

Abstract
A novel series of fully synthetic 8-azatetracyclines was prepared and evaluated for antibacterial activity. Compounds were identified that overcome both efflux (tet(K)) and ribosomal protection (tet(M)) tetracycline resistance mechanisms and are active against Gram-positive and Gram-negative organisms. Two compounds were identified that exhibit comparable efficacy to marketed tetracyclines in in vivo models of bacterial infection.
Introduction
The tetracycline class of antibacterial agents has seen widespread clinical use for over 50 years due to its broad spectrum antibacterial activity.(1) Tetracyclines inhibit bacterial growth by preventing protein biosynthesis through binding to the 30S ribosome, thereby blocking the binding of aminoacyl-tRNA to the acceptor site.(2) This class includes a number of naturally occurring compounds, such as chlorotetracycline (1)(3) and tetracycline (2),(4) as well as several semisynthetic analogues, such as doxycycline (3)(5) and minocycline (4) (Figure 1).(6) In addition to their use as antibiotics for serious infections, tetracyclines have also been prescribed for use in acne and rosacea.(7) As with many classes of antibiotics, widespread use of tetracyclines has led to resistance and has limited their use in more complicated bacterial infections.8,9
Figure 1.

Examples of naturally occurring (1 and 2) and semisynthetic (3−6) tetracyclines.
Two major bacterial resistance mechanisms have been identified for tetracyclines: (1) active transport from bacterial cells via efflux pumps(10) and (2) ribosomal protection.(11) The efflux pumps are comprised of inner membrane tetracycline efflux proteins that were first identified in Gram-negative organisms (tet(A−E))10a,10b and were later found in Gram-positive bacteria (tet(K−L)).(10c) The ribosomal protection mechanism (tet(M−O)) involves the expression of proteins that bind to the ribosome and allow protein synthesis to proceed even in the presence of tetracyclines. The exact mechanism by which this occurs remains unclear. Since these early discoveries, the number of efflux and ribosomal protection mechanisms identified has increased.(12)
The presence of the strongly activating 10-hydroxy group readily enables chemical modification of the tetracycline D-ring at the 7- and 9-positions. This strategy led to the introduction of the 7-dimethylamino group in compounds 4−6, all of which have demonstrated improved activity in bacterial strains bearing efflux pump resistance mechanisms. More recently, the 9-substituted glycylcyclines, such as tigecycline (5),(13) and the aminomethylcyclines, such as amadacycline (6),(14) have shown improved activity for resistant organisms bearing the ribosomal protection mechanism.
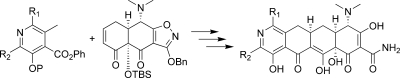
In 2005, Myers and co-workers published a fully synthetic route to the tetracyclines, the key step of which involves the Michael−Dieckmann condensation of the AB precursor 8 with structurally diverse D-ring precursors.15,16 This route provides potential access to a broad range of tetracyclines that are inaccessible via traditional semisynthesis.(17) One example of this is the introduction of a nitrogen atom into the D-ring, resulting in “azatetracyclines.” One 7-azatetracycline and one 9-azatetracycline were disclosed in Myers original work, resulting in compounds with only modest antibacterial activity. Herein we demonstrate this powerful approach using pyridyl D-ring precursors 7 to generate novel 8-azatetracyclines, 10 (Scheme 1).
Scheme 1. Fully Synthetic Approach to the 8-Azatetracyclines.

Results and Discussion
Chemistry
To explore 7- and 9-substituted-8-azatetracyclines, we required intermediates that could be readily modified either prior to or after construction of the protected compound 9. Thus, the halopyridines 15 and 16 were prepared according to Schemes 2 and 3. 3,5-Dichloroisonicotinic acid was treated with sodium benzylate to give compound 12, which was esterified to the phenyl ester 13 by conversion to the acid chloride, followed by treatment with phenol and 4-(dimethylamino)pyridine (DMAP). Suzuki coupling(18) using methylboronic acid and PdCl2Cy2 catalyst(19) gave the parent D-ring precursor 14 in good yield. N-Oxide formation with hydrogen peroxide in acetic acid followed by treatment with either phosphorousoxychloride or phosphorousoxybromide gave 15a and 15b, respectively. For 15a, the reaction gave a 4.5:1.0 ratio of the desired regioisomer, while a 25:1 ratio was observed for 15b. Both desired regioisomers were isolated in pure form by recrystallization. The regiochemistry was assigned by conversion to the dimethylamino compound 15c through a palladium-mediated amination.(20)1H NMR studies indicated an nOe between the methyl groups of the amine and the adjacent ring methyl group. The methoxy derivative 15d was prepared from 15b by treatment with sodium methoxide in 2-methylpyrrolidinone (NMP) at 120 °C. This gave the acid directly, which was re-esterified to the phenyl ester.
Scheme 2. Synthesis of D-Ring Precursors for 7-Substituted-8-azatetracyclines.

Reagents and conditions: (i) NaH (2 equiv), BnOH, NMP, 80 °C; (ii) (COCl)2, cat. DMF, CH2Cl2; (iii) PhOH, Et3N, DMAP, CH2Cl2; (iv) CH3B(OH)2, PdCl2Cy2, K3PO4, toluene, water, 100 °C; (v) H2O2, AcOH, 80 °C; (vi) POCl3, toluene, 100 °C; (vii) POBr3, toluene, 100 °C; (viii) dimethylamine in THF, Pd2dba3, Xantphos, K3PO4, 1,4-dioxane, 100 °C (sealed); (ix) NaOMe, NMP, 100−120 °C.
Scheme 3. Synthesis of D-Ring Precursors for 7,9-Disubstituted-8-azatetracyclines.

Reagents and conditions: (i) for 15a, mCPBA, CH2Cl2; for 15b, H2O2, AcOH, 80 °C; (ii) POBr3, toluene, 100 °C; (iii) BocNH2, Pd2dba3, Xantphos, Cs2CO3, 1,4-dioxane, 80 °C; (iv) HCl, 1,4-dioxane; (v) HF-pyridine, NaNO2, pyridine; (vi) dimethylamine in THF, Pd2dba3, Xantphos, Cs2CO3, 1,4-dioxane, 60 °C (sealed).
The dihalo compounds 16a and 16b were prepared by N-oxidation of 15a and 15b followed by treatment with POBr3 (Scheme 3). Yields were typically moderate (45−50%), with reduction of the N-oxide to give the starting pyridines 15 observed as major side reactions in both cases. Palladium-mediated coupling of 16b with t-butylcarbamate gave a ∼2.5:1 mixture of regioisomers, which were readily separated after conversion to the free amine 16c upon treatment with HCl. Treatment of 16c with sodium nitrite in the presence of HF-pyridine gave the fluoro derivative 16d in good yield.(21) Similarly, a 2.4:1.0 ratio of regioisomers was observed in the palladium-mediated reaction of 16b with dimethylamine to give 16e and 16f. Interestingly, the regioselectivity could be reversed by heating 16b in the presence of dimethylamine without catalyst to give 16f as the major isomer in a 3:1 ratio.
The 9-amino D-ring precursors 18a and 18b were prepared according to Scheme 4. Once again, palladium-mediated couplings using t-butylcarbamate proved to be highly effective for introduction of the amino group. Further protection of the mono-Boc intermediates 17 with Boc2O and DMAP in DMF gave the di-Boc precursors 18.
Scheme 4. Synthesis of D-Ring Precursors for 9-Amino-8-azatetracyclines.

Reagents and conditions: (i) BocNH2, Pd2dba3, Xantphos, Cs2CO3, 1,4-dioxane, 80 °C; (ii) Boc2O, DMAP, DMF.
Synthesis of the 8-azatetracyclines is outlined in Scheme 5. Most of the D-ring precursors were lithiated with lithium diisopropylamide (LDA) at −78 °C, followed by treatment with the enone 8. Warming to −20 °C yielded the fully protected tetracycline precursors 19. For D-ring precursors 16a, 16d, and 18b, a one-pot reaction was performed in which lithium bis(trimethylsilyl)amide (LHMDS) was added to a −78 °C solution of the precursor and the enone 8 followed by warming to −20 °C. Yields varied from 20 to 84% depending on substrate, with larger reaction scales generally providing better results. The LHMDS procedure also typically gave higher yields but was only possible on the more highly electron deficient D-ring precursors. The 7-bromo intermediate 19c was further derivatized at this stage via Suzuki couplings with methylboronic acid and phenylboronic acid to give 19d and 19e, respectively. Similarly, palladium-mediated amination with 19h and t-butylcarbamate provided the 7-chloro-9-amino compound 19j. Deprotection of the intermediates 19 with HF followed by hydrogenation in the presence of palladium on carbon gave the final 8-azatetracyclines 20a−k.
Scheme 5. Synthesis of 8-Azatetracyclines.

Reagents and conditions: (i) for 14, 15b−d, 16e, and 18a, LDA, TMEDA, −78 °C, THF then 8, −78 °C to −20 °C; (ii) for 16a, 16d, and 18b, LHMDS, 8, THF, −78 °C to −20 °C; (iii) CH3B(OH)2 or PhB(OH)2, PdCl2dppf2-CH2Cl2, K3PO4, toluene, 1,4-dioxane, water, 100 °C; (iv) BocNH2, Pd2dba3, Xantphos, K3PO4, 1,4-dioxane, 100 °C; (v) aqueous HF, CH3CN or 1,4-dioxane; (vi) 10% Pd/C, H2, HCl, MeOH, 1,4-dioxane.
To compare the 8-azatetracyclines directly to tigecycline, the 9-glycylamido-8-azatetracyclines 21a−c were prepared (Scheme 6). The Boc groups of compounds 19j−l were deprotected with HCl in 1,4-dioxane. Treatment with bromoacetylchloride in THF followed by excess t-butylamine gave the acylated intermediates, which were deprotected under standard conditions to give the final analogues 21a−c.
Scheme 6. Synthesis of 9-Glycylamido-8-azatetracyclines.

Reagents and conditions: (i) HCl, 1,4-dioxane; (ii) bromoacetylchloride, THF then t-BuNH2, 50 °C; (iii) aqueous HF, CH3CN or 1,4-dioxane; (iv) 10% Pd/C, H2, HCl, MeOH, 1,4-dioxane.
The bromopyridine intermediates proved to be useful for the introduction of alkylamines via palladium-mediated aminations and alkyl groups via Suzuki couplings, ultimately providing the 9-alkylamino-8-azatetracyclines 24 and 9-alkyl-8-azatetracyclines 26 (Scheme 7). Thus, 19h was treated with various amines in the presence of Pd2dba3 and 9,9-dimethyl-4,5-bis-(diphenylphosphino)xanthenes (Xantphos) in 1,4-dioxane at 100 °C. Potassium phosphate was found to be the most effective base for this transformation, with cesium carbonate and sodium t-butoxide leading to rapid decomposition. Low yields were generally observed due to decomposition of both the starting material and products upon heating. Reaction times of 2−4 h were eventually settled on as a balance between product formation and decomposition. Copper-catalyzed aminations were also explored, but rapid decomposition of the starting material was observed. Hindered amines generally gave low yields as did reactions with the 7-dimethylamino compound 19m. Alternatively, the alkylamines could be introduced prior to the Michael−Dieckmann reaction using either palladium or copper-mediated aminations(22) with 16a, 16d, and 16e. Protection of the resulting aminopyridines upon treatment with LHMDS followed by Boc2O or CbzCl gave the D-ring precursors 22. Michael−Dieckmann reaction then gave the protected intermediates 23. Suzuki reactions with 19h or 19m and various boronic acids were also carried out in moderate yields, providing the 9-alkyl intermediates 25. Deprotection of the intermediates under standard conditions gave the final 8-azatetracyclines 24 and 26.
Scheme 7. Synthesis of 7,9-Disubstituted-8-azatetracyclines.

Reagents and conditions: (i) R2R3NH2, Pd2dba3, Xantphos, K3PO4 or Cs2CO3, 1,4-dioxane, 100 °C or n-propylamine, CuI, 2-acetylcyclohexanone, K3PO4, DMF, 100 °C; (ii) LHMDS, THF then Boc2O or CbzCl; (iii) LDA, TMEDA, −78 °C, THF then 8, −78 °C to −20 °C or LHMDS, 8, THF, −78 °C to −20 °C; (iv) aqueous HF, CH3CN, or 1,4-dioxane; (v) 10% Pd/C, H2, HCl, MeOH, 1,4-dioxane; (vi) R2B(OH)2, (Ph3P)4Pd, K3PO4, toluene, 1,4-dioxane, H2O, 90 °C.
