

Abstract
Cisplatin (CDDP) is an effective anticancer agent for many solid tumors but has significant systemic toxicity limiting its use in many patients. We have designed a loco-regional delivery system to increase platinum levels in the lymphatics, where early metastasis is most likely to occur, while reducing systemic toxicities. CDDP was conjugated to a biocompatible polymer hyaluronan (HA), with a conjugation degree of approximately 20% (w/w). Conjugates were delivered via subcutaneous injection into the mammary fat pad of rats. Intravenous hyaluronan–cisplatin (HA–Pt) exhibited an increased plasma area under the curve (AUC) 2.7-fold compared to conventional CDDP but with a reduced peak plasma level (Cmax), and HA–Pt increased the ipsilateral lymph node AUC by 3.8-fold compared to CDDP. Urine creatinine was unchanged over 30 days following dosing of HA–Pt. This study demonstrates that intralymphatic drug delivery with polymer-conjugated platinum may provide greater tissue and systemic plasma concentrations of platinum than intravenous CDDP. In addition, localized particle delivery augmented distribution in the loco-regional tissue basin where tumor burden predominates, while renal toxicity compared to standard intravenous CDDP was significantly reduced.
Keywords: cancer chemotherapy, pharmacokinetics, biodegradable polymers, controlled release, lymphatic transport, polymeric drug carrier, polymeric drug delivery systems
Introduction
Since the FDA's approval of CDDP in 1977 for the treatment of metastatic ovarian cancer,1 CDDP has emerged as the principal chemotherapeutic agent for many solid tumors, despite newer platinum drugs with fewer side effects. CDDP has a high initial response rate in many cancers including head and neck squamous sarcomas, lymphomas, small cell and nonsmall cell lung, testicular, ovarian, gastric, esophagus, and pancreatic cancers.2 Although not clinically employed as a single agent, CDDP is highly active in combination with chemotherapeutic regimens for breast cancer,3 but the significant dose-limiting toxicity of CDDP has hampered its use in many patients.
Nephrotoxicity is the most dangerous and insidious of CDDP's side effects, as exhibited by renal tubular inflammation and necrosis.4 After administration, CDDP binds tightly to plasma proteins and becomes highly concentrated compared to the plasma or other organs in the kidneys and free CDDP is filtered.5 CDDP efficacy is well correlated with AUC but not with maximum plasma concentration (Cmax);6 however, toxicity is increased by high peak drug concentrations, such as immediately after bolus injection or during short i.v. infusions, further increasing the risk of nephrotoxicity.2 Fractional or metronomic dosing schedules that divide the same total dose of CDDP over several, for example, daily, smaller injections, have been shown to significantly reduce nephrotoxicity and ototoxicity6,7 due to lower peak plasma concentration. However, metronomic dosing requires a more frequent dosing schedule, longer in-hospital stays, and leads to increased care costs thereby limiting its use.
Since nonspecific organ toxicities hamper CDDP chemotherapy, oncologists have developed procedures to confine chemotherapy to the diseased areas by temporarily isolating the affected tissues or organs from the systemic circulation and perfusing them with CDDP. For example, intraarterial percutaneous pelvic perfusion of high-dose CDDP has demonstrated a therapeutic advantage in advanced uterine cervical carcinoma8 while achieving a low side-effect profile. However, these treatments are highly invasive and require specialized skills and equipment usually restricted to large medical research centers. In addition, tissue isolation is not possible in many clinical cases, including locally advanced breast cancer that has significant invasion into lymphatic tissues.
Treatment of locally advanced cancers may be improved if chemotherapy were concentrated to the draining lymphatics, while maintaining adequate systemic levels for the treatment of distant metastases. Previously we compared direct injection into rat breast tissue of CDDP with a silver-activated conjugate of CDDP;9 however, local injection of CDDP is not clinically feasible due to extensive tissue damage. Furthermore, tumor studies demonstrated that the silver activated conjugate to cause premature animal death, which promoted our development of a new conjugate that does not require silver. In this report, we examine a conjugate of the polysaccharide HA with CDDP that can concentrate CDDP to the lymphatics and reduce peak plasma concentrations. Localized conjugate chemotherapy significantly increases lymphatic tissue concentrations over systemic therapy and reduces organ toxicities including nephrotoxicity in rats.
Materials and Methods
HA from microbial fermentation was purchased from Lifecore Biomedical (Chaska, MN) as sodium hyaluronate and used without further purification. Heparin solution was purchased from Abraxis Pharmaceutical Products (Schaumburg, IL). All other reagents were purchased from Fisher Scientific (Pittsburgh, PA) or Sigma–Aldrich (St. Louis, MO) and were of ACS grade or better. Milli-Q water was used in all experiments. Animal procedures were approved by the University of Kansas Institutional Animal Care and Use Committee. Sprague–Dawley rats were purchased from Charles River Laboratories (Wilmington, MA). Cell lines were obtained from American Tissue Culture Collection (ATCC, Manassas, VA) and cultured according to the provided protocols unless noted otherwise. Caution: CDDP is extremely toxic and all chemical waste and animal tissues were treated as hazardous waste and disposed of accordingly.
