

MecA binds directly to Spo0A. (A) MecA-His6 was immobilized on a CM5 chip surface and Spo0A at the indicated concentrations was passed over the chip surface. (B) As with panel A, except that full length MecA, and twice the molar concentration each of the NTD and CTD of MecA were immobilized on separate surfaces. Spo0A (3 μM) was passed over the chip surfaces. For both panels, values from a mock-coupled chip surface without MecA-His6 were subtracted.
MecA is a two-domain protein (Persuh et al., 1999). Its N-terminal domain (NTD) establishes the major contacts when binding to ComK or ComS, whereas the C-terminal domain (CTD) contacts ClpC. We used SPR to determine which domain of MecA contacted Spo0A. For this, we expressed and purified each of the domains as His-tagged proteins and coupled them to CM5 surfaces. Fig. 8B shows that the NTD and CTD of MecA can bind separately to Spo0A. In this experiment equal masses of the full-length, NTD and CTD proteins were immobilized. Because the relative masses of MecA:CTD:NTD are 1:0.6:0.5 (Persuh et al., 1999), the immobilized molar amounts of the two domains were about 2-fold larger than that of the full-length MecA. These results suggest that although Spo0A binds to the individual domains, these interaction affinities are somewhat lower than to full-length MecA. Importantly, because the major contacts between MecA and ComK are with the NTD of ComK, it is evident that ComK and Spo0A bind differently to MecA.
We further addressed this issue by determining if a ComK-derived peptide can bind to MecA if the latter is already bound to Spo0A. For this we used K17, a 17mer peptide (CHRVPKRQEFMLYPKEER) derived from the sequence of ComK that contains its recognition sequence (underlined) for binding to MecA (Prepiak & Dubnau, 2007). This peptide and full-length ComK exhibit indistinguishable binding affinities to MecA in SPR experiments showing that all the important contacts for MecA binding are established by K17. Fig. 9 shows an SPR experiment in which a low concentration (2.4 μM) of Spo0A (see Fig. 8A) was continuously injected over a MecA surface. As the reaction approached saturation, K17 (11 μM) was co-injected. If K17 competed for binding with Spo0A, displacing it, we would detect a decreased response when it was injected because its mass is about 8% that of monomeric Spo0A. Instead, an additional response was detected, which approached a new, higher equilibrium. When buffer was then injected, the response quickly decreased to about the level of the initial Spo0A response and then continued to decline, suggesting that the off-rate for the MecA-K17 complex may be higher than that of the MecA-Spo0A complex. We conclude that K17 and Spo0A can bind at the same time to MecA and that Spo0A therefore does not obscure the ComK recognition sequence when it binds and that Spo0A binding does not depend solely on contacts with this sequence. Obviously, these results do not prove that full length ComK and Spo0A can bind simultaneously to MecA, but only that they probably use different recognition sequences. This result is consistent with the absence of a sequence within Spo0A similar to the FMLYPK motif in ComK for binding to MecA.
Fig. 9.

K17 and Spo0A bind simultaneously to MecA. MecA-His6 was immobilized on the surface of a CM5 chip. At the indicated time, Spo0A (2.4 μM) was injected. At a later time K17 peptide at a concentration of 11 μM was co-injected with Spo0A as indicated, followed about 70 seconds later by protein-free buffer.
Both domains of MecA are needed to inhibit Spo0A~P
The interaction of both the NTD and CTD of MecA with Spo0A raises the question whether both domains are needed for the inhibitory effect of MecA on the activity of Spo0A~P. We approached this question by expressing the individual domains from a multi-copy plasmid and measuring sporulation frequencies. The over-expression plasmids were described previously (Persuh et al., 1999). The NTD-over-expressing strain (BD2142) exhibited a sporulation frequency of 0.72 normalized to the wild-type strain (IS75). The normalized sporulation frequency of the CTD-over-expressing strain (PP559) was 0.76. As expected, the average normalized frequency of two different pKD93-containing strains (PP493 and PP560) was low; 4.1 × 10−5. The inability of the MecA NTD alone to inhibit spore formation stands in stark contrast to the inhibition of competence exhibited by the same NTD-over-expressing construct (Persuh et al., 1999), once again suggesting that MecA interacts differently with Spo0A and ComK. It has been established that inactivation of clpC prevents sporulation (Gerth et al., 1998, Msadek et al., 1998, Pan et al., 2001). Because the CTD of MecA binds to ClpC (Persuh et al., 1999), it is conceivable that the sequestration of ClpC by MecA contributes to the negative effect of MecA on sporulation. However, the inability of the MecA-CTD to inhibit spore formation argues against this possibility, as does our observation that the over-production of the CTD does not decrease the expression of spoIIG-luc (not shown).
Discussion
MecA and the regulation of eps and sporulation
We have shown that MecA is a pleiotropic regulator of bacterial development. Besides its role in the regulation of competence through the targeting of ComK for degradation by ClpC/ClpP, we have now shown that mecA mutant cells transition to sporulation and to eps expression more frequently than the wild-type. The most important conclusion from this study is that MecA can interfere with sporulation and eps expression by its direct interaction with Spo0A.
