

Abstract
In search of common risk alleles for prostate cancer that could contribute to high rates of the disease in men of African ancestry, we conducted a genome-wide association study (GWAS), with 1,047,986 single nucleotide polymorphism (SNP) markers examined in 3,425 African American prostate cancer cases and 3,290 African American male controls. The most significant 17 novel associations in stage 1 were followed-up in 1,844 cases and 3,269 controls of African ancestry. We identified a novel risk variant on chromosome 17q21 (rs7210100; odds ratio per allele=1.51; p=3.4×10−13). The frequency of the risk allele is ~5% in men of African descent while it is rare in other populations (<1%). Further studies are needed to investigate the biological contribution of this allele to prostate cancer risk. These findings emphasize the importance of conducting GWAS in diverse populations.
Genome-wide association studies (GWAS) of prostate cancer have identified more than 30 risk associated variants, which in aggregate are estimated to account for approximately 20% of the familial risk of prostate cancer1–12. Aside from admixture, and fine-mapping studies which identified multiple independent risk variants at 8q2413,14, and a more recent GWAS among Japanese men which identified five novel loci9, discoveries in prostate cancer have come from studies in men of European ancestry. However, prostate cancer incidence in men of African ancestry is greater than in non-African populations15, with the disparity presumably reflecting both differences in prevalence of environmental risk factors and susceptibility alleles that are shared among men of African descent. For example, the risk variants at 8q24, many of which are more common in men of African ancestry14, could contribute partly to the greater incidence of prostate cancer in this population, and provide some support for the hypothesis of a genetic contribution underlying racial/ethnic disparities in disease risk.
We assembled a consortium of prostate cancer studies that included men of African ancestry and conducted a GWAS to search for additional risk loci that may be more common in men of African descent. Stage 1 included 3,621 African American prostate cancer cases and 3,502 African American controls drawn from 11 studies (Supplementary Table 1, Online Methods). Genotyping in stage 1 was conducted using the Illumina Infinium 1M Duo. Following quality control exclusions (Online Methods), the stage 1 analysis consisted of 1,047,986 SNPs (MAF≥0.01) examined in 3,425 cases and 3,290 controls.
In comparing, for all SNPs, the observed with the expected distribution of p-values from a 1-df trend test there was evidence of inflation in the test statistic (λ=1.11). Principal components analysis highlights the high degree of admixture in this population and the over-inflation diminished following additional adjustment for ancestry (λ=1.03; Supplementary Figure 1, Online Methods). The association of four SNPs achieved genome-wide significance in the stage 1 sample with p-values between 5.4×10−9 and 5.7×10−13 (Figure 1). These SNPs were located in known prostate cancer risk regions; three at 8q24 (rs10505483, rs1456315 and rs7824364 at 128.173–128.205 Mb (NCBI36) and one at 11q13 (rs7130881 at 67.75 Mb).
Figure 1.

A plot of the −log10 P-values by chromosome.
We selected 17 SNPs (p<2×10−5) located outside of known prostate cancer risk regions to examine in a second stage. The associations of these 17 SNPs with prostate cancer risk were not influenced substantially by population stratification in the stage 1 sample, as evaluated by principal components analysis (Supplementary Table 2). The stage 2 sample included 1,396 cases and 2,383 controls of African ancestry from seven independent studies: six U.S.-based studies and one study in Ghana. Of the 17 SNPs, only marker rs7210100 at 17q21 was significantly associated with risk in the stage 2 studies (OR=1.55; p=2.5×10−5; Table 1). None of the other SNPs selected in stage 1 were significantly associated with risk in the stage 2 sample (all p-values >0.05); SNP rs13116912 was excluded due to deviating from Hardy-Weinberg Equilibrium in the majority of stage 2 studies. The results for all 17 SNPs in stage 1 and stage 2 are presented in Supplementary Table 3.
Table 1.
