

Abstract
Background
Epigenetics is a rapidly evolving field of genetic study applicable to nearly every aspect of genome-related research. The importance of epigenetics has been recognised in human hepatocellular carcinoma (HCC). Changes in DNA methylation patterns, including global hypomethylation and promoter hypermethylation, are thought to be early events in hepatocarcinogenesis.
Objectives
This review aimed to summarise the role of epigenetics in HCC, to describe the mechanisms of epigenetic changes in HCC and to examine the clinical relevance of epigenetics in HCC.
Methods
This review examines the role of CpG-rich regions and DNA methylation, and describes an epigenetic model of cancer, tumour type-specific methylation, the relationships among methylation, cirrhosis and hepatocarcinogenesis, and the role of DNA methylation in HCC. The clinical implications of epigenetics in HCC are discussed.
Results
A multivariate predictor model based on traditional clinical factors and DNA methylation profile may have important applications in the early detection of neoplastic transformation in populations at high risk for HCC. CpG methylation may be valuable in HCC prognostics. DNA methylation profiles may enable clinical prediction in pre-therapy patient biopsies, paraffin-embedded samples or plasma DNA.
Conclusions
Epigenetic changes and profiles may correlate to the biological behaviour of tumours and clinical outcome of HCC patients. The use of DNA methylation profiles as a surrogate biomarker remains an active area of clinical cancer research.
Keywords: epigenetics, hepatocellular carcinoma, methylation
Introduction
Epigenetic mechanisms of carcinogenesis are rapidly becoming important to the understanding of human malignancies. Traditionally, genetic instability and mutations are described as common events in most cancers. A stepwise progression model involving two discrete genetic pathways has been proposed to explain the progression to cancer from benign neoplasm to invasive adenocarcinoma.1–3 Oncogene and tumour suppressor gene mutations that actively control cell proliferation and death (such as APC, K-ras and p53) and mutations of DNA mismatch repair genes represent these pathways. In addition to these genetic alterations, cancer initiation and promotion can occur by way of epigenetic mechanisms.1 It is becoming increasingly clear that epigenetic events mediate carcinogenesis. Epigenetic alterations refer to aberrant gene expression profiles that are not caused by changes in the primary nucleic acid sequence (a classic mutation), but, rather, involve covalent modification of nucleotide bases in normal DNA sequences. These changes can occur as part of normal development, but can also occur as a result of age or exposure to risk factors, potentially resulting in carcinogenesis in tissues with normal DNA sequences. Hepatocellular carcinoma (HCC) typically occurs in the setting of chronic inflammation secondary to hepatitis B virus (HBV), hepatitis C virus (HCV) or alcoholism; each aetiology increases the risk for hepatocarcinogenesis. The purpose of this review is to summarise the role of epigenetics in cancer with a specific focus on HCC. Furthermore, we will describe the mechanisms of epigenetic changes in HCC and the clinical relevance of epigenetics in the prognosis and detection of HCC.
CpG islands and DNA methylation
Methylation of cytosine is the only modification of DNA in mammals known to naturally occur and cytosine-phosphate-guanine (CpG) site methylation is the best characterised epigenetic alteration.4 CpG dinucleotides are not uniformly distributed throughout the human genome. In the vast majority of the genomic sequence, CpGs occur at a frequency of approximately one per 80 dinucleotides. However, CpG-rich regions (200 basepairs to several kilobases in length), referred to as CpG islands, comprise 1–2% of the genome.5 Despite the under-representation of CpG dinucleotides in the mammalian genome, it is estimated that about 45 000 CpG islands exist in proximity to various gene promoter regions. Nearly 50–60% of all genes have an associated CpG island in the 5′ area, often encompassing the promoter and transcription start site.6 CpG islands are normally unmethylated and the molecular mechanisms maintaining normal methylation status remain under investigation. Inappropriate methylation of the cytosine bases in CpG islands interferes with promoter function of the gene. Excessive hypermethylation can lead to the transcriptional silencing of genes critical to the normal anti-neoplastic process (i.e. blocking tumour suppressor gene function).7 However, hypermethylation of promoter regions that bind transcriptional repressors can lead to increased transcription by interfering with the action of the repressor.8
Methylation of the 5′ position of cytosine leads to the formation of 5-methylcytosine (5-MC), a reaction catalysed by DNA methyltransferases (DNMTs). DNA methylation patterns are established early during development and are relatively constant afterwards, as methylated CpG dinucleotides are passed on to daughter cells.4,9 Specific proteins that bind to CpG islands and protect them from de novo methylation are postulated to be involved in this occurrence, including proteins containing a zinc (Zn) finger binding domain specific to non-methylated CpG regions and various histone demethylases that can actively demethylate CpG islands (Fig. 1). In addition, there is probably a critically important embryological function associated with DNA methylation because DNA methyltransferase knockout mice are non-viable.10 Furthermore, increased methylation of specific genes, such as MYOD1 and N33 in normal colon tissue of ageing individuals, may occur as a component of the normal ageing process.11
Figure 1.