Synthesis and Characterization of Hyaluronan–CDDP (HA–Pt) Conjugates
CDDP was conjugated to HA (35,000 g/mol) based on previously reported procedures,9,10 with and without the addition of silver nitrate as an activating agent. Typically, HA (100 mg) and CDDP (45 mg) were dissolved in H2O (20 mL) and stirred in the dark for 4 days under argon at ambient temperature (ca. 25°C). The reaction mixture was filtered (0.2-μm nylon membrane) and dialyzed against H2O (10,000 MWCO; Pierce, Rockford, IL) for 48 h at 4°C protected from light. Following dialysis, the crude product was concentrated and stored at 4°C. The degree of CDDP substitution was determined by atomic absorption spectroscopy (AAS) (Varian SpectrAA GTA-110 with graphite furnace). The furnace program was as follows: ramp to 25–80°C, hold 2 s, ramp to 120°C, hold 10 s, ramp to 1000°C, hold 5 s, ramp to 2700°C, hold 2 s, cool to 25°C over 20 s. The graphite partition tube was cleaned in every 40 samples by baking at 2800°C for 7 s. Argon was used as the injection and carrier gas.
The size of the HA–Pt conjugate was determined by field emission transmission electron microscopy (FEI, Hillsboro, OR). A concentrated solution of the HA–Pt conjugates was coated on a lacey carbon-coated copper grid (Ted Pella, Inc., Redding, CA) before imaging. Diameters were determined using ImageJ (National Institutes of Health, http://rsbweb.nih.gov/ij/) for Fert's sizing analysis (n = 40 particles).
The in vitro release rate of the active hydrated form of the cisplatin (cis-[Pt(NH3)2 (H2O)2]+) from the HA– Pt conjugate was determined in both water and phosphate-buffered saline (PBS, pH 7.4, 140 mM NaCl) solution. HA–Pt was added to a 10,000 MWCO dialysis bag (Pierce) and placed in a water or PBS bath and stirred at 37°C. The bath volume (3 L) was replaced every 4 h to maintain sink conditions. Samples were taken from the dialysis bag at predetermined time points, and the remaining Pt concentration was determined by AAS.
The lymphatically metastatic breast cancer cell line MDA-MB-468LN, kindly provided by Ann Chambers of the London Health Sciences Center, was maintained in modified Eagle's medium alpha supplemented with 10% fetal bovine serum, 1% l-glutamine, and 0.4 mg/mL G418 (geneticin).9,11 Additional cell lines MDA-MB-231, MCF-7, JMAR, and MDA-1986 were maintained in DMEM with 10% fetal bovine serum and 1% penicillin/streptomycin. Preceding proliferation studies, cells were trypsinized and seeded into 96-well plates (5000 cells/well). After 24 h, CDDP, HA–Pt, or HA was added (n = 12; seven concentrations), and 72 h postaddition, resazurin blue in 10 μL of PBS was added to each well (final concentration of 5 μM). After 4 h, well fluorescence was measured (λex 560 nm, λem 590 nm) using a fluorophotometer (SpectraMax Gemini; Molecular Devices, Sunnyvale, CA). IC50 was determined as the midpoint between saline (positive) and cell-free (negative) controls for each plate. Each experiment was repeated in triplicate.
Pharmacokinetics and Tissue Distribution
Sprague–Dawley rats (female, 200–250 g) were cannulated in the left jugular vein under isoflurane and allowed to recover overnight. Animals were then administered i.v. CDDP, i.v. HA–Pt, s.c. HA–Pt, or s.c. CDDP (1.0 or 3.3 mg/kg equivalent CDDP; n = 5 for each group) under isoflurane anesthesia. Subcutaneous injections were given in the uppermost right mammary fat pad of the animal. Whole blood was withdrawn (100 μL) from the cannula at 0, 5 min, 0.5 h, 1 h, 2 h, 4 h, 6 h, 12 h, 24 h, 48 h, and 96 h after dosing and placed into 2-mL centrifuge tubes pretreated with heparin. The cannula was washed before and after withdrawal with saline and then heparin locked. The whole blood was centrifuged at 17,000×g for 5 min, and the plasma was frozen at −80°C until analysis. In tissue distribution studies, age and weight matched animals were administered the drugs and euthanized at the stated time points. The right ipsilateral axilla nodes (treated side), left contralateral axilla nodes (control side), and major organs (liver, kidneys, heart, spleen, lungs, brain, muscle, bladder) were excised; washed with 0.9% saline; and stored at −80°C until analysis. Tissue samples were prepared using a procedure reported previously.9 Typically, 50 mg of tissue sample was digested using 1.5 mL of 6.7% nitric acid for 2 h at 80°C. After digestion, samples were homogenized (Tissue Tearor; BioSpec Products, Inc., Bartlesville, OK) and centrifuged. The supernatant and plasma samples were analyzed by AAS as described in the Synthesis and Characterization of Hyaluronan– CDDP (HA–Pt) Conjugates.
Toxicology Analysis
Sprague–Dawley rats (35 females) were randomly divided into study groups of five animals each and dosed: 1.0 mg/kg s.c. HA–Pt (with and without silver; platinum equivalent to 1.0 mg/kg CDDP), 3.3 mg/kg s.c. HA–Pt (with and without silver), i.v. CDDP at 1.0 and 3.3 mg/kg s.c.. HA (control; HA equivalent to 3.3 mg/kg HA–Pt). Each animal was administered a single bolus dose at the beginning of the 30-day study period. Urine samples were collected every day during the first 2 weeks of the study and every 4 days during the third and fourth week of the studies (except for the 3.3 mg/kg HA–Pt with silver group). In order to reduce the morbidity to animals, subjects were housed in metabolic cages for 12 h to collect approximately 5 mL of urine and then returned to cages with bedding until the next collection period. Urine samples were centrifuged at 17,000g for 5 min and stored in −80°C freezer until creatinine analysis.