Biology
The in vitro antibacterial activities of all analogues were determined for a panel of Gram-positive (e.g., Staphylococcus aureus and Streptococcus pneumoniae) and Gram-negative (e.g., Escherichia coli and Klebsiella pneumoniae) bacteria. The activities of the 7-substituted-8-azatetracycline as well as several control tetracyclines for a representative set of these organisms are found in Table 1. The parent 8-azatetracycline 20a showed comparable activity to tetracycline, with reasonable minimum inhibitory concentrations (MICs) of 0.25−1 μg/mL for the tetracycline-susceptible strains. Lacking a substituent at either C-7 or C-9, 20a demonstrated poor activity for the strains bearing tet(M), tet(K), and tet(A). Overall, the 7-position appears to be very tolerant of substitution, allowing for bulky (20e), electron withdrawing (20b and 20i), and electron donating (20f and 20g) groups while maintaining activity against the tetracycline-susceptible strains. As with minocycline, introduction of the 7-dimethylamino group in 20f led to improved activities for most strains tested. Modest (1−2 dilution) improvements in MICs were observed for the tet(M) strains, while a more significant improvement was seen for the strain bearing the tet(K) resistance mechanism. With the exception of the 7-chloro analogue, all of the 7-substituted-8-aza compounds exhibited improved MICs for the tet(K) strain, correlating well with literature reports on 7-substituted tetracyclines. Unfortunately, all of the 8-azatetracyclines appear to be substrates for the tet(A) efflux pump, exhibiting no antibacterial activity at the highest concentration tested (32 μg/mL). In general, substitution at the 7-position appears to be inefficient at overcoming the ribosomal protection resistance mechanism of the tet(M) strains. The only compound with two dilution improvements for both tet(M) strains was compound 20i, the 7-fluoro analogue. This compound was also one of the more potent antibacterial compounds in this series.
Table 1. In Vitro Antibacterial Activity of 7-Substituted-8-azatetracyclines.

| MIC (μg/mL) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
|
S. aureus |
S. pneumoniae |
E. coli |
K. pneumoniae | ||||||
| compd | R1 | wild typea | tet(M)b | tet(K)c | wild typed | tet(M)c | wild typee | tet(A)c | wild typef |
| 20a | H | 1 | 32 | 16 | 0.25 | 32 | 0.5 | >32 | 1 |
| 20b | Cl | 1 | 32 | 32 | 1 | 32 | 2 | >32 | 4 |
| 20d | CH3 | 1 | 32 | 0.5 | 0.25 | >32 | 1 | >32 | 1 |
| 20e | Ph | 0.063 | 32 | 1 | 0.031 | 4 | 0.5 | >32 | 2 |
| 20f | (CH3)2N- | 0.031 | 16 | 2 | 0.063 | 8 | 0.125 | >32 | 0.25 |
| 20g | CH3O- | 0.125 | 8 | 4 | 0.063 | 16 | 0.25 | >32 | 1 |
| 20i | F | 0.063 | 8 | 8 | 0.016 | 8 | 0.25 | >32 | 0.5 |
| tetracycline | 1 | >32 | 32 | 0.25 | 32 | 2 | >32 | 4 | |
| minocycline | 0.125 | 16 | 0.25 | <0.016 | 8 | 0.5 | 8 | 1 | |
ATCC 13709.
Obtained from Micromyx (Kalamazoo, MI).
Obtained from Marilyn Roberts’ lab at the University of Washington.
ATCC 49619.
ATCC 25922.
ATCC 13883.
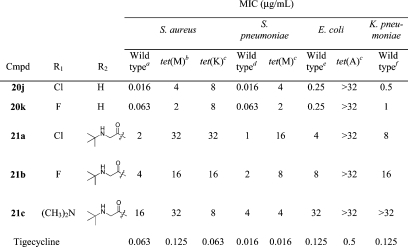
To make a direct comparison to tigecycline, the 9-glycylamido-8-azatetracyclines were prepared (Table 2). Whereas tigecycline is a very potent antibacterial agent against all of the organisms in our panel, including the four strains bearing tetracycline resistance mechanisms, the corresponding 8-azatetracycline analogues 21a−c were found to have poor overall activity. The aniline precursors 20j and 20k, however, showed significant improvements in MICs for both tet(M) strains relative to the corresponding 9-unsubstituted compounds 20b and 20i. For the 7-Cl analogue 20j, antibacterial activity also improved for the tet(K) and wild type strains. These compounds were also assessed for their ability to inhibit protein synthesis in a cell-free transcription/translation assay (Table 3). In this assay, compounds 20j and 20k were about 2-fold more potent than tetracycline, in agreement with their relative MICs for E. coli. Compounds 21a−c, on the other hand, inhibited protein synthesis with IC50s between 0.42 and 0.90 μg/mL, similar to tigecycline, despite having only modest antibacterial activity in the whole-cell assays. These data suggest that the compounds are active at the target, but that compounds 21a−c are not capable of getting to the target in the whole-cell assay, possibly due to poor cell permeability.
Table 2. In Vitro Antibacterial Activity of 9-Glycylamido-8-azatetracyclines.

 |
ATCC 13709.
Obtained from Micromyx (Kalamazoo, MI).
Obtained from Marilyn Roberts’ lab at the University of Washington.
ATCC 49619.
ATCC 25922.
ATCC 13883.
Table 3. Transcription/Translation (TnT) Assay Data.
| compd | E. colia MIC (μg/mL) | TnT IC50 (μg/mL) |
|---|---|---|
| 20f | 0.125 | 0.76 |
| 20j | 0.25 | 1.1 |
| 20k | 0.25 | 0.93 |
| 21a | 4 | 0.78 |
| 21b | 8 | 0.36 |
| 21c | 32 | 0.71 |
| 24i | 8 | 0.58 |
| 24l | 0.5 | 0.70 |
| 24c | 4 | 5.3 |
| 24n | 2 | 2.8 |
| 24o | 32 | 3.4 |
| tetracycline | 2 | 2.0 |
| tigecycline | 0.125 | 0.47 |
ATCC 25922 is a tetracycline-susceptible strain.
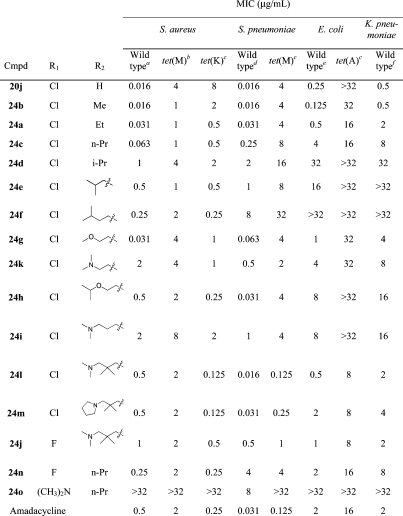
On the basis of the results of compounds 20j and 20k, a series of 9-amino-8azatetracyclines was prepared (Table 4). Compounds bearing a small, unbranched alkylamino group at the 9-position (24a−c) proved to be very potent antibacterial agents against the two tetracycline-susceptible Gram-positive strains, with MICs between 0.016 and 0.25 μg/mL. A 4-fold improvement in activity against the tet(M) S. aureus strain was observed for all of these compounds relative to 20j, while MICs for the S. pneumoniae tet(M) strain were largely unchanged. For the tet(K) S. aureus strain, antibacterial activity improved with increasing size of the alkyl group. Among the tetracycline-susceptible Gram-negative strains, however, there was a clear loss in potency as the size of the alkyl group increased, a trend that continued for the larger branched alkyl compounds 24d−f. This is not surprising, as it is known that Gram-negative antibacterial agents tend to be smaller and more polar.(23) These larger side chains also adversely affected the antibacterial activity for S. pneumoniae while having less effect on the MICs for the S. aureus strains. The 7-fluoro-9-n-propylamino (24n) and 7-dimethylamino-9-n-propylamino (24o) analogues were prepared as comparisons to 24c. While the 7-fluoro compound had MICs that tracked fairly closely to the 7-chloro analogue, the 7-dimethylamino compound exhibited no antibacterial activity for any of the strains at the highest concentration tested (32 μg/mL) with the exception of the tetracycline-susceptible S. pneumoniae strain. All three of the compounds had similar, albeit weak, activity in the transcription/translation assay, indicating the differences in MICs might be due to poor permeability of the 7-dimethylamino analogue. Introduction of a more polar heteroatom into the side chain (24g−i,k) resulted in improved antibacterial activity against the S. pneumoniae and Gram-negative strains but had minimal effect on activity toward either resistant S. aureus organisms. Interestingly, the oxygen bearing side chains (24g and 24h) yielded significantly improved antibacterial activity toward tetracycline-susceptible Gram-positive strains relative to the all carbon side chains (24e and 24f), while the nitrogen analogues (24k and 24i) were less potent against S. aureus and showed more modest improvements against S. pneumoniae. The combination of a branched side chain with a distal amino group gave the most balanced antibacterial compounds in this series (24l, 24m, and 24j). These compounds combine good antibacterial activity toward both Gram-positive and Gram-negative organisms, with modest activity against tet(A) E. coli. Both compounds 24l and 24m are less potent than tigecycline but compare favorably to amadacycline.
Table 4. In Vitro Antibacterial Activity of 9-Alkylamino-8-azatetracyclines.

 |
ATCC 13709.
Obtained from Micromyx (Kalamazoo, MI).
Obtained from Marilyn Roberts’ lab at the University of Washington.
ATCC 49619.
ATCC 25922.
ATCC 13883.
The antibacterial activity of the 9-alkyl and phenyl substituted compounds were generally modest (Table 5). Here again, a larger substituent at C-9 yielded improved activity for tet(M) and tet(K) strains but resulted in loss of activity for wild type S. pneumoniae and Gram-negative strains. Introduction of a 7-Cl or 7-dimethylamino group also gave improved activity for most of the strains in the panel. When comparing the 7-Cl-9-alkyl compounds (26b) to the 7-Cl-9-aminoalkyl compounds (24c), it is clear that the nitrogen confers significant improvements in antibacterial activity. In the 7-dimethylamino case, however, the 7-dimethylamino-9-alkyl compound 26a is a balanced Gram-positive antibacterial agent while the 9-aminoalkyl compound 24o has no significant activity.
Table 5. In Vitro Antibacterial Activity of 9-Alkyl-8-azatetracyclines.

| MIC (μg/mL) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
S. aureus |
S. pneumoniae |
E. coli |
K. pneumoniae | |||||||
| compd | R1 | R2 | wild typea | tet(M)b | tet(K)c | wild typed | tet(M)c | wild typee | tet(A)c | wild typef |
| 26a | (CH3)2N | n-Pr | 1 | 2 | 1 | 1 | 2 | 16 | >32 | 16 |
| 26b | Cl | n-Pr | 1 | 4 | 4 | 1 | 4 | >32 | >32 | 32 |
| 26c | Cl | CH3 | 2 | 8 | 16 | 0.5 | 8 | 8 | >32 | 8 |
| 26d | H | CH3 | 4 | 32 | 32 | 1 | 16 | 8 | >32 | 8 |
| 26e | Cl | Ph | 1 | 2 | 1 | 8 | 8 | >32 | >32 | >32 |
| 26f | H | Ph | 4 | 8 | 16 | 2 | 8 | 16 | >32 | 32 |
ATCC 13709.
Obtained from Micromyx (Kalamazoo, MI).
Obtained from Marilyn Roberts’ lab at the University of Washington.
ATCC 49619.
ATCC 25922.
ATCC 13883.
Compounds 20f and 24l were selected for further profiling. Both compounds inhibited protein synthesis in the transcription/translation assay with IC50s comparable to tigecycline. Susceptibility testing was performed, and MIC50 and MIC90 values were calculated for recent clinical isolates of S. aureus, S. pneumoniae, Haemophilus influenzae, and E. coli (Table 6). The panels contained both tetracycline-susceptible and resistant strains, including 12 methicillin-resistant S. aureus (MRSA) strains. As expected from the initial MIC data, compound 20f had very potent antibacterial activity for the tetracycline-susceptible strains but performed poorly against the resistant organisms. The compound did have MIC90s that were significantly lower than tetracycline. Despite having higher MICs in the primary screen relative to tigecycline, compound 24l performed similarly in the S. aureus, S. pneumoniae, and H. influenzae panels, exhibiting clinically relevant MIC90s in each case. These organisms are particularly relevant, as they are responsible for infections in a large portion of community-acquired bacterial pneumonia cases.
