

Abstract
Proteasome inhibitors are rapidly evolving as potent treatment options in cancer therapy. One of the most promising drug candidates of this type is salinosporamide A from the bacterium Salinispora tropica. This marine natural product possesses a complex, densely functionalized γ-lactam-β-lactone pharmacophore, which is responsible for its irreversible binding to its target, the β subunit of the 20S proteasome. Salinosporamide A entered phase I clinical trials for the treatment of multiple myeloma only three years after its discovery. The strong biological activity and the challenging structure of this compound have fueled intense academic and industrial research in recent years, which has led to the development of more than ten syntheses, the elucidation of its biosynthetic pathway, and the generation of promising structure–activity relationships and oncological data. Salinosporamide A thus serves as an intriguing example of the successful interplay of modern drug discovery and biomedical research, medicinal chemistry and pharmacology, natural product synthesis and analysis, as well as biosynthesis and bioengineering.
Keywords: natural products, oncology, proteasome, salinosporamides, total synthesis
1. Introduction
Natural products have a long history as agents for the treatment of human diseases.[1] Among the most prominent early examples of secondary metabolites used in human health are β-lactam antibiotics, such as the penicillins,[2] chinchona alkaloids, such as the antimalarial drug quinine,[3] and morphine,[4] a potent analgesic. The importance of these molecules for the quality of modern life has made them well-known even outside the world of science and medicine. Despite declining interest of big pharmaceutical companies in natural product drug discovery programs, the impact of nature in currently used medical agents is still impressively high.[5] Of particular importance are natural compounds in the field of cancer treatment.[6–8] Most of the established anti-cancer agents derived from nature thus far have originated from plants (for example, vinblastine, taxol, and camptothecin)[9,10] as well as terrestrial microorganisms (for example, mitomycin and doxorubicin).[7] The exploration of the marine environment in recent years has added numerous anticancer lead structures isolated from marine invertebrates such as sponges, bryozoans, and ascidians.[11,12] One in particular, ecteinascidin-743, has recently been approved for clinical use in Europe.[13] The development of such structurally complex marine metabolites as therapeutic agents, however, has been hampered by limited access to sufficient quantities of pure material. The realization that many of these compounds might be produced by microorganisms, instead of the original source,[14] has fueled recent interest in profiling the secondary metabolite spectrum of associated microbiota of chemically prolific macroorganisms. This has led to the discovery of a number of interesting microbial sources of prominent anti-cancer agents, including taxol-producing endophytic fungi[15] and potential bacterial producers of bryostatin 1.[16] In addition, obligate marine microorganisms have come into the focus of natural product researchers.[17–20] Such micro-organisms are now beginning to emerge as a new and sustainable source for novel chemical entities in academic drug-discovery programs. A particularly rich source for such compounds are marine obligate bacteria of the newly described genus Salinispora.[21,22] Chemical investigations of this young class of obligate marine bacteria have led to the discovery of various new secondary metabolites such as cyanosporaside A (1),[23] saliniketal A (2),[24] and sporolide A (3, Figure 1).[25] The most promising drug candidate derived from these actinomycetes is the highly potent proteasome inhibitor salinosporamide A (4),[26] a natural product produced by Salinispora tropica and discovered by Fenical, Jensen, and co-workers at the Scripps Institution of Oceanography.[27] This Review will highlight the impressive story of the discovery and clinical development of this new class of promising anticancer agents and give insight into the creativity of the biosynthetic and total synthetic pathways to this stimulating small molecule. Before doing so, we first review the function and inhibition of the 20S proteasome, which has recently been validated as an anticancer drug target.
Figure 1.

Selection of structurally novel natural products isolated from marine actinomycetes of the genus Salinispora: cyanosporaside A (1) from S. pacifica; saliniketal A (2) from S. arenicola; sporolide A (3) and salinosporamide A (4) from S. tropica.
2. The Proteasome—A Validated Target in Cancer Chemotherapy
Most natural anticancer drugs to date directly interfere with the cell cycle by either interacting with tubulin (for example, vinca alkaloids, taxanes, and epothilones) or by inhibiting topoisomerases (for example, camptothecins and anthracyclines).[28,29] Other main targets include histone deacetylases (HDACs), which are important for the regulation of gene expression, protein kinases, which are essential for functional changes of target proteins by phosphorylation, and the heat shock protein 90 (Hsp90), a chaperone crucial for maintaining the function of many cell-signaling proteins.[7] The proteasome has received growing interest in recent years as an additional target, with the regulatory approval of bortezomib (5; see Figure 2) in 2003.[30,31]
Figure 2.

Selection of peptide-based synthetic (5–8, 10) and natural (9, 11–12) proteasome inhibitors. Bn =benzyl.
The proteasome is responsible for nonlysosomal intra-cellular proteolysis and thus complements the function of lysosomes, which break down proteins acquired through endocytosis.[32] By regulating the homeostasis and degradation of many important proteins, the proteasome is involved in the control of a multitude of crucial cellular processes, such as the cell cycle, signal transduction, transcription, stress signaling, cell differentiation, and apoptosis.[32,33] Proteins are selected for proteasome-mediated degradation by attachment of multiple ubiquitin units to form a polyubiquitin chain.[34] The formation of this tag occurs by the action of three distinct enzymes, E1–E3.[35,36] The ubiquitin-activating enzyme E1 binds ubiquitin through a thioester bond in a reaction that consumes adenosine-5′-triphosphate (ATP; Scheme 1).[35] The substrate is then transferred to an ubiquitin-conjugating enzyme E2, which facilitates transfer of ubiquitin either directly onto an ubiquitinligase E3 bound protein substrate (RING finger ligases) or to an active-site cysteine residue on the E3 (HECT domain ligases) with subsequent attachment to the protein.[34] Ubiquitin is in most cases conjugated to an ε-NH2 function of a lysine residue of the target protein, or, sometimes, to its N-terminal group.[34] Iterative addition of further ubiquitin residues to the previously attached unit(s) results in formation of a polyubiquitin tag. The polyubiquitinated substrate is then recognized by the downstream 26S proteasome, which is a large multi-subunit complex consisting of one or two 19S caps and a 20S core unit.[37] The regulatory 19S caps of the proteasome recognize the tagged protein, cleave the ubiquitin units, and unfold the protein to allow its entry into the catalytic 20S core. The latter consists of four stacked rings arranged in a cylindrical structure as twofold symmetrical α7β7β7α7 subunits (Scheme 1).[38] Protein degradation is catalyzed by three β subunits per β7 ring. The proteolytic activities of the subunits can be categorized as caspase- (β1, CA-L), trypsin(β2, T-L), and chymotrypsin-like (β5, CT-L), and the selectivity of the subunits is dictated by the composition of their respective binding pockets.[39] The degradation of the target protein, however, follows the same mechanism in each of these proteolytic sites: the side-chain hydroxy group of the N-terminal threonine unit (Thr1) gets deprotonated by Thr1NH2 and subsequently facilitates nucleophilic cleavage of peptide bonds, thereby resulting in breakdown of the protein into small peptide fragments.[39] Inactivation of this nucleophilic process by small electrophilic molecules with affinity for the proteasome binding pockets thus allows for the inhibition of the proteolytic activity of the whole system.
Scheme 1.

Mechanism of protein degradation by the ubiquitin–proteasome system.
Most proteasome inhibitors known to date are small peptides bearing electrophilic groups, which facilitate their covalent attachment to the proteasome by nucleophilic attack of the Thr1 hydroxy groups (Thr1Oγ) of the catalytically active β subunits.[40] Suitable acceptor functions on the inhibitor are aldehydes, vinyl sulfones, and boronates. Aldehydes such as MG132 (6), which is used as a biochemical probe to study proteasome function,[41] form a hemiacetal with the proteasome and are thus relatively weak and reversible inhibitors (Figure 2). Substitution of the aldehyde function by a vinyl sulfone to give ZLVS (7), or by a boronate to yield MG262 (8), renders these inhibitors more potent, and simultaneously improves their selectivity towards the proteasome over common proteases.[36,42] This gain in selectivity and potency allowed for the development of the first proteasome inhibitor approved by the US Food and Drug Administration (FDA), the boronate bortezomib (5; Velcade, PS-341). Similar to most other peptidic proteasome inhibitors of this type, 5 shows highest affinity for the β5 subunit.[40] Natural counterparts of such compounds include peptide aldehydes such as leupeptin (N-acetyl-Leu-Leu-arginal) from various species of actinomycetes[43] and the α′,β′-epoxyketones, which include the highly selective epoxomycin (9) from Streptomyces hygroscopicus.[44,45] The epoxomycin-based synthetic carfilzomib (10) is currently in phase II clinical trials for the treatment of a series of different cancer types.[46] Other peptide-based natural products that exhibit potent proteasome inhibition are the cyclic peptide TMC-95A (11),[47] a compound which inhibits all three catalytically active β subunits through noncovalent bonding, and peptides of the belactosine type, for example, belactosine A (12).[48] The latter compound comprises a cyclic β-lactone unit that also serves as the “chemical warhead” in a different, novel class of natural proteasome inhibitors which are characterized by a γ-lactam-β-lactone bicyclic core structure.
The first nonpeptidic natural proteasome inhibitor to be isolated was lactacystin (13) from Streptomyces sp. OM-6519 (Scheme 2).[49] The structural characteristics of 13 include a densely functionalized central γ-lactam system, equipped with a hydroxylated isobutyl side chain and an N-acetylated cysteine residue bound as a thioester to the core unit. Lactacystin (13) had originally been isolated in a screening program aimed at the discovery of natural products that induce neuritogenesis for the possible treatment of nerve diseases, such as Alzheimer’s.[50] Lactacystin (13) was only later found to exhibit low nanomolar inhibitory activity against the β5 subunit of the proteasome. Interestingly, 13 undergoes a spontaneous intramolecular formation of a lactone ring by attack of the β-hydroxy function of the γ-lactam ring to cleave the thioester bond with concomitant loss of N-acetylcysteine.[51] The so-formed clasto-lactacystine-β-lactone (14, omuralide) constitutes the actual biologically active molecule. Attachment to, and thus inhibition of, the proteasome by 14 then occurs by intermolecular attack of the Thr1Oγ of the β5 subunits of the proteasome at the β-lactone under ring cleavage and formation of a covalent ester linkage.[52] These observations clearly established the novel γ-lactam-β-lactone structural motif of 14 to be well suited to selectively target the proteasome.
Scheme 2.
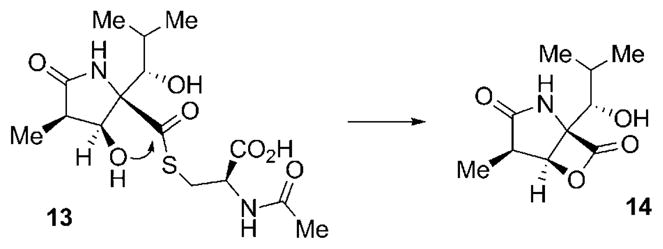
Non-enzymatic formation of omuralide (14) from its precursor, the natural product lactacystin (13).
3. Discovery of the Salinosporamide Family
The family of γ-lactam-β-lactone natural products expanded in 2003 with the discovery of salinosporamide A (4) from S. tropica isolated from marine sediment from the Bahamas.[27] This compound has the same core structure as omuralide (14), but with significant differences in the substitution pattern: 4 possesses an unusual cyclohexenyl substituent, a methyl group situated at the β-lactone system, and a chloroethyl side chain at the α position. These structural variations led to a significant increase in the proteasome inhibition potency of 4 against all three proteolytic subunits of the proteasome when compared to 14. The pronounced biological activity of 4, with IC50 values in the low nanomolar range, prompted a closer examination of the producing organism, which yielded a series of related compounds, among them the deschloro analogue salinosporamide B (15), the tricyclic γ-lactam analogue salinosporamide C (16), and a number of salinosporamide A derived decomposition products 17–21 (Scheme 3).[53] A probable pathway for the decomposition of 4 to 17–19 is initiated by cleavage of the lactone ring (either in a concerted way as shown or by aqueous hydrolysis of the lactone followed by dehydration) to give carboxylate 22, which upon decarboxylation delivers pyrrole 23, a tautomer of the diasteromers 17 and 18. Additional loss of H2O then leads to 19. The formation of 20 and 21 can be explained by nucleophilic attack of MeOH on the lactone ring to give 20, and subsequent intramolecular formation of a THF ring by displacement of the chlorine substituent to furnish 21. Interestingly, a similar reaction sequence is responsible for the pronounced biological activity of 4, in which the inhibitor gets attached to Thr1Oγ of the proteasome by nucleophilic cleavage of its lactone ring, thus initiating chlorine displacement with subsequent formation of a THF ring. The newly generated cyclic ether effectively blocks the hydrolytically active H2O from cleaving the proteasome–inhibitor ester bond, and thus renders 4 irreversibly bound. This molecular mode of action is discussed in more detail in Section 6.
Scheme 3.

Structures and mechanisms of formation of several salinosporamide-type compounds from S. tropica.
Another set of highly related secondary metabolites produced by S. tropica was identified by Nereus Pharmaceuticals, in the course of purifying multigram quantities of 4 from large-scale fermentations of the strain.[54] Most of these derivatives showed differences in the C2 side chain, either being shorter as in methyl-substituted salinosporamide D (24), longer as in propyl analogue salinosporamide E (25), or epimeric as in salinosporamides F–H (26–28; Figure 3). In addition, the C3 ethyl-substituted salinosporamide I (29) and the deshydroxy derivative salinosporamide J (30) were isolated. Most interestingly, substitution of NaCl by NaBr in the fermentation broth allowed for the production of bromosalinosporamide (31). Meanwhile, closely related γ-lactam-β-lactone natural products, cinnabaramides A–G (32–38), were identified from a terrestrial Streptomyces strain.[55] Cinnabaramides A–C (32–34) all contain a hexyl substituent at C2 but have different oxygenation patterns at C5 and C12. Hydrolysis of the β-lactone of 32 and 33 leads to cinnabaramides D (35) and E (36). In addition, two thioester derivatives 37 and 38 of the main congener 32 could be identified. Interestingly, despite lacking the chlorine substituent responsible for the strong activity of salinosporamide A (4), cinnabaramide A (32) shows a strong inhibition of the proteasome. A more detailed discussion of structure–activity relationships and their interpretation is presented in Section 6.
Figure 3.