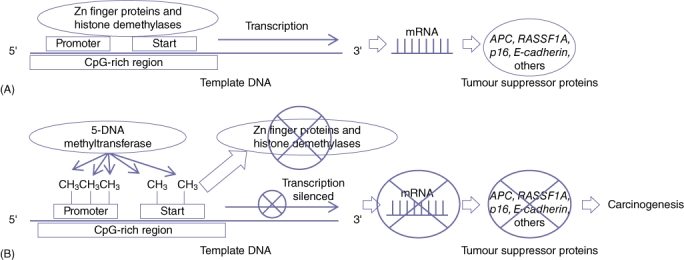
Schematic demonstrating how epigenetic DNA methylation blocks gene promoter function. (A) Areas rich in cytosine-phosphate-guanine (CpG) are preferentially located at or near promoters of genes. Normal gene expression is promoted by zinc finger proteins (Zn fingers) and histone demethylases inhibiting methylation of the CpG sites. (B) Loss of Zn finger and histone demethylase proteins and/or increased 5-DNA methyltransferase activity results in increased CpG methylation and subsequent gene silencing. Silencing of tumour suppressor genes in this manner can lead to carcinogenesis
Epigenetic model of cancer
This model suggests that the predominant role of epigenetic changes involves tissue-specific alterations, ultimately leading to carcinogenesis. Epigenetics can play a role as one of the ‘first hits’ in the development of cancer referred to in the Knudson ‘two-hit’ model.12 Along this line, Feinberg et al. described the epigenetic progenitor model of cancer as involving three steps in tumour development beginning with early epigenetic changes.13 These epigenetic changes, when occurring in tissue progenitor or stem cells, result in either increased expansion of this population or alterations increasing susceptibility to neoplastic changes. The second step involves genetic mutation within this cell population, resulting in cancer. Once cancer is developed, there is enhanced genetic and epigenetic plasticity (an increased ability for further phenotype changes brought on by DNA and chromatin instability). This contributes to tumour heterogeneity and the development of metastatic and drug-resistant phenotypes (Fig. 2).13
Figure 2.
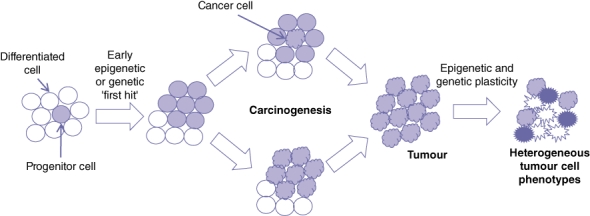
Progenitor cell model of DNA methylation and hepatocarcinogenesis. The development of cancer is initiated by a genetic or epigenetic ‘first hit’, which increases progenitor cell proliferation. A genetic and epigenetic ‘second hit’ results in neoplastic transformation of a progenitor cell or a ‘colour shift’ of a population of cells. These transformed cancer cells proliferate to form a tumour and, through increased genetic and epigenetic instability, develop additional phenotypes resulting in heterogeneous tumour cell populations
Additional models describing the role of epigenetics in cancer have been described. Kopp-Schneider et al. used mathematical modelling to suggest that the epigenetic changes resulting in cancer follow a model of tumorigenesis called a ‘colour shift’, in which an entire colony of cells shifts to cancer, as opposed to the clonal model, in which cancer begins from a single cell transformation.14 Their analysis showed evidence for this model in HCC development. A shift in an entire colony of cells could be explained by epigenetic alterations that prime a group of cells for the ultimate inciting insult that results in cancer development. Whether HCC is generated from a single cellular transformation or multiple cellular changes, there is strong evidence for the creation of ‘epigenetic fields’ that predispose to cancer development. Epigenetic silencing of a DNA repair gene (MGMT) has been defined as one such field defect, in which the resulting altered methylation of genes creates a pre-neoplastic state in liver, colon, oesophageal, lung, breast and renal cancers.15,16
There has been increasing interest in stem cells in the development and progression of cancer. A stem cell niche has been described in the liver, composed primarily of clonal cells in the canal of Hering near the periportal space. When activated, these cells proliferate in a cord-like fashion towards the hepatic veins to regenerate the liver.17–19 These cells are particularly important in severe and chronic injury in which hepatocyte replication is insufficient and progenitor cells are primarily responsible for regenerating the parenchyma. In diseased liver, these progenitor populations are surrounded by inflammatory cells, such as myofibroblasts and macrophages.20 Knudson acknowledged that the proliferation of stem cells is a critical phenomenon in the development of cancer because proliferating stem cells can obtain neoplastic mutations at a higher rate. This theory is supported in tumour development.12 The dependence on progenitor cell activation in chronic liver injury makes it highly likely that progenitor cells are involved in the development of HCC and are primed for neoplastic development through epigenetic modification.
Characterising the epigenetic modulation of the cancer stem cell (CSC) population in HCC requires further investigation, as the markers are still to be clearly defined. Side populations isolated using CD133 have been described; however, the resulting reference standard tumorgenicity studies in mice have not consistently confirmed an HCC stem cell population.19 CD90 expression has been associated with an increasingly tumorigenic population, particularly when associated with CD44 co-expression in HCC. Yamashita et al. used the classic CSC marker, EpCAM, to isolate a CSC-enriched HCC population.21 These cells showed increased sphere formation and were more tumorigenic in vivo compared with EpCAM- cells.22 Lingala et al. showed that staining for CD133+/ALDH+ cells alone, or in combination with CD44 and CD90, was able to identify putative CSCs in HCC specimens; however, non-malignant specimens also expressed these markers.23 Novel markers continue to be identified, including Transgelin, which is over-expressed 25-fold in tumorigenic HCC cells from the Huh7 line.24 Marquardt et al. directly implicated epigenetic changes with HCC CSCs by inhibiting methylation in human cell lines using a DNMT1 inhibition. The reduced methylation resulted in increased tumorgenicity of the cells in the isolated CSC side population.22 Further study is needed to definitively identify the CSC population in HCC, which will aid in the characterisation of the genetic and epigenetic changes involved in these progenitor cells and possibly open the door to better clinical and therapeutic prognostic indicators and targets.