Creatinine Analysis
Urine creatinine was analyzed using the Quanti-Chrom™ Creatinine Assay Kit according to the manufacturer's instructions (BioAssay Systems, Hayward, CA). Creatinine concentration of the sample was calculated as (ODSAMPLE 5 − ODSAMPLE 1)/(ODSTD 5 − ODSTD 1) × [STD] (mg/dL). ODSAMPLE 5, ODSAMPLE 1, ODSTD 5, and ODSTD 1 are OD510 nm values of sample and standard at 5 and 1 min, respectively. [STD] is 50 mg/dL for the urine assay.
Results
Synthesis and Analysis of HA–Pt Conjugates
The structure of CDDP lends itself to complex formation with polycarboxylic polymers, since one or more of the chlorides can be displaced allowing formation of a labile ester linkage with the polymer.12 CDDP was highly conjugated to HA with typical conjugations of 0.20 (w/w) platinum/complex (approximately 65% CDDP conjugation efficiency). In previous studies, we synthesized CDDP conjugates by first activating HA with AgNO3, but we found eliminating this step does not significantly reduce conjugation and eliminates potential silver toxicity. The AAS produced a linear curve in the range of 10– 450 ng/mL (R2 = 0.999) with a limit of detection of 5 ng/mL. Concentrated samples were diluted with 0.1% nitric acid into the linear analytical range prior to analysis.
The HA–Pt conjugates formed individual particles of approximately 7.55 ± 1.72 nm (n = 40 particles), with some larger clusters (ca. 25.20 ± 4.43 nm, n ± 40 groups) of conjugates. Clustering was likely due to the formation of alternating hydrophobic regions created by the eight adjacent CH groups that are present for every three disaccharides (Fig. 1). Overall, the formed particles were within the scopic size range shown to have good lymphatic uptake and nodal retention.13
Figure 1.

TEM image of HA-Pt conjugates.
The in vitro release of platinum from the complexes exhibited pseudo-first-order release kinetics with a release half-life of 42 h in water and 10 h in PBS (Fig. 2). The release rate constant was determined to be 0.0148 s−1 in water and 0.0700 s−1 in PBS, respectively. The chloride ion in PBS was expected to more rapidly displace the carboxylate group bound to the platinum, increasing the release rate of the hydrated platinum.
Figure 2.
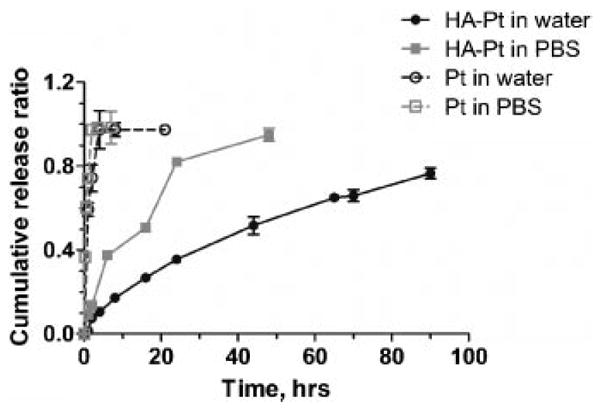
In vitro release of CDDP from HA-Pt conjugate. Experiments were conducted in both water (black) and PBS (grey).
Cell toxicity was determined as the reduction in cell proliferation over 72 h. No appreciable difference in toxicity was detected between CDDP and HA–Pt in three human breast cancer cell lines and two head and neck squamous cell carcinoma lines (Tab. 1). HA showed no toxicity at 10 mg/mL, the upper limit of testing in all cell lines compared with saline controls (data not shown).
Table 1. IC50 Values of CDDP and HA–Pt in Human Cancer Cells.
| Cell Lines/IC50, μg/mL (μM) | CDDP | HA–Pt |
|---|---|---|
| MDA-MB-468LN | 5.0 μg/mL (17 μM) | 10 μg/mL (33 μM) |
| MDA-MB-231 | 6.0 μg/mL (20 μM) | 10 μg/mL (33 μM) |
| MCF-7 | 6.0 μg/mL (20 μM) | 11 μg/mL (37 μM) |
| JMAR | 2.0 μg/mL (6.8 μM) | 1.9 μg/mL (6.3 μM) |
| MDA-1986 | 2.4 μg/mL (8.1 μM) | 1.9 μg/mL (6.2 μM) |
Doses are given on a relative Pt basis and cells were exposed to drugs for 72 h followed by the resazurin blue assay for cell proliferation (n = 12 wells × 3 assays).
Pharmacokinetics and Tissue Distribution
The pharmacokinetic parameters of HA–Pt and CDDP were determined by fitting individual animals' data to a two-compartment model with SAAM II (version 1.2, University of Washington; Tab. 2). The AUC of s.c. HA–Pt was 660% greater than i.v. CDDP. The plasma half life (t1/2) of s.c. HA–Pt was 200% greater than i.v. CDDP and 490% greater than i.v. HA–Pt. Clearance rates (Cl) of s.c. HA–Pt and i.v. HA–Pt were not significantly different, but the total body clearance (Cl) of i.v. CDDP was 590% greater than s.c. HA–Pt. The steady-state volume of distribution (Vss) of platinum was greater for the i.v. CDDP than s.c. or i.v. HA–Pt.