The association of variant rs7210100 at 17q21 with prostate cancer risk in men of African ancestry.
| Stage 1 Studies | Cases/Controlsa | RAF in controls | OR(95% CI)b | P-valuec |
|---|---|---|---|---|
| MEC | 1060/1055 | 0.04 | 1.58(1.21–2.08) | 8.8×10−4 |
| SCCS | 201/412 | 0.05 | 1.40(0.85–2.31) | 0.19 |
| PLCO | 227/239 | 0.05 | 1.44(0.82–2.52) | 0.21 |
| CPS-II | 64/112 | 0.07 | 0.66(0.24–1.78) | 0.41 |
| MDA | 527/437 | 0.05 | 1.39(0.95–2.02) | 0.089 |
| IPCG | 354/157 | 0.05 | 1.54(0.84–2.82) | 0.17 |
| LAAPC | 288/287 | 0.06 | 0.94(0.57–1.56) | 0.81 |
| CaP Genes | 71/85 | 0.06 | 1.72(0.78–3.82) | 0.18 |
| DCPD | 263/341 | 0.07 | 1.14(0.75–1.75) | 0.54 |
| KCPCS | 141/75 | 0.05 | 0.95(0.42–2.16) | 0.90 |
| GECAP | 224/89 | 0.05 | 2.47(1.14–5.34) | 0.022 |
| Combined | 3,420/3,289 | 1.40(1.21–1.62) | 5.2×10−6 | |
| PHet=0.89d | ||||
| Stage 2 Studies | ||||
| SFPCS | 86/36 | 0.04 | 1.86(0.53–6.55) | 0.34 |
| FMHS | 125/339 | 0.06 | 1.70(0.98–2.93) | 0.058 |
| MEC-LAC | 551/555 | 0.04 | 1.92(1.30–2.83) | 9.7×10−4 |
| NCPCS | 214/249 | 0.06 | 0.92(0.51–1.66) | 0.79 |
| WFPCS | 58/65 | 0.04 | 1.90(0.56–6.42) | 0.30 |
| WUPCS | 73/153 | 0.04 | 1.96(0.76–5.03) | 0.16 |
| GHS | 264/964 | 0.07 | 1.37(0.94–2.01) | 0.11 |
| Combined | 1,371/2,361 | 1.55(1.26–1.89) | 2.5×10−5 | |
| PHet=0.25d | ||||
| Stage 3 Studies | ||||
| SCORE | 146/267 | 0.05 | 1.58(0.88–2.83) | 0.13 |
| PROGRÈS | 79/395 | 0.05 | 2.64(1.36–5.10) | 4.0×10−3 |
| PCBP | 246/242 | 0.05 | 2.02(1.20–3.39) | 7.9×10−3 |
| Combined | 471/904 | 2.07(1.49–2.88) | 1.5×10−5 | |
| PHet=0.51d | ||||
| Stages 1+2+3 | 5,262/6,554 | 1.51(1.35–1.69) | 3.4×10−13 | |
| PHet=0.58d |
Number of cases and controls with genotype data for rs7210100.
Adjusted for age and eigenvectors 1–10 in stage 1 (and study in pooled analysis). Adjusted for age in stage 2 and stage 3. Adjusted for age and study in stage 1+2+3 analysis.
P for trend (1-d.f.).
Test of heterogeneity. RAF: risk allele frequency.
We further examined the association with rs7210100 in a third stage that included three studies among men of African descent, a study from the U.S (SCORE), a study in Senegal (PROGRÈS), and a study in Barbados (PCBP). SNP rs7210100 was found to be positively associated with risk in all three studies (stage 3: 471 cases and 904 controls; combined OR= 2.07, p=1.5×10−5; Table 1).