Tumour type-specific methylation
Unique epigenetic changes have been identified in different genes in different tumour types, revealing site-specific methylation profiles. Esteller et al. used a candidate gene approach to look for aberrant promoter methylation profiles in 12 genes in 600 primary tumour samples from 15 major tumour sites.25 They found at least one or more genes to be hypermethylated in each tumour type and site-specific profiles began to emerge. Gastrointestinal tumours had increased methylation of p16INK4a, p14ARF, MGMT, APC and hMLH1, whereas head, neck and lung tumours were hypermethylated at DAPK, MGMT and p16INK4a, but not at hMLH1 or p14ARF. Breast and ovary cancer methylated genes included BRCA1, GSTP1 and p16INK4a.25 Additionally, there appear to be tissue-specific methylation profiles that develop over time as individuals age. Autopsy studies by Waki et al. showed that there was specific methylation in organs, similar to the trends in the corresponding site-specific tumours.26 In patients over 42 years of age, methylated promoters were found with increased frequency in: APC in the stomach, duodenum, liver, pancreas and lung; E-cadherin and DAP-kinase in the stomach and intestines; RASSF1A in the rectum, liver, pancreas and kidney, and p16 and RUNX3 in the stomach.26
Specific epigenetic alteration patterns in the key oncogenes and tumour suppressor genes in HCC have been described. Many of these sites are common cancer initiators, involving changes to TP53, B-catenin, ErbB receptors, MET, hepatocyte growth factor (HGF), p16INK4a, E-cadherin and cyclooxygenase 2 (COX-2).27 Hypermethylation of p16INK4a can occur in early HCC. Maeta et al. observed that when HCC cells were treated with demethylating agents, there was increased expression of p16INK4a and subsequent cell growth inhibition, suggesting that modulation of the epigenetic control of these genes may be an important target in HCC therapeutics.28 Lee et al. examined 60 paired HCC and non-HCC human liver samples to screen for hypermethylation.29 The most frequent hypermethylated genes in HCC were found to be APC (81.7%), GTP1 (76.7%), RASSF1a (66.7%), p16 (48.3%), COX-2 (35.0%) and E-cadherin (33.3%). COX-2, p16, RassF1A and TIMP-3 were specifically associated with HCC and not methylated in cirrhosis or chronic hepatitis.29
Methylation, cirrhosis and hepatocarcinogenesis
Epigenetic changes in normal tissue may be precursor events that increase the likelihood of developing cancer. As cirrhosis is often a precursor to HCC, it is possible that cirrhosis, caused by chronic hepatocyte injury and inflammation, leads to global DNA hypermethylation. The data to support this theory appear to be conflicting. Kondo et al. found no difference in DNA methylation between normal and cirrhosis liver.16 However, other investigators demonstrated increased methylation and loss of tumour suppressor gene function in patient samples of cirrhosis and/or chronic hepatitis.30 Increased methylation of the p16 promoter appears to be involved in the pathogenesis of HCC, and hypermethylation of p16 gene is thought to be an inactivating mechanism.31 This observation clearly supports a multistep hepatocarcinogenesis from cirrhotic nodules (CNs) through dysplastic nodules (DNs). They thus examined the methylation status of p16 in HCCs surrounded by DNs and CNs to define the significance of p16 hypermethylation in the early stage of hepatocarcinogenesis. In addition, the p16 methylation status of HCC cell lines (with or without HBV infection) was tested and HBV-infected cell lines were found to be exclusively methylation-positive. Other investigators demonstrated a stepwise progression (from normal liver to HCC) in the methylation of APC, GSTP1, RASSF1A, p16, COX-2 and E-cadherin.29 Thus, there appears to be a growing body of evidence that would suggest that cirrhosis appears to induce or is associated with DNA methylation and that these epigenetic changes are early events in hepatocarcinogenesis (Fig. 3).
Figure 3.

The role of cirrhosis as it relates to DNA methylation. Cirrhosis plays an important role in the development of hepatocellular carcinoma (HCC). This can result from epigenetic changes and can also cause epigenetic and genetic changes that ultimately lead to HCC. HCV, hepatitis C virus; HBV, hepatitis B virus
DNA methylation in HCC
The contribution of DNA methylation to the development of HCC is not well understood. The study of methylation in HCC is also challenging as there are several well-known risk factors for HCC, such as alcohol-induced cirrhosis and chronic viral hepatitis B or C infection. Whereas colorectal cancer (CRC) typically develops in otherwise ‘normal’ colonic tissue, HCC routinely arises in a distinctly abnormal and often cirrhotic liver. Thus, the cirrhotic liver may be thought of as an epigenetic field suited for hepatocarcinogenesis. It is possible that differential promoter methylation occurs in: (i) normal liver; (ii) cirrhotic liver, and (iii) HCC. Because cirrhosis is in the causal pathway (from normal liver to HCC), it is by definition not a confounding factor, but induces interactions resulting in carcinogenesis (Fig. 4). Thus, high-throughput analysis of global methylation must take into account the appropriate control group because the comparison of methylation in HCC with that in completely normal liver tissue may not be valid.
Figure 4.