A series of epistasis experiments and the bypass of the MecA overproduction phenotype by expression of the Sad67 mutant form of Spo0A strongly suggested that the negative effects of MecA on eps expression and on spore formation are exerted via Spo0A (Figs. 3–5). The failure of MecA to impact the amount of Spo0A protein in growing cells (Fig. 6) and the failure of spo0A expression from the Pspac promoter to bypass pKD93 (Fig. 5) both show that MecA does not act on the synthesis of Spo0A and is therefore likely to act post-translationally. Consistent with this, the SPR results show that MecA binds directly to Spo0A (Fig. 8). Finally, both in vitro and in vivo data 21 suggest that MecA does not increase the rate of degradation of Spo0A (Figs. 6, 7). Fig. 7D shows that MecA by itself does not prevent phosphoryl flux through the phosphorelay. This may seem to contradict the sad67 results, but we suspect that this bypass of pKD93 is due to a failure of MecA to bind effectively to the Sad67 protein, which contains a 19-residue deletion.
The most likely explanation for the inhibiting effect of MecA seems to be that it prevents Spo0A~P from acting as a transcription factor (Fig. 10). It may do this by preventing it from binding to DNA, similarly to the action of RapH, RapF and RapC with another response regulator, ComA (Smits et al., 2007, Core & Perego, 2003, Bongiorni et al., 2005), or by binding to Spo0A~P on the DNA, preventing it from interacting with RNA polymerase as TorI does with the response regulator TorR (Ansaldi et al., 2004). Other possibilities exist, but appear less likely. For example, MecA may potentiate the activity of Spo0A~P as a substrate for Spo0E or some other phosphatase.
Fig. 10.

Two modes of developmental regulation by MecA. In the case of competence (A), MecA binds to the transcriptional activator ComK and targets it for degradation by a complex of MecA, ClpC and ClpP (Turgay et al., 1998). These proteins exist as dimers, hexamers and double heptamer rings respectively. When quorum-sensing results in the synthesis of ComS, this small protein competes with ComK for binding to MecA (Prepiak & Dubnau, 2007). Free ComK can then bind to competence promoters. As shown here, MecA can also interact with Spo0A (B) either preventing it from binding to its target promoters or from acting as a transcription factor once bound.
Although it appears that MecA does not target Spo0A for degradation by ClpC/ClpP, a clpC knockout strain does over-express eps (Fig. 3C). We did not attempt to explore equivalent effects on spore expression because a clpC loss-of-function mutation prevents sporulation (Gerth et al., 1998, Msadek et al., 1998, Pan et al., 2001). As noted above, the in vivo behavior observed for the present system differs from that of ComK regulation. ClpC is not needed for the down-regulation of comK when mecA is over-expressed, because binding of ComK by excess MecA is sufficient to prevent it from binding to PcomK even in the absence of ClpC (Turgay et al., 1997). In contrast, when pKD93 is present a clpC mutant still over-expresses eps (Fig. 3C). The failure of clpC inactivation to yield as high a level of eps transcription as does inactivation of mecA (Fig. 3) also differentiates this system from competence regulation and leads to the hypothesis that MecA and ClpC regulate eps transcription using distinct pathways. Recent work with the SlrR protein suggests a candidate mechanism for the effect of ClpC (Chai et al., 2010b). SlrR binds to SinR, and its over-expression titrates this repressor, resulting in over-expression of eps. Interestingly, SlrR accumulates in a clpC mutant and in such a mutant more robust biofilms are formed, consistent with the results of Fig. 3C (Chai et al., 2010a).
Interactions of MecA with its partners
ComK and ComS compete for binding to MecA, interacting with overlapping binding sites (Prepiak & Dubnau, 2007). Binding to this site on MecA leads to the degradation of either substrate by ClpC/ClpP. The SPR results in Fig. 9 suggest strongly that Spo0A binding to MecA does not require the ComK/ComS site on MecA. Because MecA-ClpC does not target Spo0A for degradation, we suspect that distinct binding sites for ComK and Spo0A may facilitate different outcomes; either degradation or inactivation as a transcription factor, but this hypothesis remains to be proved. It has been shown that the small protein Spx also binds to MecA, increasing its affinity for ComK (Nakano et al., 2002a). Spx and ComK can bind together to MecA, and therefore interact with different surfaces on this versatile protein. We do not know if the Spx and Spo0A binding sites are distinct. Defining these sites and their interactions and the identification of additional binding partners for MecA will be an important task for the future.
The biological roles of MecA
MecA controls premature transitions to the competent state by directing the degradation of ComK and also controls transitions to eps expression and spore formation by a non-degradative mechanism. In addition, it has been reported that MecA regulates motility by read-through from a ComK-dependent promoter, but also by an uncharacterized ComK-independent mechanism (Liu & Zuber, 1998, Rashid et al., 1996).
By interacting with Spo0A, MecA acts at a critical point to regulate several developmental adaptations, all bimodally expressed. We propose that MecA acts as a buffer during exponential growth, ensuring that most cells thriving in a nutrient-rich environment do not commit valuable resources to an unnecessary physiological adaptation. Nevertheless, we suggest that noise in the accumulation of MecA, or of a pathway-specific effector protein, provides a bet-hedging mechanism, ensuring that a few cells escape and enter one or another of these pathways.
In contrast to this situation obtaining during growth, cells entering stationary phase may undergo programmed changes that alter the barrier imposed by MecA. These represent deterministic mechanisms that are superimposed on stochastic systems of decision-making. For example, the MecA buffer system for ComK is overcome because the quorum-sensing pathway leads to the production of ComS and also because the mean basal expression of ComK is adjusted upward and then downward as a culture arrives in stationary phase (Leisner et al., 2007, Maamar et al., 2007).