Adjustment for global ancestry or local ancestry (African versus European) in the stage 1 studies did not influence the results for rs7210100 (OR= 1.41 without adjustment for ancestry; OR=1.40 adjusted for global ancestry; OR=1.43 adjusted for global and local ancestry. The effect estimate for rs7210100 was also similar in men with <15% global European ancestry (1,251 cases and 1,325 controls; OR=1.41) as well as in cases and controls estimated to have 2 chromosomes of African ancestry at this location (2,214 cases and 2,080 controls; OR=1.47). We observed no evidence of heterogeneity of the association by study for this variant in the stage 1 (phet=0.89), stage 2 (phet=0.25), or stage 3 studies (phet=0.51), or among all studies (phet=0.58). Results for all SNPs examined in the replication stages were also unaffected when adjusting for European ancestry in studies in which information on global ancestry was available (Supplementary Tables 4 and 5).
In combining the results across all three stages (5,262 cases and 6,554 controls), rs7210100 was strongly and significantly associated with risk (OR = 1.51; 95% CI, 1.35–1.69; p=3.4×10−13). The risk for heterozygote and homozygote carriers was 1.49 (95 % CI, 1.32–1.68) and 2.73 (95% CI, 1.50–4.96), respectively. We did not find any stronger signal with imputed SNPs to the Phase 2 HapMap populations in the surrounding region at chromosome 17q21 (Figure 2, Supplementary Figure 2).
Figure 2.
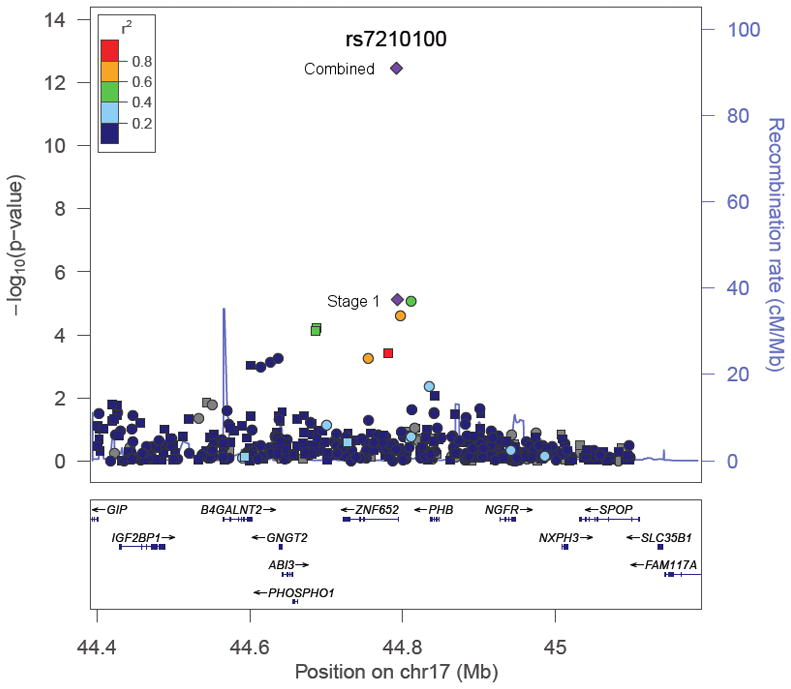
A regional plot of the −log10 P-values for genotyped (squares) and imputed (circles) SNPs at the chromosome 17q21 risk locus in the stage 1 African American sample. The shading depicts the strength of the correlation (r2) between SNP rs7210100 and the SNPs tested in the region. The correlation is estimated in the YRI population from the 1000 Genomes Project (June 2010). Also shown are human genome build 18 coordinates (Mb), recombination rates in centimorgans (cM) per megabase (Mb) and genes in the region. The plot was generate using LocusZoom.
The association with rs7210100 was similar when stratifying on age (p=0.72) and first-degree family history of prostate cancer (p=0.36). We also observed no significant difference in the association of rs7210100 with prostate cancer stage (p=0.94) or tumor grade (p=0.11) at diagnosis. However, the association with rs7210100 was greater for non-advanced disease when classified based on stage and grade (Gleason Score <8 and localized stage, 2,433 cases and 6,554 controls: OR=1.67, p=8.6×10−12) than for advanced disease (Gleason Score ≥8 or non-localized disease, 1,719 cases and 6,554 controls: OR=1.27, P=5.0×10−3: phet = 6.0×10−3).