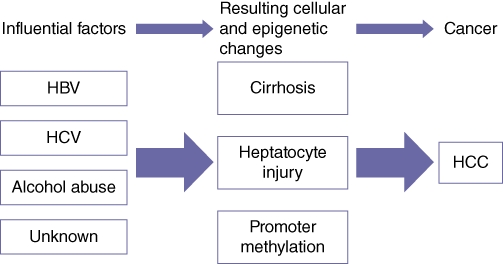
Proposed pathway relating epidemiological exposures to the development of hepatocellular carcinoma (HCC). Acquired disease results in chronic hepatic insults, which accumulate and result in the development of HCC. HCV, hepatitis C virus; HBV, hepatitis B virus
Currently, the role of DNA methylation in hepatocarcinogenesis is under more scientific scrutiny. Hepatocellular carcinoma promoter methylation and the subsequent loss of protein expression have been demonstrated to involve the tumour suppressor p16, E-cadherin, p15, SOCS1 and RIZ1.32–37 However, not all of these studies demonstrated the combination of increased DNA methylation and decreased protein expression.36,37 Recently, several studies have examined global CpG promoter methylation in HCC patient samples. Archer et al. recently published HCC methylation data for normal liver tissue, HCV-infected liver tissue and HCC.38 In this report, a paired analysis was performed between HCV–HCC and adjacent HCV–cirrhotic liver tissue. A total of 56 CpG sites (corresponding to 49 genes) were differentially methylated, with the APC gene located on chromosome 5 demonstrating significant promoter hypermethylation. Ingenuity Pathways Analysis software was also used to examine genomic pathways that appeared to be affected by promoter methylation. Two signalling pathways identified included transforming growth factor-β (TGF-β) and hepatic fibrosis/hepatic stellate cell activation.38 It is well known that HCV infection leads to chronic inflammation and possibly to hepatic stellate cell activation/fibrosis. Therefore, continued hepatocyte damage as a result of HCV infection appears to lead to promoter methylation and subsequent HCC.
Several reports have described APC gene promoter hypermethylation in HCC. However, conflicting reports have detailed APC promoter methylation in adjacent non-cancerous liver tissue. Archer et al. found no evidence of hypermethylation, whereas Csepregi et al. found hypermethylation of APC in 82% of non-cancerous specimens.38,39 However, Csepregi et al. found that APC protein expression was significantly reduced in HCC samples compared with samples of the adjacent non-tumour tissue.39 These results imply that promoter methylation of the APC gene is a key component of carcinogenesis in HCC because the product of the gene encodes a tumour suppressor that has been extensively studied in CRC. Other candidate genes have also been studied. He et al. showed hypermethylation of the tumour suppressor small heterodimer partner (SHP) in 10 matched pairs of liver and HCC tissue, which corresponded to decreased SHP mRNA in HCC tissues.40 Thus, they concluded that SHP hypermethylation may be a common event in the development of HCC.40 Although these studies provide important information as to potential mechanisms of epigenetic events in HCC formation, they have not been used to correlate methylation with clinical variables such as stage, response to treatment or outcome.
Clinical implications of the epigenetics of HCC
The accumulating evidence for epigenetic defects in cancer may be potentially useful in cancer diagnosis and treatment. First, as described above, aberrant promoter methylation occurs very early during carcinogenesis and can be detected in the normal epithelium of some patients (i.e. in patients with chronic HBV or HCV). High degrees of methylation are thought to be associated with an increased risk for neoplastic transformation. Indeed, aberrant CpG island methylation in non-cancerous tissues has been detected in the colons of older people, patients with ulcerative colitis, in the livers of patients with chronic viral hepatitis, in the lungs of heavy smokers and in Barrett's oesophagus, all conditions of increased risk for cancer development.41–45 DNA methylation status will also be valuable as a prognostic indicator. Cheng et al. showed that the high CpG island methylator phenotype CIMP (defined as having four or five of 10 commonly methylated genes in HCC) correlated with worse prognosis, increased tumour–node–metastasis (TNM) stage, increased metastasis and increased gamma-glutamyl transferase (GGT) levels in HCC patients compared with those with less hypermethylated candidate genes.46 Further understanding of the commonly methylated genes in HCC may be very valuable in guiding treatment options based on an improved understanding of patient prognosis.
Ongoing studies will address whether DNA methylation measurement (and, importantly, which gene and which technique) may be useful in predicting an individual patient's risk for tumour development. Second, it is apparent that, in a growing list of specimens, such as sputum, serum, urine and breast ductal fluid, promoter hypermethylation can be detected in early-stage cancers. The limitations of current detection methods using bisulphate treatment followed by methylation-specific polymerase chain reaction (MSP) can lead to degradation of potentially already scarce methylated DNA in serum and body fluids. Sakamoto et al. proposed one novel detection method utilising immunoprecipitation with anti-histone antibodies to improve isolation of methylated DNA.47 Using this technique, they were able to improve detection of serum p16 methylation in p16 methylation-positive colon cancer patients, with overall detection rates of 81% vs. 59% and detection rates of 91% vs. 45% in stage II patients.47 Techniques like this, which improve the sensitivity of detection, may be of significant value to patients, particularly those at the vital stages of disease in which early detection may have drastic results on outcome. Marker selection will also be key to providing appropriately sensitive assays and screens using epigenetic markers. Glockner et al. described methylated tissue factor pathway inhibitor 2 (TFPI2), which was detectable in stool from stage I–III CRC patients with a sensitivity of 76–89% and a specificity of 79–93%.48 Markers such as these can potentially augment current detection methods and improve the early diagnosis of various tumours.