Table 2. Pharmacokinetics of Formulations.
| Parameters | Unit | CDDP i.v.a | HA–Pt i.v.b | HA–Pt s.c.a,b,c | CDDP s.c.c |
|---|---|---|---|---|---|
| Vss | L/kg | 8.310 ± 3.508 | 0.154 ± 0.038 | 3.628 ± 0.760 | 3.585 ± 2.420 |
| AUC0–inf | (μg h)/mL | 14.104 ± 4.570 | 141.176 ± 38.878 | 107.122 ± 59.062 | 56.707 ± 27.102 |
| Cl | L/(kg h) | 0.257 ± 0.097 | 0.030 ± 0.007 | 0.037 ± 0.028 | 21.790 ± 13.749 |
| Cmax | μg/mL | 4.025 ± 0.265 | 59.864 ± 5.504 | 1.035 ± 0.688 | 2.713 ± 0.381 |
| MRT | h | 5.148 ± 2.510 | 2.640 ± 1.153 | 15.497 ± 4.943 | 15.263 ± 7.554 |
| t1/2 (β) | h | 3.568 ± 1.740 | 1.830 ± 0.800 | 10.742 ± 3.426 | 10.580 ± 5.236 |
Data were modeled using a two-compartment model for individual animals and then averaged for each group (n = 5, average ± std).
Study groups, i.v. CDDP and s.c. HA–Pt, differed significantly for all parameters.
Study groups, i.v. HA–Pt and s.c. HA–Pt, differed significantly for Vss, Cmax, MRT, and t1/2. Significance defined as p < 0.05 using Student's t-test (n = 5).
Study groups, s.c. CDDP and s.c. HA–Pt differed significantly for Cl and Cmax.
The s.c. HA–Pt preferentially accumulated in the draining ipsilateral axillary lymph nodes as compared to the i.v. CDDP control (Fig. 3); preferential accumulation was still evident at 48 h postinjection despite an in vitro disassociation half-life of Pt from HA of 10 h.9 The ipsilateral axillary node AUC of HA–Pt when injected locally was 279% greater than i.v. CDDP node levels (p < 0.001), and the peak node concentration (Cmax) of HA–Pt was 520% greater than i.v. CDDP. The relative organ AUCs for i.v. CDDP were kidneys > axilla nodes > spleen > liver > bladder > brain > muscle > lungs > heart; for s.c. HA–Pt, ipsilateral axilla nodes > kidneys > liver > heart > contralateral axilla nodes > spleen > lungs > brain > muscle > bladder. The ratio of tissue AUCs to plasma AUCs (i.e., tissue clearance) for i.v. CDDP were kidneys > axilla nodes > spleen > liver > brain > bladder > muscle > lungs > heart; for s.c. HA–Pt, ipsilateral axilla nodes > kidneys > liver > heart > contralateral axilla nodes > spleen > lungs > brain > muscle > bladder. CDDP was injected s.c. as a comparison to the carrier HA–Pt. There was no apparent increase in concentration in the draining lymph nodes, but the muscle tissue near the injection site had the highest platinum concentrate relative to the other tissues.
Figure 3.
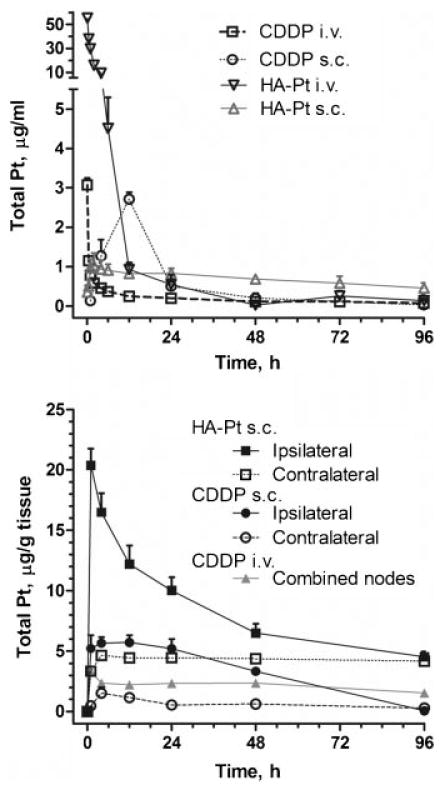
Plasma concentration of Pt after i.v. or s.c. injection of CDDP or HA-Pt (3.3 mg/kg Pt-basis) (top). Concentration of Pt in ipsilateral (right) and axillary nodes and contralateral (left) axillary nodes after i.v. or s.c. injection of CDDP or s.c. injection of HA-Pt conjugates (3.3 mg/kg Pt-basis) into the right mammary fatpad.