Is there a ComS equivalent that alters the buffering effect of MecA on Spo0A? Perhaps the programmed activation of the phosphorelay is sufficient, so that when the concentration of Spo0A~P in a given cell exceeds a threshold set by the MecA concentration, the excess Spo0A~P initiates biofilm and spore formation. The kinetics of spoIIG-luc expression in the mecA strain hints that more than this passive mechanism may be at play. The timing of the initial rise in spoIIG expression is not perturbed in the mecA mutant, suggesting that in the wild-type strain, neither the increase in Spo0A~P that occurs at this time nor its activity, are limited by MecA. In contrast, the later rise in spoIIG expression is augmented in the mutant, suggesting that MecA does limit this increase. We suggest that during exponential growth there is enough MecA to lower the probability of transition to sporulation. When the phosphorelay becomes active, the increased amount of Spo0A~P overwhelms the MecA barrier and spore formation initiates in many cells. Later during this process, the MecA barrier may be enhanced, limiting the activity of Spo0A~P. The nature of this enhancement is completely unknown.
The control of energy-intensive stress response pathways by MecA-dependent mechanisms is complex, varied and delicately balanced; cells faced with environmental stress enter developmental pathways at high rates when appropriate and at rates that maximize fitness and survival of a shared genotype. When times are good, MecA serves to dampen but not eliminate the expression of these developmental pathways, which relies on stochastic fluctuations in the levels of regulatory proteins. MecA appears to be an important hub protein in the B. subtilis regulatory network for development.
EXPERIMENTAL PROCEDURES
Microbiological procedures
The B. subtilis strains used are all derivatives of strain 168 and are listed in Table 1. Bacteria were grown in liquid competence minimal medium (Albano et al., 1987), in Luria-Bertani (LB) medium or in DSM (Schaeffer et al., 1965). When required for selection, the media were supplemented with chloramphenicol, erythromycin or kanamycin (each at 5 μg/ml), phleomycin (2 μg/ml) or spectinomycin (100 μg/ml). B. subtilis competent cells were prepared as described previously (Albano et al., 1987). E. coli DH5α (Invitrogen) was used for cloning. Strain construction was by transformation or by transduction with bacteriophage PBS1. Sporulation was measured after growth for 24 and 48 hours at 37° C in DSM medium by plating for viable counts before and after heating the cells at 80° C for 30 min.
Table 1.
Strains
| Strain | Genotype1 | Source |
|---|---|---|
| B. subtilis strains: | ||
| BD630 | his leu met | Lab strain |
| BD1512 | comG-lacZ (erm) | |
| BD2091 | mecA::erm | (Kong et al., 1993) |
| BD2142 | amyE::spo0A-lacZ (cat) pMecA-NTD | (Persuh et al., 1999) |
| BD2148 | mecA::spc comK::kan spo0A-lacZ (cat) | Lab strain |
| BD2149 | comK::kan spo0A-lacZ (cat) | |
| BD2200 | comG-lacZ (erm) pKD93 (Phl) | Lab strain |
| BD3980 | mecA::erm epsG::cat eps-lacZ::tet | This work |
| BD4498 | eps-lacZ (tet) | D. Kearns |
| BD4538 | eps-lacZ (tet) mecA::erm | This work |
| BD4544 | eps-lacZ (tet) sinR::cat | This work |
| BD4549 | eps-lacZ (tet) pKD93 (phl)2 sinR::cat | This work |
| BD4555 | sinI-lacZ (cat) | I. Smith |
| BD4557 | sinI-lacZ (cat) pKD93 (phl) | This work |
| BF4568 | sinI-lacZ (cat) mecA::erm | This work |
| BD4580 | eps-lacZ (cat) clpC::tet | This work |
| BD4615 | eps-lacZ (tet) abrB::cat sinR::kan | This work |
| BD4621 | eps-cfp (cat) | M. Dias |
| BD4623 | eps-lacZ (tet) abrB::cat | This work |
| BD4624 | eps-lacZ (tet) abrB::cat sinR::kan pKD93 (phl) | This work |
| BD4626 | spo0A::kan amyE::Pspac-sad67 (cat) | (Ireton et al., 1993) |
| BD4628 | spo0A::kan amyE::Pspac-sad67 pKD93 (phl) | This work |
| BD4642 | eps-cfp (cat) mecA::erm | This work |
| BD4643 | eps-cfp (cat) pKD93 (phl) | This work |
| BD4644 | eps-lacZ (tet) pKD93 (phl) | This work |
| BD4692 | Pspac-spo0A (cat)4 pKD93 (phl) | This work |
| BD4928 | eps-lacZ (tet) spo0A::kan | This work |
| BD4929 | eps-lacZ (tet) spo0A::kan sinR::cat | This work |
| BD4930 | eps-lacZ (tet) spo0A::kan abrB::cat | This work |
| BD4931 | eps-lacZ (tet) spo0A::kan abrB::cat sinR::kan3 | This work |
| BD5113 | eps-lacZ (tet) pKD93 (phl) abrB::cat | This work |
| BD5114 | eps-lacZ (cat) clpC::tet mc mecA (kan) | This work |
| BD5589 | eps-lacZ (tet) spo0A::kan mecA::erm | This work |
| PP479 | spoIIE-gfp (spc) mecA::erm comK::kan | (Fujita & Losick, 2003) |