Table 6. MIC90 Analysis.
| species | MIC (μg/mL) | 20f | 24l | tigecycline | tetracycline |
|---|---|---|---|---|---|
| S. aureus (n = 31) | MIC50 | ≤0.016 | 0.125 | 0.063 | 0.125 |
| MIC90 | 16 | 2 | 1 | >64 | |
| MIC range | ≤0.016−32 | 0.063−4 | ≤0.016−1 | 0.063→64 | |
| S. pneumoniae (n = 19) | MIC50 | 2 | ≤0.016 | ≤0.016 | 32 |
| MIC90 | 4 | 0.063 | ≤0.016 | >32 | |
| MIC range | ≤0.016−8 | ≤0.016−0.063 | ≤0.016-≤0.016 | 0.125→32 | |
| H. influenzae (n = 11) | MIC50 | 0.5 | 1 | 0.5 | 4 |
| MIC90 | 2 | 1 | 0.5 | 16 | |
| MIC range | 0.063−4 | 0.25−2 | 0.25−1 | 0.25−16 | |
| E. coli (n = 20) | MIC50 | 0.25 | 1 | 0.25 | 2 |
| MIC90 | >32 | 4 | 0.5 | >64 | |
| MIC range | ≤0.016−32 | 0.5−8 | 0.125−2 | 0.25→64 | |
The pharmacokinetic profiles of the two 8-azatetracyclines were determined in Sprague−Dawley rats (Table 7). Compound 20f had very similar AUC, clearance, and volume of distribution profiles to both tetracycline and tigecycline, although the half-life was somewhat shorter. Low oral bioavailability (%F) of 12.7% was observed but was comparable to tetracycline (12.1%). In humans, tetracycline is generally reported to have oral bioavailability of 50−70%.(24) We and others have found the oral bioavailability of several tetracycline derivatives to be significantly lower in rat than reported data in humans. Tigecycline, administered only intravenously in humans, had very low oral bioavailability. Compound 24l had a 4-fold higher AUC and 5-fold lower clearance than the other three compounds as well as a significantly lower volume of distribution. Unfortunately, oral bioavailability was comparable to tigecycline.
Table 7. Pharmacokinetic Properties of 20f and 24l in Sprague−Dawley Ratsa.
| compd | AUC (h·ng/mL) | Clobs (mL/h/kg) | Vzobs (mL/kg) | t1/2 (hr) | %F |
|---|---|---|---|---|---|
| 20f | 1087 | 919 | 4177 | 3.1 | 12.7 |
| 24l | 4872 | 202 | 1256 | 4.3 | 0.8 |
| tetracycline | 802 | 1230 | 3676 | 4.5 | 12.1 |
| tigecycline | 1052 | 929 | 6119 | 4.6 | 1.1 |
Compounds were dosed at 1 mg/kg IV and 10 mg/kg PO with sterile water as vehicle.
The compounds were also investigated in mouse S. aureus and E. coli septicemia models (Table 8). Both 8-azatetracyclines performed well in the S. aureus model following IV administration, with PD50 values comparable to the two marketed control tetracyclines. 20f, the most potent of the four compounds, also provided the best protection, with 100% survival at the lowest dose at which it was administered (0.30 mg/kg). Despite having an 8-fold higher MIC, compound 24l provided equivalent protection relative to tigecycline, likely due to its higher AUC and lower clearance.(25) The E. coli model proved to be more challenging, requiring higher doses for protection. Compound 20f had a PD50 of 4.3 mg/kg compared to 2.1 mg/kg for tigecycline. Compound 24l was 4-fold less potent than 20f and was comparable to tetracycline in this model.
Table 8. IV Efficacy in Mouse Septicemia Modela.
|
S. aureus |
E. coli |
|||||
|---|---|---|---|---|---|---|
| compd | MIC (μg/mL) | PD50 (mg/kg) | 95% C.I. | MIC (μg/mL) | PD50 (mg/kg) | 95% CI |
| 20f | 0.031 | <0.30b | 0.13 | 4.3 | 4.1−4.6 | |
| 24l | 0.5 | 0.36 | 0.36−0.56 | 0.5 | 17 | 4.1−30 |
| tetracycline | 0.25 | 0.35 | 0.34−0.37 | 1 | 17 | 7.3−27 |
| tigecycline | 0.063 | 0.35 | 0.24−0.47 | 0.13 | 2.1 | 1.8−2.4 |
S. aureus ATCC 13709 (Smith) or E. coli ATCC 25922 was mixed with 5% mucin and inoculated by intraperitoneal injection at 2.1 × 106 cfu/mouse (S. aureus) or 2.0 × 107 cfu/mouse (E. coli). One hour postchallenge, mice received IV treatment (vehicle = sterile water) with the test article at concentrations ranging from 0.05−10 mg/kg (S. aureus) or 0.30−30 mg/kg (E. coli). The PD50 in mg/kg was calculated as survival after 48 h.
Lowest dose was 0.30 mg/kg with 100% survival at all doses.
Conclusions
A novel, fully synthetic series of 8-azatetracycline analogues was prepared and optimized. Through modification of the 7- and 9-positions, compounds were found that overcome both tet(K) efflux and tet(M)-mediated ribosomal protection. The 7-chloro-9-alkylamino-8-azatetracycline 24l exhibits clinically relevant MIC90 profiles against both Gram-positive and Gram-negative organisms that include these resistance mechanisms. This compound also demonstrated in vivo efficacy in murine models of infection that is equivalent to currently marketed tetracycline antibacterial agents. The 7-dimethylamino-8-azatetracycline 20f was efficacious when administered intravenously and had oral bioavailability in rat equivalent to tetracycline.
The current work demonstrates the ability of this chemistry platform to generate antibacterial compounds that are inaccessible through traditional semisynthesis, providing the medicinal chemist a broader array of tools through which essential antibacterial properties can be modulated. Importantly, this includes the ability to overcome ever evolving resistance mechanisms and to identify structure−activity relationships to deliver compounds with desired pharmacokinetic properties.
Experimental Procedures
General
Air and moisture sensitive liquids and reagents were transferred via syringe or cannula and were introduced into flame-dried glassware under a positive pressure of dry nitrogen through rubber septa. All reactions were stirred magnetically. Commercial reagents were used without further purification. Analytical thin-layer chromatography was performed on EM Science precoated glass-backed silica gel 60 Å F-254 250 μm plates. Visualization of the plates was effected by ultraviolet illumination and/or immersion of the plate in a basic solution of potassium permanganate in water followed by heating. Column chromatography was performed on a FlashMaster Personal system using ISOLUTE Flash Si II silica gel prepacked cartridges (available from Biotage). Preparative reversed-phase HPLC chromatography (HPLC) was accomplished using a Waters Autopurification system with mass-directed fraction collection. All intermediate compounds were purified with a Waters Sunfire Prep C18 OBD column (5 μm, 19 mm × 50 mm; flow rate = 20 mL/min) using a mobile phase mixture of H2O (A) and CH3CN (B) containing 0.1% HCO2H. The final 8-azatetracycline compounds were purified using a Phenomenex Polymerx 10 μ RP 100A column (10 μm, 30 mm × 21.2 mm; flow rate = 20 mL/min) using a mobile phase mixture of 0.05N HCl in H2O (A) and CH3CN (B). All final 8-azatetracycline compounds were isolated as mono-, di-, or trihydrochloride salts following freeze-drying. 1H NMR spectra were recorded on a JEOL ECX-400 (400 MHz) spectrometer and are reported in ppm using residual solvent as the internal standard (CDCl3 at 7.24 ppm, DMSO-d6 at 2.50 ppm, or CD3OD at 3.31 ppm). High performance liquid chromatography−electrospray mass spectra (LC-MS) were obtained using a Waters Alliance HPLC system equipped with a binary pump, a diode array detector, a Waters Sunfire C18 (5 μm, 4.6 mm I.D. × 50 mm) column, and a Waters 3100 series mass spectrometer with electrospray ionization. Spectra were scanned from 100 to 1200 amu. The eluent was a mixture of H2O (A) and CH3CN (B) containing 0.1% HCO2H at a flow rate of 1 mL/min. Purity of the final compounds was assessed by the following method: (a) time = 0, 100% A; (b) time = 0.5 min, 100% A; (c) time = 3.5 min, 100% B; (d) time = 5 min, 100% B (e) time = 6 min, 100% A; (f) time = 7 min, 100% A. All final products were ≥95% purity as assessed by this method.
3-(Benzyloxy)-5-chloroisonicotinic Acid (12)
Sodium hydride (60% dispersion in mineral oil, 4.37 g, 109 mmol) was added portionwise to a solution of 3,5-dichloroisonicotinic acid (10.2 g, 53.3 mmol) in NMP (100 mL). After gas evolution ceased, benzyl alcohol (5.52 mL, 53.3 mmol) was added dropwise. After gas evolution ceased, the reaction mixture was heated to 80 °C. After 1 h, the reaction was complete and was allowed to cool to room temperature and stand overnight. The reaction mixture was diluted with water (300 mL) and was washed with Et2O (2 × 100 mL, discarded). The aqueous layer was brought to pH ∼2 with conc HCl, causing a precipitate to form. The mixture was diluted with brine (100 mL) and was allowed to stand for 30 min. The solid was collected by filtration, was washed with water (3×), and was dried in a 45 °C vacuum oven overnight. This gave 9.36 g (67%) of the title compound as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 14.30−14.10 (bs, 1 H), 8.52 (s, 1H), 8.34 (s, 1 H), 7.50−7.30 (m, 5 H), 5.33 (s, 2 H). MS (ESI) m/z 264.20 (M + H).
Phenyl 3-(Benzyloxy)-5-chloroisonicotinate (13)
Oxalyl chloride (4.9 mL, 56 mmol) was added to a suspension of 12 (3.71 g, 14.1 mmol) in CH2Cl2 (50 mL) over ∼2 min. DMF was added dropwise in ∼20 μL portions every 5 min until complete solution was achieved. After stirring for an additional 30 min, the reaction mixture was concentrated under reduced pressure. The resulting solid was dissolved in CH2Cl2 (50 mL), and phenol (2.65 g, 28.1 mmol), DMAP (0.172 g, 1.41 mmol), and Et3N (9.76 mL, 70.4 mmol) were added. After 1 h, the reaction mixture was diluted with CH2Cl2 (50 mL) and was washed with water (2 × 100 mL) and NaHCO3 (saturated, aqueous, 100 mL). The organics were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by column chromatography (Biotage 50 g column, 0−25% EtOAc in hexanes gradient), yielding 4.20 g (88%) of the product as an off-white solid. Rf = 0.43 in 30% EtOAc/hexanes. 1H NMR (400 MHz, DMSO-d6) δ 8.70 (s, 1H), 8.48 (s, 1 H), 7.52−7.30 (m, 8 H), 7.15−7.08 (m, 2 H), 5.44 (s, 2 H). MS (ESI) m/z 340.25 (M + H).
Phenyl 3-(Benzyloxy)-5-methylisonicotinate (14)
Compound 13 (2.01 g, 5.92 mmol), methyl boronic acid (1.06 g, 17.7 mmol), dichlorobis(tricyclohexylphosphine)palladium(II) (102 mg, 0.296 mmol), and K3PO4 (3.76 g, 17.7 mmol) were heated to 100 °C in toluene (20 mL) and water (2 mL). After 16 h, the reaction mixture was allowed to cool to room temperature and was diluted with EtOAc (20 mL). This was washed with water (20 mL) and brine (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by column chromatography (Biotage 50 g column, 0−40% EtOAc in hexanes gradient), yielding 1.66 g (88%) of the product as a white solid. Rf = 0.18 in 30% EtOAc/hexanes. 1H NMR (400 MHz, DMSO-d6) δ 8.50 (s, 1H), 8.26 (s, 1 H), 7.52−7.30 (m, 8 H), 7.20−7.10 (m, 2 H), 5.37 (s, 2 H), 2.38 (s, 3 H). MS (ESI) m/z 320.27 (M + H).
Phenyl 5-(Benzyloxy)-2-chloro-3-methylisonicotinate (15a)
Compound 14 (9.66 g, 30.3 mmol) was heated to 80 °C in acetic acid (50 mL) and hydrogen peroxide (30% aqueous solution, 17 mL). After 6 h, LC/MS indicated that the starting material was consumed and the N-oxide present. Most of the acetic acid and water were removed under reduced pressure. The material was then diluted with EtOAc (200 mL), and the mixture was washed with NaHCO3 (saturated aqueous solution, 3 × 50 mL). The organics were dried over Na2SO4, filtered, and concentrated under reduced pressure, yielding 9.37 g (92%) of the crude N-oxide. The material was dissolved in toluene (60 mL), phosphorousoxychloride (7.69 g, 50.1 mmol) was added, and the reaction mixture was heated to 100 °C. After 2 h, LC/MS indicated that the reaction was complete. Upon cooling to room temperature, the reaction mixture was poured into a solution of K2CO3 (40 g) in water (200 mL). This mixture was extracted with EtOAc (3 × 150 mL), and the combined extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. 1H NMR indicated a mixture of regioisomers (4.5:1). The material was redissolved in EtOAc (30 mL) with heating and was then allowed to cool to room temperature and stand overnight. A white crystalline solid was obtained (4.99 g, 51%). Another 1.92 g (19%) of the white crystalline solid was obtained from the mother liquor after standing in a refrigerator overnight. Both were the desired pure regioisomer 15a by 1H NMR. 1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.43−7.30 (m, 7 H), 7.29−7.20 (m, 1 H), 7.07 (d, J = 8.2 Hz, 2 H), 5.21 (s, 2 H), 2.46 (s, 3 H). MS (ESI) m/z 398.22, 400.22 (M + H).