Toxicology
Urine creatinine levels are an indirect indicator of glomerular filtration rate which can indicate changes in renal function and renal toxicity. A decrease in creatinine excretion corresponds to a decreased renal function and possible renal toxicity or damage. Significant renal toxicity was observed in animals administered our initial HA–Pt silver conjugate (3.3 mg/kg), with a 30% decrease in creatinine excretion apparent at 3 days and a 70% decrease at 4 days (Fig. 4). All animals in this group died within 1 week of treatment due to drug-related cachexia. In contrast, the HA–Pt did not demonstrate significant toxicity, and creatinine levels remained near predosing levels throughout the study's duration. Toxicity as measured by creatinine excretion was compared to our previous silver formulation, which had not been previously assessed. The silver-free formulations reported here demonstrate significantly less renal toxicity. The higher levels of platinum in neural tissues may potentiate toxicity, brain sections of the treated animals were preserved in 80% alcoholic formalin solution, stained with hematoxylin and eosin, and then examined and graded by an independent blinded veterinarian pathologist (Veterinary Lab Resources; University of Kansas Medical Center). The brain tissues of the CDDP and HA–Pt treated groups had no microscopic lesions or abnormal findings in any of the animals (n = 4/treatment) (data not shown).
Figure 4.
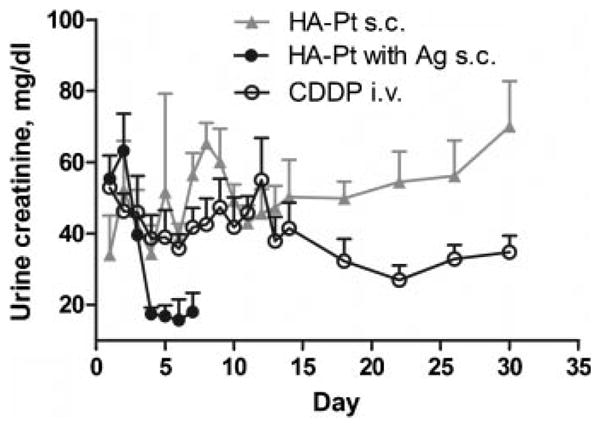
Urine creatinine concentration of animals that received 3.3 mg/kg s.c. HA-Pt with or without silver or i.v. CDDP. Student t-test analysis revealed urine creatinine level for study groups, i.v. CDDP and s.c. HA-Pt without silver, differed significantly at day 22 and 30, and urine creatinine level for study groups, s.c. HA-Pt with and without silver, differed significantly at day 4, 6 and 7 (n = 5, p < 0.05).
Discussion
CDDP is one of the most widely used chemotherapeutic agents for solid tumors; however, its toxicity and chemotherapeutic tumor resistance severely limits its dose and use in many patients. Penetration of systemic CDDP into the lymphatics may be suboptimal, and alternatives treatments for localized cancers, that is, surgical removal or radiation, can lead to serious side effects such as lymphedema.14,15 Our objective in this study was to determine if platinum therapy delivered to the loco-regional tissues and lymphatics, using a FDA-approved biocompatible carrier, can increase drug exposure in the most tissues prone to toxicity and decrease renal toxicity compared to traditional i.v. therapy with CDDP. A delivery system for CDDP with reduced toxicity and ability to be administered through intralymphatic routes would have important implications in the treatment of many CDDP responsive cancers, for example, breast, nonsmall cell lung, ovarian, and head and neck squamous cell carcinoma, as the lymphatics are involved in early-to-late diseases.
Numerous targeting and carrier strategies have been reported to increase the dosage of CDDP reaching tumor-bearing tissues, while sparing normal tissues from toxic doses. These technologies can be categorized into untargeted or passively targeted carriers and actively targeted carriers, for example, antibodies. Among the untargeted carriers, polymeric micelle formulations, that is, NC-6004, have demonstrated reduced nonspecific toxicities in preclinical studies and have progressed to early clinical trials.16 In phase I trials, an untargeted linear polymer conjugate of N-(2-hydroxypropyl)methacrylamide (HMPA) and platinum, AP5280, had demonstrated higher sustained plasma concentrations of platinum in humans with minimum toxicity compared to i.v. CDDP.17 These untargeted carriers rely on the enhanced permeability and retention effect (EPR) to improve tumor accumulation of the drug,18 but in tumors that are not highly vascularized, the EPR effect is greatly reduced and untargeted carriers have less advantages.19 We sought to improve treatment of tumors, regardless of tumor vascularity, by selectively confining treatment to the loco-regional lymphatics of the tumor via a subcutaneous injection at the site where the primary tumor would exist, allowing the drug to be delivered along the lymphatic pathway where tumors are most likely to initially metastasize.
HA is a natural polysaccharide, of alternating d-glucuronic acid and N-acetyl d-glucosamine, found endogenously in the extracellular matrix of connective tissues. HA is also a ligand for CD44 receptor and is cleared primarily by the lymphatic system where it is catabolized in the nodes by CD44 receptor-mediated endocytosis followed by lysosomal degradation. Coupled with rapid uptake and a short half-life of 12–72 h,20 HA was seen as an ideal carrier for the treatment of lymphatic tumors. HA has a known safety profile unlike other possible carriers such as polyamino acids.21 Each disaccharide unit of HA contains a carboxylic acid, facilitating coupling of CDDP.10–12 In a previous report, we first activated the HA with silver nitrate prior to conjugation with CDDP, as this has been reported to improve conjugation efficiency.12 However, we found it extremely difficult to remove all traces of silver from the resulting conjugates, even after multiple rounds of extended dialysis against water. In our initial pharmacokinetic and toxicity studies, a significant number of animals succumbed to silver-induced toxicity as determined by pathological examination. This led us to change our formulation scheme to eliminate silver from the conjugation procedure and reoptimize the reaction for highest conjugation efficiency, which ultimately did not impair formation of the HA–Pt conjugates. This is a significant advancement in terms of clinical development as CDDP and HA are both approved by the FDA for use in humans and no additional substances are required for formation of the complex. The resulting complexes still had excellent antiproliferation activity against multiple breast cancer lines in vitro, indicating the change in formulation method does not affect cell-based drug efficacy.