| PP480 | spoIIE-gfp (spc) comK::kan | (Fujita & Losick, 2003) |
| PP485 | thrC::spoIIE-lacZ (erm) amyE::Pspac-sad67(cat) | (Ireton et al., 1993) |
| PP487 | thrC::spoIIE-lacZ (erm) pKD93 (phl) | This work |
| PP488 | thrC::spoIIE-lacZ (erm) amyE::Pspac-sad67(cat) pKD93 (phl) | This work |
| PP493 | amyE::spo0A-lacZ (cat) pKD93 (phl) | Lab strain |
| PP510 | thrC::spoIIE-lacZ (erm) amyE::Pspac-spo0A (cat)) | (Fujita et al., 2005) |
| PP512 | thrC::spoIIE-lacZ (erm) amyE::Pspac-spo0A (cat) pKD93 (phl) | This work |
| PP516 | thrC::spoIIE-lacZ (erm) amyE::Pspac-sad67(cat)) spo0A::kan | This work |
| PP522 | thrC::spoIIE-lacZ (erm) amyE::Pspac-sad67(cat)) spo0A::kan pKD93 | This work |
| PP533 | spoIIG-luc (cat) | This work |
| PP551 | spoIIG-luc (cat) mecA::erm comK::spc | This work |
| PP559 | amyE::comK-lacZ (cat) pMecA-CTD | (Persuh et al., 1999) |
| PP560 | amyE::comK-lacZ (cat) pKD93 | This work |
| PP565 | spoIIG-luc (cat) pKD93(phl) | This work |
| E. coli strain: | ||
| PP494 | B. subtilis spo0A in pET26b(+) in E. coli B834 | (Muchova et al., 2004) |
All strains were constructed in the his leu met background of IS75, except for PP485, PP487, PP488, PP479 and PP480 which are in the prototrophic PY79 background and PP510, PP512, PPP516 and PP522 which are in a trp phe background.
The plasmid pKD93, consists of pUB110, into which was inserted mecA under control of a constitutive promoter (Kong et al., 1993). This plasmid has been shown by Western blotting to overproduce MecA. The plasmid expressed resistance to both phleomycin (phl) and kanamycin (kan).
BD4931 was constructed by congression with selection for Kan and Cat, picking up the spo0A, sinR and abrB markers at the same time, as verified by.
Pspac-spo0A is a Campbell-like integrant, which inactivates the native spo0A gene.
β-galactosidase and luciferase assays
β-galactosidase assays were carried out as previously described (Albano et al., 1987), modified for use in a plate reader. For the detection of luciferase activity, strains were first grown in DSM medium to an optical density at 600 nm (OD600) of 2. Cells were then centrifuged and resuspended in fresh DSM, adjusting all the cultures to an OD600 of 2. These pre-cultures were then diluted 20 fold in fresh DSM and 200 μl was distributed in each of two wells in a 96-well black plate (Corning). 10 μl of luciferin were added to each well to reach a final concentration of 1.5 mg/ml (4.7 mM). The cultures were incubated at 37°C with agitation in a PerkinElmer Envision™ 2104 Multilabel Reader equipped with an enhanced sensitivity photomultiplier for luminometry. The temperature of the clear plastic lid was maintained at 38°C to avoid condensation. Relative Luminescence Unit (RLU) and OD600 were measured at 1.5 min intervals.
Western blot analysis
Preparation of whole cell extracts and Western blotting was carried out by standard methods. Protein extracts were made by pelleting 1 ml of cells, washing in STM (50 mM NaCl, 25% sucrose, 50 mM Tris-HCl, pH 8.5, 5 mM MgCl2), lysing by sonication, followed by mixing with 5X glycerol sample buffer (0.225 M Tris-HCl pH 6.8, 50% glycerol, 5% SDS, 0.05% Bromophenol Blue, 1% β-mercaptoethanol). Proteins were resolved by SDS-PAGE (12% Tris-tricine) (Schagger & von Jagow, 1987) at a constant amperage (25 mA) and then transferred to nitrocellulose membranes for 1 hour at 12 V in a semi-dry transfer apparatus (Bio-Rad). Transferred proteins were detected using appropriate antibodies, all raised in rabbits and used with the following dilutions: 1:500 for anti-SinI, 1:2,000 for anti-MecA and 1:5,000 for anti-Spo0A and anti-ComK, followed by secondary anti-rabbit antibodies conjugated to horseradish peroxidase (Zymed #401315, 1:10,000). Secondary antibodies were detected using enhanced chemiluminescence ECL+ (Amersham). The anti-SinI antiserum was kind gift from D. Kearns.
Microscopy
Cultures were grown in LB or DSM until the indicated times and samples were attached to poly-L-lysine-coated slides, mounted in Slowfade (Molecular Probes). Either staining with propidium iodide (10 μg/ml) or DIC imaging was used to visualize the cell bodies. Microscopy was performed with an upright Nikon Eclipse 90i microscope equipped with an Orca-ER Digital Camera (Hamamatsu), and a Nikon TIRF 1.45 NA Plan Neo-Fluor 100 X oil immersion objective. Velocity software (Improvision) was used for image acquisition and processing. Two images fluorescence images for CFP and PI were captured for each field of cells. Appropriate Semrock optical filter sets were used for each fluorophore.