Among controls with PSA levels measured and ≤4 ng/ml (n=2,383) we found no significant association between PSA levels and rs7210100 genotype (p=0.58). Limiting the analysis to controls with PSA levels (<4 ng/ml) and cases from these studies did not change the association between rs7210100 and prostate cancer risk (n=3,157 cases and 2,383 controls; OR=1.62, p=4.5×10−8).
The variant rs7210100 is located in intron 1 of the ZNF652 gene on chromosome 17q21.32. ZNF652 encodes a zinc-finger protein transcription factor that has been shown to interact with the Eight-Twenty-One (ETO) protein, CBFA2T3, which acts as a transcriptional repressor by forming complexes with corepressor proteins and HDACs16. Co-expression of ZNF652 and the androgen receptor in prostate tumors has been associated with a decrease in relapse-free survival17. A common variant just upstream of the ZNF652 gene has also been associated with blood pressure in a GWAS of men and women of European ancestry18. Sequencing of the 5 coding exons of ZNF652 in 48 subjects (with over-sampling of risk allele carriers; Online Methods) did not reveal a coding variant strongly correlated with rs7210100. Further work is needed to map this locus in order to nominate optimal candidate markers, in addition to rs7210100, for functional studies in pursuit of regulatory effects of one or more variants in the region.
The risk allele of rs7210100 is relatively uncommon in men of African ancestry (4–7%), and is extremely rare (<1%) in non-African populations as reported by the 1000 Genomes Project. The frequency of the risk allele in men of West African ancestry (Ghana and Senegal) is very similar to that observed in African Americans as well as men from East Africa (Uganda, n=111; RAF=0.04). GWAS in populations of European ancestry have not pointed to this region of 17q21 as a risk locus for prostate cancer (Supplemental Figure 3). Together these observations suggest that the underlying biologically relevant allele may be limited to populations of African descent. As reported by the National Cancer Institute’s, Surveillance, Epidemiology and End Results (SEER) Program, prostate cancer incidence in African American men is 1.56-times higher than the incidence of non-Hispanic Whites. Since approximately 10% of African American men carry this variant that increases their risk 1.50-fold over non-carriers, we estimate that this locus may be responsible for as much as 9% (95% CI, 6–12%) of the greater incidence of prostate cancer to African American men (Online Methods).
In summary, we detected a marker of risk for prostate cancer that appears specific to men of African descent, who have an increased incidence and mortality of this disease. These findings provide strong support for conducting GWAS in diverse populations to identify markers of risk that may be population-specific and which could contribute to racial and ethnic disparities in disease incidence. Further work is needed to characterize the 17q21 region and conduct the functional studies required to understand the role of this germ-line variation in prostate cancer susceptibility.
Online Methods
Studies
The studies included in stage 1 were drawn from 11 epidemiological studies of prostate cancer among African American men. These studies included: The Multiethnic Cohort (MEC; 1,094 cases /1,096 controls), The Southern Community Cohort Study (SCCS, 212/419), The Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial (PLCO, 286/269), The Cancer Prevention Study II Nutrition Cohort (CPS-II, 76/152), Prostate Cancer Case-Control Studies at MD Anderson (MDA, 543/474), Identifying Prostate Cancer Genes (IPCG, 368/172), The Los Angeles Study of Aggressive Prostate Cancer (LAAPC, 296/303), Prostate Cancer Genetics Study (CaP Genes, 75/85), Case-Control Study of Prostate Cancer among African Americans in Washington, DC (DCPC, 292/359), King County (Washington) Prostate Cancer Study (KCPCS, 145/81), and The Gene-Environment Interaction in Prostate Cancer Study (GECAP, 234/92). These studies provided DNA samples for 3,621 cases and 3,502 controls.