The pharmacokinetics of s.c. HA–Pt was greatly altered compared to the same carrier administered i.v. or the standard i.v. CDDP formulation. The greatly increased AUC of s.c. HA–Pt is consistent with sustained release of platinum by the carrier. This effect may be due both to the delayed diffusion of platinum from the s.c. injection site and the slow release of conjugated platinum from the carrier as i.v. administration of HA–Pt also resulted in relatively large AUC. In a previous study, we determined that CDDP administered s.c. gives rise to a comparable AUC, although this known vesicant can be highly inflammatory to tissues, which may alter the diffusion rate. The relatively small Vss and Cl and large AUC of i.v. HA–Pt indicates platinum release from the carrier may be slow. CDDP is readily eliminated renally but the highly molecular weight of HA–Pt precludes rapid renal clearance. Pharmacokinetic modeling suggests the carrier also slowed the rate constant of distribution transfer of platinum from the plasma compartment to the tissues, which was approximately 10-fold slower for i.v. HA–Pt compared to i.v. CDDP. This was possible due to the ionization state and high molecular weight of HA, which is naturally cleared from the body by lymphatic transport into the systemic circulation followed by enzymatic degradation in the liver. CDDP administered s.c. did not significantly improve node concentrations, giving slightly lower exposure than i.v. CDDP. Indeed, the majority of CDDP administered s.c. was retained in the surrounding muscle tissue. Since CDDP is a known vesicant, subcutaneous administration is not a safe and feasible method in the absence of a drug carrier such as the HA conjugate we have described.
Clinical studies have indicated that CDDP's severe dose-limiting side effects are likely due to the high peak plasma levels (Cmax) experienced immediately after i.v. administration. Recent applications of metronomic dosing regimens have been shown to decrease toxicity although they are clinically cumbersome and inconvenient and resulted in increased costs to patients.6,7 Based on our preliminary experience with this delivery system, locally administered carrier chemotherapeutics may avoid high peak concentrations due to slow release of platinum from the carrier, with a half-life in saline of around 11 h compared to 4 h for the standard CDDP formulation,9 and also as a result of slow platinum diffusion from the tissues or lymph into the systemic circulation after release from the carrier.
We expected lymphatic delivery of platinum would reduce systemic tissue exposure compared to i.v. CDDP, but s.c. HA–Pt had a much greater AUC than i.v. CDDP in most tissues including the kidneys (Tab. 3). Vd is affected by partition coefficient and tissue and plasma protein binding which may be different between the parent CDDP and our conjugate HA–Pt. However, the urine creatinine studies did not indicate an increase in toxicity. The reduction in toxicity could be due to protection of tissues by the carrier; HA frequently is used in the clinic as a rescue therapy after accidental venipuncture by intravenous lines. The predominant chemical form of platinum in the kidneys may differ significantly between the two formulations as AAS measures only total elemental platinum, so it is unknown if platinum from HA–Pt was detoxified before renal uptake. This could provide an explanation as why the higher accumulated AUC in the kidney seen with HA–Pt was not as destructive as i.v. CDDP. CDDP is believed to become nephrotoxic after displacement of a single chloride leads to conjugation of the positively charged aqua ligand and glutathione, followed by a series of metabolic reactions leading to cysteine conjugates that can produce highly reactive thiol species in the proximal tubule cells.22 Since the mechanism of platinum release and metabolism from HA is still under investigation, it is unknown if platinum reaching the kidneys from HA–Pt complexes is significantly detoxified unlike the free drug in i.v. CDDP. On-going studies are examining the distribution of carrier-bound and free platinum at the cellular level in the tissues and tumors of xenografts by transmission electron microscopy.
Table 3. Tissue AUC (Average ± Std) and Cmax (Average ± Std) of 3.3 mg/kg i.v. CDDP and s.c. HA–Pt Study Groups.
| AUC0–96 h (μg/g h) | Cmax (μg/g) (Tmax) | |||||
|---|---|---|---|---|---|---|
| Tissue | CDDP i.v. | HA–Pt s.c. | CDDP s.c. | CDDP i.v.a | HA–Pt s.c. | CDDP s.c. |
| Heart | 128 ± 8 | 465 ± 23 | 422 ± 120 | 1.7 ± 0.1 (1 h) | 14.7 ± 1.1 (1 h) | 13.6 ± 8.5 (4 h) |
| Lungs | 139 ± 7 | 347 ± 13 | 170 ± 92 | 2.2 ± 0.1 (1 h) | 7.5 ± 0.7 (1 h) | 9.0 ± 6.7 (4 h) |
| Kidneys | 291 ± 8 | 669 ± 28 | 517 ± 76 | 4.6 ± 0.4 (1 h) | 12.3 ± 2.5 (4 h) | 10.5 ± 3.1 (4 h) |
| Brain | 152 ± 11 | 344 ± 22 | 341 ± 57 | 2.3 ± 0.1 (1 h) | 7.5 ± 2.0 (4 h) | 9.4 ± 4.2 (4 h) |
| Liver | 178 ± 5 | 495 ± 20 | 359 ± 33 | 2.9 ± 0.2 (4 h) | 10.6 ± 2.2 (4 h) | 10.8 ± 2.5 (12 h) |
| Spleen | 201 ± 7 | 384 ± 15 | 299 ± 34 | 3.0 ± 0.4 (1 h) | 6.6 ± 0.6 (4 h) | 8.9 ± 1.6 (4 h) |
| Muscle | 151 ± 9 | 262 ± 15 | 367 ± 27 | 2.2 ± 0.2 (1 h) | 8.5 ± 2.1 (4 h) | 11.9 ± 5.9 (4 h) |
| Bladder | 162 ± 10 | 194 ± 6 | 50 ± 38 | 3.1 ± 0.4 (1 h) | 2.9 ± 0.4 (4 h) | 2.0 ± 1.1 (1 h) |
| Ipsilateral axilla nodes | 205 ± 12b | 776 ± 9 | 312 ± 19 | 3.3 ± 0.3 (1 h) | 20.4 ± 1.4 (1 h) | 1.7 ± 1.0 (4 h) |
| Contralateral axilla nodes | 205 ± 12b | 413 ± 17 | 60 ± 41 | 3.3 ± 0.3 (1 h) | 4.6 ± 0.3 (4 h) | 5.7 ± 1.3 (12 h) |
Two-way ANOVA analysis revealed study groups (i.v. CDDP and s.c. HA–Pt) differed significantly for all tissues AUCs, p < 0.001 (n = 5).