The effects of chemotherapeutic agents that induce hypomethylation may be beneficial in the short-term, but they may allow progression and recurrence from cancer cells that survive or are even enhanced by DNA hypomethylation (an independent factor in cancer progression and formation).49 As described in the epigenetic progenitor model of cancer proposed by Feinberg et al,13 although early epigenetic changes can prime cells for neoplastic transformation, they also continue to contribute to the heterogeneity of tumours and thus may represent a mechanism of chemoresistance. Lu et al. recently described the role of Tip30 as a putative tumour suppressor gene, which is frequently downregulated in several human tumour cells and was identified as a novel metastasis suppressor gene in small cell lung cancer.50 Its expression has been detected in many normal human tissues; however, in HCC, melanoma, breast cancer and CRC, its expression was found to be decreased.50–55 Methylation of Tip30 was positively associated with tumour recurrence and poor prognosis in patients with HCC, which suggests that Tip30 may be a novel marker for predicting prognosis in patients with HCC and opens the door to the potential development of Tip30-targeted therapy.50
The availability of methylation array chips has allowed global CpG analysis. Recently, Hernandez-Vargas et al. published a rather unique paper in which the global methylation profile (methylome) was studied in 30 HCC tumours.56 This study employed the Illumina GoldenGate methylation assay and probed 1505 CpG sites, which correspond to 807 cancer-related genes.56 After filtering for a P-value of < 0.001 and correcting for a false discovery rate of < 0.1, 124 CpG sites among 94 genes were differentially methylated when compared with surrounding non-cancerous liver tissue. In addition, there appeared to be differentially methylated promoters when comparing: (i) early-stage with later-stage tumours; (ii) well-differentiated with poorly differentiated tumours, and (iii) cirrhotic with non-cirrhotic liver tissue. Interestingly, DNMT1 was hypermethylated in poorly differentiated tumours. Another unique finding of this study was the ‘survival risk prediction’ function data using BRB-ArrayTools.56 This analysis yielded 10 CpG sites (FAT, MYLK, FLT1, CDKN1C, TFPI2, PDE1B, MME, IGF1R, COL1A2 and TP73) with the highest ability to predict survival (high-risk vs. low-risk). However, these very interesting findings should be viewed and interpreted with caution for several reasons. Firstly, the overall sample size (n = 30) was relatively small. Secondly, the aetiology of HCC was HBV (n = 9), HCV (n = 5), alcohol-related (n = 8) and unknown (n = 8). Currently, in the USA, HCV, not HBV, is the primary epidemiological factor associated with HCC. Thirdly, other specific treatment and co-morbidity data are lacking. Lastly, overall survival would appear to be much longer than expected based upon recent data from the USA, which may limit the external validity of the study data.57 The important expansion of studies like this will need to identify effective epigenetic markers for HCC in a larger cohort. Furthermore, the clinical application of this panel may be particularly limited in the setting of acute or chronic inflammation and cirrhosis, as these states have all been shown to share epigenetic markers with HCC. Focusing on COX-2, p16, RassF1A, TIMP-3 and RUNX3 may increase the ability to make this distinction because, as discussed above, these are not epigenetically modified in non-malignant tissue.58,59 Further studies should validate these and other markers to clearly distinguish HCC from non-malignant hepatic pathology commonly present in patients. This specificity would increase the potential to develop effective serum or blood tests to identify advancement to early HCC and offer the best chance of cure.
Future directions
Although some may argue that early detection of HCC has improved, there are no widely accepted and utilised screening protocols in the USA. The US Preventative Services Task Force (USPSTF) has no recommendation for HCC screening and the American Association for the Study of Liver Disease (AASLD) guidelines60 may be too general. Most studies from the western hemisphere would indicate that the diagnosis is established only in advanced stages. Thus, there is a pressing need for additional modes of providing earlier diagnosis, establishing a response to therapy and predicting recurrence. This is problematic in HCC because numerous staging systems have been developed for prognostic information.61 It is possible that biomarkers and/or methylation profiles may be used as prognostic factors as an adjunct to staging systems for future clinical trials. The strength of DNA hypermethylation profiles is underscored by their validation and the stability of DNA relative to RNA, making methylation profiling a tool better suited to clinical settings.62 A multivariate predictor model based upon traditional clinical factors and DNA methylation profile may have important applications in the early detection of neoplastic transformation in populations at high risk for HCC. CpG methylation is already being linked to prognosis in oesophageal cancer, outcome in CRC following surgery and fluorouracil (5-FU), neuroblastoma, ovarian cancer, acute lymphoblastic leukaemia and gastric cancer,63 and thus may be valuable in HCC prognostics. In addition, the availability of DNA methylation profiles to predict survival outcome and response to treatment may enable clinical prediction in pre-therapy patient biopsies, paraffin-embedded samples or plasma DNA. The ideal methylation signatures for HCC remain to be defined and there is great heterogeneity amongst the different systems that use epigenetic targets as prognostic or therapeutic markers. Therefore, currently the role of epigenetics in HCC is very limited for the hepatobiliary surgeon and these multivariate signatures require prospective validation with larger cohorts before their clinical application can be considered.
Conflicts of interest
None declared.