Protein Purification
Spo0A protein purification was performed as previously described by Muchová et al (Muchova et al., 2004). Briefly, four one liter cultures of the E. coli strain PP494, which carries spo0A cloned into the pET26b(+) vector (Novagene), were grown in LB with 30 μg/μl kanamycin until the cultures reached an OD600 of 0.6. IPTG (1 mM final concentration) was added and the cultures were incubated at 30°C for 4.5 hours. PP494 was a kind gift from I. Barak. The cells were harvested by centrifugation and resuspended in Buffer A (250 mM NaCl, 20 mM Tris-HCl pH 7.5, 2 mM EDTA, 2 mM DTT, 1 mM PMSF). The cells were lysed with an EmulsiFlex-C5 (Avestin) disruptor and centrifuged for 20 minutes at 25,000 rpm. The supernatant was collected and loaded onto a pre-equilibrated 5 ml Heparin Hi Trap column for FPLC. Protein fractions were eluted with a gradient of Buffer A (up to 1M NaCl). Fractions with Spo0A were collected, pooled, concentrated to 1 mg/ml, and stored at −80°C until use.
MecA-His6 was purified as described previously (Turgay et al., 1997). One liter cultures of E. coli M15 harboring pQE60-mecA were grown in LB with Ampicillin (100 μg/ml) at 37° C until OD600 = 500 – 800, at which time expression of the protein was induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). After three hours of induction, cells were harvested by centrifugation at 4° C for 10 min at 5,000 rpm. The pellets were resuspended in cold lysis buffer (25 mM HEPES, pH 7.5, 500 mM NaCl, 10 mM imidazole) and centrifuged again using the same conditions. The pellets were resuspended in 25 ml cold lysis buffer and treated with one tablet of protease inhibitor cocktail (Roche #1873580). Cells were broken by passage through a French Press three times at 1,200 psi. Cell debris was pelleted by centrifugation at 4° C for 35 min at 20,000 rpm. The supernatants were mixed with 1 ml of Ni2+ resin (Qiagen), equilibrated in lysis buffer in a 50 ml conical tube and then diluted with lysis buffer to approximately 45 ml. Incubation with the nickel beads was continued for one hour on a rotary shaker at 4° C to facilitate binding. The mixture was then poured into a column (BioRad), allowed to flow through and washed extensively (~50 column volumes) with wash buffer (25 mM HEPES, pH 7.5, 500 mM NaCl, 25 mM imidazole). The protein was then eluted with 75 mM imidazole. Total protein was assayed with the BioRad protein determination reagent and purity was determined by 12% SDS-PAGE and Coomassie blue staining. Protein samples were dialyzed into storage buffer (25 mM HEPES, pH 7.5, 400 mM NaCl, 1 mM EDTA, 0.5 mM DTT). Protein concentrations were determined by dilution into 7.4 M guanidinium hydrochloride, 50 mM Na phosphate, pH 7.5 using extinction coefficients at 280 nm determined from amino acid compositions (http:://au.expasy.org/tools/protparam.html).
For the purification of ClpC and ClpP, one liter cultures of E. coli M15 harboring plasmids that express either clpC or clpP fused to self-cleaving intein tags. The clpC-intein construct was a gift from M. Nakano (Nakano et al., 2002b). The strains were grown in LB with ampicillin (100 μg/ml) at 37° C until OD600 = 500 – 800, at which time expression of the protein was induced with 4 mM IPTG. These constructs, kind gifts from M. Nakano, were under IPTG-inducible control (IMPACT system, New England Biolabs). Following addition of IPTG, cultures were incubated overnight with shaking at 15° C. Cells were harvested by centrifugation at 4° C for 10 min at 5,000 rpm. The pellet was resuspended in cold column buffer (20 mM Tris, pH 8.0, 500 mM NaCl, 1mM EDTA) and centrifuged again under the same conditions. The pellet was then resuspended in 25 ml cold column buffer. The cells were broken by passing through a French Press three times at 1,200 psi. Cell debris was pelleted by centrifuging at 4° C for 35 min at 20,000 rpm. The supernatants were mixed with 0.5 ml of chitin resin (New England Biolabs), equilibrated in column buffer in a 50 ml conical tube and then diluted with column buffer to approximately 45 ml. The protein fraction with chitin beads was incubated on a rotary shaker for 1 hour at 4° C to facilitate binding. The mixture was poured into a column (BioRad), allowed to flow through and was washed extensively (~50 column volumes) with column buffer. Cleavage of the intein tag was induced by quickly flushing the column with 3 column volumes of cleavage buffer (20 mM Tris, pH 8.0, 500 mM NaCl, 1 mM EDTA, 50 mM DTT), closing the column and leaving it overnight at 4 C. The flow-through was collected the following day. Protein concentrations were determined by BioRad protein assay and protein purity was determined using 12% SDS-PAGE and Coomassie blue staining. Protein samples were dialyzed into storage buffer (50 mM HEPES, pH 7.5, 300 mM NaCl, 5 mM MgCl2). Protein concentration was determined spectrophotometrically as described above.
SPR experiments
A Biacore 2000 instrument was used for all experiments. MecA-His6 was covalently coupled to the surface of CM5 sensor chips using amine-coupling chemistry. Solutions containing the indicated concentrations of the analyte proteins in HBS-EP buffer (0.01 M HEPES, pH 7.4, 0.15 M NaCl, 3 mM EDTA, 0.005% v/v surfactant P20) were passed over the sensor chip surface at a constant flow rate 20 μl/min and at 25° C. The response of the same solutions on a mock-coupled surface was subtracted from all sensograms.