Structures of further secondary metabolites of the salinosporamide family.
4. The Biosynthesis of Salinosporamide A
The strong biological activity of many γ-lactam-β-lactone natural products together with their intriguing chemical structures made studies on their biosynthesis highly rewarding. Several questions, in particular concerning the biosynthesis of salinosporamide A (4), had to be answered, for example, about the timing and mechanism of the chlorination reaction and the biosynthesis of the unusual cyclohexenyl moiety. The molecular structures of the isolated salinosporamide-type compounds strongly suggested a biosynthesis of these metabolites from three building blocks: an unusual cyclohexenyl-alanine derivative 39, acetate (or propionate in the case of 29), and a set of promiscuously incorporated short-chain carboxylic acids, which facilitates generation of the observed structural diversity of the substitution pattern at C2. The introduction of the chlorine substituent in 4 was initially anticipated to occur by late-stage oxidative halogenation of 15. In the course of fermentation studies by Nereus Pharmaceuticals to improve the titer of the pharmaceutically more relevant 4 and to decrease the formation of 15, however, feeding of butyric acid surprisingly resulted in a decreased formation of 4 but an increased production of 15.[56] The application of [13C4]butyric acid revealed the incorporation of this building block into C1-C2-C12-C13 of 15 only, while the presumably acetate-derived unit C3–C14 was labeled in both molecules. This observation suggested different biosynthetic origins for the C1-C2-C12-C13 portions of salinosporamides A (4) and B (15). Further evidence for this unexpected result was obtained by a more comprehensive isotopic labeling study carried out simultaneously in our laboratory.[57] In a set of experiments using [13C2]acetate, [1-13C1]butyrate, and [13C6]glucose, the C1-C2-C12-C13 unit of 4 was clearly identified to be derived from a tetrose precursor biosynthetically connected to erythrose 4-phosphate (40), while the same carbon atoms in 15 originated from two acetate units, consistent with the intermediacy of a butyric acid derivative (Scheme 4). The [13C6]glucose feeding experiment additionally revealed that the cyclohexenyl residue is also derived from a tetrose unit. The northern part of the salinosporamides was thus assumed to be shunted from the shikimate pathway. The observed nonsymmetric labeling pattern in the cyclohexenyl group thereby suggested a divergence of the biosynthetic precursor of the salinosporamides from the shikimate pathway at a stage prior to aromatization (which in turn would have led to a randomization of the labeling pattern such as in phenylalanine). These assumptions were further corroborated by application of [1,7-13C2]shikimic acid (41) and [1-13C1]phenylalanine to S. tropica. As expected, labeling of 4 and 15 was only achieved with [1,7-13C2]-(41). A plausible route to the formation of the amino acid building block 39 would thus start with phosphoenyl pyruvate (42) and erythrose 4-phosphate (40), which would be transformed into 3-deoxy-D-arabinoheptulosonate-7-phosphate (43, DAHP) by action of DAHP synthase, and further to shikimic (41) and prephenic acid (44) by primary metabolism. Compound 44 could then be reduced to give 45, which upon decarboxylative dehydration would yield 46. Final reduction and transamination would lead to 39, which after oxidative installation of the side-chain hydroxy group serves as a building block for 4 and 15.[57]
Scheme 4.

Results of the stable isotope labeling experiments of 4 and 15, as well as the proposed mechanism of formation of the unusual amino acid precursor 39.
These extensive labeling studies thus shed light on the biosynthetic origin of all the molecular building blocks of 4 and 15. Together, the results were suggestive of an unprecedented mixed polyketide synthase (PKS)/nonribosomal peptide synthetase (NRPS) pathway towards the salinosporamides, which contrasts that of lactacystin/omuralide, which is assembled through the PKS-independent condensation between valine-derived 2-methyl-3-oxopropionic acid (methylmalonic semialdehyde) and leucine.[58,59] The identification of the complete salinosporamide biosynthesis gene cluster (sal) in the course of the whole-genome sequencing of the producing strain S. tropica CNB-440 comprising 5183331 base pairs further corroborated the conclusions drawn from the isotope experiments.[60,61] The central element of the 41000 base pair sal gene cluster is comprised of the PKS-coding gene salA and the NRPS-coding gene salB (Scheme 5a). The bimodular gene product of salA harbors six domains—two acyltransferases (ATs), two acyl carrier proteins (ACPs), a ketosynthase (KS), and a condensation domain (C)—organized in a noncanonical fashion: ACPL-KS1-ATL-AT1-ACP1-C2 (Scheme 5b).[60] The domain architecture thus deviates from typical PKSs (ATL-ACPL-KS1-AT1-ACP ),[62] but resembles the organization found in a number of myxobacterial megasynthases, such as those encoding the biosyntheses of stigmatellin,[63] soraphen,[64] and aurofuranone.[65] The contiguous AT domains are thought to be responsible for the selection of the PKS starter (ATL) and extender (AT1) units, as well as their attachment to ACPL and ACP1, respectively (Scheme 5b). Condensation of the PKS building blocks in the presence of a KS1 catalyst results in an ACP1-bound β-ketothioester. The central role of the PKS gene salA in the biosynthesis of salinosporamide was confirmed by targeted inactivation by single-crossover homologous recombination, after which the resulting mutant was void of all salinosporamide chemistry.[66] The amino acid precursor 39 is then selected by the adenylation domain (A) and attached to the peptidyl carrier protein (PCP) of the SalB didomain. PCP-bound 39 is subsequently oxidized by the cytochrome P450 hydroxylase SalD. Fusion of the PKS- and the NRPS-derived precursors by the C-terminal condensation (C) domain of SalA leads to a PCP-bound linear intermediate, which after bicyclization and concurrent release from the synthetase yields the fully assembled salinosporamide molecules by an as yet unknown process.
Scheme 5.

a) Organization of the salinosporamide biosynthetic gene cluster (sal); b) proposed mechanism of salinosporamide assembly by the PKS-NRPS machinery SalA-SalB.
The observed structural diversity of the salinosporamide family is predominantly caused by the relaxed substrate specificity of the AT domains of SalA. Although selection of an acetate starter unit by ATL leads to the typical methyl substituent at C3 of the salinosporamides, an alternative priming with propionate would allow for the production of the only C3-ethyl derivative isolated so far, salinosporamide I (29). The promiscuity of AT1 in turn facilitates the formation of the observed variability in the substitution pattern at C2. For the assembly of salinosporamide A (4), AT1 incorporates the novel halogenated PKS extender unit chloroethylmalonyl-CoA (47; CoA=coenzyme A), which is unique to the salinosporamide family.[66] Interestingly, the sal cluster lacks any previously described oxidative chlorinases. but instead contains the gene salL,[67] whose protein product shows 35% amino acid identity with the fluorinase FlA from Streptomyces cattleya.[68] FlA catalyzes the nucleophilic addition of fluoride to S-adenosyl-L-methionine (48) with displacement of L-methionine as the initial step in the biosynthetic pathway to the mammalian toxin fluoroacetate and the antibiotic 4-fluorothreonine.[68,69] In vivo and in vitro characterization of SalL demonstrated its ability to instead act as a chlorinase on 48 to yield 49 (Scheme 6).[67] In addition, SalL accepts bromide and iodide as substrates to form the respective halodeoxyadenosines, albeit at significantly reduced rates in vitro. The incorporation of fluorine, however, is not possible with SalL because of subtle rearrangements of the halide-binding pocket, as observed in the high-resolution crystal structures of SalL and active-site mutants. Together with the fluorinase FlA[68] and the (S)-adenosyl-L-methionine (SAM) hydroxide adenosyltransferases (duf-62) recently discovered,[70–72] SalL forms a novel class of SAM-metabolizing enzymes.[73] The formation of 5′-chloro-5′-deoxyadenosine (49, 5-ClDA) initiates a complex biosynthetic route to 47, with all the crucial genes present in the sal cluster, as deduced by gene inactivation and chemical complementation experiments (Scheme 5a, colored in blue).[66] The second step in this pathway, phosphorolytic cleavage of 49 to give 5-chloro-5-deoxy-D-ribose-1-phosphate (50, 5-ClRP), is facilitated by SalT, a homologue of FlB, which catalyzes the respective reaction on the fluorinated substrate in S. cattleya.[69]
Scheme 6.

Biosynthetic pathway to the novel salinosporamide A PKS extender unit chloroethylmalonyl-CoA (47) from (S)-adenosyl-L-methionine (48).
Divergence from the established fluoroacetate pathway occurs by cleavage of the phosphate group by the phosphatase SalN to give 5-chloro-5-deoxy-D-ribose (51, 5-ClRP) and subsequent oxidation by the dehydrogenase/reductase SalM to yield 5-chloro-5-deoxy-D-ribono-1,4-lactone (52, ClRL) and finally 5-chlororibonate (53, 5-ClRI). The dihydroxyacid dehydratase homologue SalH converts 53 into 5-chloro-4-hydroxy-2-oxopentanoate (54), which is oxidatively decarboxylated to 4-chloro-3-hydroxybutyryl-CoA (55) and reduced by the putative dehydratase SalS to give 4-chlorocrotonyl-CoA (56). Compound 56 is further converted by SalG. This enzyme shares more than 60% sequence identity to crotonyl-CoA carboxylases/reductases (CCRs), which have recently been shown to catalyze the direct formation of ethylmalonyl-CoA from crotonyl-CoA.[74,75] Similarly, in vitro studies with recombinant SalG showed that it acts as a 5-chlorocrotonyl-CoA carboxylase/reductase, transforming 56 into the salinosporamide A specific PKS extender unit 47.[66]
In vitro characterization of SalG revealed its preference for the chlorinated substrate 56 over crotonyl-CoA by a factor of seven, thus suggesting it to predominantly form 47, the PKS extender unit necessary for the biosynthesis of salinosporamide A (4).[66] The S. tropica genome harbors a second CCR gene (Strop_3612), which was thus thought to be employed for the biosynthesis of ethylmalonyl-CoA. Inactivation of Strop_3612 indeed led to a drop in the formation of salinosporamide B (15) to about 50% of the wild-type level, while production of 4 was not significantly affected. Further investigations on both S. tropica CCR deletion mutants revealed that production of salinosporamide E (25) was exclusively lost in the salG-deficient mutant.[76] This finding suggests that SalG has evolved for the production of 2-alkylmalonyl-CoA substrates larger than ethylmalonyl-CoA. A combination of in vitro and in vivo experiments verified this assumption, by showing that SalG facilitates the formation of the novel PKS extender units propyl-, 4-bromo-, and 4-fluoromalonyl-CoA. It is interesting to note that 2-alkenoates with C6–C8 chains were not accepted as substrates by SalG.[76] The isolation of the cinnabaramides 32–38,[55] whose biosynthesis would necessitate incorporation of a 2-hexylmalonyl-CoA unit, however, strongly suggests the existence of a CCR homologue capable of accommodating longer chain 2-alkenyl-CoAs. Alternatively, the biosynthetic pathway to longer chain 2-alkylmalonates might occur in an orthogonal manner by α-carboxylation of fatty acids, as observed in the biosynthesis of mycolic acid.[77] In summary, these investigations identified a number of previously unknown PKS extender units accessible by reductive carboxylation of α,β-unsaturated acyl-CoA thioesters by action of a CCR—a mechanism which might prove useful for the bioengineering of other novel polyketides.[66,76]
Besides the genes involved in biosynthesis of the PKS extender unit 47 and the PKS-NRPS machinery SalA-SalB, the results of the labeling studies[57] suggest that many of the genes associated with the proposed biosynthesis of the novel nonproteinogenic amino acid 39 are present in the sal cluster (Scheme 5a, colored in green). These include coding for a dedicated DAHP synthase (salU), thought to initiate the biosynthesis of amino acids by formation of 43 from 40 and 42, a prephenate dehydratase homologue (salX), presumably catalyzing reductive decarboxylation of 45 to give 46, and an aminotransferase (salW), which could install the amino function in the transformations involved in generating 39 from 46 (see Scheme 4).[60] A similar pathway has recently been identified in Bacillus subtilis, which produces tetrahydrotyrosine as part of the biosynthesis of anticapsin.[78] Deletion of salX abolished the biosynthesis of all the salinosporamides and allowed for the directed mutagenetic production of a series of unnatural salinosporamide derivatives (see Section 6.2).[79,80] The seemingly missing enzymes (that is, those involved in formation 44 from 43) as well as the two missing reductases have not been identified and may be located outside the sal locus. The hydroxy group is finally installed in the side chain by the P450 hydroxylase SalD, as evidenced by inactivation of the respective gene leading to production of salinosporamide J (30). Based on precedence with related P450s, this likely takes place while the amino acid 39 is bound to the PCP domain of SalB.[79]
In addition, the sal locus harbors genes responsible for regulation and resistance (Scheme 5a, colored in yellow), and a number of genes with diverse other functions (Scheme 5a, colored in black), among those salF, which codes for a type-II thioesterase thought to remove misprimed precursor molecules from the PKS-NRPS module, and genes putatively involved in the biscylization processes leading to the final products. The functions of these genes and their protein products are currently under investigation in our laboratory.
5. Total Syntheses of Salinosporamide A
The challenging structure of the salinosporamides, with five contiguous stereogenic centers and a complex bicyclic framework, has resulted in this natural product family attracting tremendous interest from the synthetic community. Of particular interest has been salinosporamide A (4) with its most pronounced inhibitory activity against the 20S proteasome and the resulting biomedical importance of this secondary metabolite. Key features of all the total synthetic efforts towards 4 are summarized in Table 1 and are discussed in more detail below.
Table 1.
Total syntheses of salinosporamide A (4).
| Group | Year | Steps (yield) | Key features |
|---|---|---|---|
| E. J. Corey | 2004 | 17 steps from 57 (13.6%) | shortest stereoselective route; highest chemical yield and ee value; development of key reactions also used in later syntheses |
| S. J. Danishefsky | 2005 | 28 steps from 71 (1.8%) | regioselective lactone ring opening using a selenium species; cationic hemiacetal selenocyclization to give C4 configuration |
| G. Pattenden | 2006 | 14 steps from 82 (9.6%) | synthesis of racemic 4; biomimetic diastereoselective acid-catalyzed cyclization of the γ-lactam ring |
| D. Romo | 2007 | 7 steps from 89 (3.3%) | synthesis of racemic 4; still shortest approach; biomimetic bicyclization reaction; also applied to the synthesis of 32 |
| Neureus Pharma. | 2007 | 18 steps from 96 (0.8%) | intramolecular aldol cyclization generating three stereocenters; new method for the introduction of the cyclohexenyl moiety |
| S. Hatakeyama | 2008 | 21 steps from 101 (3.5%) | Coniaene reaction as the key step in the γ-lactam formation |
| S. Omura | 2008 | 36 steps from 106 (2.0%) | early stage, stereoselective construction of the cyclohexene moiety |
The first total synthesis of 4 was reported in 2004, only one year after its structure was published.[81] The synthetic access elaborated by Corey and co-workers set the standard for later synthetic approaches by many other research groups. This milestone study, although reviewed earlier,[82] is discussed in detail. The synthesis commenced with (S)-threonine derivative 57, which was cyclized to give oxazoline 58 (Scheme 7).[81] The quaternary stereocenter at C4 was installed by alkylation of 58 with chloromethyl benzyl ether to furnish 59. Reductive cleavage of the oxazoline moiety afforded N-PMB-amine 60, which was selectively N-acylated with acrylyl chloride, facilitated by intermediate in situ protection of the secondary hydroxy group as a TMS ether, to finally give 61. Oxidation of the hydroxy group with Dess–Martin periodane delivered ketone 62, the key precursor for the generation of the γ-lactam moiety. Ring closure was achieved by an internal Baylis–Hillman-aldol reaction in the presence of quinuclidine as the catalytic base, which delivered a 9:1 mixture of the diastereomeric lactams 63 and 64 in favor of the desired stereoisomer 63. In a subsequent study, the Corey research group developed an unusual and highly efficient alternative cyclization reaction of 62 by using the Kulinkovich reagent,[83] which permitted complete diastereoselectivity to give 63 in significantly reduced reaction times.[84,85] Silylation of 63 by using bromomethyldimethylsilyl chloride gave 65, which was cyclized in a radical-chain reaction mediated by tri-n-butyltin hydride to deliver only the desired cis-fused lactam 66. O-Debenzylation and oxidation of the primary alcohol thus obtained yielded aldehyde 67, the substrate for the following stereoselective installation of the salinosporamide side chain. The addition of 2-cyclohexenylzinc chloride (68)[81,86] to the aldehyde function in 67 allowed for the introduction of the cyclohexenyl moiety with impressive control over the newly formed stereocenters at C5 and C6 (20:1 in favor of 69). A subsequent Tamao–Fleming oxidation and CAN-mediated N-deprotection resulted in triol 70, which was transformed into the natural product 4 by saponification of the methyl ester, lactone formation, and final OH-Cl exchange. Overall, this synthesis route by Corey and co-workers gave access to 4 in an impressive 16.5% yield from 57 with excellent stereocontrol.[81] The synthetic route stands out because of the conservative use of protective groups and the lack of any protective-group interchange within the sequence. The high impact of a number of synthetic features developed during this study, in particular the installation of the cyclohexenyl residue and the late-stage introduction of the chlorine substituent, is evidenced by their recurring use in later synthetic studies towards 4 by other research groups (see other syntheses below).
Scheme 7.