References
- 1.Soreide K, Nedrebo BS, Knapp JC, Glomsaker TB, Soreide JA, Korner H. Evolving molecular classification by genomic and proteomic biomarkers in colorectal cancer: potential implications for the surgical oncologist. Surg Oncol. 2009;18:31–50. doi: 10.1016/j.suronc.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 3.Smith G, Carey FA, Beattie J, Wilkie MJ, Lightfoot TJ, Coxhead J, et al. Mutations in APC, Kirsten-ras, and p53– alternative genetic pathways to colorectal cancer. Proc Natl Acad Sci U S A. 2002;99:9433–9438. doi: 10.1073/pnas.122612899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zingg JM, Jones PA. Genetic and epigenetic aspects of DNA methylation on genome expression, evolution, mutation and carcinogenesis. Carcinogenesis. 1997;18:869–882. doi: 10.1093/carcin/18.5.869. [DOI] [PubMed] [Google Scholar]
- 5.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 6.Smiraglia DJ, Rush LJ, Fruhwald MC, Dai Z, Held WA, Costello JF, et al. Excessive CpG island hypermethylation in cancer cell lines versus primary human malignancies. Hum Mol Genet. 2001;10:1413–1419. doi: 10.1093/hmg/10.13.1413. [DOI] [PubMed] [Google Scholar]
- 7.Costello JF, Plass C. Methylation matters. J Med Genet. 2001;38:285–303. doi: 10.1136/jmg.38.5.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nabilsi NH, Broaddus RR, Loose DS. DNA methylation inhibits p53-mediated survivin repression. Oncogene. 2009;28:2046–2050. doi: 10.1038/onc.2009.62. [DOI] [PubMed] [Google Scholar]
- 9.Sun L, Hui AM, Kanai Y, Sakamoto M, Hirohashi S. Increased DNA methyltransferase expression is associated with an early stage of human hepatocarcinogenesis. Jpn J Cancer Res. 1997;88:1165–1170. doi: 10.1111/j.1349-7006.1997.tb00345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 11.Horii J, Hiraoka S, Kato J, Harada K, Kuwaki K, Fujita H, et al. Age-related methylation in normal colon mucosa differs between the proximal and distal colon in patients who underwent colonoscopy. Clin Biochem. 2008;41:1440–1448. doi: 10.1016/j.clinbiochem.2008.08.089. [DOI] [PubMed] [Google Scholar]
- 12.Knudson AG. Hereditary cancer: two hits revisited. J Cancer Res Clin Oncol. 1996;122:135–140. doi: 10.1007/BF01366952. [DOI] [PubMed] [Google Scholar]
- 13.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 14.Kopp-Schneider A, Portier C, Bannasch P. A model for hepatocarcinogenesis treating phenotypical changes in focal hepatocellular lesions as epigenetic events. Math Biosci. 1998;148:181–204. doi: 10.1016/s0025-5564(97)10007-4. [DOI] [PubMed] [Google Scholar]
- 15.Ushijima T. Epigenetic field for cancerization. J Biochem Mol Biol. 2007;40:142–150. doi: 10.5483/bmbrep.2007.40.2.142. [DOI] [PubMed] [Google Scholar]
- 16.Kondo Y, Kanai Y, Sakamoto M, Mizokami M, Ueda R, Hirohashi S. Genetic instability and aberrant DNA methylation in chronic hepatitis and cirrhosis – a comprehensive study of loss of heterozygosity and microsatellite instability at 39 loci and DNA hypermethylation on eight CpG islands in microdissected specimens from patients with hepatocellular carcinoma. Hepatology. 2000;32:970–979. doi: 10.1053/jhep.2000.19797. [DOI] [PubMed] [Google Scholar]
- 17.Fellous TG, Islam S, Tadrous PJ, Elia G, Kocher HM, Bhattacharya S, et al. Locating the stem cell niche and tracing hepatocyte lineages in human liver. Hepatology. 2009;49:1655–1663. doi: 10.1002/hep.22791. [DOI] [PubMed] [Google Scholar]
- 18.Gaudio E, Carpino G, Cardinale V, Franchitto A, Onori P, Alvaro D. New insights into liver stem cells. Dig Liver Dis. 2009;41:455–462. doi: 10.1016/j.dld.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 19.Alison MR, Islam S, Lim S. Stem cells in liver regeneration, fibrosis and cancer: the good, the bad and the ugly. J Pathol. 2009;217:282–298. doi: 10.1002/path.2453. [DOI] [PubMed] [Google Scholar]
- 20.Lorenzini S, Bird TG, Boulter L, Bellamy C, Samuel K, Aucott R, et al. Characterization of a stereotypical cellular and extracellular adult liver progenitor cell niche in rodents and diseased human liver. Gut. 2010;59:645–654. doi: 10.1136/gut.2009.182345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, et al. EpCAM positive hepatocellular carcinoma cells are tumor initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012–1024. doi: 10.1053/j.gastro.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marquardt JU, Factor VM, Thorgeirsson SS. Epigenetic regulation of cancer stem cells in liver cancer: current concepts and clinical implications. J Hepatol. 2010;53:568–577. doi: 10.1016/j.jhep.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lingala S, Cui YY, Chen X, Ruebner BH, Qian XF, Zern MA, et al. Immunohistochemical staining of cancer stem cell markers in hepatocellular carcinoma. Exp Mol Pathol. 2010;89:27–35. doi: 10.1016/j.yexmp.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee EK, Han GY, Park HW, Song YJ, Kim CW. Transgelin promotes migration and invasion of cancer stem cells. J Proteome Res. 2010;9:5108–5117. doi: 10.1021/pr100378z. [DOI] [PubMed] [Google Scholar]