Stage 2 included 1,396 cases and 2,383 controls from 7 studies: San Francisco Bay Area Prostate Cancer Study (SFPCS, 86/37), The Flint Men’s Health Study (FMHS, 135/353), The Multiethnic Cohort/Los Angeles County (MEC-LA, 554/557), North Carolina Prostate Cancer Study (NCPCS, 214/249), Wake Forest University Prostate Cancer Study (WFPCS, 59/66), Washington University Prostate Cancer Study (WUPCS, 75/153), and The Ghana Men’s Health Study (GHS, 271/968). Stage 3 included 484 cases and 947 controls from 3 studies: The Study of Clinical Outcomes, Risk and Ethnicity (SCORE, 152/280), Prostate-Genetique-Recherche-Senegal (PROGRÈS, 86/414) and Prostate Cancer in a Black Population (PCBP, 246/253). Detailed information about the design and organization of each study is provided in the Supplementary Note.
Genotyping and Quality Control
Genotyping in stage 1 (3,621 cases and 3,502 controls) was conducted using the Illumina Infinium Human1M-Duo. Samples (n=408) were removed based on the following exclusion criteria: 1) unknown replicates across studies, 2) call rates <95%; 3) >10% mean heterozygosity on the X chromosome and/or <10% mean intensity on the Y chromosome, 4) ancestry outliers, and; 5) samples that were related (discussed below). The concordance rate for 158 replicate samples was 99.99%. Starting with 1,153,397 SNPs, we removed SNPs with <95% call rate, MAFs <1%, or >1 QC mismatch based on sample replicates (n=105,411). The analysis included 1,047,986 SNPs among 3,425 cases and 3,290 controls. We used PLINK to calculate the probabilities of sharing 0, 1, and 2 alleles (Z = Z0, Z1, Z2) across all possible pairs of samples to determine individuals who were likely to be related to others within and across studies. We identified 167 pairs of related subjects (MZ twin, parent-offspring, full and half-sibling pairs), based on the values of their observed probability vector Z being within 1 SD of the expected values of Z for their respective relationship. The criterion for removal was such that individuals that were connected with a higher number of pairs were chosen for removal. In all other cases, one of the two members was randomly selected for removal. A total of 141 subjects were removed.
The EIGENSTRAT software was used to calculate eigenvectors that explained genetic differences in ancestry among samples in the study19. We included data from both HapMap populations (CEPH (Utah residents with ancestry from northern and western Europe) (CEU), Japanese in Tokyo, Japan (JPT), Yoruba in Ibadan, Nigeria (YRI), and African ancestry in Southwestern U.S. (ASW)) and our study, so that comparisons to reference populations of known ethnicity could be made. A total of 2,546 ancestry-informative SNPs from the Illumina array were selected based on low inter-marker correlation and ability to differentiate between samples of African and European descent. An individual was subject to filtering from the analysis if his value along eigenvector 1 or 2 was outside of 4 SDs of the mean of each respective eigenvector. We identified 108 individuals who met this criterion. Eigenvector 1 was highly correlated (ρ=0.997, p<1 × 10−16) with percentage of European ancestry, estimated in HAPMIX20. Together the top 10 eigenvectors explain 21% of the global genetic variability among subjects.
Genotyping in the stage 2 and 3 studies was conducted using the TaqMan allelic discrimination assay. In stage 2, we removed samples missing data for >3 SNPs (n=36). To assess genotyping reproducibility each study included replicate samples; the concordance was >98% for each SNP within each study. SNP rs13116912 deviated from HWE in all but one of the stage 2 studies and was removed from the stage 2 analysis. No other SNP deviated from HWE (i.e. P<0.01 in >2 studies) in stage 1 or 2. The call rate for rs7210100 was very high in stage 1 (99.9%) and similar in cases (99.9%) and controls (99.9%). The call rate for this SNP was also very high in stages 2 (99.8% overall, 99.9% in cases and 99.8% in controls) and 3 (96.1% overall, 97.3% in cases and 95.5% in controls).
Sequencing
Bi-directional sequencing of rs7210100 and the 5 coding exons of ZNF652 was performed in 48 subjects (20 homozygous for the risk variant, 20 heterozygous for the risk variant and 8 homozygous for the wild-type allele.) Primers were designed at least 50 bases upstream and downstream from each exon.