Total synthesis of salinosporamide A (4) by the Corey research group. AIBN=2,2′-azobisisobutyronitrile, Bn =benzyl, BIP =1-benzotriazolyloxytris(dimethylamino)phosphonium hexafluorophosphate, CAN=cerium ammonium nitrite, DMAP =4-dimethylaminopyridine, LDA =lithium diisopropylamide, PMB =para-methoxybenzyl, py =pyridine, TMS =trimethylsilyl, Ts =toluene-4-sulfonyl (tosyl).
An entirely different approach for the stereoselective construction of the chiral centers situated on the γ-lactam ring of 4 was pursued by Endo and Danishefsky.[87] Starting with the known pyroglutamate derivative 71 derived from the chiral pool, the C3 substituent was introduced by 1,4-addition of a vinyl cuprate to deliver solely compound 72 (Scheme 8). Subsequent alkylation of 72 led to diastereoselective (14:1) installation of the C2 side chain to give 73. Ozonolysis, carbonate formation, and removal of the N,O-acetal furnished primary alcohol 74, which was transformed into imidate ester 75 by oxidation, ester formation, and treatment with Meerwein salt. The quaternary stereocenter at C4 was then elegantly established by internal acylation with the ethyl carbonate positioned as the C3 substituent to give 76. Acidic deprotection of the γ-lactam, followed by protection of the nitrogen atom with a PMB group and hydrogenolytic cleavage of the O-benzyl group gave alcohol 77. The lactone ring was then cleaved with phenylselenium anion and the liberated acid group was benzylated to afford 78, which was converted into aldehyde 79 by selenide oxidation/elimination and Dess–Martin oxidation. The following acetal-mediated cationic cyclization, by treatment of 79 with phenylselenyl bromide and AgBF4, allowed for the construction of the stereocenter at C3 with perfect stereocontrol. Product 80 was subjected to radical deselenenylation, thus leading to the desired methyl group at C3, and the benzyl ester at C4 chemoselectively converted into an aldehyde to give 81. The synthesis of 4 from 81 was concluded in seven more steps, including the cyclohexenylzinc addition and lactonization/chlorination sequence developed by Corey and co-workers.
Scheme 8.

Synthesis route to salinosporamide A (4) by Endo and Danishefsky. HMDS =hexamethyldisilazide, TfOH =trifluoromethane-sulfonic acid.
A first biosynthetically inspired approach towards racemic 4 was presented by Pattenden and co-workers in 2006 (Scheme 9).[88,89] The starting material in this synthesis, the α-substituted β-ketoester 82[90] is structurally highly related to the PKS extension units employed by nature in the biosynthesis of 4. Protection of the keto group of 82 and saponification of the methyl ester delivered acid 83, which was coupled with dimethyl 2-aminomalonate to form amide 84. Compound 84, in turn, resembles the proposed linear PKS-NRPS intermediate of the salinosporamide biosynthesis (see Scheme 5b). Acidic removal of the dioxalan led to a biomimetic intramolecular cyclization to give γ-lactam 85 as a single diastereomer. Formation of the TMS ether of the newly formed tertiary alcohol followed by PMB protection of the amide to furnish 86 set the stage for chemoselective reduction of the desired methyl ester group to give aldehyde 87. From there, the synthesis of racemic 4 was concluded in seven additional steps, in analogy to the synthesis of Corey and coworkers.
Scheme 9.

First biomimetic total synthesis of racemic salinosporamide A [rac-(4)] as elaborated by Pattenden and co-workers. EDC =N′-(3-dimethylaminopropyl)-N-ethylcarbodiimide, HOBt = 1-hydroxy-1H-benzotriazole.
Another, even shorter biomimetic synthesis of racemic 4 was published shortly thereafter by Romo and co-workers.[91] In this route, the bicyclic framework of 4 was generated in a single step from an easily available linear precursor molecule 88, which in turn was obtained by treating the orthogonally protected N-PMB-serine allyl ester 89 with the heteroketene dimer rac-90 in a single step (Scheme 10).[91] As opposed to all the routes described previously, the chlorine substituent was already introduced at this early stage of the synthesis. Cleavage of the allyl ester under mild conditions set the stage for biscyclization using 2-bromo-1-isopropylpyridinium triflate (91) and 4-pyrrolidinopyridine (92, 4-PPY) to give a diastereomeric mixture (d.r. = 2:1–3:1) of the desired γ-lactam-β-lactones 93 and 94, in favor of the isomer with the relative configuration found in 4. Reductive removal of the O-benzyl protecting group in 93 allowed for the enrichment of the desired diastereomer 95 (to 6:1–10:1) after purification. The synthesis of rac-4 was completed by oxidation of the primary alcohol to the aldehyde, addition of Corey’s cyclohexenylzinc 68, and final N-deprotection. It is important to note that the addition of 68 proceeded with relatively low diastereoselectivity (d.r. = 3.5:1) when compared to the other syntheses in which this method was applied. However, Endo and Danishefsky likewise observed a reduced diastereoselectivity when employing N-unprotected substrates,[87] thus illustrating the strong substrate dependence of this addition reaction. Although the synthesis by Romo and co-workers suffers from relatively low chemical yield and enantioselectivity in key transformations, its extremely short and innovative route by a biomimetic biscyclization is highly remarkable. In addition, this approach was also utilized for the first and so far only total synthesis of racemic cinnabaramide A (32).[91]
Scheme 10.

Short biomimetic total synthesis of racemic salinosporamide A [rac-(4)] by Romo and co-workers.
The first total synthetic route to 4 that used a slightly different approach for the introduction of the cyclohexenyl moiety than used by Corey and co-workers was presented by Nereus Pharmaceuticals.[92] Furthermore, the installation of the stereocenters in the γ-lactam ring system followed a novel strategy. For that, the key precursor, β-keto amide 96, was subjected to an intramolecular aldol reaction, taking advantage of the concept of self-regeneration of stereocenters developed by Seebach et al.[93] to furnish oxazolidine-γ-lactam 97, which already contained all the required stereogenic elements (Scheme 11). Compound 97 was obtained in d.r. = 17:3 and converted into the pure desired enantiomer by crystallization. Remarkably, this step (and all preceding transformations not shown here) could be scaled to generate 100 g of 97. A series of oxidation/reduction steps delivered aldehyde 98, which was initially subjected to the cyclohexenylzinc method of Corey and co workers. In this case, however, the reaction delivered an anti addition product with the undesired configurations at C5 and C6. The installation of the side chain was finally accomplished by using the allylboration chemistry developed by Kramer and Brown: coupling of 98 with B-2-cyclohexen-1-yl-9-borabicyclo-[3.3.1]nonane (99)[94] led to the expected syn addition product 100, and this time with the correct configuration at C6. After several functional-group conversions, including lactone formation and OH-Cl exchange as elaborated by Corey and coworkers, and protective-group alterations, the undesired configuration in the obtained C5-epi-salinosporamide A (C5-epi-4) had to be corrected. This was achieved by oxidation of C5-epi-4 to keto-salinosporamide with subsequent stereoselective reduction. Interestingly, the reduction step turned out to be the most problematic, as all the chemical methods tested failed to give the desired product 4 in satisfying yields. The goal was ultimately attained by an enzymatic reduction with the NADH-dependent ketoreductase KRED-EXP-B1Y and glucose dehydrogenase (GDH) for in situ regeneration of the cofactor.[92,95]
Scheme 11.
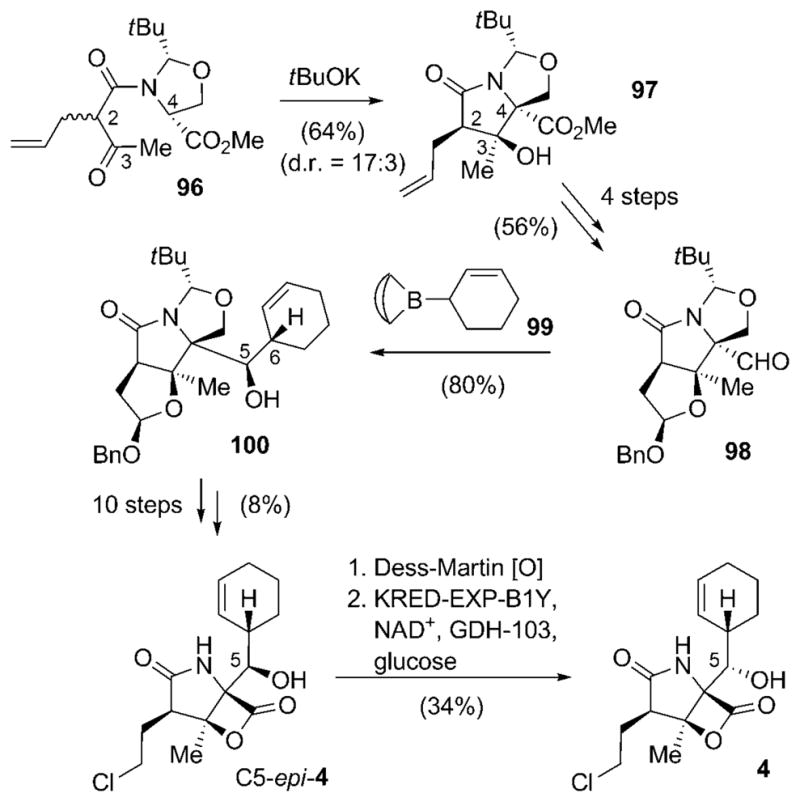
Total synthetic pathway towards salinosporamide A (4) by Nereus Pharmaceuticals. NAD+=nicotinamide adenine dinucleotide (oxidized form).
A novel access to the γ-lactam portion of 4 by utilizing an indium-catalyzed Coniaene reaction[96] as the key step was elaborated by Hatakeyama and co-workers (Scheme 12).[97] The chiral propargyl alcohol 101[98] was converted in a five-step sequence into diol 102. Oxidation of 102 and activation of the carboxylic acid thus obtained as an acid chloride followed by condensation with dimethyl 2-(PMB-amino)malonate gave the key precursor 103. Column chromatography resulted in 103 already partially cyclizing to an inseparable 72:28 mixture of 103 and the desired product 104. An In(Otf)3-mediated Coniaene reaction of this mixture led to a complete conversion into γ-lactam 104 with 90% ee. The observed high enantiomeric purity of 104 is suggestive of a Coniaene reaction promoted by silica gel during the work-up of 103, instead of cyclization through the achiral allenylamide, which would in turn have led to a loss of enantiomeric purity. The instability of 104 under basic conditions resulted in the acetyl protecting group being removed under mild conditions with a lipase, and the released primary alcohol subsequently oxidized to give aldehyde 105. Compound 105 is nearly identical to Danishefsky’s intermediate 79, only differing in the ester functions (two methyl groups in 105 versus tert-butyl and benzyl in 79; see Scheme 8), and was consequently transformed into the natural product 4 by a similar route.
Scheme 12.