- 25.Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61:3225–3229. [PubMed] [Google Scholar]
- 26.Waki T, Tamura G, Sato M, Motoyama T. Age-related methylation of tumour suppressor and tumour-related genes: an analysis of autopsy samples. Oncogene. 2003;22:4128–4133. doi: 10.1038/sj.onc.1206651. [DOI] [PubMed] [Google Scholar]
- 27.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 28.Maeta Y, Shiota G, Okano J, Murawaki Y. Effect of promoter methylation of the p16 gene on phosphorylation of retinoblastoma gene product and growth of hepatocellular carcinoma cells. Tumour Biol. 2005;26:300–305. doi: 10.1159/000089288. [DOI] [PubMed] [Google Scholar]
- 29.Lee S, Lee HJ, Kim JH, Lee HS, Jang JJ, Kang GH. Aberrant CpG island hypermethylation along multistep hepatocarcinogenesis. Am J Pathol. 2003;163:1371–1378. doi: 10.1016/S0002-9440(10)63495-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaneto H, Sasaki S, Yamamoto H, Itoh F, Toyota M, Suzuki H, et al. Detection of hypermethylation of the p16(INK4A) gene promoter in chronic hepatitis and cirrhosis associated with hepatitis B or C virus. Gut. 2001;48:372–377. doi: 10.1136/gut.48.3.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shim YH, Yoon GS, Choi HJ, Chung YH, Yu E. p16 Hypermethylation in the early stage of hepatitis B virus-associated hepatocarcinogenesis. Cancer Lett. 2003;190:213–219. doi: 10.1016/s0304-3835(02)00613-4. [DOI] [PubMed] [Google Scholar]
- 32.Nishida N, Nagasaka T, Nishimura T, Ikai I, Boland CR, Goel A. Aberrant methylation of multiple tumour suppressor genes in ageing liver, chronic hepatitis, and hepatocellular carcinoma. Hepatology. 2008;47:908–918. doi: 10.1002/hep.22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuda Y, Ichida T, Matsuzawa J, Sugimura K, Asakura H. p16(INK4A) is inactivated by extensive CpG methylation in human hepatocellular carcinoma. Gastroenterology. 1999;116:394–400. doi: 10.1016/s0016-5085(99)70137-x. [DOI] [PubMed] [Google Scholar]
- 34.Kanai Y, Ushijima S, Hui AM, Ochiai A, Tsuda H, Sakamoto M, et al. The E-cadherin gene is silenced by CpG methylation in human hepatocellular carcinomas. Int J Cancer. 1997;71:355–359. doi: 10.1002/(sici)1097-0215(19970502)71:3<355::aid-ijc8>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 35.Wong IH, Lo YM, Yeo W, Lau WY, Johnson PJ. Frequent p15 promoter methylation in tumour and peripheral blood from hepatocellular carcinoma patients. Clin Cancer Res. 2000;6:3516–3521. [PubMed] [Google Scholar]
- 36.Yoshikawa H, Matsubara K, Qian GS, Jackson P, Groopman JD, Manning JE, et al. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat Genet. 2001;28:29–35. doi: 10.1038/ng0501-29. [DOI] [PubMed] [Google Scholar]
- 37.Du Y, Carling T, Fang W, Piao Z, Sheu JC, Huang S. Hypermethylation in human cancers of the RIZ1 tumour suppressor gene, a member of a histone/protein methyltransferase superfamily. Cancer Res. 2001;61:8094–8099. [PubMed] [Google Scholar]
- 38.Archer KJ, Mas VR, Maluf DG, Fisher RA. High-throughput assessment of CpG site methylation for distinguishing between HCV-cirrhosis and HCV-associated hepatocellular carcinoma. Mol Genet Genomics. 2010;283:341–349. doi: 10.1007/s00438-010-0522-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Csepregi A, Rocken C, Hoffmann J, Gu P, Saliger S, Muller O, et al. APC promoter methylation and protein expression in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2008;134:579–589. doi: 10.1007/s00432-007-0321-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He N, Park K, Zhang Y, Huang J, Lu S, Wang L. Epigenetic inhibition of nuclear receptor small heterodimer partner is associated with and regulates hepatocellular carcinoma growth. Gastroenterology. 2008;134:793–802. doi: 10.1053/j.gastro.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 41.Arasaradnam RP, Khoo KT, Bradburn M, Mathers JC, Kelly SB. DNA methylation of ESR-1 and N-33 in colorectal mucosa of patients with ulcerative colitis (UC) Epigenetics. 2010;5:422–426. doi: 10.4161/epi.5.5.11959. [DOI] [PubMed] [Google Scholar]
- 42.Jin Z, Cheng Y, Gu W, Zheng Y, Sato F, Mori Y, et al. A multicentre, double-blinded validation study of methylation biomarkers for progression prediction in Barrett's oesophagus. Cancer Res. 2009;69:4112–4115. doi: 10.1158/0008-5472.CAN-09-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clement G, Guilleret I, He B, Yagui-Beltran A, Lin YC, You L, et al. Epigenetic alteration of the Wnt inhibitory factor-1 promoter occurs early in the carcinogenesis of Barrett's oesophagus. Cancer Sci. 2008;99:46–53. doi: 10.1111/j.1349-7006.2007.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu H, Zhou Y, Boggs SE, Belinsky SA, Liu J. Cigarette smoke induces demethylation of prometastatic oncogene synuclein-gamma in lung cancer cells by downregulation of DNMT3B. Oncogene. 2007;26:5900–5910. doi: 10.1038/sj.onc.1210400. [DOI] [PubMed] [Google Scholar]