Phenyl 5-(Benzyloxy)-2-bromo-3-methylisonicotinate (15b)
Compound 14 (25.9 g, 81.1 mmol) was heated to 80 °C in acetic acid (160 mL) and hydrogen peroxide (30% aqueous solution, 40 mL). After stirring overnight, LC/MS indicated that the starting material was consumed and the N-oxide present. The reaction mixture was concentrated under reduced pressure, was diluted with EtOAc, and was washed with NaHCO3 (saturated aqueous solution, 3×). The organics were dried over Na2SO4, filtered, and concentrated under reduced pressure, yielding 25.4 g (93% crude) of the N-oxide as a tan solid. The material was suspended in toluene (200 mL) and was heated to 80 °C, at which point most of the solid had dissolved. Phosphorousoxybromide (25 g, 87 mmol) was added in one portion. After 30 min, LC/MS indicated the reaction was complete. Upon cooling to room temperature, the reaction mixture was poured into a solution of K2CO3 (83 g) in water (300 mL). This was stirred for 30 min at room temperature. The layers were separated, and the aqueous layer was extracted with EtOAc. The combined extracts were dried over MgSO4, filtered through Celite, and concentrated under reduced pressure. The material was recrystallized from EtOAc/hexanes (1:1), yielding 20.6 g (64%) of the product as an off-white solid. Single isomer by 1H NMR. 1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.43−7.30 (m, 7 H), 7.29−7.20 (m, 1 H), 7.07 (d, J = 8.2 Hz, 2 H), 5.21 (s, 2 H), 2.46 (s, 3 H). MS (ESI) m/z 398.22, 400.22 (M + H).
Phenyl 5-(Benzyloxy)-2-(dimethylamino)-3-methylisonicotinate (15c)
Compound 15b (1.06 g, 2.66 mmol), dimethylamine (2.0 M solution in THF, 4.0 mL, 8.0 mmol), K3PO4 (1.7 g, 8.0 mmol), tris(dibenzylideneacetone)dipalladium (120 mg, 0.13 mmol), and Xantphos (220 mg, 0.40 mmol) were heated to 100 °C in 1,4-dioxane (10 mL) in a sealed pressure vessel. After heating overnight, the reaction mixture was cooled to room temperature, was diluted with EtOAc (100 mL), and was washed with water (3 × 50 mL) and brine (50 mL). The organics were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by column chromatography (Biotage 50 g column, 0−15% EtOAc in hexanes gradient), yielding 388 mg (40%) of the title compound as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.96 (s, 1H), 7.45−7.20 (m, 8 H), 7.08 (d, J = 8.2 Hz, 2 H), 5.16 (s, 2 H), 2.76 (s, 6 H), 2.37 (s, 3 H). MS (ESI) m/z 363.45 (M + H).
Phenyl 5-(Benzyloxy)-2-methoxy-3-methylisonicotinate (15d)
Sodium methoxide (38 mg, 0.70 mmol) was added to a solution of compound 15b (118 mg, 0.351 mmol) in NMP (2 mL), and the reaction mixture was heated to 100 °C. After heating overnight, additional sodium methoxide (57 mg, 1.1 mmol) was added and heating was continued at 100 °C. After 1 h, the reaction mixture was heated to 120 °C. After an additional 1 h, the reaction mixture was cooled to room temperature, water (20 mL) was added, and the pH was adjusted to 2 with 6 N aqueous HCl. This was extracted with EtOAc (3 × 20 mL), and the combined extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude acid intermediate was dissolved in CH2Cl2 (10 mL), and oxalyl chloride (0.123 mL, 1.40 mmol) was added. After 1 h, the reaction mixture was concentrated under reduced pressure. The material was dissolved in CH2Cl2 (10 mL), and phenol (66 mg, 0.70 mmol), DMAP (4 mg, 0.04 mmol), and Et3N (0.245 mL, 1.76 mmol) were added. After 1 h, the reaction mixture was diluted with EtOAc (50 mL) and was washed with water (2 × 30 mL) and brine (30 mL). The organics were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by column chromatography (Biotage 10 g column, 0−12% EtOAc in hexanes gradient), yielding 20.3 mg (17%) of clean product. An additional 29.1 mg of product contaminated with some residual phenol was also isolated. Rf = 0.39 in 20% EtOAc/hexanes. 1H NMR (400 MHz, CDCl3) δ 7.75 (s, 1H), 7.45−7.24 (m, 9 H), 7.10 (d, J = 7.3 Hz, 2 H), 5.13 (s, 2 H), 3.91 (s, 3 H), 2.67 (s, 3 H). MS (ESI) m/z 336.1, 338.1 (M + H).
Phenyl 3-(Benzyloxy)-2-bromo-6-chloro-5-methylisonicotinate (16a)
Compound 15a (4.99 g, 14.1 mmol) was dissolved in CH2Cl2 (30 mL), and 3-chloroperbenzoic acid (77%, 6.33 g, 28.2 mmol) was added in one portion. After 6 h, an additional portion of 3-chloroperbenzoic acid (3.15 g, 14.1 mmol) was added, and the reaction was stirred at room temperature overnight. The reaction mixture was diluted with CH2Cl2 (200 mL) and was washed with Na2CO3 (saturated aqueous solution, 100 mL) and brine. The organics were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by column chromatography (Biotage 70 g column, 5−50% EtOAc in hexanes gradient), yielding 3.90 g (75%) of the pure N-oxide as an off-white solid (Rf = 0.33 in 66% EtOAc/hexanes). The N-oxide was dissolved in toluene (30 mL), phosphorousoxybromide (6.36 g, 22.1 mmol) was added, and the reaction mixture was heated to 80 °C. After 1 h, the reaction mixture was allowed to cool to room temperature and was poured into a solution of K2CO3 (10 g) in water (50 mL). This mixture was extracted with EtOAc (3 × 100 mL), and the combined extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by column chromatography (Biotage 50 g column, 0−10% EtOAc in hexanes gradient), yielding 2.06 g (45%) of the desired product 16a as an oil which slowly solidified on standing overnight. In addition, 1.01 g (20%) of compound 15a was recovered. 1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.43−7.30 (m, 7 H), 7.29−7.20 (m, 1 H), 7.07 (d, J = 8.2 Hz, 2 H), 5.21 (s, 2 H), 2.46 (s, 3 H). MS (ESI) m/z 398.22, 400.22 (M + H).
Phenyl 3-(Benzyloxy)-2,6-dibromo-5-methylisonicotinate (16b)
Compound 15b (8.54 g, 21.4 mmol) was heated to 80 °C in acetic acid (120 mL) and hydrogen peroxide (30% aqueous solution, 30 mL). After stirring overnight, LC/MS indicated that the starting material was mostly consumed and the N-oxide present. The reaction mixture was cooled to room temperature and was diluted with brine (150 mL). The mixture was cooled in a −20 °C freezer for 30 min. The resulting precipitate was collected by filtration and was washed with water/brine (1:1, 2×). The solid was dissolved in CH2Cl2 and was dried over Na2SO4, filtered, and concentrated under reduced pressure. This gave 7.83 g (88% crude) of the N-oxide as a tan solid. The material was suspended in toluene (200 mL) and was heated to 100 °C, at which point most of the solid had dissolved. A solution of phosphorousoxybromide (10.8 g, 37.8 mmol) in toluene (10 mL) was added rapidly. After 45 min, the reaction mixture was allowed to cool to room temperature and was poured into a solution of K2CO3 (40 g) in water (150 mL). After stirring for 30 min at room temperature, the layers were separated and the aqueous layer was extracted with EtOAc. The combined extracts were dried over MgSO4, filtered, and concentrated under reduced pressure. The material was purified by column chromatography (Biotage 50 g column, 0−8% EtOAc in hexanes gradient to isolate the product then 15% EtOAc in hexanes to recover 15b), yielding 5.02 g (49%) of the product as an off-white solid. Compound 15b was also recovered. 1H NMR (400 MHz, DMSO-d6) δ 7.49−7.32 (m, 8 H), 7.10 (d, J = 7.3 Hz, 2 H), 5.15 (s, 2 H), 2.41 (s, 3 H). MS (ESI) m/z 476.18, 478.16, 480.16 (M + H).
Phenyl 3-(Benzyloxy)-2-bromo-6-[(t-butoxycarbonyl)amino]-5-methylisonicotinate
Compound 16b (2.15 g, 4.50 mmol), t-butylcarbamate (633 mg, 5.40 mmol), Cs2CO3 (2.93 g, 9.00 mmol), tris(dibenzylideneacetone)dipalladium (206 mg, 0.225 mmol), and Xantphos (380 mg, 0.673 mmol) were weighed into a flask. This was evacuated and backflushed with nitrogen (3×), and 1,4-dioxane (15 mL) was added. The reaction mixture was heated to 80 °C. After 3 h, the reaction mixture was cooled to room temperature, was diluted with EtOAc (100 mL), and was washed with water (2 × 50 mL). The organics were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by column chromatography (Biotage 50 g column, 0−12% EtOAc in hexanes gradient), yielding 1.43 g (62%) of a 4:1 mixture of the two regioisomeric compounds. 1H NMR (400 MHz, CDCl3) δ 7.51−7.46 (m, 2 H), 7.42−7.32 (m, 7 H), 7.30−7.24 (m, 1 H), 7.12 (d, J = 8.2 Hz, 0.5 H), 7.03 (d, J = 8.2 Hz, 2 H), 6.88 (s, 0.25 H), 6.71 (s, 1 H), 5.12 (s, 2 H), 5.02 (s, 0.5 H), 2.43 (s, 0.75 H), 2.34 (s, 3 H), 1.50 (s, 9 H), 1.47 (s, 2.25 H). MS (ESI) m/z 535.10, 537.10 (M + Na).
Phenyl 2-Amino-5-(benzyloxy)-6-bromo-3-methylisonicotinate (16c)
The above regioisomeric mixture (1.43 g, 2.78 mmol) was stirred in 4 M HCl in 1,4-dioxane (20 mL) and 1,4-dioxane (5 mL) overnight. The reaction mixture was diluted with EtOAc (100 mL) and was washed with NaHCO3 (saturated aqueous solution, 2 × 100 mL). The organics were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by column chromatography (Biotage 20 g column, 0−30% EtOAc in hexanes gradient), yielding 805 mg (70%) of the major regioisomer 16c and 270 mg (24%) of the minor regioisomer. Data for 16c: Rf = 0.39 in 40% EtOAc/hexanes. 1H NMR (400 MHz, CDCl3) δ 7.50−7.42 (m, 2 H), 7.40−7.30 (m, 5 H), 7.30−7.24 (m, 1 H), 7.04 (d, J = 8.2 Hz, 2 H), 5.06 (s, 2 H), 4.56 (s, 2 H), 2.16 (s, 3 H). MS (ESI) m/z 413.11, 415.01 (M + H). Data for phenyl 2-amino-3-(benzyloxy)-6-bromo-5-methylisonicotinate: Rf = 0.72 in 40% EtOAc/hexanes; 1H NMR (400 MHz, CDCl3) δ 7.42−7.34 (m, 8 H), 7.13−7.10 (m, 2 H), 5.01 (s, 2 H), 4.64 (s, 2 H), 2.35 (s, 3 H); MS (ESI) m/z 413.09, 415.09 (M + H).
Phenyl 3-(Benzyloxy)-2-bromo-6-fluoro-5-methylisonicotinate (16d)
HF-Pyridine solution (∼70% HF, 2 mL) was added to a 0 °C solution of compound 16c (884 mg, 2.14 mmol) in pyridine (1 mL). Sodium nitrite (177 mg, 2.57 mmol) was added (bubbling observed), and the reaction mixture was allowed to slowly warm to room temperature. After 3 days, the reaction mixture was poured into Na2CO3 (saturated aqueous solution, 30 mL) and was extracted with EtOAc (3 × 30 mL). The combined extracts were washed with HCl (1 N aqueous solution, 2 × 50 mL) and were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by column chromatography (Biotage 20 g column, 0−6% EtOAc in hexanes gradient), yielding 687 mg (77%) of the product as a colorless oil that slowly solidified on standing. Rf = 0.39 in 10% EtOAc/hexanes. 1H NMR (400 MHz, CDCl3) δ 7.50−7.42 (m, 2 H), 7.40−7.30 (m, 5 H), 7.30−7.24 (m, 1 H), 7.08−7.02 (m, 2 H), 5.14 (s, 2 H), 2.33 (s, 3 H). MS (ESI) m/z 416.17, 418.15 (M + H).