Total synthetic access to salinosporamide A (4) according to Hatakeyama and co-workers. DDQ =2,3-dichloro-5,6-dicyano-1,4-benzoquinone, Ms =methanesulfonyl (mesyl), TBAF =tetrabutylammonium fluoride, TBS =tert-butyldimethylsilyl.
A completely different approach for the construction of salinosporamide A (4), involving the stereoselective synthesis of the cyclohexene ring at an early stage of the synthesis, was presented by Omura and co-workers.[99] Starting from aldehyde 106,[100] Wittig olefination and deprotection delivered diol 107, which was transformed into chiral building block 108 in 97% ee by lipase-catalyzed enzymatic desymmetrization followed by O-silylation (Scheme 13). Compound 108 already comprises the later C4 quaternary stereocenter of 4. Reprotection of the O-acetate as an O-MEM ether, removal of the TBDPS protecting group, intramolecular carbamation, and N-PMB protection furnished 109, which was oxidatively converted into aldehyde 110. The following key step stereo-selectively installed a cyclohexanone group by an LDA-mediated chelation-controlled aldol reaction. In situ O-benzylation of the resulting secondary hydroxy group afforded benzoate 111 with d.r. = 20:1. The required transformation of the cyclohexanone into the desired cyclohexene portion proved to be challenging and required a series of reactions. Luche reduction and solvolysis resulted in diol 112, which was converted in two steps into sulfate 113. Treatment of 113 with DBU and subsequent exposure of the resulting O-sulfonic acid to TsOH delivered cyclohexene 114, which was subjected to intramolecular transcarbamation and Swern oxidation to give aldehyde 115. Addition of methylmagnesium bromide, Dess–Martin oxidation, and N-PMB deprotection furnished 116. The γ-lactam moiety was subsequently installed by N-acetylation of 116 and an intramolecular aldol reaction to give 117 as a single isomer. The still-missing C2 side chain was incorporated next. For that, 117 was subjected to a SmI2-mediated Reformatsky-type reaction with benzyloxyacetaldehyde to give 118. As direct deoxygenation of 118 proved to be unsuccessful, removal of the undesired hydroxy group was achieved by mesylation and elimination via an intermediate alkene, which was diastereoselectively reduced to afford γ-lactam 119 with a moderate d.r. value of 4.2:1. The natural product 4 was obtained from 119 in nine more steps, predominantly involving protective-group manipulations, as well as the method of Corey and co-workers for the closure of a β-lactone and introduction of the chlorine substituent.[99]
Scheme 13.

Total synthesis of 4 by Omura and co-workers with an early introduction of the cyclohexenyl moiety. Boc =tert-butyloxycarbonyl, Bz =benzoyl, DBU =1,6-diazabicyclo[5.4.0]undec-7-ene, imid. =imidazole, MEM=(2-methoxyethoxy)methyl, NMO =methylmorpholine N-oxide, TBDPS=tert-butyldiphenylsilyl.
Yet another method for the construction of the γ-lactam moiety of salinosporamide A (4) was recently published in a formal total synthesis of 4 by Chida and co-workers.[101] In this route the chiral information was derived from protected furanose derivative 120,[102,103] which was transformed into benzoate 121 in five steps (Scheme 14). After a series of reprotection reactions and glycol cleavage to give pyranose 122, the anomeric hydroxy group was protected as an O-PMB-β-glycoside, the O-formyl group hydrolyzed, and the liberated secondary alcohol oxidized to give ketone 123. This set the stage for a substrate-controlled, highly stereoselective installation of the future C3 quaternary stereocenter to give 124. Removal of the O-Tos group and oxidation of the alcohol thus obtained allowed for a Horner–Wadsworth–Emmons reaction to obtain alkene 125 selectively. O-Silylation, reduction of the ethyl ester, and installation of the trichloroacetimidate furnished 126, the key precursor for the generation of the C4 quaternary stereocenter. This was achieved by Overman rearrangement of 126, which resulted in 127 with moderate d.r. = 4.3:1. Replacement of the N-trichloroacetyl group with an N-Cbz protecting group delivered 128. Treatment of 128 with aqueous TFA facilitated rearrangement to the desired γ-lactam 129. A two-step oxidation, followed by methyl ester formation, O-TMS protection of the tertiary alcohol at C3, and oxidative cleavage of the exocyclic double bond afforded aldehyde 130. The natural product 4 can then be obtained in five additional steps by using the approach elaborated by Corey and co-workers.
Scheme 14.

Synthesis of 4 from D-glucose-derived precursor 120 according to Chida and co-workers. Cbz =benzyloxycarbonyl, DIBAL= diisobutylaluminum hydride, TFA =trifluoroacetic acid.
A few other research groups have likewise embarked upon formal total syntheses of salinosporamide A (4), in which they devised diverse synthetic routes to compounds resembling intermediates of Corey’s pathway. Langlois and co-workers generated the chirality on the γ-lactam system by a regioselective N-methylnitrone cycloaddition (Scheme 15).[104,105] This step was performed on pyrrolinone 131 to give the diastereomeric cycloaddition products 132 and 133 in yields of 54 and 14%, respectively. The product with the desired configuration, 132, was converted into Corey’s intermediate 63 in two more steps and 58% yield. Lam and co-workers used a nickel-catalyzed reductive aldol cyclization/lactonization to assemble the lactam moiety of 4 by formation of a C2–C3 bond, strategically similar to the synthesis by Corey and co-workers.[106] In the key step of this route, substrate 134, which in turn resembles intermediate 62 of the Corey pathway and prepared in a similar way, was cyclized using [(Me3P)2NiCl2] and Et2Zn as a precatalyst and reductant to give pyrrolinones 135 and 136 in 72% yield and in a 1.4:1 ratio in favor of the desired product 135. Compound 135 was transformed into late intermediate 137 of the Corey synthesis in four additional steps. Struble and Bode succeeded in developing an intramolecular lactonization method catalyzed by a N-heterocyclic carbene for the generation of Lam’s intermediate 135.[107] Cyclization of key precursor 138 was achieved using DBU and precatalyst 139 to yield a mixture of 135 and 136 in 88% yield and a 1:1.1 ratio in favor of the undesired diastereomer. In addition, Mosey and Tepe elaborated an oxazoline-mediated ene-type reaction starting from 140 to give 141 (88% yield, d.r. = 3:1), which was converted into (here racemic) amine 60 of the Corey synthesis.[108]
Scheme 15.

Diverse approaches to intermediates of Corey’s total synthesis of 4 as developed by the research groups of Langlois, Lam, Bode, and Tepe. Mes =2,4,6-trimethylphenyl.
The impressive number of approaches and the creativity of the diverse synthetic methods developed to tackle the highly complex framework of salinosporamide A (4) clearly demonstrate the uniqueness of this small molecule. The desire to understand the molecular mode of action and to reveal the structural features of 4 that render this compound superior in terms of biological activity when compared to other members of the group of γ-lactam-β-lactone proteasome inhibitors furthermore led to in-depth investigations of the mechanistic and structure–activity relationships (SARs).
6. Creating Molecular Diversity on the Salinosporamide Scaffold and Studies on their Mechanism of Action
To fully understand the unique mode of proteasome inhibition of 4, a more detailed discussion of the molecular mode of action of the salinosporamides is needed (Scheme 16). This process was beautifully illuminated by X-ray crystallographic studies of 4 and 15 in complexes with the 20S proteasome.[109] In the first step of inhibition, Thr1Oγ cleaves the lactone ring of 4 or 15, thereby forming a salinosporamide–proteasome adduct (A), which resembles the mode of action by covalent attachment of the inhibitor to the protein as previously established for other molecules,[39] such as the structurally highly related omuralide (14).[110] In the case of 14 and 15, the released tertiary alcohol displaces water molecules situated in the active site of the proteasome, thus hampering subsequent undesired saponification of the inhibitor–protein ester linkage (A→B). However, the molecular architecture of 4 facilitates a secondary reaction: Catalyzed by deprotonation of the tertiary hydroxy group of 4 by Thr1NH2, nucleophilic displacement of the side-chain chlorine atom by the respective alcoholate leads to intra-molecular formation of a THF ring (C). Generation of this cyclic ether likewise causes displacement of hydrolytically active water molecules, but furthermore prevents re-formation of the β-lactone of 4 (D) and any catalytic involvement in ester cleavage by the now fully protonated Thr1NH3+.[109] This arrangement renders the inhibitor firmly and irreversibly bound to the proteasome. Consequently, the biological activity of 15 (which is only missing the chlorine atom compared to 4 and thus lacks the additional protection of the ester bond after formation of the cyclic ether) drops significantly.[111]
Scheme 16.

Molecular mechanisms of binding/cleavage of 4 and 15 to/from the proteasome.
In addition, the opposite side of the protein–inhibitor ester bond is sterically shielded from hydrolysis by the cyclohexenyl ring (P1) of the salinosporamides in the S1 binding pocket and by the protein backbone itself. Besides the shielding effect of the P1 residue, favorable additional interactions of this moiety with hydrophobic residues in the S1 pocket are thought to increase the residence time of the salinosporamides in the active site of the proteasome. This additional binding contributes to the pronounced biological activity of this compound when compared to other γ-lactam-β-lactones such as 14, and might be the reason for the observed inhibition of all three proteasome subunits. These valuable mechanistic insights obtained by crystallographic studies,[109] which had previously been anticipated merely on the basis of preliminary SAR data,[111] thus suggest structural variations of the C2 and C5 substituents of the salinosporamide core structure to be most beneficial to probe SARs.
6.1. Effect of Structural Changes at C2 on the Biological Activity of the Salinosporamides
The natural salinosporamide analogues with structural diversity at C2 produced by S. tropica include the most active salinosporamide—salinosporamide A (4)—its deschloro analogue salinosporamide B (15), as well as the methyl- and ethyl-substituted derivatives D (24) and E (25), respectively.[27,53,54] Furthermore, the C2 epimers F–H (26–28) of 4, 15, and 25 can also be isolated. As described in Section 3, directed biosynthesis can be utilized to trigger the formation of bromosalinosporamide 31.[54,112] To further broaden the diversity of C2 substituents and to probe the effect of substituting the mechanistically crucial chlorine atom of 4, Nereus Pharmaceuticals used several (semi)synthetic manipulations to introduce different halogen and non-halogen leaving groups (LGs) as well as non-LGs into the salinosporamide scaffold (Scheme 17).[111,113] Subjecting 4 or 31 to Finkelstein conditions allowed for the production of iodosalinosporamide A (142), the central intermediate for further modifications of the C2-position. Treatment of 142 with NaN3 delivered azide 143. The propyl derivative salinosporamide E (25), which at the time of the initial SAR study had not been isolated, was accessible by the addition of 142 to lithium dimethyl cuprate. Formation of hydroxy-substituted 144 was achieved by exposing 142 to NaOH, but only in low yields (not shown). A more effective route to 144 was discovered during attempts to generate the fluorinated analogue 145 from 142 by utilizing AgF. Although only trace amounts of 145 were obtained under these conditions, 144 was produced in 15% yield. Having 144 in hand paved the way to produce the respective mesylated, dansylated, and tosylated derivatives 146–148. Compound 148 was also accessed by total synthesis.[113] Remarkably, the synthesis of the most interesting halogenated derivative 145, which was expected to be a valuable addition to SAR investigations because of the poor leaving group properties of fluorine and the generally favorable effect of introducing fluorine in medical agents,[114] could not be improved despite testing a wide variety of conditions.
Scheme 17.