The first tissue sampling was at 1 h so a Cmax prior to this would not be detected. The first plasma sampling was at 5 min.
For i.v. CDDP, the bilateral axilla nodes were assayed together.
The increased tissue concentrations of platinum achieved with s.c. HA–Pt compared to i.v. CDDP carries two potential advantages: (1) this could result in a lower dose of platinum being required to achieve the same tissue effect such that the HA–Pt dose may be reduced by several fold and still maintain equivalent tissue levels; and (2) maintaining therapeutic systemic concentrations of drug is important for utilizing this drug as an adjuvant therapy since it is well known that most patients with cancers which have metastasized to the loco-regional lymph nodes likely have micro in addition to systemic macrometastases. Therefore, this treatment if extendable to the clinical situation could potentially replace daily systemic intravenous therapy utilizing a less invasive and less-frequent dosing schedule, for example, weekly subcutaneous dosing as compared to current therapy which is daily intravenous infusion, while simultaneously providing augmented local drug delivery and exposure over time to the loco-regional tumor basin and lymphatics. The larger tissue AUC of s.c. HA–Pt may also increase rates of tumor apoptosis since a recent study found that a prolonged subtoxic level of CDDP can substantial improve tumor cell apoptosis compared to a single high dose.23 Overall, assuming linear pharmacokinetics, the total platinum dose for local HA–Pt therapy could be reduced by 75% while maintaining the same systemic and node concentrations of platinum as i.v. CDDP, possibly with lower concentrations in the other tissues with the exception of the heart.
Conclusion
We have synthesized a conjugate of HA and CDDP that concentrates platinum in the breast lymphatics after subcutaneous injection into the mammary fat pad with excellent tolerability. These conjugates increased the plasma and tissue AUC of platinum and increased the mean residence time and terminal elimination half-life. No injection site or lymph node toxicities were observed; neither were any systemic toxicities observed in the kidneys or brain on pathological examination. Ongoing xenograft studies are examining HA–Pt conjugate efficacy in locally aggressive metastatic cancers, including xenograft tumor models of breast cancer and head and neck squamous cell carcinomas. We believe this intralymphatic delivery platform may offer significant advantages for the use of platinum medicines in the management of locally advanced cancers, treating microscopic lymph node disease with better efficacy than current high-dose systemic chemotherapy without additional toxicities.
Acknowledgments
This work was supported by awards from the National Institutes of Health (R21 CA132033 and P20 RR015563) and the American Cancer Society (RSG-08-133-01-CDD).
Abbreviations
- AUC
area under the curve
- CDDP
cis-diamminedichloroplatinum(II) or cisplatin
- Cmax
peak measured concentration of drug
- HA
hyaluronan
- HA–Pt
hyaluronan–CDDP conjugate
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.FDA oncology tools approval summary for cisplatin for metastatic ovarian tumors. [cited December 12, 2008];2008 Available from: http://www.accessdata.fda.gov/scripts/cder/onctools/summary.cfm?ID=73.