Statistical Analysis
In stage 1, we tested the association of each SNP and prostate cancer risk using a 1-d.f. χ2 likelihood ratio test from a logistic regression analysis adjusted for age, study and the first 10 eigenvectors estimated by principal components analysis19. Over-inflation of the test statistic was examined with and without adjustment for ancestry and visualized with quantile-quantile plots. Lambdas were estimated as the median of the test statistics divided by 0.456 (the median of the 1-d.f. χ2 null distribution). Age-adjusted odds ratios (OR) and 95% confidence intervals (95% CI) for each SNP were estimated from the same logistic regression model. At each locus and for each participant, local ancestry was defined as the estimated number of European chromosomes (continuous between 0–2) carried by the participant, estimated via the HAPMIX program20. Local ancestry at the 17q21 locus was evaluated as a confounder in the analysis of rs7210100.
Phased haplotype data from the founders of the CEU and YRI HapMap Phase 2 samples were used to infer LD patterns in order to impute untyped markers. We carried out genome-wide imputation using the software MACH21. The Rsq metric was used as a threshold in determining which SNPs to filter from analysis (Rsq<0.3). Imputed SNPs in the 17q21 risk region, as shown in Figure 2, were examined in association with prostate cancer risk as described for typed SNPs above.
In stage 2, the SNPs were analyzed using logistic regression controlling for age and study (in the pooled analysis). Information regarding European ancestry was available for 7 studies included in stages 2 and 3. As observed in stage 1 (Supplementary Table 2) the OR for rs7210100 was similar with and without adjustment for estimated European ancestry in these studies (Supplementary Table 4). The results for rs7210100 in stage 2, stage 3 and stages 1+2+3 are presented without adjustment for ancestry.
Association testing in the stage 2 and stage 3 studies was performed using logistic regression, adjusting for age and study. For seven of the replication studies, information about global European ancestry was available and examined as a confounding factor for variant rs7210100. For rs7210100, a combined analysis of all stages was performed adjusted for age and study. Heterogeneity of the OR across studies was evaluated using a likelihood ratio test.
Effect modification by age and first-degree family history of prostate cancer was assessed in stratified analyses, and significance determined comparing the model with and without the cross-product term using a likelihood ratio test. We also examined the association of rs7210100 genotype with stage, Gleason Score as well as the combination of stage and grade, with advanced disease defined as Gleason Score≥8 or stage ≥2 (non-localized disease) and non-advanced disease defined as Gleason Score<8 and stage=1 (localized disease). Case-only analysis was used to test for differences in the association of rs7210100 with disease phenotypes. The association of rs7210100 with least-squares geometric mean PSA levels was examined using multiple linear regression adjusting for age, body mass index and study.
We estimated the risk ratio between populations of different ancestral origin (African / European) due to rs7210100 as RR= [(1-pA)2+2pA(1-pA)RR1+pA2RR2]/(1-pE)2+2pE(1-pE)RR1+pE2RR2]. Here pA is the risk allele frequency in African origin populations, pE is the risk allele frequency in European populations and RR1 is the relative risk associated with carrying 1 copy of the risk allele (compared to none) and RR2 is the relative risk associated with carrying 2 copies of the risk allele. We used values pA = 0.05, pE = 0, RR1 = 1.5, and RR2 = 1.52 so that the risk ratio between populations due to the influence of this risk allele was estimated to be equal to 1.050625. Using the SEER incidence rates of prostate cancer in African Americans (234.6 per 100,000) and non-Hispanic Whites (150.4 cases per 100,000), we estimated the ratio of risks between these populations as 234.6/150.4 = 1.56. The percentage of greater risk to African Americans that may be associated with rs7210100 was estimated as 1-[(1.56–1.050625)/(1.56-1)] × 100.