Phenyl 3-(Benzyloxy)-2-bromo-6-(dimethylamino)-5-methylisonicotinate (16e) and Phenyl 3-(Benzyloxy)-6-bromo-2-(dimethylamino)-5-methylisonicotinate (16f)
Compound 16b (1.51 g, 3.16 mmol), Cs2CO3 (3.1 g, 9.5 mmol), tris(dibenzylideneacetone)dipalladium (145 mg, 0.158 mmol), and Xantphos (267 mg, 0.474 mmol) were weighed into a vial equipped with a septum. This was evacuated and flushed with nitrogen (3×), and 1,4-dioxane (7 mL) and dimethylamine (2.0 M solution in THF, 4.75 mL, 9.50 mmol) were added. The reaction mixture was heated to 60 °C and was stirred for 4 h. The reaction mixture was allowed to cool to room temperature, was filtered through Celite, and was concentrated under reduced pressure. The material was purified by column chromatography (Biotage 50 g column, 0−10% EtOAc in hexanes gradient), yielding 868 mg (58%) of the major regioisomer 16e and 354 mg (24%) of the minor regioisomer 16f. Data for 16e: 1H NMR (400 MHz, CDCl3) δ 7.50−7.46 (m, 2 H), 7.41−7.30 (m, 5 H), 7.30−7.24 (m, 1 H), 7.08−7.02 (m, 2 H), 5.08 (s, 2 H), 2.85 (s, 6 H), 2.32 (s, 3 H); MS (ESI) m/z 441.19, 443.18 (M + H); Rf = 0.29 in 10% EtOAc/hexanes. The regiochemistry was confirmed by an NOE between the methyl protons and the N,N-dimethylamino protons (0.61%). Data for 16f: 1H NMR (400 MHz, CDCl3) δ 7.41−7.31 (m, 7 H), 7.30−7.20 (m, 1 H), 7.01 (d, J = 7.3 Hz, 2 H), 4.94 (s, 2 H), 3.04 (s, 6 H), 2.33 (s, 3 H); MS (ESI) m/z 441.19, 443.18 (M + H); Rf = 0.40 in 10% EtOAc/hexanes. The regiochemistry was confirmed by NOE between the benzylic protons of the O-benzyl group and the N,N-dimethylamino protons (0.44%).
Phenyl 5-Benzyloxy-6-[(t-butoxycarbonyl)amino]-2-fluoro-3-methylisonicotinate (17a)
Compound 16d (538 mg, 1.29 mmol), t-butylcarbamate (302 mg, 2.58 mmol), Cs2CO3 (840 mg, 2.58 mmol), tris(dibenzylideneacetone)dipalladium (59 mg, 0.065 mmol), and Xantphos (109 mg, 0.190 mmol) were weighed into a flask. This was evacuated and backflushed with nitrogen (3×), and 1,4-dioxane (3 mL) was added. The reaction mixture was heated to 80 °C. After 4 h, the reaction mixture was cooled to room temperature, was diluted with EtOAc (50 mL), and was washed with water (2 × 25 mL). The organics were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by column chromatography (Biotage 20 g column, 0−12% EtOAc in hexanes gradient), yielding 451 mg (77%) of the product. Rf = 0.35 in 20% EtOAc/hexanes. 1H NMR (400 MHz, CDCl3) δ 7.42−7.24 (m, 8 H), 7.04−6.99 (m, 2 H), 4.99 (s, 2 H), 2.38 (s, 3 H), 1.40 (s, 18 H). MS (ESI) m/z 453.14 (M + H).
Phenyl 3-Benzyloxy-2-[bis(t-butoxycarbonyl)amino]-6-fluoro-5-methylisonicotinate (18a)
Compound 17a (451 mg, 1.00 mmol) was treated with di-t-butyldicarbonate (1.09 g, 5.00 mmol) and DMAP (24 mg, 0.20 mmol) in DMF (15 mL). After 30 min, the reaction mixture was diluted with EtOAc (100 mL) and was washed with water (3 × 50 mL) and brine (50 mL). The organics were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by column chromatography (Biotage 10 g column, 0−8% EtOAc in hexanes gradient), yielding 481 mg (87%) of the title compound. Rf = 0.16 in 10% EtOAc/hexanes. 1H NMR (400 MHz, CDCl3) δ 7.42−7.24 (m, 8 H), 7.04−6.99 (m, 2 H), 4.99 (s, 2 H), 2.38 (s, 3 H), 1.40 (s, 18 H). MS (ESI) m/z 575.25 (M + Na).
Phenyl 5-Benzyloxy-6-[(t-butoxycarbonyl)amino]-2-(dimethylamino)-3-methylisonicotinate (17b)
Compound 16e (467 mg, 1.06 mmol), t-butylcarbamate (372 mg, 3.17 mmol), Cs2CO3 (1.04 g, 3.17 mmol), tris(dibenzylideneacetone)dipalladium (48 mg, 0.053 mmol), and Xantphos (89.3 mg, 0.159 mmol) were weighed into a vial. This was evacuated and backflushed with nitrogen (3×), and 1,4-dioxane (3 mL) was added. The reaction mixture was heated to 100 °C. After 4 h, the reaction mixture was cooled to room temperature and was filtered through Celite. The filtrate was concentrated under reduced pressure, and the material was purified by column chromatography (Biotage 20 g column, 0−16% EtOAc in hexanes gradient), yielding 493 mg (98%) of the product containing ∼20% of an unidentified impurity. Rf = 0.45 in 30% EtOAc/hexanes. 1H NMR (400 MHz, CDCl3) δ 7.43−7.32 (m, 8 H), 7.30−7.24 (m, 1 H), 7.15−7.10 (m, 2 H), 6.80 (s, 1 H), 4.94 (s, 2 H), 2.85 (s, 6 H), 2.31 (s, 3 H), 1.48 (s, 9 H). MS (ESI) m/z 478.27 (M + H).
Phenyl 3-Benzyloxy-2-[bis(t-butoxycarbonyl)amino]-6-(dimethylamino)-5-methylisonicotinate (18b)
Compound 17b (490 mg, 1.03 mmol) was treated with di-t-butyldicarbonate (672 mg, 3.08 mmol) and DMAP (12.5 mg, 0.103 mmol) in DMF (5 mL). After stirring overnight, the reaction mixture was diluted with EtOAc (25 mL) and was washed with water (3 × 20 mL) and brine (20 mL). The organics were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by column chromatography (Biotage 10 g column, 0−10% EtOAc in hexanes gradient), yielding 424 mg (72%) of the product. Rf = 0.22 in 15% EtOAc/hexanes. 1H NMR (400 MHz, CDCl3) δ 7.42−7.24 (m, 8 H), 7.06−7.00 (m, 2 H), 4.92 (s, 2 H), 2.79 (s, 6 H), 2.36 (s, 3 H), 1.39 (s, 18 H). MS (ESI) m/z 578.33 (M + H).
(4aS,11aR,12aS,13S)-13-(Dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-11a,12,12a,13-tetrahydro-5-hydroxy-3,7-bis(phenylmethoxy)-isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinoline-4,6(4aH,11H)-dione (19a)
LDA was prepared at −78 °C from n-butyllithium (1.6 M solution in hexane, 0.495 mL, 0.792 mmol) and diisopropylamine (0.112 mL, 0.792 mmol) in THF (5 mL). TMEDA (0.318 mL, 2.11 mmol) was added, followed by dropwise addition of a solution of compound 14 (169 mg, 0.529 mmol) in THF (2 mL). This resulted in a deep-red colored solution. After 5 min, a solution of 8 (128 mg, 0.264 mmol) in THF (2 mL) was added. After complete addition, the reaction mixture was allowed to warm to −20 °C over 1 h. The reaction was quenched by the addition of ammonium chloride (saturated, aqueous solution, 20 mL) and was extracted with EtOAc (2 × 20 mL). The combined extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by preparative HPLC (Sunfire Prep C18 column, 80−100% B gradient), yielding 65.5 mg (35%) of the title compound as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 15.77 (s, 1 H), 8.36 (s, 1 H), 8.16 (s, 1 H), 7.55−7.24 (m, 10 H), 5.40−5.25 (m, 4 H), 3.93 (d, J = 11.0 Hz, 1 H), 3.16−3.04 (m, 1 H), 2.98−2.90 (m, 1 H), 2.76−2.64 (m, 1 H), 2.60−2.40 (m, 8 H), 2.12 (d, J = 14 Hz, 1 H), 0.82 (s, 9 H), 0.26 (s, 3 H), 0.13 (s, 3 H). MS (ESI) m/z 708.72 (M + H).
The following compounds were synthesized by methods similar to 19a.
(4aS,11aR,12aS,13S)-10-Chloro-13-(dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-11a,12,12a,13-tetrahydro-5-hydroxy-3,7-bis(phenylmethoxy)-isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinoline-4,6(4aH,11H)-dione (19b)
Prepared from 15a in 38% yield, yellow solid. 1H NMR (400 MHz, CDCl3) δ 15.6 (s, 1 H), 8.12 (s, 1 H), 7.56−7.49 (m, 4 H), 7.42−7.32 (m, 6 H), 5.37 (s, 2 H), 5.07 (s, 2 H), 3.91 (d, J = 11.0 Hz, 1 H), 3.13−3.02 (m, 1 H), 2.97 (dd, J = 15.6 4.9 Hz, 1 H), 2.65−2.45 (m, 3 H), 2.50 (s, 6 H), 2.14 (d, J = 15.6 Hz, 1 H), 0.82 (s, 9 H), 0.27 (s, 3 H), 0.13 (s, 3 H). MS (ESI) m/z 742.56 (M + H).
(4aS,11aR,12aS,13S)-10-Bromo-13-(dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-11a,12,12a,13-tetrahydro-5-hydroxy-3,7-bis(phenylmethoxy)-isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinoline-4,6(4aH,11H)-dione (19c)
Prepared from 15b in 71% yield, yellow solid. 1H NMR (400 MHz, CDCl3) δ 15.55 (s, 1 H), 8.12 (s, 1 H), 7.55−7.24 (m, 10 H), 5.40−5.22 (m, 4 H), 3.90 (d, J = 11.0 Hz, 1 H), 3.25 (dd, J = 16.5 Hz, J = 4.88 Hz, 1 H), 3.12−3.02 (m, 1 H), 2.62−2.42 (m, 9 H), 2.14 (d, J = 14.0 Hz, 1 H), 0.82 (s, 9 H), 0.26 (s, 3 H), 0.13 (s, 3 H). MS (ESI) m/z 786.63, 788.63 (M + H).
(4aS,11aR,12aS,13S)-10,13-Bis(dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-11a,12,12a,13-tetrahydro-5-hydroxy-3,7-bis(phenylmethoxy)-isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinoline-4,6(4aH,11H)-dione (19f)
Prepared from 15c in 50% yield, yellow solid. 1H NMR (400 MHz, CDCl3) δ 15.65 (s, 1 H), 8.01 (s, 1 H), 7.52−7.22 (m, 10 H), 5.36 (s, 2 H), 5.17 (q, J = 11.0 Hz, 2 H), 4.02 (d, J = 11.0 Hz, 1 H), 3.05 (dd, J = 17.2 Hz, J = 11.6 Hz, 1 H), 2.98−2.86 (m, 1 H), 2.73 (s, 6 H), 2.62−2.38 (m, 9 H), 2.12 (d, J = 13.4 Hz, 1 H), 0.81 (s, 9 H), 0.26 (s, 3 H), 0.13 (s, 3 H). MS (ESI) m/z 751.79 (M + H).
(4aS,11aR,12aS,13S)-13-(Dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-11a,12,12a,13-tetrahydro-5-hydroxy-10-methoxy-3,7-bis(phenylmethoxy)-isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinoline-4,6(4aH,11H)-dione (19g)
Prepared from 15d in 27% yield, yellow solid. 1H NMR (400 MHz, CDCl3) δ 15.70 (s, 1 H), 7.83 (s, 1 H), 7.55−7.24 (m, 10 H), 5.36 (s, 2 H), 5.17 (q, J = 13.4 Hz, 2 H), 3.98 (d, J = 11.0 Hz, 1 H), 3.72 (s, 3 H), 3.18 (d, J = 16.5 Hz, 1 H), 3.04 −2.96 (m, 1 H), 2.60−2.30 (m, 9 H), 2.15 (d, J = 14 Hz, 1 H), 0.82 (s, 9 H), 0.26 (s, 3 H), 0.13 (s, 3 H). MS (ESI) m/z 738.66 (M + H).