Semisynthetic preparation of salinosporamide C2 analogues. Ds =5-(dimethylamino)-1-naphthalinesulfonyl (dansyl).
Prior to the synthetic efforts, a mutasynthetic access to fluorosalinosporamide (145) was explored in our group. Previous experiments aimed at the directed biosynthesis of 145 by S. tropica by substitution of NaCl in the production medium with NaF had shown the fluoride to be toxic effect to the bacterium.[112] In addition, in vitro experiments with the pathway-specific chlorinase SalL demonstrated that fluoride is not accepted as a substrate by this enzyme.[67] These issues were solved by genetic manipulations of the producing organism.[115] Deletion of the chlorinase salL-coding gene in the sal biosynthesis gene cluster, which catalyzes the initial step from 48 to 49 in the chlorination pathway towards 4 (see Scheme 6), abolished production of salinosporamide A (4) by the mutant strain. Chemical complementation of the fermentation broth with synthetic 5′-fluorodesoxyadenosine (5′-FDA),[116] the fluorinated analogue of the intermediate 5′-CDA (49, see Scheme 6) in the chlorination pathway,[66] then allowed for the production of 145 by fermentation (Scheme 18).[115] This result demonstrated the flexibility of downstream biosynthetic transformations to incorporate fluorine into the salinosporamide framework. Similar results were consequently also obtainable by supplementing salL− mutant fermentations with the later-stage chlorination pathway analogues 4-fluorocrotonic (149) and 4-bromochrotonic acid (150) to obtain 145 and 31, respectively.[76] Most recently, chromosomal replacement of the chlorinase gene salL in S. tropica by the fluorinase flA from S. cattleya was achieved, thus creating a genetically engineered organism capable of assembling 145 directly from an inorganic fluoride salt.[117]
Scheme 18.
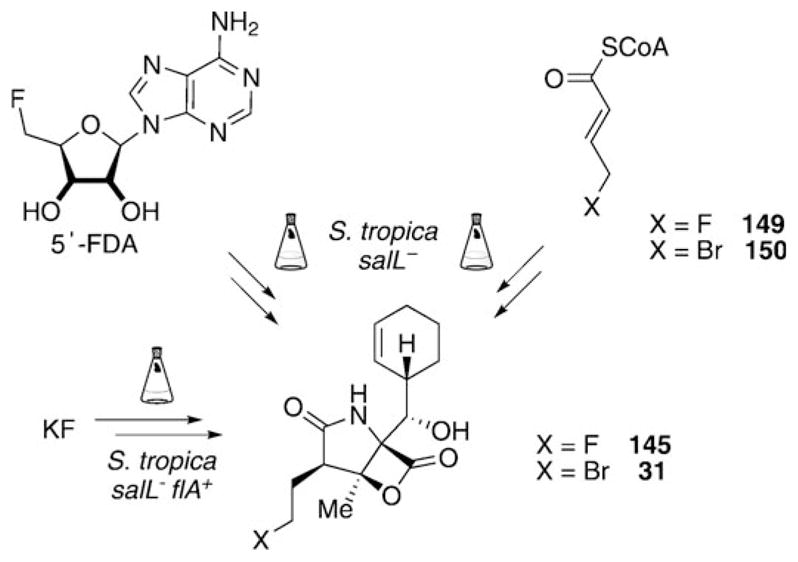
Methods for the production of 145 and 31 by genetic manipulations of S. tropica and chemical complementation of the respective mutant strains.
Analysis of the biological activity of all C2 side-chain derivatives led to several important findings. Generally, all the compounds of the salinosporamide class showed the same ranking order in the inhibition of proteasome subunits: CT-L > T-L > CA-L (Table 2).[111,113] Compounds bearing a good leaving group at C2 (namely, Cl, Br, I, OMs, ODs, OTs) showed the strongest activity across all the subunits. The size of the leaving group did not decrease the inhibition potential of the respective derivatives. This finding can be rationalized by the large open cavity of the S2 binding pocket in the proteasome,[109] which can easily accommodate large groups such as tosyl or dansyl. On the contrary, the dansylate 147 and the tosylate 148 even showed enhanced activity in T-L and CA-L inhibition assays, thus suggesting favorable interactions of these large substituents in the respective binding pockets. Remarkably 26, the C2 epimer of 4, had an about 100-fold attenuated activity, thus highlighting the importance of the natural C2 stereochemistry for formation of the THF ring and overall fit of the inhibitor into the active sites of the proteasome.
Table 2.
Salinosporamide analogues with altered C2 substituents: Biological activity against the rabbit 20S proteasome.[a]
 | ||||
|---|---|---|---|---|
| R | Inhibition of rabbit 20S proteasome [nm] | |||
| CT-L[b] | T-L[b] | CA-L[b] | ||
| 4 | C2H4Cl | 2.6 ± 0.2 | 21 ± 3 | 430 ± 60 |
| 145 | C2H4F | 9.2 ± 10.2 | ND[c] | ND[c] |
| 31 | C2H4Br | 2.6 ± 0.4 | 14 ± 2 | 290 ± 60 |
| 142 | C2H4I | 2.8 ± 0.5 | 13 ± 3 | 410 ± 230 |
| 146 | C2H4OMs | 4.3 ± 0.8 | 65 ± 8 | 870 ± 32 |
| 147 | C2H4ODs | 3.0 ± 0.5 | 12 ± 2.3 | 90 ± 11 |
| 148 | C2H4OTs | 2.4 ± 0.4 | 9.9 ± 0.2 | 127 ± 5 |
| 26 | epi-C2H4Cl | 330 ± 20 | 2500 ± 500 | >20000 |
| 143 | C2H4N3 | 7.7 ± 2.5 | 210 ± 40 | 560 ± 60 |
| 144 | C2H4OH | 14.0 ± 1.5 | 1200± 150 | 1200 ± 57 |
| 24 | CH3 | 7.5 ± 0.6 | 370 ± 44 | 460 ± 49 |
| 15 | C2H5 | 26 ± 6.7 | 610 ± 35 | 1200 ± 110 |
| 25 | C3H7 | 24 ± 5 | 1100 ± 200 | 1200 ± 200 |
The biological activity against the three proteolytic subunits was assessed by incubation of the inhibitor-pretreated proteasome with fluorogenic substrates with a high affinity to a certain subunit (Suc-LLVY-AMC for CT-L, Z-LLE-AMC for C-L, Bz-VGR-AMC for T-L) and subsequent quantification of the amount of proteolytically cleaved AMC (7-amino-4-methylcoumarin).[111,113, 115, 118]
CA-L = caspase-like activity, T-L = trypsin-like activity, CT-L = chymotrypsin-like activity.
Not determined.
The next most effective group (2–3 times less active than 4 in the CT-L assay) of proteasome inhibitors comprises the fluorine- (145), azide- (143), and methyl-functionalized (24) analogues with poor/no LG properties. The ethyl- (15), hydroxyethyl- (144), and propyl-substituted (25) derivatives were about 5–10 times less effective CT-L inhibitors than 4. The decrease in the activity of these non-LG-substituted derivatives in comparison to methyl analogue 24 can be explained by increases destabilizing effects on binding interactions because of the “wagging” of the longer side chain in the active site.[109] In addition, non-LG derivatives in general possessed a much lower cytotoxic effect in cell-based assays (not shown).[111,113]
It is interesting to note that the cinnabaramides, although not bearing a LG in the C2 side chain, are strong nanomolar inhibitors of the human 20S proteasome.[55] This questions the importance of β-lactone re-formation as a possible side reaction that removes non-LG inhibitors from the active site of the proteasome (see Scheme 16, D). Furthermore, it points to a mechanistic importance of the hexyl substituent at C2 of these molecules, which still needs to be investigated.
Of particular interest among all the C2 derivatives, however, is the fluorinated compound 145. Wash-out assays with 145 bound to the proteasome showed a recovery of proteasome activity, thus indicating a reversible mode of binding.[115] Similar results were obtained by dialysis of proteasome–inhibitor complexes.[113] While non-LG substrates were shown to be removed from the proteasome in these assays over time—ultimately leading to complete recovery of proteasome activity—and LG-bearing analogues retained their inhibitory effect, proteasome treated with 145 partially regained its activity. The rationale for this observation was again delivered by crystallographic studies.[119] Analysis of the β5 subunit of the proteasome after one hour crystal soak time revealed that 145 bound to the protein through an ester bond, but with a still intact fluoroethyl side chain. Nevertheless, the inhibitor seemed to be perfectly prearranged in the active site for subsequent displacement of the fluorine atom: the fluoroethyl side chain was in a well-defined position with the tertiary hydroxy group in van der Waals distance to the C2 side chain, Thr1NH2 within hydrogen-bonding distance to the hydroxy group, and the fluorine atom in proximity to a network of water molecules that might play a role in solvation of the fluoride after its elimination.[119] Crystals analyzed after a 24 h soak time indeed showed complete displacement of the fluorine atom during formation of a THF ring, analogous to derivatives with a good LG in the C2 substituent. The recovery of the proteasome activity after treatment with 145 can, therefore, be explained by competing partial hydrolytic cleavage (or β-lactone reformation) of the still-fluorinated inhibitor from the protein facilitated by the slow displacement of the fluorine atom, while the cyclized, THF-bearing portion of the complexes contribute to the partially retained inhibitory effect, even after dialysis.
The SAR data on C2 analogues of the salinosporamide class clearly validates that leaving groups in the ethyl side chain are mechanistically important and lead to prolonged inhibition of the proteasome.[113] The observations made with fluorosalinosporamide (145), furthermore, shows that alterations at this position facilitates the fine-tuning of the potency and duration of the inhibition. This might ultimately be useful in designing novel salinosporamide-type compounds with well-defined pharmacologic properties that allow for fine-regulation of the effect of the respective inhibitor on proteasome downstream processes.
6.2. Effect of Structural Changes at C5 on the Biological Activity of the Salinosporamides
In contrast to the structural diversity at the C2-position, the cyclohexenyl residue at C5 is conserved among all the natural salinosporamides. Furthermore, feasible semisynthetic modifications at this position are limited due to the high reactivity of the lactone system in this class of compounds, thus limiting easy access to SAR data. A small series of C5 derivatives was generated by manipulation of the cyclohexenyl double bond (Scheme 19).[111] Catalytic hydrogenation at this position delivered the fully saturated cyclohexyl analogue 151. Oxidation of the double bond using mCPBA gave rise to a mixture of epoxides 152 and 153 (d.r. = 1:11). Subsequent ring opening of the epoxide in 153 using HCl yielded chlorohydrin 154. In addition, Corey and co-workers accessed the salinosporamide–omuralide hybrid antiprotealide (155; Scheme 20), bearing an isopropyl residue at C5, by total synthesis.[85]
Scheme 19.
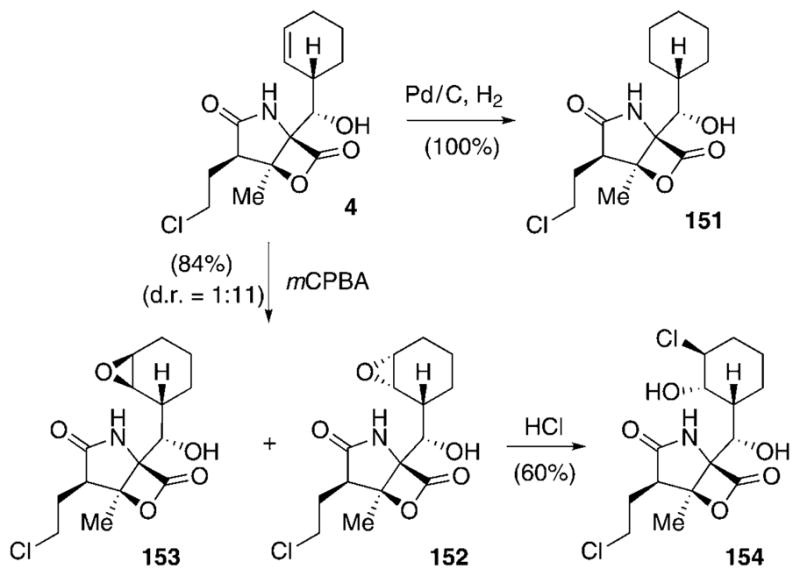
Semisynthesis of salinosporamide C5 analogues. mCPBA=meta-chloroperbenzoic acid.
Scheme 20.
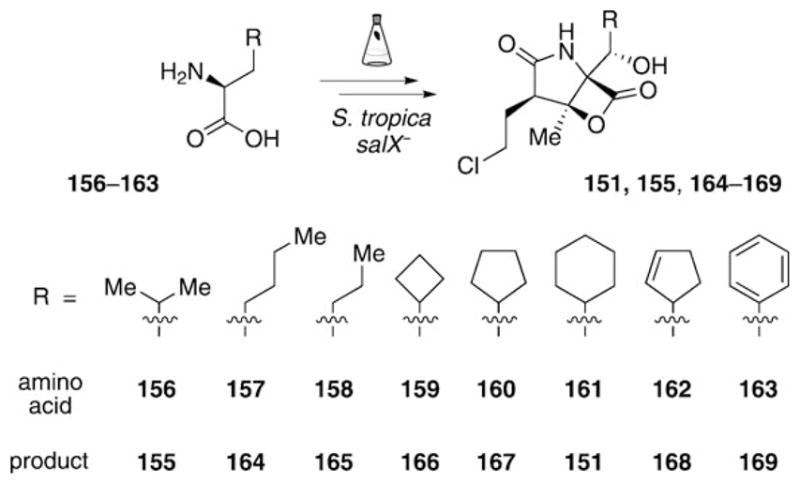
Generation of a focused library of salinosporamide C5 analogues by gene inactivation in combination with mutasynthesis.
A more straightforward approach for the generation of a focused library of C5 derivatives was again developed by utilizing a combination of gene inactivation and mutasynthesis (Scheme 20).[79,80] To eliminate the production of the natural amino acid precursor and, in consequence, of all known salinosporamides by S. tropica, the deletion of a pathway-specific gene involved in cyclohexenylalanine biosynthesis was targeted. The gene of choice was the prephenate dehydrogenase homologue coding gene salX, which is thought to convert dihydroprephenic acid 45 into 46 (see Scheme 4). Chemical analysis of extracts of the salX− mutant strain did not only reveal the expected complete abolishment of the production of any known salinosporamide, but also the presence of a metabolite not previously generated by the wild-type strain.[79] This new secondary metabolite was shown to be antiprotealide (155), which arises by incorporation of a leucine unit into the salinosporamide framework. Compound 155 was later verified as a minor natural product in large-scale fermentations of S. tropica.[120] The occurrence of 155 demonstrated the flexibility of the sal biosynthetic machinery to incorporate amino acids other than the natural precursor, and set the stage for a systematic exploration of in vivo substrate flexibility. Chemical complementation of cultures of the S. tropica salX− mutant strain with a series of noncyclic alkyl (156–158), alicyclic (159–162), and aromatic (163) amino acids indeed led to the production of a small library of salinosporamide C5 analogues bearing the respective substitutions (151, 155, 164–169).[79,80]