URLs
SEER: http://seer.cancer.gov/
LocusZoom: http://csg.sph.umich.edu/locuszoom/
Supplementary Material
Acknowledgments
The MEC and the genotyping in this study were supported by NIH grants CA63464, CA54281, CA1326792, CA148085 and HG004726. Genotyping of the PLCO and Ghana samples was funded by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics, NCI, NIH. LAAPC was funded by grant 99-00524V-10258 from the Cancer Research Fund, under Interagency Agreement #97-12013 (University of California contract #98-00924V) with the Department of Health Services Cancer Research Program. Cancer incidence data for the MEC and LAAPC studies have been collected by the Los Angeles Cancer Surveillance Program of the University of Southern California with Federal funds from the NCI, NIH, Department of Health and Human Services, under Contract No. N01-PC-35139, and the California Department of Health Services as part of the statewide cancer reporting program mandated by California Health and Safety Code Section 103885, and grant number 1U58DP000807-3 from the Centers for Disease Control and Prevention. KCPCS was supported by NIH grants CA056678, CA082664 and CA092579, with additional support from the Fred Hutchinson Cancer Research Center and the Intramural Program of the National Human Genome Research Institute. MDA was support by grants, CA68578, ES007784, DAMD W81XWH-07-1-0645, and CA140388. GECAP was supported by NIH grant ES011126. CaP Genes was supported by CA88164 and CA127298. IPCG was support by DOD grant W81XWH-07-1-0122. DCPC was supported by NIH grant S06GM08016 and DOD grants DAMD W81XWH-07-1-0203 and DAMD W81XWH-06-1-0066. SCCS is funded by NIH grant CA092447. SCCS sample preparation was conducted at the Epidemiology Biospecimen Core Lab that is supported in part by the Vanderbilt-Ingram Cancer Center (CA68485). Data on SCCS cancer cases used in this publication were provided by the Alabama Statewide Cancer Registry; Kentucky Cancer Registry; Tennessee Department of Health, Office of Cancer Surveillance; Florida Cancer Data System; North Carolina Central Cancer Registry, North Carolina Division of Public Health; Georgia Comprehensive Cancer Registry; Louisiana Tumor Registry; Mississippi Cancer Registry; South Carolina Central Cancer Registry; Virginia Department of Health, Virginia Cancer Registry; Arkansas Department of Health, Cancer Registry. The Arkansas Central Cancer Registry is fully funded by a grant from National Program of Cancer Registries, Centers for Disease Control and Prevention (CDC). Data on SCCS cancer cases from Mississippi were collected by the Mississippi Cancer Registry which participates in the National Program of Cancer Registries (NPCR) of the Centers for Disease Control and Prevention (CDC). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the CDC or the Mississippi Cancer Registry. We thank Mr. Christopher Edlund from the USC Genomics Core for his technical and informatics assistance. The authors thank Drs. Christine Berg and Philip Prorok, Division of Cancer Prevention, NCI, the screening center investigators and staff of the PLCO Cancer Screening Trial, Mr. Thomas Riley and staff at Information Management Services, Inc., and Ms. Barbara O’Brien and staff at Westat, Inc. for their contributions to the PLCO Cancer Screening Trial. We also acknowledge the technical support of Marta Gielzak and Guifang Yan. The authors would like to thank Ms. Evelyn Tay and Ms. Vicky Okyne for their expert help in coordinating the Ghana study; Drs. Angelo DeMarzo and George Netto of the Johns Hopkins University for pathology review: Ms. Violet Devairakkam, Ms. Norma Kim, and Mr. John Heinrich of Research Triangle Institute (RTI) for their expert study management; Ms. Shelley Niwa and Ms. Ann Truelove of Westat for study support and data management. WUPCS was supported by CA112028, the St. Louis Men’s Group Against Cancer, and the David H. Nickerson Foundation. SFPCS was funded by grant 99-00527V-10182 from the California Cancer Research Program. SCORE was supported by CA085074 and CA105641, as well as a grant from the Commonwealth of Pennsylvania. CPS-II is supported by the American Cancer Society.