- 45.Tang M, Xu W, Wang Q, Xiao W, Xu R. Potential of DNMT and its epigenetic regulation for lung cancer therapy. Curr Genomics. 2009;10:336–352. doi: 10.2174/138920209788920994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng Y, Zhang C, Zhao J, Wang C, Xu Y, Han Z, et al. Correlation of CpG island methylator phenotype with poor prognosis in hepatocellular carcinoma. Exp Mol Pathol. 2010;88:112–117. doi: 10.1016/j.yexmp.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 47.Sakamoto J, Fujiya M, Okamoto K, Nata T, Inaba Y, Moriichi K, et al. Immunoprecipitation of nucleosomal DNA is a novel procedure to improve the sensitivity of serum screening for the p16 hypermethylation associated with colon cancer. Cancer Epidemiol. 2010;34:194–199. doi: 10.1016/j.canep.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 48.Glockner SC, Dhir M, Yi JM, McGarvey KE, Van Neste L, Louwagie J, et al. Methylation of TFPI2 in stool DNA: a potential novel biomarker for the detection of colorectal cancer. Cancer Res. 2009;69:4691–4699. doi: 10.1158/0008-5472.CAN-08-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21:5400–5413. doi: 10.1038/sj.onc.1205651. [DOI] [PubMed] [Google Scholar]
- 50.Lu B, Ma Y, Wu G, Tong X, Guo H, Liang A, et al. Methylation of Tip30 promoter is associated with poor prognosis in human hepatocellular carcinoma. Clin Cancer Res. 2008;14:7405–7412. doi: 10.1158/1078-0432.CCR-08-0409. [DOI] [PubMed] [Google Scholar]
- 51.Jackson-Grusby L, Beard C, Possemato R, Tudor M, Fambrough D, Csankovszki G, et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat Genet. 2001;27:31–39. doi: 10.1038/83730. [DOI] [PubMed] [Google Scholar]
- 52.Zhao J, Zhang X, Shi M, Xu H, Jin J, Ni H, et al. TIP30 inhibits growth of HCC cell lines and inhibits HCC xenografts in mice in combination with 5-FU. Hepatology. 2006;44:205–215. doi: 10.1002/hep.21213. [DOI] [PubMed] [Google Scholar]
- 53.Zhou Y, Zhong Y, Wang Y, Zhang X, Batista DL, Gejman R, et al. Activation of p53 byMEG3 non-coding RNA. J Biol Chem. 2007;282:24731–24742. doi: 10.1074/jbc.M702029200. [DOI] [PubMed] [Google Scholar]
- 54.Zheng S, Chen P, McMillan A, Lafuente A, Lafuente MJ, Ballesta A, et al. Correlations of partial and extensive methylation at the p14(ARF) locus with reduced mRNA expression in colorectal cancer cell lines and clinicopathological features in primary tumours. Carcinogenesis. 2000;21:2057–2064. doi: 10.1093/carcin/21.11.2057. [DOI] [PubMed] [Google Scholar]
- 55.Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, et al. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1in human cancers. Nat Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 56.Hernandez-Vargas H, Lambert MP, Le Calvez-Kelm F, Gouysse G, McKay-Chopin S, Tavtigian SV, et al. Hepatocellular carcinoma displays distinct DNA methylation signatures with potential as clinical predictors. PLoS ONE. 2010;5:e9749. doi: 10.1371/journal.pone.0009749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Artinyan A, Mailey B, Sanchez-Luege N, Khalili J, Sun CL, Bhatia S, et al. Race, ethnicity, and socioeconomic status influence the survival of patients with hepatocellular carcinoma in the United States. Cancer. 2010;116:1367–1377. doi: 10.1002/cncr.24817. [DOI] [PubMed] [Google Scholar]
- 58.Ng EK, Tsang WP, Ng SS, Jin HC, Yu J, Li JJ, et al. MicroRNA-143 targets DNA methyltransferases 3A in colorectal cancer. Br J Cancer. 2009;101:699–706. doi: 10.1038/sj.bjc.6605195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saito Y, Kanai Y, Nakagawa T, Sakamoto M, Saito H, Ishii H, et al. Increased protein expression of DNA methyltransferase (DNMT) 1 is significantly correlated with the malignant potential and poor prognosis of human hepatocellular carcinomas. Int J Cancer. 2003;105:527–532. doi: 10.1002/ijc.11127. [DOI] [PubMed] [Google Scholar]
- 60.Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology. 2005;42:1208–1236. doi: 10.1002/hep.20933. [DOI] [PubMed] [Google Scholar]
- 61.Huitzil-Melendez FD, Capanu M, O'Reilly EM, Duffy A, Gansukh B, Saltz LL, et al. Advanced hepatocellular carcinoma: which staging systems best predict prognosis? J Clin Oncol. 2010;28:2889–2895. doi: 10.1200/JCO.2009.25.9895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359:1995–2004. doi: 10.1056/NEJMoa0804525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Teodoridis JM, Hardie C, Brown R. CpG island methylator phenotype (CIMP) in cancer: causes and implications. Cancer Lett. 2008;268:177–186. doi: 10.1016/j.canlet.2008.03.022. [DOI] [PubMed] [Google Scholar]