Comparison of the proteasome inhibition data of these compounds revealed that alicyclic C5 analogues are generally more active than linear ones (Table 3).[79,80] Among the cycloalkyl derivatives 151, 166, and 167, the cyclopentane residue in 167 seems to provide a ring size that offers an ideal compromise between steric repulsion and favorable hydrophobic interactions in the S1 binding pocket of the proteasome, as suggested by a tenfold drop of activity in the cyclobutyl analogue 166 and a threefold decreased potency of cyclohexanyl 151. The introduction of a cyclopentenyl residue into the salinosporamide scaffold was thus anticipated to lead to pronounced biological activity when compared to 4. In fact, 168 showed equal potency in proteasome inhibition, combined with slightly increased cytotoxicity in cell-based assays (not shown).[80] In the group of noncyclic, alkyl-substituted derivatives, the branched antiprotealide 155 exhibited the strongest proteasome inhibition (50-fold lower than 4). The two- to threefold decreased potency of its propyl analogue 165 might be explained by destabilizing “wagging” of the more flexible side chain, which is in part compensated in butanyl 164 by the additional van der Waals attractions of the longer hydrocarbon chain within the S1 binding pocket. The introduction of an aromatic C5 residue, as in 169, led to a significant drop in the biological activity. This is in agreement with previous SAR studies on analogues of omuralide (14)[121] and might be explained by the rigid, planar nature of the phenyl ring, which cannot align to the shape of the S1 binding pocket and thus impairs van der Waals interactions with the protein. Interestingly, oxidation of the double bond of 4 to the (2S,3R)-epoxycyclohexane 152 was well tolerated, while the 2R,3S-configured analogue 153 showed strongly reduced biological activity (3- versus 45-fold reduced when compared to 4).[111] This can most likely be attributed to steric repulsion between the epoxide in 153 (with the oxygen atom on the opposite face as in 152) and the protein. The assumption was confirmed by the 4000-fold decrease in the activity of 154, which bears the even larger chlorine substituent on the same side of the molecule as the epoxy group in 153.[111]
Table 3.
Salinosporamide analogues with altered C5 substituents: Biological activity against the [a] yeast and [b] rabbit 20S proteasome.[c]
 | ||
|---|---|---|
| R | CT-L [nm] | |
| 4 | cyclohex-2-enyl | 1.9 ± 0.2[a] |
| 2.6 ± 0.2[b] | ||
| 168 | cyclopent-2-enyl | 2.2 ± 0.1[a] |
| 151 | cyclohexyl | 27.5 ± 3.7[a] |
| 167 | cyclopentyl | 9.3 ± 1.6[a] |
| 166 | cyclobutyl | 93.4 ± 4.3[a] |
| 164 | butyl | 132 ± 19[a] |
| 155 | isopropyl | 101 ± 15[a] |
| 165 | propyl | 245 ± 38[a] |
| 152 | (2S,3R)-epoxycyclohexyl | 6.3 ± 0.6[b] |
| 153 | (2R,3S)-epoxycyclohexyl | 91 ± 8[b] |
| 154 | (3S)-chloro-(2S)-hydroxycyclohexyl | 8200 ± 3000[b] |
| 169 | phenyl | 1029 ± 419[a] |
With the exception of cyclopentenyl derivative 168,[80] all the other C5 analogues exhibit lower biological activity than the parent compound 4. As the C5 residue is crucial for favorable interactions with the S1 binding pocket in the proteasome, alterations at this position, however, might lead to distinct selectivity of the new inhibitor towards the three proteolytic units of this protein. This hypothesis remains to be tested, but might ultimately prove useful in the design of inhibitors with a desired affinity for a certain catalytic subunit.
6.3. Further Alterations of the Salinosporamide Core Structure
Structural changes of the substituents R and X at C3 and C5, respectively (see Table 4), of the natural salinosporamide family are rare. The only known C3 analogue, natural compound 29, bears a larger ethyl chain at this position.[54] Furthermore, the non-hydroxylated derivative 30 can be found in small quantities in extracts of S. tropica. The yield of 30 can be largely increased by deletion of the cytochrome P450 hydroxylase coding gene salD.[79] In addition to these two metabolites, the C5 keto analogue 170 was produced by oxidation of 4 (Scheme 21). Reduction of 170 at low temperature yielded a 9.5:0.5 diastereomeric mixture of the respective secondary alcohols C5-epi-4 and 4.[111]
Table 4.
C3-R and C5-X salinosporamide analogues: Biological activity against the [a] yeast and [b] rabbit 20S proteasome.[c]
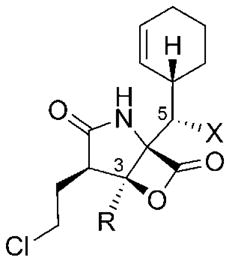 | ||
|---|---|---|
| R/X | CT-L [nM] | |
| 4 | Me/OH | 1.9 ± 0.2[a] |
| 2.6 ± 0.2[b] | ||
| 30 | Me/H | 20.8 ± 0.8[a] |
| 52 ± 2[b] | ||
| 170 | Me/O | 8200 ± 600[b] |
| C5-epi-4 | Me/epi-OH | >20000[b] |
| 29 | Et/OH | 2100 ± 100[b] |
See footnote [c] in Table 3.
Scheme 21.
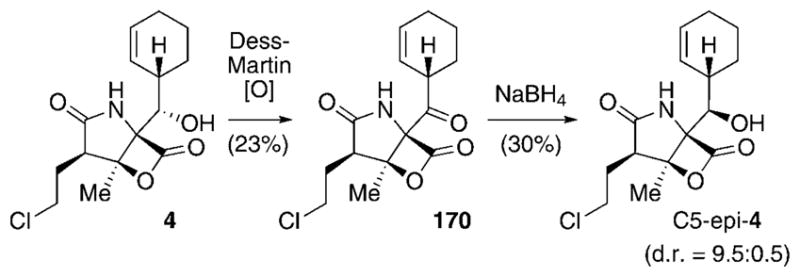
Semisynthesis of X analogues of 4.
The biological data of the C3-R and C5-X analogues of 4 revealed that structural changes at either of these positions are not well tolerated. Removal of the C5-hydroxy group in 30 led to a 10–20-fold decrease in proteasome inhibition (Table 4).[79,111] The activity of the oxidized derivative 170 dropped even more significantly, and the epimeric alcohol C5-epi-4 did not show any proteasome inhibition.[111] These observations can be rationalized by complex, stabilizing hydrogen bonds between the C5-OH group in 4 and the proteasome, as seen by X-ray crystallographic analysis.[109] Such interactions are missing in 30 and are substituted by unfavorable steric repulsion between the oxygen atoms in 170, and in particular in C5-epi-4, and the protein backbone. The C3-ethyl derivative 29 likewise exhibited a 1000-fold decreased activity, which can be attributed to a steric clash of the ethyl group pointing towards a small protein pocket that can only accommodate hydrogen or methyl groups.[109]
In summary, the SAR data obtained for the salinosporamides suggest that substitutions at the ethyl side chain at C2 allow for the development of novel derivatives with altered dynamics of proteasome inhibition. In addition, changes at the C5-position might provide a handle for producing new analogues with defined selectivity for certain proteasome subunits. Nevertheless, the biological activity of the strongest natural inhibitor 4 could not be improved substantially by these investigations. This indicates the excellent design of this small-molecule proteasome inhibitor achieved by nature, where every single atom is strategically placed to contribute to the overall activity of the compound. Salinosporamide A (4) thus serves as one of the few recent examples of a genuine natural product that entered clinical trials without any chemical optimization.
7. Salinosporamide A in Cancer Therapy
Salinosporamide A (4) is currently being developed by Nereus Pharmaceuticals under the name NPI-0052 (Marizomib) as a new agent for the treatment of human cancer.[26,122] Despite the numerous total synthetic routes towards 4 (see Section 4), the compound is still obtained by fermentation of the natural producer S. tropica. The initial cultivation conditions of S. tropica to obtain 4[27] had to be adjusted to allow the manufacture of larger quantities in sufficient quality for application to humans. The optimization of production titers was explored in a variety of studies,[112,123–126] which led: 1) to the establishment of a fermentation protocol based on an XAD (hydrophobic cross-linked polystyrene copolymer absorbent) resin[127] which helps to overcome the loss of yield caused by the instability of 4 in aqueous media,[128] 2) the discovery of more efficient producing organisms by strain screening and selection, and 3) the development of the first industrial-scale saline fermentation process to meet current Good Manufacturing Practice (cGMP) guidelines. Taken together, these efforts towards the enhanced production of 4 led to an approximately 100-fold increase in isolated 4, thus rendering the process suitable for the biotechnological production on an industrial scale and clinical evaluation of 4.[26,122]
The previously established efficacy of the proteasome inhibitor bortezomib (5) in multiple myeloma,[129,130] led to salinosporamide A (4) initially being tested in multiple myeloma xenograft models in mice.[131] Further biological evaluation of 4 in a series of nonclinical studies have meanwhile assessed its potential in multiple myeloma,[131,132] Waldenstrom’s macroglobulinemia,[133] acute lymphocytic and myeloid leukemia,[134] and chronic lymphoid leukemia,[135] as well as colon,[136] pancreatic carcinomas,[137] and non-Hodgkin’s lymphoma.[138] Besides the established activity of 4 as a single agent against solid tumors and hematologic malignancies, this compound in combination with other chemotherapeutics and biologics,[134,136,137,139] in particular when applied together with a low-dose of 5,[132,133] have been shown to broaden the therapeutic potential. Additional recent studies indicate that 4 has an effect on the inhibition of NF-κB activation[140] and interferes with the NF-κB-Snail-RKIP circuity in tumors,[141,142] which might ultimately prove useful to reverse the resistance of tumor cells to conventional anticancer agents. Overall these data suggest the high potential of 4 against a variety of human cancer indications. Currently, salinosporamide A (4) is evaluated in dose escalations in a series of phase I clinical trials as a single agent for the treatment of multiple myeloma, solid tumors, and lymphomas.[26,122]
8. Concluding Remarks and Outlook
The isolation of salinosporamide A in 2003 was a landmark discovery that has already contributed significantly to the basic and applied sciences of natural product drug discovery. Foremost was its rapid preclinical development and entry into human clinical trials as a promising anticancer agent with a novel mechanism of proteasome inhibition. As a consequence, many “firsts” were developed during these studies, including the first oncological trials of a marine bacterial natural product, the first GMP production of a natural drug by the saline fermentation of a marine microbe, and the first report of an irreversible proteasome inhibitor based on a γ-lactam-β-lactone skeleton. The evaluation of the biosynthesis of the salinosporamide family furthermore led to a number of new and unexpected discoveries, including novel biochemical chlorination and polyketide synthase reactions, which led to new methods to bioengineer derivatives, including the first genetic engineering of a fluorometabolite. The total syntheses of the salinosporamides have also highlighted the creativity and practicality of synthetic organic chemists, who developed numerous routes to assemble the densely functionalized structure.
While it remains to be seen if salinosporamide A will ultimately be successful in the clinic as an anticancer agent, proteasome inhibitors such as the salinosporamides also hold promise in other disease states, including inflammation,[143] anthrax,[144] tuberculosis,[145] and malaria.[146] Further pharmacological research with the salinosporamides will help ascertain the relevance of proteasome targeting associated with these maladies. Additional challenges and opportunities await in the biosynthesis of the salinosporamides, where numerous unanswered questions remain: How is the novel bicyclic γ-lactam-β-lactone core produced? How is the biosynthetic pathway regulated, and can it be genetically altered to increase biosynthetic titers? What is the role of the sal-specific proteasome β subunit SalI, and is it involved in a new mechanism of proteasome resistance in bacteria? Lastly, in the synthetic arena, can new approaches be developed to improve upon existing methods to salinosporamide A (4) to provide an alternative GMP source of the drug molecule? All these questions await creative solutions.
9. Addendum (August 1, 2010)
During the preparation of the manuscript, Romo and coworkers published an enantioselective version of their biomimetic route depicted in Scheme 10. The key step of this impressively short, nine-step total synthesis of 4 involves the bicyclization of an enantiomerically pure linear precursor (obtained by chromatographic separation of the 1:1 diastereomeric mixture of this compound) with retention of its configuration through A1,3 strain.[147] Nereus Pharmaceuticals likewise elaborated another total synthesis of 4, which is based on their previous synthetic experiences. In this study, the stereocenters at C2 and C3 of 4 are installed by regio- and stereoselective epoxidation of a precursor molecule with a C2–C3 double bond, followed by regioselective reductive opening of the epoxide ring.[148] In addition, further studies on the pharmacodynamics of 4 in human plasmacytoma xenograft murine models[149] as well as investigations on potential combination therapies of 4 with lenalidomide[150] have appeared in the literature.
Acknowledgments
The work on the biosynthesis and bioengineering of salinosporamide metabolites in B.S.M.’s laboratory is generously supported by a NIH grant from the NCI (CA127622), and a DAAD postdoctoral fellowship to T.A.M.G. We thank Dr. Tanja Gulder for valuable discussions.