2-[(4aS,11aR,12aS,13S)-13-(Dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-10-fluoro-4,4a,6,11,11a,12,12a,13-octahydro-5-hydroxy-4,6-dioxo-3,7-bis(phenylmethoxy)isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinolin-8-yl]-imidodicarbonic Acid, 1,3-Bis(1,1-dimethylethyl) Ester (19k)
Prepared from 18a in 56% yield, yellow solid. 1H NMR (400 MHz, CDCl3) δ 15.59 (s, 1 H), 7.56−7.24 (m, 10 H), 5.36 (s, 2 H), 4.92 (q, J = 67.8 Hz, J = 9.16 Hz, 2 H), 3.91 (d, J = 11.0 Hz, 1 H), 3.20−3.02 (m, 2 H), 2.62−2.45 (m, 9 H), 2.19−2.12 (m, 1 H), 1.37, (s, 18 H), 0.82 (s, 9 H), 0.26 (s, 3 H), 0.13 (s, 3 H). MS (ESI) m/z 941.59 (M + H).
(4aS,11aR,12aS,13S)-8-Bromo-10,13-bis(dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-11a,12,12a,13-tetrahydro-5-hydroxy-3,7-bis(phenylmethoxy)-isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinoline-4,6(4aH,11H)-dione (19m)
Prepared from 16e in 54% yield, yellow solid. 1H NMR (400 MHz, CDCl3) δ 15.65 (s, 1 H), 7.52−7.22 (m, 10 H), 5.36 (s, 2 H), 5.17 (q, J = 11.0 Hz, 2 H), 4.01 (d, J = 11.0 Hz, 1 H), 3.05 (dd, J = 17.2 Hz, J = 11.6 Hz, 1 H), 2.98−2.86 (m, 1 H), 2.73 (s, 6 H), 2.62−2.38 (m, 9 H), 2.12 (d, J = 13.4 Hz, 1 H), 0.82 (s, 9 H), 0.26 (s, 3 H), 0.13 (s, 3 H). MS (ESI) m/z 829.49, 831.48 (M + H).
(4aS,11aR,12aS,13S)-8-Bromo-10-chloro-13-(dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-11a,12,12a,13-tetrahydro-5-hydroxy-3,7-bis(phenylmethoxy)-isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinoline-4,6(4aH,11H)-dione (19h)
Compound 16a (793 mg, 1.84 mmol) and compound 8 (885 mg, 1.84 mmol) were dissolved in THF (16 mL), and the solution was cooled to −78 °C. LHMDS (1.0 M solution in THF, 5.5 mL, 5.5 mmol) was added slowly via syringe. After 10 min, the reaction mixture was allowed to slowly warm to 0 °C over 1 h. A phosphate buffer solution (pH 7.0, 20 mL) was added, followed by the addition of ammonium chloride (saturated, aqueous solution, 50 mL). The resulting mixture was extracted with CH2Cl2 (3 × 50 mL), and the combined extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting brown solid was washed with cold methanol (3 × 5 mL) to afford 1.11 g (74%) of the title compound as a yellow−brown powder. The organics were concentrated under reduced pressure and purified by column chromatography (Biotage 20 g column, 5−20% EtOAc in hexanes gradient), yielding an additional 150 mg (10%) of the product. 1H NMR (400 MHz, CDCl3) δ 15.45 (br, 1 H), 7.54−7.48 (m, 4 H), 7.40−7.30 (m, 6 H), 5.36 (s, 2 H), 5.03 (abq, J = 10.4 Hz, 2 H), 3.87 (d, J = 11.0 Hz, 1 H), 3.27−3.23 (m, 1 H), 3.10−3.00 (m, 1 H), 2.65−2.57 (m, 1 H), 2.57−2.43 (m, 8 H), 2.16 (d, J = 11.0 Hz, 1 H), 0.81 (s, 9 H), 0.26 (s, 3 H), 0.12 (s, 3 H). MS (ESI) m/z 820.37, 822.37 (M + H).
The following compounds were prepared by methods similar to 19h.
(4aS,11aR,12aS,13S)-8-Bromo-13-(dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-10-fluoro-11a,12,12a,13-tetrahydro-5-hydroxy-3,7-bis(phenylmethoxy)-isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinoline-4,6(4aH,11H)-dione (19i)
Prepared from 16d in 45% yield, yellow solid. 1H NMR (400 MHz, CDCl3) δ 15.55 (s, 1 H), 7.56−7.26 (m, 10 H), 5.36 (s, 2 H), 5.02 (s, 2 H), 3.88 (d, J = 10.4 Hz, 1 H), 3.18−3.04 (m, 2 H), 2.62−2.58 (m, 1 H), 2.58−2.40 (m, 8 H), 2.17 (d, J = 14.6 Hz, 1 H), 0.81 (s, 9 H), 0.25 (s, 3 H), 0.12 (s, 3 H). MS (ESI) m/z 804.34, 806.34 (M + H).
2-[(4aS,11aR,12aS,13S)-10,13-Bis(dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4,4a,6,11,11a,12,12a,13-octahydro-5-hydroxy-4,6-dioxo-3,7-bis(phenylmethoxy)isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinolin-8-yl]-imidodicarbonic Acid, 1,3-Bis(1,1-dimethylethyl) Ester (19l)
Prepared from 18b in 53% yield, yellow solid. 1H NMR (400 MHz, CDCl3) δ 15.50 (s, 1 H), 7.52−7.24 (m, 10 H), 5.36 (s, 2 H), 4.85 (q, J = 86.1 Hz, J = 9.76 Hz, 2 H), 4.02 (d, J = 10.4 Hz, 1 H), 3.08−3.00 (m, 1 H), 3.00−2.82 (m, 1 H), 2.77 (s, 6 H), 2.62−2.38 (m, 9 H), 2.20−2.12 (m, 1 H), 1.35, (br s, 18 H), 0.79 (s, 9 H), 0.26 (s, 3 H), 0.12 (s, 3 H). MS (ESI) m/z 966.59 (M + H).
(4aS,11aR,12aS,13S)-13-(Dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-11a,12,12a,13-tetrahydro-5-hydroxy-10-methyl-3,7-bis(phenylmethoxy)-isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinoline-4,6(4aH,11H)-dione (19d)
A solution of compound 19c (52 mg, 0.067 mmol), methylboronic acid (40 mg, 0.67 mmol), dichloro[1,1′-bis(diphenylphosphino)ferrocene]palladium(II) dichloromethane adduct (3 mg, 0.003 mmol), and K3PO4 (142 mg, 0.667 mmol) in toluene (1 mL), 1,4-dioxane (1 mL), and water (0.2 mL) was heated to 70 °C. After 2 h, the reaction mixture was heated to 100 °C. After an additional 2 h, the reaction mixture was diluted with EtOAc (20 mL) and was washed with water (15 mL) and brine (15 mL). The organics were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by preparative HPLC (Sunfire Prep C18 column, 80−100% B gradient), yielding 18.3 mg (38%) of the title compound as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 15.71 (s, 1 H), 8.24 (s, 1 H), 7.55−7.24 (m, 10 H), 5.36 (s, 2 H), 5.30−5.20 (m, 2 H), 3.95 (d, J = 10.4 Hz, 1 H), 3.10−2.92 (m, 2 H), 2.62−2.42 (m, 12 H), 2.12 (d, J = 14.0 Hz, 1 H), 0.82 (s, 9 H), 0.26 (s, 3 H), 0.14 (s, 3 H). MS (ESI) m/z 722.72 (M + H).
(4aS,11aR,12aS,13S)-13-(Dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-11a,12,12a,13-tetrahydro-5-hydroxy-10-phenyl-3,7-bis(phenylmethoxy)-isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinoline-4,6(4aH,11H)-dione (19e)
A solution of compound 19c (31 mg, 0.040 mmol), phenylboronic acid (24.5 mg, 0.201 mmol), dichloro[1,1′-bis(diphenylphosphino)ferrocene]palladium(II) dichloromethane adduct (1.6 mg, 0.002 mmol), and sodium carbonate (21.3 mg, 0.201 mmol) in toluene (1 mL), 1,4-dioxane (1 mL), and water (0.2 mL) was heated to 100 °C via microwave reactor for 10 min. The reaction mixture was diluted with EtOAc (10 mL) and was washed with water (5 mL) and brine (5 mL). The organics were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by preparative HPLC (Sunfire Prep C18 column, 80−100% B gradient), yielding 16.9 mg (54%) of the title compound as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 15.66 (s, 1 H), 8.48 (s, 1 H), 7.52−7.24 (m, 15 H), 5.42−5.30 (m, 4 H), 3.97 (d, J = 10.4 Hz, 1 H), 3.00−2.86 (m, 2 H), 2.78−2.62 (m, 1 H), 2.58−2.30 (m, 8 H), 2.00 (d, J = 14.0 Hz, 1 H), 0.80 (s, 9 H), 0.25 (s, 3 H), 0.14 (s, 3 H). MS (ESI) m/z 784.75 (M + H).
N-[(4aS,11aR,12aS,13S)-10-Chloro-13-(dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4,4a,6,11,11a,12,12a,13-octahydro-5-hydroxy-4,6-dioxo-3,7-bis(phenylmethoxy)isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinolin-8-yl]-carbamic Acid, 1,1-Dimethylethyl Ester (19j)
Compound 19h (100 mg, 0.122 mmol), t-butylcarbamate (42.9 mg, 0.366 mmol), K3PO4 (77.7 mg, 0.366 mmol), tris(dibenzylideneacetone)dipalladium (5.6 mg, 0.006 mmol), and Xantphos (10.3 mg, 0.018 mmol) were weighed into a vial equipped with a septum. The vial was evacuated and flushed with nitrogen (3×), and 1,4-dioxane (0.5 mL) was added. The reaction mixture was heated to 100 °C and stirred for 2 h. The reaction mixture was allowed to cool to room temperature, was filtered through Celite, and was concentrated under reduced pressure. The material was purified by preparative HPLC (Sunfire Prep C18 column, 80−100% B gradient), yielding 41.7 mg (40%) of the title compound as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 15.65−15.45 (br s, 1 H), 7.56−7.50 (m, 2 H), 7.43−7.34 (m, 7 H), 7.29−7.20 (m, 1 H), 5.38 (s, 2 H), 4.94 (dd, J = 29.3 Hz, J = 11.0 Hz, 2 H), 3.90 (d, J = 11.0 Hz, 1 H), 3.27 (dd, J = 16.4 Hz, J = 11.6 Hz, 1 H), 3.12−3.03 (m, 1 H), 2.68−2.60 (m, 1 H), 2.60−2.45 (m, 8 H), 2.18 (d, J = 14.6 Hz, 1 H), 1.47 (s, 9 H), 0.85 (s, 9 H), 0.29 (s, 3 H), 0.15 (s, 3 H). MS (ESI) m/z 857.67 (M + H).
6-Demethyl-6-deoxy-8-azatetracycline Dihydrochloride (20a)
Aqueous HF (48%, 0.4 mL) was added to a solution of 19a (65.5 mg, 0.093 mmol) in CH3CN (0.6 mL) in a plastic vial. After 18 h, the reaction mixture was poured into a solution of K2HPO4 (4.8 g) in water (20 mL). The mixture was extracted with EtOAc (3 × 20 mL). The combined extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was dissolved in MeOH (1 mL) and 1,4-dioxane (1 mL), and palladium on carbon (Degussa, 10 wt %, ∼5 mg) was added. An atmosphere of hydrogen was introduced, and the reaction mixture was stirred for 2 h. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. The material was purified by preparative HPLC (Phenomenex Polymerx column, 0−100% B gradient). Fractions with the desired MW were collected and freeze-dried to yield 18.4 mg (41%, 2 steps) of the title compound as a yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 8.55 (s, 1 H), 8.36 (s, 1 H), 4.19 (s, 1 H), 3.26−2.98 (m, 9 H), 2.71 (t, J = 14.2 Hz, 1 H), 2.39−2.32 (m, 1 H), 1.75−1.63 (m, 1 H). MS (ESI) m/z 416.44 (M + H).
The following compounds were synthesized by similar methods to 20a.
7-Chloro-6-demethyl-6-deoxy-8-azatetracycline Hydrochloride (20b)
Prepared from 19b in 24% yield, 2 steps, yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.08 (d, J = 6.0 Hz, 1 H), 4.09 (s, 1 H), 3.08−2.92 (m, 9 H), 2.30−2.15 (m, 2 H), 1.70 −1.58 (m, 1 H). MS (ESI) m/z 450.18 (M + H).