Footnotes
C.A.H., D.O.S, and B.E.H. contributed to the study concept and design. L.C.P., E.N.C., E.R.D., L.Y.X., D.V.D.B., S.J.C., C.A.H. and X.S supervised the laboratory analyses and quality control. G.K.C., P.W., and D.O.S. contributed to the statistical analysis. C.A.H. wrote the manuscript. W.J.B., S.S.S., S.I.B., R.A.K., B.A.R., W.B.I., S.A.I., J.L.S., W.R.D., J.S.W., A.W.H., B.N., T.R.R., K.A.C., J.X., A.S.K., J.J.H., E.M.J., S.M.G., S.W., L.B.S., R.B.H., Z.W., E.Y., Y.T., Q.C., S.K., E.A.O., C.Z.-J., Y.Y., C.N.-D., J. H.-M., W.W., V.T., G.O.A., A.M., B.-L.C., S.L.Z., M.C.L., S.-Y.W., A.M.R., A.J.M.H., M.J.T., J.C., G.C., I.C., K.R.M., F.S., L.L.M., L.N.K., B.E.H. conducted the epidemiologic studies that contributed to the scan. All authors helped in the interpretation and discussion of the findings and approved the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Al Olama AA, et al. Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat Genet. 2009;41:1058–60. doi: 10.1038/ng.452. [DOI] [PubMed] [Google Scholar]
- 2.Amundadottir LT, et al. A common variant associated with prostate cancer in European and African populations. Nat Genet. 2006;38:652–8. doi: 10.1038/ng1808. [DOI] [PubMed] [Google Scholar]
- 3.Eeles RA, et al. Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat Genet. 2009;41:1116–21. doi: 10.1038/ng.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eeles RA, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40:316–21. doi: 10.1038/ng.90. [DOI] [PubMed] [Google Scholar]
- 5.Gudmundsson J, et al. Genome-wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nat Genet. 2009;41:1122–6. doi: 10.1038/ng.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gudmundsson J, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39:631–7. doi: 10.1038/ng1999. [DOI] [PubMed] [Google Scholar]
- 7.Gudmundsson J, et al. Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate cancer. Nat Genet. 2008;40:281–3. doi: 10.1038/ng.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gudmundsson J, et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet. 2007;39:977–83. doi: 10.1038/ng2062. [DOI] [PubMed] [Google Scholar]
- 9.Takata R, et al. Genome-wide association study identifies five new susceptibility loci for prostate cancer in the Japanese population. Nat Genet. 42:751–4. doi: 10.1038/ng.635. [DOI] [PubMed] [Google Scholar]
- 10.Thomas G, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008;40:310–5. doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- 11.Yeager M, et al. Identification of a new prostate cancer susceptibility locus on chromosome 8q24. Nat Genet. 2009;41:1055–7. doi: 10.1038/ng.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yeager M, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645–9. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- 13.Freedman ML, et al. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc Natl Acad Sci U S A. 2006;103:14068–73. doi: 10.1073/pnas.0605832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haiman CA, et al. Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet. 2007;39:638–44. doi: 10.1038/ng2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.American Cancer Society. Cancer Facts & Figures for African Americans 2009–2010. American Cancer Society; Atlanta, GA: 2009. [Google Scholar]
- 16.Amann JM, et al. ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol Cell Biol. 2001;21:6470–83. doi: 10.1128/MCB.21.19.6470-6483.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Callen DF, et al. Co-expression of the androgen receptor and the transcription factor ZNF652 is related to prostate cancer outcome. Oncol Rep. 23:1045–52. doi: 10.3892/or_00000731. [DOI] [PubMed] [Google Scholar]
- 18.Newton-Cheh C, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41:666–76. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 20.Price AL, Tandon A, Patterson N, Barnes KC, Rafaels N, Ruczinski I, Beaty TH, Mathias R, Reich D, Myers S. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet. 2009;5:e1000519. doi: 10.1371/journal.pgen.1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Willer C, Sanna S, Abecasis G. Genotype imputation. Annu Rev Genomics Hum Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.