Biographies
 Bradley Moore is Professor of Oceanography and Pharmaceutical Sciences at the University of California, San Diego. He studied chemistry at the Universities of Hawaii (BS 1988) and Washington (PhD 1994 with Prof. H. G. Floss), carried out postdoctoral research with Prof. J. A. Robinson at the University of Zurich (1994–1995), and held prior faculty appointments at the Universities of Washington (1996–1999) and Arizona (1999–2005). He explores and exploits marine microbial genomes to discover new biosynthetic enzymes, secondary metabolic pathways, and natural products for drug discovery and development.
Bradley Moore is Professor of Oceanography and Pharmaceutical Sciences at the University of California, San Diego. He studied chemistry at the Universities of Hawaii (BS 1988) and Washington (PhD 1994 with Prof. H. G. Floss), carried out postdoctoral research with Prof. J. A. Robinson at the University of Zurich (1994–1995), and held prior faculty appointments at the Universities of Washington (1996–1999) and Arizona (1999–2005). He explores and exploits marine microbial genomes to discover new biosynthetic enzymes, secondary metabolic pathways, and natural products for drug discovery and development.
 Tobias A. M. Gulder studied Chemistry at the University of Würzburg in 2004, followed by a PhD with Prof. G. Bringmann in analytical and synthetic natural product chemistry, for which he received the 2009 DECHEMA PhD award. In 2008, he joined Prof. Moore at Scripps Institution of Oceanography (UCSD) as a DAAD postdoctoral fellow where he worked on the elucidation and exploitation of marine natural product biosynthetic pathways. He returned to Germany in the middle of 2010, supported by a DAAD reintegration fellowship, to start his independent research at the University of Bonn.
Tobias A. M. Gulder studied Chemistry at the University of Würzburg in 2004, followed by a PhD with Prof. G. Bringmann in analytical and synthetic natural product chemistry, for which he received the 2009 DECHEMA PhD award. In 2008, he joined Prof. Moore at Scripps Institution of Oceanography (UCSD) as a DAAD postdoctoral fellow where he worked on the elucidation and exploitation of marine natural product biosynthetic pathways. He returned to Germany in the middle of 2010, supported by a DAAD reintegration fellowship, to start his independent research at the University of Bonn.
References
- 1.Li J, Vederas JC. Science. 2009;325:161. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 2.Bentley R. J Ind Microbiol Biotechnol. 2009;36:775. doi: 10.1007/s10295-009-0553-8. [DOI] [PubMed] [Google Scholar]
- 3.Kaufman TS, Rúveda EA. Angew Chem. 2005;117:876. doi: 10.1002/anie.200400663. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:854. [Google Scholar]
- 4.Blakemore PR, White JD. Chem Commun. 2002:1159. doi: 10.1039/b111551k. [DOI] [PubMed] [Google Scholar]
- 5.Newman DJ, Cragg GM. J Nat Prod. 2007;70:461. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 6.Cragg GM, Newman DJ. Phytochem Rev. 2009;8:313. [Google Scholar]
- 7.Cragg GM, Grothaus PG, Newman DJ. Chem Rev. 2009;109:3012. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- 8.Bailly C. Biochem Pharmacol. 2009;77:1447. doi: 10.1016/j.bcp.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 9.Amin A, Gali-Muhtasib H, Ocker M, Schneider-Stock R. Int J Biomed Sci. 2009;5:1. [PMC free article] [PubMed] [Google Scholar]
- 10.Dholwani KK, Saluja AK, Gupta AR, Shah DR. Indian J Pharmacol. 2008;40:49. doi: 10.4103/0253-7613.41038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newman DJ, Cragg GM. Curr Med Chem. 2004;11:1693. doi: 10.2174/0929867043364982. [DOI] [PubMed] [Google Scholar]
- 12.Molinski TF, Dalisay DS, Lievens SL, Saludes JP. Nat Rev Drug Discovery. 2009;8:69. doi: 10.1038/nrd2487. [DOI] [PubMed] [Google Scholar]
- 13.Cuevas C, Francesch A. Nat Prod Rep. 2009;26:322. doi: 10.1039/b808331m. [DOI] [PubMed] [Google Scholar]
- 14.König GM, Kehraus S, Seibert SF, Abdel-Lateff A, Müller D. ChemBioChem. 2006;7:229. doi: 10.1002/cbic.200500087. [DOI] [PubMed] [Google Scholar]
- 15.Guo B, Wang Y, Sun X, Tang K. Appl Biochem Microbiol. 2008;44:136. [Google Scholar]
- 16.Sudek S, Lopanik NB, Waggoner LE, Hildebrand M, Anderson C, Liu H, Patel A, Sherman DH, Haygood MG. J Nat Prod. 2007;70:67. doi: 10.1021/np060361d. [DOI] [PubMed] [Google Scholar]
- 17.Tan LT. Phytochemistry. 2007;68:954. doi: 10.1016/j.phytochem.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 18.Bull AT, Stach JEM. Trends Microbiol. 2007;15:491. doi: 10.1016/j.tim.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 19.Williams PG. Trends Biotechnol. 2009;27:45. doi: 10.1016/j.tibtech.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 20.Gulder TAM, Moore BS. Curr Opin Microbiol. 2009;12:252. doi: 10.1016/j.mib.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fenical W, Jensen PR. Nat Chem Biol. 2006;2:666. doi: 10.1038/nchembio841. [DOI] [PubMed] [Google Scholar]
- 22.Jensen PR, Williams PG, Oh D-C, Zeigler L, Fenical W. Appl Environ Microbiol. 2007;73:1146. doi: 10.1128/AEM.01891-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oh D-C, Williams PG, Kauffman CA, Jensen PR, Fenical W. Org Lett. 2006;8:1021. doi: 10.1021/ol052686b. [DOI] [PubMed] [Google Scholar]
- 24.Williams PG, Asolkar RN, Kondratyuk T, Pezzuto JM, Jensen PR, Fenical W. J Nat Prod. 2007;70:83. doi: 10.1021/np0604580. [DOI] [PubMed] [Google Scholar]
- 25.Buchanan GO, Williams PG, Feling RH, Kauffman CA, Jensen PR, Fenical W. Org Lett. 2005;7:2731. doi: 10.1021/ol050901i. [DOI] [PubMed] [Google Scholar]
- 26.Fenical W, Jensen PR, Palladino MA, Lam KS, Lloyd GK, Potts BC. Bioorg Med Chem. 2009;17:2175. doi: 10.1016/j.bmc.2008.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Angew Chem. 2003;115:369. doi: 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2003;42:355. [Google Scholar]
- 28.Kingston DGI. J Nat Prod. 2009;72:507. doi: 10.1021/np800568j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nobili S, Lippi D, Witort E, Donnini M, Bausi L, Mini E, Capaccioli S. Pharmacol Res. 2009;59:365. doi: 10.1016/j.phrs.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 30.Orlowski RZ, Kuhn DJ. Clin Cancer Res. 2008;14:1649. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- 31.Meiners S, Ludwig A, Stangl V, Stangl K. Med Res Rev. 2008;28:309. doi: 10.1002/med.20111. [DOI] [PubMed] [Google Scholar]
- 32.Goldberg AL. Biochem Soc Trans. 2007;35:12. doi: 10.1042/BST0350012. [DOI] [PubMed] [Google Scholar]
- 33.Goldberg AL. Nature. 2003;426:895. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 34.Ciechanover A. Neurology. 2006;66:S7. doi: 10.1212/01.wnl.0000192261.02023.b8. [DOI] [PubMed] [Google Scholar]
- 35.Pickart CM. Annu Rev Biochem. 2001;70:503. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 36.Verdoes M, Florea BI, van der Marel GA, Overkleeft HS. Eur J Org Chem. 2009:3301. [Google Scholar]
- 37.Fuchs D, Berges C, Naujokat C. Biol Unserer Zeit. 2008;38:168. [Google Scholar]
- 38.Groll M, Huber R. Int J Biochem Cell Biol. 2003;35:606. doi: 10.1016/s1357-2725(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 39.Borissenko L, Groll M. Chem Rev. 2007;107:687. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- 40.Moore BS, Eustáquio AS, McGlinchey RP. Curr Opin Chem Biol. 2008;12:434. doi: 10.1016/j.cbpa.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim KB, Crews CM. J Med Chem. 2008;51:2600. doi: 10.1021/jm070421s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang L, Chen CH. Curr Med Chem. 2009;16:931. doi: 10.2174/092986709787581860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aoyagi T, Miyata S, Nanbo M, Kojima F, Matsuzaki M, Ishizuka M, Takeuchi T, Umezawa H. J Antibiot. 1969;22:558. doi: 10.7164/antibiotics.22.558. [DOI] [PubMed] [Google Scholar]
- 44.Meng L, Mohan R, Kwok BHB, Elofsson M, Sin N, Crews CM. Proc Natl Acad Sci USA. 1999;96:10403. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Groll M, Kim KB, Kairies N, Huber R, Crews CM. J Am Chem Soc. 2000;122:1237. [Google Scholar]
- 46.Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennet MK, van Leeuwen FWB, Chanan-Khan AA, Orlowski RZ. Blood. 2007;110:3281. doi: 10.1182/blood-2007-01-065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koguchi Y, Kohno J, Nishio M, Takahashi K, Okuda T, Ohnuki T, Komatsubara S. J Antibiot. 2000;53:105. doi: 10.7164/antibiotics.53.105. [DOI] [PubMed] [Google Scholar]
- 48.Asai A, Hasegawa A, Ochiai K, Yamashita Y, Mizukami T. J Antibiot. 2000;53:81. doi: 10.7164/antibiotics.53.81. [DOI] [PubMed] [Google Scholar]
- 49.Omura S, Matsuzaki K, Fujimoto T, Kosuge K, Furuya T, Fujita S, Nakagawa A. J Antibiot. 1991;44:117. doi: 10.7164/antibiotics.44.117. [DOI] [PubMed] [Google Scholar]
- 50.Omura S, Fujimoto T, Otoguro K, Matsuzaki K, Moriguchi R, Tanaka H, Sasaki Y. J Antibiot. 1991;44:113. doi: 10.7164/antibiotics.44.113. [DOI] [PubMed] [Google Scholar]
- 51.Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Science. 1995;268:726. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 52.Groll M, Berkers CR, Ploegh HL, Ovaa H. Structure. 2006;14:451. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 53.Williams PG, Buchanan GO, Feling RH, Kauffman CA, Jensen PR, Fenical W. J Org Chem. 2005;70:6196. doi: 10.1021/jo050511+. [DOI] [PubMed] [Google Scholar]
- 54.Reed KA, Manam RR, Mitchell SS, Xu J, Teisan S, Chao T-H, Deyanat-Yazdi GC, Neuteboom ST, Lam KS, Potts BCM. J Nat Prod. 2007;70:269. doi: 10.1021/np0603471. [DOI] [PubMed] [Google Scholar]
- 55.Stadler M, Bitzer J, Mayer-Bartschmid A, Müller H, Benet-Buchholz J, Gantner F, Tichy H-V, Reinemer P, Bacon KB. J Nat Prod. 2007;70:246. doi: 10.1021/np060162u. [DOI] [PubMed] [Google Scholar]
- 56.Tsueng G, McArthur KA, Potts BCM, Lam KS. Appl Microbiol Biotechnol. 2007;75:999. doi: 10.1007/s00253-007-0899-7. [DOI] [PubMed] [Google Scholar]
- 57.Beer LL, Moore BS. Org Lett. 2007;9:845. doi: 10.1021/ol063102o. [DOI] [PubMed] [Google Scholar]
- 58.Nakagawa A, Takahashi S, Uchida K, Matsuzaki K, Omura S, Nakamura A, Kurihara N, Nakamatsu T, Miyake Y, Take K, Kainosho M. Tetrahedron Lett. 1994;35:5009. [Google Scholar]
- 59.Takahashi S, Uchida K, Nakagawa A, Miyake Y, Kainosho M, Matsuzaki K, Omura S. J Antibiot. 1995;48:1015. doi: 10.7164/antibiotics.48.1015. [DOI] [PubMed] [Google Scholar]
- 60.Udwary DW, Zeigler L, Asolkar RN, Singan V, Lapidus A, Fenical W, Jensen PR, Moore BS. Proc Natl Acad Sci USA. 2007;104:10376. doi: 10.1073/pnas.0700962104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Penn K, Jenkins C, Nett M, Udwary DW, Gontang EA, McGlinchey RP, Foster B, Lapidus A, Podell S, Allen EE, Moore BS, Jensen PR. ISME J. 2009;3:1193. doi: 10.1038/ismej.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hertweck C. Angew Chem. 2009;121:4782. [Google Scholar]; Angew Chem Int Ed. 2009;48:4688. [Google Scholar]
- 63.Gaitatzis N, Silakowski B, Kunze B, Nordsiek G, Blöcker H, Höfle G, Müller R. J Biol Chem. 2002;277:13082. doi: 10.1074/jbc.M111738200. [DOI] [PubMed] [Google Scholar]
- 64.Ligon J, Hill S, Beck J, Zirkle R, Molnár I, Zawodny J, Money S, Schupp T. Gene. 2002;285:257. doi: 10.1016/s0378-1119(02)00396-7. [DOI] [PubMed] [Google Scholar]
- 65.Frank B, Wenzel SC, Bode HB, Scharfe M, Blöcker H, Müller R. J Mol Biol. 2007;374:24. doi: 10.1016/j.jmb.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 66.Eustáquio AS, McGlinchey RP, Liu Y, Hazzard C, Beer LL, Florova G, Alhamadsheh MM, Lechner A, Kale AJ, Kobayashi Y, Reynolds KA, Moore BS. Proc Natl Acad Sci USA. 2009;106:12295. doi: 10.1073/pnas.0901237106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eustáquio AS, Pojer F, Noel JP, Moore BS. Nat Chem Biol. 2008;4:69. doi: 10.1038/nchembio.2007.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dong C, Huang F, Deng H, Schaffrath C, Spencer JB, O’Hagan D, Naismith JH. Nature. 2004;427:561. doi: 10.1038/nature02280. [DOI] [PubMed] [Google Scholar]