In vitro degradation assay
In vitro degradation assays were based on published procedures (Turgay et al., 1998) and contained the necessary components for degradation; ClpC, ClpP and ATP, the adaptor protein MecA and a substrate, either ComK or Spo0A. ClpC, ClpP and ComK were added at 1.5 μM, MecA was added at 0.5 μM unless otherwise indicated and Spo0A was added at 0.3 μM. An ATP regeneration system was used containing 10 mM ATP, pH 7, 0.033 mg/ml creatine phosphokinase and 1.6 mM creatine phosphate. A preincubation step with ADP (5 mM) was employed to allow for assembly of the proteolytic complex. The reaction components were mixed in Buffer A (100 mM KCl, 25 mM MOPS, pH 7, 5 mM MgCl2) in the following order: ClpC, MecA, ComK or Spo0A, ClpP, ADP. After addition of ADP the reactions were incubated at 30° C for 30 min after which the reactions were moved to 37° C and the ATP and regenerating system were added. Reactions were incubated for one hour at 37° C. Samples of 45 μl were collected and the reaction was stopped by adding glycerol samples buffer (see Western blot protocol). Western blotting was used to detect ComK, Spo0A or MecA as described above.
Phosphorelay reaction
Phosphorylation reactions were done as described in Burbulys at. al. (1991) Reaction mixtures (total 20 μl) contained 1 μM KinA-His6, 0.2 μM Spo0F-His6, 0.2 μM Spo0B-His6, 1 μM Spo0A and 0–120 μM MecA-His6. The reactions were initiated by addition of ATP, incubated 1 hour at 25°C and stopped by addition of SDS Sample buffer. Just before loading, bovine serum albumin was added to equalize the total protein loaded per lane and the samples were immediately loaded on a 16% SDS polyacrylamide gel. Gels were exposed using a Phosphorimager Screen, which was scanned using a Typhoon™ scanner (GE Healthcare).
Acknowledgments
We thank all members of our lab and the Neiditch lab as well as R. Grau, K. Muchova and I Barak for useful advice and discussions. We also thank M. Nakano and D. Kearns for the kind gifts of strains and anti-sera and Miguel Dias for constructing the eps-cfp fusion. This work was supported by NIH grant GM057720.
References
- Albano M, Hahn J, Dubnau D. Expression of competence genes in Bacillus subtilis. J Bacteriol. 1987;169:3110–3117. doi: 10.1128/jb.169.7.3110-3117.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansaldi M, Theraulaz L, Mejean V. TorI, a response regulator inhibitor of phage origin in Escherichia coli. Proc Natl Acad Sci U S A. 2004;101:9423–9428. doi: 10.1073/pnas.0401927101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai U, Mandic-Mulec I, Smith I. SinI modulates the activity of SinR, a developmental switch protein of Bacillus subtilis, by protein-protein interaction. Genes Dev. 1993;7:139–148. doi: 10.1101/gad.7.1.139. [DOI] [PubMed] [Google Scholar]
- Banse AV, Chastanet A, Rahn-Lee L, Hobbs EC, Losick R. Parallel pathways of repression and antirepression governing the transition to stationary phase in Bacillus subtilis. Proc Natl Acad Sci U S A. 2008;105:15547–15552. doi: 10.1073/pnas.0805203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongiorni C, Ishikawa S, Stephenson S, Ogasawara N, Perego M. Synergistic regulation of competence development in Bacillus subtilis by two Rap-Phr systems. J Bacteriol. 2005;187:4353–4361. doi: 10.1128/JB.187.13.4353-4361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branda SS, Chu F, Kearns DB, Losick R, Kolter R. A major protein component of the Bacillus subtilis biofilm matrix. Mol Microbiol. 2006;59:1229–1238. doi: 10.1111/j.1365-2958.2005.05020.x. [DOI] [PubMed] [Google Scholar]
- Branda SS, Gonzalez-Pastor JE, Dervyn E, Ehrlich SD, Losick R, Kolter R. Genes involved in formation of structured multicellular communities by Bacillus subtilis. J Bacteriol. 2004;186:3970–3979. doi: 10.1128/JB.186.12.3970-3979.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbulys D, Trach KA, Hoch JA. Initiation of sporulation in B. subtilis is controlled by a multicomponent phosphorelay. Cell. 1991;64:545–552. doi: 10.1016/0092-8674(91)90238-t. [DOI] [PubMed] [Google Scholar]
- Chai Y, Chu F, Kolter R, Losick R. Bistability and biofilm formation in Bacillus subtilis. Mol Microbiol. 2008;67:254–263. doi: 10.1111/j.1365-2958.2007.06040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Kolter R, Losick R. Reversal of an epigenetic switch governing cell chaining in Bacillus subtilis by protein instability. Mol Microbiol. 2010a;78:218–229. doi: 10.1111/j.1365-2958.2010.07335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Norman T, Kolter R, Losick R. An epigenetic switch governing daughter cell separation in Bacillus subtilis. Genes Dev. 2010b;24:754–765. doi: 10.1101/gad.1915010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu F, Kearns DB, McLoon A, Chai Y, Kolter R, Losick R. A novel regulatory protein governing biofilm formation in Bacillus subtilis. Mol Microbiol. 2008;68:1117–1127. doi: 10.1111/j.1365-2958.2008.06201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core L, Perego M. TPR-mediated interaction of RapC with ComA inhibits response regulator-DNA binding for competence development in Bacillus subtilis. Mol Microbiol. 2003;49:1509–1522. doi: 10.1046/j.1365-2958.2003.03659.x. [DOI] [PubMed] [Google Scholar]