- 2.Launay-Vacher V, Rey JB, Isnard-Bagnis C, Deray G, Daouphars M. Prevention of cisplatin nephrotoxicity: State of the art and recommendations from the European Society of Clinical Pharmacy Special Interest Group on Cancer Care. Cancer Chemother Pharmacol. 2008;61:903–909. doi: 10.1007/s00280-008-0711-0. [DOI] [PubMed] [Google Scholar]
- 3.Stemmler HJ, Kahlert S, Brudler O, Beha M, Muller S, Stauch B, Heinemann V. High efficacy of gemcitabine and cisplatin plus trastuzumab in patients with HER2-overexpressing metastatic breast cancer: A phase II study. Clin Oncol (R Coll Radiol) 2005;17:630–635. doi: 10.1016/j.clon.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 4.Pabla N, Dong Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008;73:994–1007. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- 5.Safirstein R, Miller P, Guttenplan JB. Uptake and metabolism of cisplatin by rat kidney. Kidney Int. 1984;25:753–758. doi: 10.1038/ki.1984.86. [DOI] [PubMed] [Google Scholar]
- 6.Shimizu Y, Hasumi K. Chemotherapy with consecutive low-dose CDDP combined with 5-FU for gynecologic malignancies. Gan To Kagaku Ryoho. 1999;26:1564–1574. [PubMed] [Google Scholar]
- 7.Zuur CL, Simis YJ, Verkaik RS, Schornagel JH, Balm AJ, Dreschler WA, Rasch CR. Hearing loss due to concurrent daily low-dose cisplatin chemoradiation for locally advanced head and neck cancer. Radiother Oncol. 2008;89:38–43. doi: 10.1016/j.radonc.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 8.Maruo T, Motoyama S, Hamana S, Yoshida S, Ohara N, Yamasaki M, Ku Y. Percutaneous pelvic perfusion with extracorporeal chemofiltration for advanced uterine cervical carcinoma. Surg Oncol Clin N Am. 2008;17:843–856. doi: 10.1016/j.soc.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Cai S, Xie Y, Bagby TR, Cohen MS, Forrest ML. Intralymphatic chemotherapy using a hyaluronan-cisplatin conjugate. J Surg Res. 2008;147:247–252. doi: 10.1016/j.jss.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeong YI, Kim ST, Jin SG, Ryu HH, Jin YH, Jung TY, Kim IY, Jung S. Cisplatin-incorporated hyaluronic acid nanoparticles based on ion-complex formation. J Pharm Sci. 2008;97:1268–1276. doi: 10.1002/jps.21103. [DOI] [PubMed] [Google Scholar]
- 11.Vantyghem SA, Allan AL, Postenka CO, Al-Katib W, Keeney M, Tuck AB, Chambers AF. A new model for lymphatic metastasis: Development of a variant of the MDA-MB-468 human breast cancer cell line that aggressively metastasizes to lymph nodes. Clin Exp Metastasis. 2005;22:351–361. doi: 10.1007/s10585-005-0745-1. [DOI] [PubMed] [Google Scholar]
- 12.Maeda M, Takasuka N, Suga T, Uehara N, Hoshi A. Antitumor activity of a new series of platinum complexes: Trans(+/−)-1,2-cyclohexanediammineplatinum(II) conjugated to acid polysaccharides. Anticancer Drugs. 1993;4:167–171. doi: 10.1097/00001813-199304000-00006. [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi H, Kawamoto S, Choyke PL, Sato N, Knopp MV, Star RA, Waldmann TA, Tagaya Y, Brechbiel MW. Comparison of dendrimer-based macromolecular contrast agents for dynamic micro-magnetic resonance lymphangiography. Magn Reson Med. 2003;50:758–766. doi: 10.1002/mrm.10583. [DOI] [PubMed] [Google Scholar]
- 14.Tejler G, Aspegren K. Complications and hospital stay after surgery for breast cancer: A prospective study of 385 patients. Br J Surg. 1985;72:542–544. doi: 10.1002/bjs.1800720714. [DOI] [PubMed] [Google Scholar]
- 15.Vinton AL, Traverso LW, Jolly PC. Wound complications after modified radical mastectomy compared with tylectomy with axillary lymph node dissection. Am J Surg. 1991;161:584–588. doi: 10.1016/0002-9610(91)90905-s. [DOI] [PubMed] [Google Scholar]
- 16.Uchino H, Matsumura Y, Negishi T, Koizumi F, Hayashi T, Honda T, Nishiyama N, Kataoka K, Naito S, Kakizoe T. Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br J Cancer. 2005;93:678–687. doi: 10.1038/sj.bjc.6602772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rademaker-Lakhai JM, Terret C, Howell SB, Baud CM, De Boer RF, Pluim D, Beijnen JH, Schellens JH, Droz JP. A phase I and pharmacological study of the platinum polymer AP5280 given as an intravenous infusion once every 3 weeks in patients with solid tumors. Clin Cancer Res. 2004;10:3386–3395. doi: 10.1158/1078-0432.CCR-03-0315. [DOI] [PubMed] [Google Scholar]
- 18.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J Control Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 19.Ogawara K, Un K, Minato K, Tanaka K, Higaki K, Kimura T. Determinants for in vivo anti-tumor effects of PEG liposomal doxorubicin: Importance of vascular permeability within tumors. Int J Pharm. 2008;359:234–240. doi: 10.1016/j.ijpharm.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 20.Fraser JR, Laurent TC. Turnover and metabolism of hyaluronan. Ciba Found Symp. 1989;143:41–53. discussion 53–59, 281–285. [PubMed] [Google Scholar]
- 21.Dünne AA, Boerner HG, Kukula H, Schlaad H, Wiegand S, Werner JA, Antonietti M. Block copolymer carrier systems for translymphatic chemotherapy of lymph node metastases. Anticancer Res. 2007;27:3935–3940. [PubMed] [Google Scholar]
- 22.Townsend DM, Deng M, Zhang L, Lapus MG, Hanigan MH. Metabolism of cisplatin to a nephrotoxin in proximal tubule cells. I. Am Soc Nephrol. 2003;14:1–10. doi: 10.1097/01.asn.0000042803.28024.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kishimoto S, Kawazoe Y, Ikeno M, Fukushima S, Takeuchi Y. Continuous exposure to low-dose cisplatin and apoptosis. Biol Pharm Bull. 2005;28:1954–1957. doi: 10.1248/bpb.28.1954. [DOI] [PubMed] [Google Scholar]