- 69.Huang F, Haydock SF, Spiteller D, Mironenko T, Li T-L, O’Hagan D, Leadley PF, Spencer JB. Chem Biol. 2006;13:475. doi: 10.1016/j.chembiol.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 70.Deng H, Botting CH, Hamilton JTG, Russel RJM, O’Hagan D. Angew Chem. 2008;120:5437. doi: 10.1002/anie.200800794. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:5357. [Google Scholar]
- 71.Deng H, McMahon SA, Eustáquio AS, Moore BS, Naismith JH, O’Hagan D. ChemBioChem. 2009;10:2455. doi: 10.1002/cbic.200900369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eustáquio AS, Härle J, Noel JP, Moore BS. ChemBioChem. 2008;9:2215. doi: 10.1002/cbic.200800341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Deng H, O’Hagan D. Curr Opin Chem Biol. 2008;12:582. doi: 10.1016/j.cbpa.2008.06.036. [DOI] [PubMed] [Google Scholar]
- 74.Erb TJ, Berg IA, Brecht V, Müller M, Fuchs G, Alber BE. Proc Natl Acad Sci USA. 2007;104:10631. doi: 10.1073/pnas.0702791104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Erb TJ, Brecht V, Fuchs G, Müller M, Alber BE. Proc Natl Acad Sci USA. 2009;106:8871. doi: 10.1073/pnas.0903939106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu Y, Hazzard C, Eustáquio AS, Reynolds KA, Moore BS. J Am Chem Soc. 2009;131:10376. doi: 10.1021/ja9042824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gande R, Dover LG, Krumbach K, Besra GS, Sahm H, Oikawa T, Eggeling L. J Bacteriol. 2007;189:5257. doi: 10.1128/JB.00254-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mahlstedt SA, Walsh CT. Biochemistry. 2010;49:912. doi: 10.1021/bi9021186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McGlinchey RP, Nett M, Eustáquio AS, Asolkar RN, Fenical W, Moore BS. J Am Chem Soc. 2008;130:7822. doi: 10.1021/ja8029398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nett M, Gulder TAM, Kale AJ, Hughes CC, Moore BS. J Med Chem. 2009;52:6163. doi: 10.1021/jm901098m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reddy LR, Saravanan P, Corey EJ. J Am Chem Soc. 2004;126:6230. doi: 10.1021/ja048613p. [DOI] [PubMed] [Google Scholar]
- 82.Shibasaki M, Kanai M, Fukuda N. Chem Asian J. 2007;2:20. doi: 10.1002/asia.200600310. [DOI] [PubMed] [Google Scholar]
- 83.Kulinkovich OG. Chem Rev. 2003;103:2597. doi: 10.1021/cr010012i. [DOI] [PubMed] [Google Scholar]
- 84.Reddy LR, Fournier J-F, Reddy BVS, Corey EJ. Org Lett. 2005;7:2699. doi: 10.1021/ol0508734. [DOI] [PubMed] [Google Scholar]
- 85.Reddy LR, Fournier J-F, Reddy BVS, Corey EJ. J Am Chem Soc. 2005;127:8974. doi: 10.1021/ja052376o. [DOI] [PubMed] [Google Scholar]
- 86.Miyake H, Yamamura K. Chem Lett. 1992;21:507. [Google Scholar]
- 87.Endo A, Danishefsky SJ. J Am Chem Soc. 2005;127:8298. doi: 10.1021/ja0522783. [DOI] [PubMed] [Google Scholar]
- 88.Mulholland NP, Pattenden G, Walters IAS. Org Biomol Chem. 2006;4:2845. doi: 10.1039/b607109k. [DOI] [PubMed] [Google Scholar]
- 89.Mulholland NP, Pattenden G, Walters IAS. Org Biomol Chem. 2008;6:2782. doi: 10.1039/b803818j. [DOI] [PubMed] [Google Scholar]
- 90.Lee M, Kim DH. Bioorg Med Chem. 2002;10:913. doi: 10.1016/s0968-0896(01)00340-6. [DOI] [PubMed] [Google Scholar]
- 91.Ma G, Nguyen H, Romo D. Org Lett. 2007;9:2143. doi: 10.1021/ol070616u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ling T, Macherla VR, Manam RR, McArthur KA, Potts BCM. Org Lett. 2007;9:2289. doi: 10.1021/ol0706051. [DOI] [PubMed] [Google Scholar]
- 93.Seebach D, Sting AR, Hoffmann M. Angew Chem. 1996;108:2881. [Google Scholar]; Angew Chem Int Ed Engl. 1996;35:2708. [Google Scholar]
- 94.Kramer GW, Brown HC. J Organomet Chem. 1977;132:9. [Google Scholar]
- 95.Manam RR, Macherla VR, Potts BCM. Tetrahedron Lett. 2007;48:2537. [Google Scholar]
- 96.Conia JM, Le Perchec P. Synthesis. 1975:1. [Google Scholar]
- 97.Takahashi K, Midori M, Kawano K, Ishihara J, Hatakeyama S. Angew Chem. 2008;120:6340. doi: 10.1002/anie.200801967. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:6244. [Google Scholar]
- 98.Kiyotsuka Y, Igarashi J, Kobayashi Y. Tetrahedron Lett. 2002;43:2725. [Google Scholar]
- 99.Fukuda T, Sugiyama K, Arima S, Harigaya Y, Nagamitsu T, Omura S. Org Lett. 2008;10:4239. doi: 10.1021/ol8016066. [DOI] [PubMed] [Google Scholar]
- 100.Ooi H, Ishibashi N, Iwabuchi Y, Ishihara J, Hatakeyama S. J Org Chem. 2004;69:7765. doi: 10.1021/jo048817o. [DOI] [PubMed] [Google Scholar]
- 101.Momose T, Kaiya Y, Hasegawa J-i, Sato T, Chida N. Synthesis. 2009:2983. doi: 10.1002/asia.201000602. [DOI] [PubMed] [Google Scholar]
- 102.Rosenthal A, Nguyen LB. J Org Chem. 1969;34:1029. [Google Scholar]
- 103.Fleet GWJ, James K, Lunn RJ, Mathews CJ. Tetrahedron Lett. 1986;27:3057. [Google Scholar]
- 104.Caubert V, Langlois N. Tetrahedron Lett. 2006;47:4473. [Google Scholar]
- 105.Caubert V, Massé J, Retailleau P, Langlois N. Tetrahedron Lett. 2007;48:381. [Google Scholar]
- 106.Margalef IV, Rupnicki L, Lam HW. Tetrahedron. 2008;64:7896. [Google Scholar]
- 107.Struble JR, Bode JW. Tetrahedron. 2009;65:4957. doi: 10.1016/j.tet.2009.03.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mosey RA, Tepe JJ. Tetrahedron Lett. 2009;50:295. [Google Scholar]
- 109.Groll M, Huber R, Potts BCM. J Am Chem Soc. 2006;128:5136. doi: 10.1021/ja058320b. [DOI] [PubMed] [Google Scholar]
- 110.Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik HD, Huber R. Nature. 1997;386:463. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 111.Macherla VR, Mitchell SS, Manam RR, Reed KA, Chao T-H, Nicholson B, Deyanat-Yazdi G, Mai B, Jensen PR, Fenical WF, Neuteboom STC, Lam KS, Palladino MA, Potts BCM. J Med Chem. 2005;48:3684. doi: 10.1021/jm048995+. [DOI] [PubMed] [Google Scholar]
- 112.Lam KS, Tsueng G, McArthur KA, Mitchell SS, Potts BCM, Xu J. J Antibiot. 2007;60:13. doi: 10.1038/ja.2007.2. [DOI] [PubMed] [Google Scholar]
- 113.Manam RR, McArthur KA, Chao T-H, Weiss J, Ali JA, Palombella VJ, Groll M, Lloyd GK, Palladino MA, Neuteboom STC, Macherla VR, Potts BCM. J Med Chem. 2008;51:6711. doi: 10.1021/jm800548b. [DOI] [PubMed] [Google Scholar]
- 114.Müller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 115.Eustáquio AS, Moore BS. Angew Chem. 2008;120:4000. [Google Scholar]; Angew Chem Int Ed. 2008;47:3936. [Google Scholar]
- 116.Ashton TD, Scammells PJ. Bioorg Med Chem Lett. 2005;15:3361. doi: 10.1016/j.bmcl.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 117.Eustáquio AS, O’Hagan D, Moore BS. J Nat Prod. 2010;73:378. doi: 10.1021/np900719u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kimura Y, Takaoka M, Tanaka S, Sassa H, Tanaka K, Polevoda B, Sherman F, Hirano H. J Biol Chem. 2000;275:4635. doi: 10.1074/jbc.275.7.4635. [DOI] [PubMed] [Google Scholar]
- 119.Groll M, McArthur KA, Macherla VR, Manam RR, Potts BC. J Med Chem. 2009;52:5420. doi: 10.1021/jm900559x. [DOI] [PubMed] [Google Scholar]
- 120.Manam RR, Macherla VR, Tsueng G, Dring CW, Weiss J, Neuteboom STC, Lam KS, Potts BC. J Nat Prod. 2009;72:295. doi: 10.1021/np800578e. [DOI] [PubMed] [Google Scholar]
- 121.Corey EJ, Li W-DZ. Chem Pharm Bull. 1999;47:1. doi: 10.1248/cpb.47.1. [DOI] [PubMed] [Google Scholar]
- 122.Lam KS, Lloyd GK, Neuteboom STC, Palladino MA, Sethna KM, Spear MA, Potts BC. In: Natural Product Chemistry for Drug Discovery. Buss AD, Butler MS, editors. Royal Society of Chemistry; Cambridge: 2010. p. 355. [Google Scholar]
- 123.Tsueng G, Lam KS. Appl Microbiol Biotechnol. 2008;78:821. doi: 10.1007/s00253-008-1357-x. [DOI] [PubMed] [Google Scholar]
- 124.Tsueng G, Teisan S, Lam KS. Appl Microbiol Biotechnol. 2008;78:827. doi: 10.1007/s00253-008-1358-9. [DOI] [PubMed] [Google Scholar]
- 125.Tsueng G, Lam KS. Appl Microbiol Biotechnol. 2008;80:873. doi: 10.1007/s00253-008-1614-z. [DOI] [PubMed] [Google Scholar]
- 126.Tsueng G, Lam KS. J Antibiot. 2009;62:213. doi: 10.1038/ja.2009.7. [DOI] [PubMed] [Google Scholar]
- 127.Tsueng G, Lam KS. J Antibiot. 2007;60:469. doi: 10.1038/ja.2007.61. [DOI] [PubMed] [Google Scholar]
- 128.Denora N, Potts BCM, Stella VJ. J Pharm Sci. 2007;96:2037. doi: 10.1002/jps.20835. [DOI] [PubMed] [Google Scholar]
- 129.Bross PF, Kane R, Farrell AT, Abraham S, Benson K, Brower ME, Bradley S, Gobburu JV, Goheer A, Lee S-L, Leighton J, Liang CY, Lostritto RT, McGuinn WD, Morse DE, Rahman A, Rosario LA, Verbois SL, Williams G, Wang Y-C, Pazdur R. Clin Cancer Res. 2004;10:3954. doi: 10.1158/1078-0432.CCR-03-0781. [DOI] [PubMed] [Google Scholar]
- 130.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, Siegel D, Orlowski RZ, Kuter D, Limentani SA, Lee S, Hideshima T, Esseltine D-L, Kauffman M, Adams J, Schenkein DP, Anderson KC. N Engl J Med. 2003;348:2609. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 131.Chauhan D, Catley L, Li G, Podar K, Hideshima T, Velankar M, Mitsiades C, Mitsiades N, Yasui H, Letai A, Ovaa H, Berkers C, Nicholson B, Chao T-H, Neuteboom STC, Richardson P, Palladino MA, Anderson KC. Cancer Cell. 2005;8:407. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 132.Chauhan D, Singh A, Brahmandam M, Podar K, Hideshima T, Richardson P, Munshi N, Palladino MA, Anderson KC. Blood. 2008;111:1654. doi: 10.1182/blood-2007-08-105601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Roccaro AM, Leleu X, Sacco A, Jia X, Melhem M, Moreau A-S, Ngo HT, Runnels J, Azab A, Azab F, Burwick N, Farag M, Treon SP, Palladino MA, Hideshima T, Chauhan D, Anderson KC, Ghobrial IM. Blood. 2008;111:4752. doi: 10.1182/blood-2007-11-120972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Miller CP, Ban K, Dujka ME, McConkey DJ, Munsell M, Palladino M, Chandra J. Blood. 2007;110:267. doi: 10.1182/blood-2006-03-013128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ruiz S, Krupnik Y, Keating M, Chandra J, Palladino M, McConkey D. Mol Cancer Ther. 2006;5:1836. doi: 10.1158/1535-7163.MCT-06-0066. [DOI] [PubMed] [Google Scholar]
- 136.Cusack JC, Jr, Liu R, Xia L, Chao T-H, Pien C, Niu W, Palombella VJ, Neuteboom STC, Palladino MA. Clin Cancer Res. 2006;12:6758. doi: 10.1158/1078-0432.CCR-06-1151. [DOI] [PubMed] [Google Scholar]
- 137.Sloss CM, Wang F, Liu R, Xia L, Houston M, Ljungman D, Palladino MA, Cusack JC., Jr Clin Cancer Res. 2008;14:5116. doi: 10.1158/1078-0432.CCR-07-4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Baritaki S, Suzuki E, Umezawa K, Spandidos DA, Berenson J, Daniels TR, Penichet ML, Jazirehi AR, Palladino M, Bonavida B. J Immunol. 2008;180:6199. doi: 10.4049/jimmunol.180.9.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Miller CP, Rudra S, Keating MJ, Wierda WG, Palladino M, Chandra J. Blood. 2009;113:4289. doi: 10.1182/blood-2008-08-174797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ahn KS, Sethi G, Chao T-H, Neuteboom STC, Chaturvedi MM, Palladino MA, Younes A, Aggarwal BB. Blood. 2007;110:2286. doi: 10.1182/blood-2007-04-084996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Baritaki S, Yeung K, Palladino M, Berenson J, Bonavida B. Cancer Res. 2009;69:8376. doi: 10.1158/0008-5472.CAN-09-1069. [DOI] [PubMed] [Google Scholar]
- 142.Baritaki S, Chapman A, Yeung K, Spandidos DA, Palladino M, Bonavida B. Oncogene. 2009;28:3573. doi: 10.1038/onc.2009.214. [DOI] [PubMed] [Google Scholar]
- 143.Elliot PJ, Zollner TM, Böhncke W-H. J Mol Med. 2003;81:235. doi: 10.1007/s00109-003-0422-2. [DOI] [PubMed] [Google Scholar]
- 144.Tang G, Leppla SH. Infect Immun. 1999;67:3055. doi: 10.1128/iai.67.6.3055-3060.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lin G, Li D, de Carvalho LPS, Deng H, Tao H, Vogt G, Wu K, Schneider J, Chidawanyika T, Warren JD, Li H, Nathan C. Nature. 2009;461:621. doi: 10.1038/nature08357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Prudhomme J, McDaniel E, Ponts N, Bertani S, Fenical W, Jensen P, Le Roch K. PLoS One. 2008;3:e2335. doi: 10.1371/journal.pone.0002335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nguyen H, Ma G, Romo D. Chem Commun. 2010;46:4803. doi: 10.1039/c0cc00607f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ling T, Potts BC, Macherla VR. J Org Chem. 2010;75:3882. doi: 10.1021/jo100432g. [DOI] [PubMed] [Google Scholar]
- 149.Singh AV, Palladino MA, Lloyd GK, Potts BC, Chauhan D, Anderson KC. Br J Haematol. 2010;149:550. doi: 10.1111/j.1365-2141.2010.08144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chauhan D, Singh AV, Ciccarelli B, Richardson PG, Palladino MA, Anderson KC. Blood. 2010;115:834. doi: 10.1182/blood-2009-03-213009. [DOI] [PMC free article] [PubMed] [Google Scholar]