- Dubnau D, Losick R. Bistability in bacteria. Mol Microbiol. 2006;61:564–572. doi: 10.1111/j.1365-2958.2006.05249.x. [DOI] [PubMed] [Google Scholar]
- Fujita M, Gonzalez-Pastor JE, Losick R. High- and low-threshold genes in the Spo0A regulon of Bacillus subtilis. J Bacteriol. 2005;187:1357–1368. doi: 10.1128/JB.187.4.1357-1368.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M, Losick R. The master regulator for entry into sporulation in Bacillus subtilis becomes a cell-specific transcription factor after asymmetric division. Genes Dev. 2003;17:1166–1174. doi: 10.1101/gad.1078303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M, Sadaie Y. Feedback loops involving Spo0A and AbrB in in vitro transcription of the genes involved in the initiation of sporulation in Bacillus subtilis. J Biochem. 1998;124:98–104. doi: 10.1093/oxfordjournals.jbchem.a022103. [DOI] [PubMed] [Google Scholar]
- Gaur NK, Cabane K, Smith I. Structure and expression of the Bacillus subtilis sin operon. J Bacteriol. 1988;170:1046–1053. doi: 10.1128/jb.170.3.1046-1053.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerth U, Krüger E, Derré I, Msadek T, Hecker M. Stress induction of the Bacillus subtilis clpP gene encoding a homologue of the proteolytic component of the Clp protease and the involvement of ClpP and ClpX in stress tolerance. Mol Microbiol. 1998;28:787–802. doi: 10.1046/j.1365-2958.1998.00840.x. [DOI] [PubMed] [Google Scholar]
- Hahn J, Roggiani M, Dubnau D. The major role of Spo0A in genetic competence is to downregulate abrB, an essential competence gene. J Bacteriol. 1995;177:3601–3605. doi: 10.1128/jb.177.12.3601-3605.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamon MA, Stanley NR, Britton RA, Grossman AD, Lazazzera BA. Identification of AbrB-regulated genes involved in biofilm formation by Bacillus subtilis. Mol Microbiol. 2004;52:847–860. doi: 10.1111/j.1365-2958.2004.04023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireton K, Rudner DZ, Jaacks-Siranosian K, Grossman AD. Integration of multiple developmental signals in Bacillus subtilis through the SpoOA transcription factor. Genes Dev. 1993;7:283–294. doi: 10.1101/gad.7.2.283. [DOI] [PubMed] [Google Scholar]
- Kearns DB, Chu F, Branda SS, Kolter R, Losick R. A master regulator for biofilm formation by Bacillus subtilis. Mol Microbiol. 2005;55:739–749. doi: 10.1111/j.1365-2958.2004.04440.x. [DOI] [PubMed] [Google Scholar]
- Kong L, Dubnau D. Regulation of competence-specific gene expression by Mec-mediated protein-protein interaction in Bacillus subtilis. Proc Natl Acad Sci USA. 1994;91:5793–5797. doi: 10.1073/pnas.91.13.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Siranosian KJ, Grossman AD, Dubnau D. Sequence and properties of mecA, a negative regulator of genetic competence in Bacillus subtilis. Mol Microbiol. 1993;9:365–373. doi: 10.1111/j.1365-2958.1993.tb01697.x. [DOI] [PubMed] [Google Scholar]
- Leisner M, Stingl K, Radler JO, Maier B. Basal expression rate of comK sets a ‘switching-window’ into the K-state of Bacillus subtilis. Mol Microbiol. 2007;63:1806–1816. doi: 10.1111/j.1365-2958.2007.05628.x. [DOI] [PubMed] [Google Scholar]
- Liu J, Zuber P. A molecular switch controlling competence and motility: competence regulatory factors ComS, MecA, and ComK control sigmaD-dependent gene expression in Bacillus subtilis. J Bacteriol. 1998;180:4243–4251. doi: 10.1128/jb.180.16.4243-4251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losick R, Desplan C. Stochasticity and cell fate. Science. 2008;320:65–68. doi: 10.1126/science.1147888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maamar H, Raj A, Dubnau D. Noise in gene expression determines cell fate in Bacillus subtilis. Science. 2007;317:526–529. doi: 10.1126/science.1140818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molle V, Fujita M, Jensen ST, Eichenberger P, Gonzalez-Pastor JE, Liu JS, Losick R. The Spo0A regulon of Bacillus subtilis. Mol Microbiol. 2003;50:1683–1701. doi: 10.1046/j.1365-2958.2003.03818.x. [DOI] [PubMed] [Google Scholar]
- Msadek T, Dartois V, Kunst F, Herbaud ML, Denizot F, Rapoport G. ClpP is required for competence development, motility, degradative enzyme synthesis, growth at high temperature and sporulation. Mol Microbiol. 1998;27:899–914. doi: 10.1046/j.1365-2958.1998.00735.x. [DOI] [PubMed] [Google Scholar]