6-Demethyl-6-deoxy-7-methyl-8-azatetracycline Dihydrochloride (20d)
Prepared from 19d in 34% yield, 2 steps, yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 8.35 (s, 1 H), 4.19 (s, 1 H), 3.25−2.95 (m, 9 H), 2.80−2.25 (m, 5 H), 1.80−1.63 (m, 1 H). MS (ESI) m/z 430.46 (M + H).
6-Demethyl-6-deoxy-7-phenyl-8-azatetracycline Dihydrochloride (20e)
Prepared from 19e in 24% yield, 2 steps, yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 8.53 (s, 1 H), 7.62 (s, 5 H), 4.18 (s, 1 H), 3.14−2.82 (m, 9 H), 2.81−2.66 (m, 1 H), 2.24−2.14 (m, 1 H), 1.66−1.52 (m, 1 H). MS (ESI) m/z 492.48 (M + H).
6-Demethyl-6-deoxy-7-(dimethylamino)-8-azatetracycline Dihydrochloride (20f)
Prepared from 19f in 96% yield, 2 steps, yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 8.22 (s, 1 H), 4.19 (s, 1 H), 3.38−3.30 (m, 2 H), 3.24 (s, 6 H), 3.12−2.95 (m, 7 H), 2.60 (t, J = 14.2 Hz, 1 H), 2.42−2.34 (m, 1 H), 1.75−1.63 (m, 1 H). MS (ESI) m/z 459.50 (M + H).
6-Demethyl-6-deoxy-7-methoxy-8-azatetracycline Dihydrochloride (20g)
Prepared from 19g in 51% yield, 2 steps, yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 7.80 (s, 1 H), 4.15 (s, 1 H), 3.97 (s, 3 H), 3.40−2.98 (m, 9 H), 2.32−2.20 (m, 2 H), 1.72−1.56 (m, 1 H). MS (ESI) m/z 446.39 (M + H).
6-Demethyl-6-deoxy-7-fluoro-8-azatetracycline Hydrochloride (20i)
Prepared from 19i in 68% yield, 2 steps, yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 7.81 (s, 1 H), 4.17 (s, 1 H), 3.26−2.96 (m, 9 H), 2.42−2.25 (m, 2 H), 1.70−1.58 (m, 1 H). MS (ESI) m/z 434.27 (M + H).
9-Amino-7-chloro-6-demethyl-6-deoxy-8-azatetracycline (20j)
Prepared from 19j in 68% yield, 2 steps, yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 4.19 (s, 1 H), 3.20−2.98 (m, 8 H), 2.35−2.22 (m, 2 H), 1.68−1.56 (m, 1 H). MS (ESI) m/z 465.32 (M + H).
9-Amino-6-demethyl-6-deoxy-7-fluoro-8-azatetracycline Dihydrochloride (20k)
Prepared from 19k in 49% yield, 2 steps, yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 4.20 (s, 1 H), 3.40−2.90 (m, 9 H), 2.42−2.22 (m, 2 H), 1.70−1.54 (m, 1 H). MS (ESI) m/z 449.27 (M + H).
N-[(4aS,11aR,12aS,13S)-10-Chloro-13-(dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4,4a,6,11,11a,12,12a,13-octahydro-5-hydroxy-4,6-dioxo-3,7-bis(phenylmethoxy)isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinolin-8-yl]-2-[(1,1-dimethylethyl)amino]-acetamide
Compound 19j (68.3 mg, 0.080 mmol) was stirred in 4 M HCl in 1,4-dioxane for 2 h. The reaction mixture was concentrated under reduced pressure. The material was dissolved in THF (2 mL), and bromoacetylchloride (12.5 mg, 0.080 mmol) was added. After stirring overnight, an additional portion of bromoacetylchloride (0.020 mL) was added. After 4 h, the reaction mixture was concentrated under reduced pressure. MS (ESI) m/z 879.42 (M + H). t-Butylamine (0.063 mL, 0.60 mmol) was added to a solution of the intermediate (35 mg, 0.040 mmol) in THF (1 mL), and the reaction mixture was heated to 50 °C. After stirring overnight, the reaction mixture was concentrated under reduced pressure and was purified by preparative HPLC (Sunfire Prep C18 column, 50−100% B gradient). This gave 16.8 mg (49%) of the title compound as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.20 (br s, 1 H), 7.52−7.46 (m, 2 H), 7.43−7.30 (m, 8 H), 5.36 (s, 2 H), 4.95 (dd, J = 40.6 Hz, J = 11.0 Hz, 2 H), 3.88 (d, J = 11.0 Hz, 1 H), 3.32−3.24 (m, 1 H), 3.12−3.03 (m, 1 H), 2.66−2.59 (m, 1 H), 2.58−2.42 (m, 8 H), 2.18 (d, J = 14.6 Hz, 1 H), 1.30−1.10 (br s, 9 H), 0.82 (s, 9 H), 0.27 (s, 3 H), 0.12 (s, 3 H). MS (ESI) m/z 870.60 (M + H).
9-[2-(tert-Butylamino)acetamido]-7-chloro-6-demethyl-6-deoxy-8-azatetracycline Dihydrochloride (21a)
Aqueous HF (48%, 0.4 mL) was added to a solution of the preceding intermediate (16.8 mg, 0.019 mmol) in CH3CN (0.6 mL) in a plastic vial. After 18 h, the reaction mixture was poured into a solution of K2HPO4 (7.8 g) in water (15 mL). The mixture was extracted with EtOAc (3×). Sodium chloride (10 g) was added to the aqueous layer, and this was extracted with EtOAc (2×). The combined extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was dissolved in MeOH (1 mL) and 1,4-dioxane (1 mL), and palladium on carbon (Degussa, 10 wt %, ∼5 mg) was added. An atmosphere of hydrogen was introduced, and the reaction mixture was stirred for 1 h. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. The material was purified by preparative HPLC (Phenomenex Polymerx 10 μ RP 100A column, 0−100% B gradient). Fractions with the desired MW were collected and freeze-dried to yield 1.2 mg (10%, 2 steps) of the title compound as a yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 4.30−4.20 (s, 2 H), 4.19 (s, 1 H), 3.25−2.96 (m, 9 H), 2.48−2.28 (m, 2 H), 1.72−1.58 (m, 1 H), 1.43 (s, 9 H). MS (ESI) m/z 578.48 (M + H).
The following compounds were prepared according to methods similar to 21a:
9-[2-(tert-Butylamino)acetamido]-6-demethyl-6-deoxy-7-fluoro-8-azatetracycline Dihydrochloride (21b)
Prepared from 19k in 68% yield for the acylation/amine displacement and 58% yield for the deprotections, yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 4.24−4.65 (m, 3 H), 3.26−2.97 (m, 9 H), 2.38−2.28 (m, 2 H), 1.69−1.56 (m, 1 H), 1.44 (s, 9 H). MS (ESI) m/z 562.37 (M + H).
9-[2-(tert-Butylamino)acetamido]-6-demethyl-6-deoxy-7-(dimethylamino)-8-azatetracycline Trihydrochloride (21c)
Prepared from 19l in 43% yield for the acylation/amine displacement and 55% yield for the deprotections, yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 4.30−4.40 (m, 3 H), 3.40−3.25 (m, 8 H), 3.12−3.04 (m, 4 H), 2.99 (s, 3 H), 2.62−2.52 (m, 1 H), 2.41−2.35 (m, 1 H), 1.73−1.60 (m, 1 H), 1.45 (s, 9 H). MS (ESI) m/z 587.43 (M + H).
(4aS,11aR,12aS,13S)-10-Chloro-13-(dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-8-(ethylamino)-11a,12,12a,13-tetrahydro-5-hydroxy-3,7-bis(phenylmethoxy)-isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinoline-4,6(4aH,11H)-dione
Compound 19h (50 mg, 0.061 mmol), K3PO4 (39 mg, 0.18 mmol), tris(dibenzylideneacetone)dipalladium (2.8 mg, 0.003 mmol), and Xantphos (5.1 mg, 0.009 mmol) were added to a small vial equipped with a septum. After the flask was evacuated and flushed with nitrogen (3×), 1,4-dioxane (0.5 mL) and ethylamine (2.0 M solution in THF, 0.091 mL, 0.18 mmol) were added. The reaction mixture was heated to 100 °C and stirred for 3 h. The reaction mixture was allowed to cool to room temperature and was filtered through Celite. The material was purified by preparative HPLC (Sunfire Prep C18 column, 90−100% B gradient). Fractions with the desired MW were collected and freeze-dried to yield 11 mg (23%) of the title compound as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 15.6 (br, 1 H), 7.56−7.49 (m, 4 H), 7.42−7.30 (m, 6 H), 5.37 (s, 2 H), 5.03 (s, 2 H), 3.89 (d, J = 11.0 Hz, 1 H), 3.24−3.04 (m, 3 H), 2.97 (dd, J = 15.6, 4.9 Hz, 1 H), 2.64−2.43 (m, 3 H), 2.49 (s, 6 H), 2.14 (d, J = 15.6 Hz, 1 H), 1.25 (t, J = 6.8 Hz, 3 H), 0.81 (s, 9 H), 0.25 (s, 3 H), 0.11 (s, 3 H). MS (ESI) m/z 785.57 (M + H).
7-Chloro-6-demethyl-6-deoxy-9-(ethylamino)-8-azatetracycline Dihydrochloride (24a)
Aqueous HF (48%, 0.3 mL) was added to a solution of the preceding intermediate (11 mg, 0.014 mmol) in CH3CN (7 mL) in a plastic vial. After 18 h, the reaction mixture was poured into a solution of K2HPO4 (2 g) in water (10 mL). The mixture was extracted with EtOAc (3×). The combined EtOAc extracts were dried over Na2SO4 and concentrated under reduced pressure. The material was dissolved in MeOH (2 mL) and 1,4-dioxane (2 mL), and palladium on carbon (Degussa, 10 wt %, 5.6 mg) was added. An atmosphere of hydrogen was introduced, and the reaction mixture was stirred for 1 h. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. The material was purified by preparative HPLC (Phenomenex Polymerx 10 μ RP 100A column, 0−100% B gradient). Fractions with the desired MW were collected and freeze-dried to yield 3.2 mg (46%, 2 steps) of the title compound as a yellow solid. 1H NMR (400 MHz, CD3OD) δ 4.08 (s, 1 H), 3.43 (q, J = 7.4 Hz, 2 H), 3.08−2.92 (m, 9 H), 2.30−2.15 (m, 2 H), 1.67−1.55 (m, 1 H), 1.22 (t, J = 7.4 Hz, 3H). MS (ESI) m/z 493.24 (M + H).
The following compounds were prepared by similar methods to 24a:
7-Chloro-6-demethyl-6-deoxy-9-(methylamino)-8-azatetracycline Dihydrochloride (24b)
Prepared from 19h and methylamine (1.0 M solution in THF) in 21% yield for the amination step, 19% for the 2 deprotection steps, yellow solid. 1H NMR (400 MHz, CD3OD) δ 4.08 (s, 1 H), 3.41 (s, 3 H), 3.08−2.92 (m, 9 H), 2.30−2.15 (m, 2 H), 1.67−1.55 (m, 1 H). MS (ESI) m/z 479.22 (M + H).
7-Chloro-6-demethyl-6-deoxy-9-(propylamino)-8-azatetracycline Dihydrochloride (24c)
Prepared from 19h and n-propylamine in 24% yield for the amination step, 40% for the 2 deprotection steps, yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 4.16 (s, 1H), 3.47 (t, J = 7.3 Hz, 2 H), 3.15−2.96 (m, 9 H), 2.32−2.20 (m, 2 H), 1.78−1.55 (m, 3 H), 1.01 (t, J = 7.4 Hz, 3 H). MS (ESI) m/z 507.29 (M + H).
7-Chloro-6-demethyl-6-deoxy-9-(propan-2-ylamino)-8-azatetracycline Dihydrochloride (24d)
Prepared from 19h and 2-propylamine in 11% yield for the amination step, 16% for the 2 deprotection steps, yellow solid. 1H NMR (400 MHz, CD3OD) δ 4.21−4.13 (m, 1 H), 4.08 (s, 1 H), 3.14−2.92 (m, 9 H), 2.31−2.15 (m, 2 H), 1.67−1.56 (m, 1 H), 1.23 (dd, J = 6.4, 1.8 Hz, 6 H). MS (ESI) m/z 507.24 (M + H).