- Muchova K, Lewis RJ, Perecko D, Brannigan JA, Ladds JC, Leech A, Wilkinson AJ, Barak I. Dimer-induced signal propagation in Spo0A. Mol Microbiol. 2004;53:829–842. doi: 10.1111/j.1365-2958.2004.04171.x. [DOI] [PubMed] [Google Scholar]
- Murray EJ, Strauch MA, Stanley-Wall NR. SigmaX is involved in controlling Bacillus subtilis biofilm architecture through the AbrB homologue Abh. J Bacteriol. 2009;191:6822–6832. doi: 10.1128/JB.00618-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano MM, Nakano S, Zuber P. Spx (YjbD), a negative effector of competence in Bacillus subtilis, enhances ClpC-MecA-ComK interaction. Mol Microbiol. 2002a;44:1341–1349. doi: 10.1046/j.1365-2958.2002.02963.x. [DOI] [PubMed] [Google Scholar]
- Nakano S, Zheng G, Nakano MM, Zuber P. Multiple pathways of Spx (YjbD) proteolysis in Bacillus subtilis. J Bacteriol. 2002b;184:3664–3670. doi: 10.1128/JB.184.13.3664-3670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Garsin DA, Losick R. Self-reinforcing activation of a cell-specific transcription factor by proteolysis of an anti-sigma factor in B. subtilis. Mol Cell. 2001;8:873–883. doi: 10.1016/s1097-2765(01)00362-8. [DOI] [PubMed] [Google Scholar]
- Persuh M, Turgay K, Mandic-Mulec I, Dubnau D. The N- and C-terminal domains of MecA recognize different partners in the competence molecular switch. Mol Microbiol. 1999;33:886–894. doi: 10.1046/j.1365-2958.1999.01544.x. [DOI] [PubMed] [Google Scholar]
- Prepiak P, Dubnau D. A peptide signal for adapter protein-mediated degradation by the AAA+ protease ClpCP. Mol Cell. 2007;26:639–647. doi: 10.1016/j.molcel.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid HR, Tamakoshi A, Sekiguchi J. Effects of mecA and mecB (clpC) mutations on expression of sigD, which encodes an alternative sigma factor, and autolysin operons and on flagellin synthesis in Bacillus subtilis. J Bacteriol. 1996;178:4861–4869. doi: 10.1128/jb.178.16.4861-4869.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer P, Millet J, Aubert J-P. Catabolic repression of bacterial sporulation. Proc Natl Acad Sci USA. 1965;54:704–711. doi: 10.1073/pnas.54.3.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schagger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- Shafikhani SH, Leighton T. AbrB and Spo0E control the proper timing of sporulation in Bacillus subtilis. Curr Microbiol. 2004;48:262–269. doi: 10.1007/s00284-003-4186-2. [DOI] [PubMed] [Google Scholar]
- Shafikhani SH, Mandic-Mulec I, Strauch MA, Smith I, Leighton T. Postexponential regulation of sin operon expression in Bacillus subtilis. J Bacteriol. 2002;184:564–571. doi: 10.1128/JB.184.2.564-571.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits WK, Bongiorni C, Veening JW, Hamoen LW, Kuipers OP, Perego M. Temporal separation of distinct differentiation pathways by a dual specificity Rap-Phr system in Bacillus subtilis. Mol Microbiol. 2007;65:103–120. doi: 10.1111/j.1365-2958.2007.05776.x. [DOI] [PubMed] [Google Scholar]
- Strauch M, Webb V, Spiegelman G, Hoch JA. The SpoOA protein of Bacillus subtilis is a repressor of the abrB gene. Proc Natl Acad Sci USA. 1990;87:1801–1805. doi: 10.1073/pnas.87.5.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauch MA, Trach KA, Day J, Hoch JA. Spo0A activates and represses its own synthesis by binding at its dual promoters. Biochimie. 1992;74:619–626. doi: 10.1016/0300-9084(92)90133-y. [DOI] [PubMed] [Google Scholar]
- Suel GM, Kulkarni RP, Dworkin J, Garcia-Ojalvo J, Elowitz MB. Tunability and noise dependence in differentiation dynamics. Science. 2007;315:1716–1719. doi: 10.1126/science.1137455. [DOI] [PubMed] [Google Scholar]
- Turgay K, Hahn J, Burghoorn J, Dubnau D. Competence in Bacillus subtilis is controlled by regulated proteolysis of a transcription factor. EMBO J. 1998;17:6730–6738. doi: 10.1093/emboj/17.22.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgay K, Hamoen LW, Venema G, Dubnau D. Biochemical characterization of a molecular switch involving the heat shock protein ClpC, which controls the activity of ComK, the competence transcription factor of Bacillus subtilis. Genes Dev. 1997;11:119–128. doi: 10.1101/gad.11.1.119. [DOI] [PubMed] [Google Scholar]
- van Sinderen D, Luttinger A, Kong L, Dubnau D, Venema G, Hamoen L. comK encodes the competence transcription factor, the key regulatory protein for competence development in Bacillus subtilis. Mol Microbiol. 1995;15:455–462. doi: 10.1111/j.1365-2958.1995.tb02259.x. [DOI] [PubMed] [Google Scholar]
- Veening JW, Smits WK, Kuipers OP. Bistability, epigenetics, and bet-hedging in bacteria. Annu Rev Microbiol. 2008;62:193–210. doi: 10.1146/annurev.micro.62.081307.163002. [DOI] [PubMed] [Google Scholar]
- Winkelman JT, Blair KM, Kearns DB. RemA (YlzA) and RemB (YaaB) regulate extracellular matrix operon expression and biofilm formation in Bacillus subtilis. J Bacteriol. 2009;191:3981–3991. doi: 10.1128/JB.00278-09. [DOI] [PMC free article] [PubMed] [Google Scholar]