7-Chloro-6-demethyl-6-deoxy-9-[(2-methylpropyl)amino]-8-azatetracycline Dihydrochloride (24e)
Prepared from 19h and 2-methylpropan-1-amine in 15% yield for the amination step, 57% for the 2 deprotection steps, yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 4.18 (s, 1H), 3.38 (d, J = 7.3 Hz, 2 H), 3.20−2.96 (m, 9 H), 2.34−2.20 (m, 2 H), 2.10−1.98 (m, 1H), 1.68−1.54 (m, 1 H), 1.00 (d, J = 6.4 Hz, 6 H). MS (ESI) m/z 521.40 (M + H).
7-Chloro-6-demethyl-6-deoxy-9-[(3-methylbutyl)amino]-8-azatetracycline Dihydrochloride (24f)
Prepared from 19h and 3-methyl-butan-1-amine in 25% yield for the amination step, 66% for the 2 deprotection steps, yellow solid. 1H NMR (400 MHz, CD3OD) δ 4.09 (s, 1 H), 3.45−3.40 (m, 2 H), 3.20 (s, 1 H), 3.08−2.93 (m, 9 H), 2.30−2.16 (m, 2 H), 1.72−1.58 (m, 2 H), 1.55−1.48 (m, 2 H), 0.96 (d, J = 6.4 Hz, 6 H). MS (ESI) m/z 535.30 (M + H).
7-Chloro-6-demethyl-6-deoxy-9-[(2-methoxyethyl)amino]-8-azatetracycline Dihydrochloride (24g)
Prepared from 19h and 2-methoxyethylamine in 19% yield for the amination step, 44% for the 2 deprotection steps, yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 4.13 (s, 1H), 3.72−3.60 (m, 4H), 3.40 (s, 3 H), 3.12−2.95 (m, 9 H), 2.32−2.21 (m, 2 H), 1.69−1.56 (m, 1 H). MS (ESI) m/z 523.32 (M + H).
7-Chloro-6-demethyl-6-deoxy-9-[[2-(propan-2-yloxy)ethyl]amino]-8-azatetracycline Dihydrochloride (24h)
Prepared from 19h and 2-(propan-2-yloxy)ethan-1-amine in 9% yield for the amination step, 30% for the 2 deprotection steps, yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 4.19 (s, 1H), 3.82−3.69 (m, 5H), 3.20−2.98 (m, 9 H), 2.35−2.22 (m, 2 H), 1.68−1.56 (m, 1 H), 1.18 (d, J = 6.4 Hz, 6H). MS (ESI) m/z 551.41 (M + H).
7-Chloro-6-demethyl-6-deoxy-9-[[3-(dimethylamino)propyl]amino]-8-azatetracycline Trihydrochloride (24i)
Prepared from 19h and (3-aminopropyl)dimethylamine in 9% yield for the amination step, 21% for the 2 deprotection steps, yellow solid. 1H NMR (400 MHz, CD3OD) δ 4.09 (s, 1 H), 3.70−3.46 (m, 4 H), 3.10−2.90 (m, 15 H), 2.31−2.17 (m, 2 H), 2.10−2.00 (m, 2 H), 1.68−1.56 (m, 1 H). MS (ESI) m/z 550.34 (M + H).
Phenyl 3-(Benzyloxy)-2-[[(benzyloxy)carbonyl][3-(dimethylamino)-2,2-dimethylpropyl]amino]-6-fluoro-5-methylpyridine-4-carboxylate (22a)
Compound 16d (100 mg, 0.24 mmol), Cs2CO3 (235 mg, 0.720 mmol), tris(dibenzylideneacetone)dipalladium (11 mg, 0.012 mmol), and Xantphos (20 mg, 0.036 mmol) were weighed into a flask. This was evacuated and backflushed with nitrogen (3×), and 1,4-dioxane (0.5 mL) and N,N-dimethylneopentanediamine (0.057 mL, 0.36 mmol) were added. The reaction mixture was heated to 70 °C. After 1.5 h, the reaction mixture was cooled to room temperature and was filtered through Celite. The filtrate was concentrated under reduced pressure and was purified by column chromatography (Biotage 10 g column, 0−3% MeOH in CH2Cl2 gradient), yielding 76.9 mg (69%) of the intermediate. Rf = 0.25 in 5% MeOH/CH2Cl2. MS (ESI) m/z 466.28 (M + H). LHMDS (1.0 M in THF, 0.22 mL, 0.22 mmol) was added dropwise to a −78 °C solution of the above intermediate (93 mg, 0.20 mmol) in THF (5 mL). After 5 min, benzyl chloroformate (0.084 mL, 0.60 mmol) was added. After 15 min, the reaction mixture was quenched by the addition of NH4Cl (saturated, aqueous solution, 15 mL) and was extracted with EtOAc. The organics were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by column chromatography (Biotage 10 g column, 0−7% MeOH in CH2Cl2 gradient), yielding 111 mg (93%) of the product. Rf = 0.42 in 10% MeOH/CH2Cl2. 1H NMR (400 MHz, CDCl3) δ 7.45−7.20 (m, 13 H), 7.06−6.98 (m, 2 H), 5.16 (s, 2 H), 4.87 (s, 2 H), 3.78 (br s, 2 H), 2.38 (s, 3 H), 2.18 (br s, 6 H), 1.62 (br s, 2 H), 0.90 (br s, 6 H). MS (ESI) m/z 600.28 (M + H).
N-[(4aS,11aR,12aS,13S)-13-(Dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-10-fluoro-4,4a,6,11,11a,12,12a,13-octahydro-5-hydroxy-4,6-dioxo-3,7-bis(phenylmethoxy)isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinolin-8-yl]-N-[3-(dimethylamino)-2,2-dimethylpropyl]-carbamic Acid, Phenylmethyl Ester (23a)
A solution of compound 22a (54 mg, 0.090 mmol) in THF (0.5 mL) was added dropwise to a −78 °C solution of LDA (2.0 M solution in THF, 0.112 mL, 0.224 mmol) and TMEDA (0.081 mL, 0.54 mmol) in THF (2 mL), resulting in an orange-colored solution. After 10 min, a solution of 8 (43 mg, 0.090 mmol) in THF (0.5 mL) was added dropwise over ∼30 s. After complete addition, the reaction mixture was allowed to warm to −10 °C over 1 h. The reaction was quenched by the addition of ammonium chloride (saturated, aqueous solution), was diluted with water, and was extracted with EtOAc (2×). The combined extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by preparative HPLC (Sunfire Prep C18 column, 20−100% B gradient), yielding 22.9 mg (26%) of the desired product as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 15.55 (br s, 1 H), 7.52−7.20 (m, 15 H), 5.36 (s, 2 H), 5.20−5.00 (m, 2 H), 4.80 (s, 2 H), 3.90 (d, J = 11.0 Hz, 1 H), 3.19−3.05 (m, 2 H), 2.62−2.57 (m, 1 H), 2.56−2.12 (m, 19 H), 0.94−0.74 (m, 15 H), 0.26 (s, 3 H), 0.12 (s, 3 H). MS (ESI) m/z 988.59 (M + H).
6-Demethyl-6-deoxy-9-[[3-(dimethylamino)-2,2-dimethylpropyl]amino]-7-fluoro-8-azatetracycline Trihydrochloride (24j)
Aqueous HF (48%, 0.4 mL) was added to a solution of 23a (22.9 mg, 0.0232 mmol) in CH3CN (0.8 mL) in a plastic vial. After 18 h, the reaction mixture was poured into a solution of K2HPO4 (4.8 g) in water. The mixture was extracted with EtOAc, and the combined extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was dissolved in MeOH (1 mL), 0.5 M HCl in MeOH (0.2 mL), and 1,4-dioxane (1 mL), and palladium on carbon (Degussa, 10 wt %, ∼2 mg) was added. An atmosphere of hydrogen was introduced, and the reaction mixture was stirred for 3 h. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. The material was purified by preparative HPLC (Phenomenex Polymerx column, 0−100% B gradient). Fractions with the desired MW were collected and freeze-dried to yield 9.9 mg (67%, 2 steps) of the desired product as a yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 4.16 (s, 1 H), 3.43 (s, 2 H), 3.15−2.90 (m, 17 H), 2.30−2.22 (m, 1 H), 2.22−2.12 (m, 1 H), 1.65−1.54 (m, 1 H), 1.17, (s, 6 H). MS (ESI) m/z 562.43 (M + H).
The following compounds were prepared according to methods similar to 24j:
7-Chloro-6-demethyl-6-deoxy-9-[[2-(dimethylamino)ethyl]amino]-8-azatetracycline Trihydrochloride (24k)
Prepared from 16a and N,N-dimethylethylenediamine. Yellow solid. 1H NMR (400 MHz, CD3OD) δ 4.13 (s, 1 H), 3.84−3.77 (m, 2 H), 3.44−3.38 (m, 2 H), 3.10−2.92 (m, 15 H), 2.32−2.20 (m, 2 H), 1.68−1.55 (m, 1 H). MS (ESI) m/z 536.25 (M + H).
7-Chloro-6-demethyl-6-deoxy-9-[[3-(dimethylamino)-2,2-dimethylpropyl]amino]-8-azatetracycline Trihydrochloride (24l)
Prepared from 16a and N,N-dimethylneopentanediamine. Yellow solid. 1H NMR (400 MHz, CD3OD) δ 4.10 (s, 1 H), 3.51−3.36 (m, 4 H), 3.10−2.92 (m, 15 H), 2.33−2.18 (m, 2 H), 1.68−1.57 (m, 1 H), 1.174 (s, 3 H), 1.169 (s, 3 H). MS (ESI) m/z 578.35 (M + H).
7-Chloro-6-demethyl-6-deoxy-9-[[2-methyl-2-(pyrrolidin-1-ylmethyl)propyl]amino]-8-azatetracycline Trihydrochloride (24m)
Prepared from 16a and 2,2-dimethyl-3-(pyrrolidin-1-yl)propan-1-amine. Yellow solid. 1H NMR (400 MHz, CD3OD) δ 4.09 (s, 1 H), 3.88−3.78 (m, 2 H), 3.50−3.39 (m, 2 H), 3.18 −2.92 (m, 9 H), 2.32−2.08 (m, 6 H), 1.67−1.56 (m, 1 H), 1.15 (s, 6 H). MS (ESI) m/z 604.29 (M + H).
6-Demethyl-6-deoxy-7-fluoro-9-(propylamino)-8-azatetracycline Dihydrochloride (24n)
Prepared from 16d and n-propylamine. Yellow solid. 1H NMR (400 MHz, CD3OD) δ 4.09 (s, 1 H), 3.33 (t, J = 6.9 Hz, 2 H) 3.42−3.38 (m, 2 H), 3.20 (s, 1 H), 3.07−2.94 (m, 8 H), 2.91 (dd, J = 14.6, 4.6 Hz, 1 H), 2.23−2.12 (m, 2 H), 1.68−1.55 (m, 3 H), 0.96 (t, J = 7.3 Hz, 3 H). MS (ESI) m/z 491.23 (M + H).
Phenyl 5-Benzyloxy-2-(dimethylamino)-3-methyl-6-(propylamino)isonicotinate
Copper(I) iodide (4.7 mg, 0.025 mmol) and K3PO4 (209 mg, 0.984 mmol) were added to a vial equipped with a septum. After the vial was evacuated and flushed with nitrogen (3×), a solution of compound 16e (217 mg, 0.492 mmol) in DMF (2 mL), 1-propylamine (0.162 mL, 1.97 mmol), and 2-acetylcyclohexanone (0.013 mL, 0.098 mmol) were added. The reaction mixture was heated to 100 °C for 3 h. The reaction mixture was allowed to cool to room temperature, was diluted with EtOAc (50 mL), and was washed with water (3 × 40 mL). The organics were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by column chromatography (Biotage 10 g column, 0−12% EtOAc in hexanes gradient), yielding 74.6 mg (36%) of the title compound as a thick oil. Rf = 0.32 in 10% EtOAc/hexanes. 1H NMR (400 MHz, CDCl3) δ 7.50−7.46 (m, 2 H), 7.41−7.30 (m, 5 H), 7.30−7.24 (m, 1 H), 7.08−7.02 (m, 2 H), 5.08 (s, 2 H), 2.85 (s, 6 H), 2.32 (s, 3 H). MS (ESI) m/z 420.34 (M + H).
Phenyl 5-Benzyloxy-6-[(t-butoxycarbonyl)(propyl)amino]-2-(dimethylamino)-3-methylisonicotinate (22b)
LHMDS (1.0 M solution in THF, 0.167 mL, 0.167 mmol) was added to a solution of phenyl 5-benzyloxy-2-(dimethylamino)-3-methyl-6-(propylamino)isonicotinate (63.5 mg, 0.152 mmol) in THF (2 mL), resulting in a bright-yellow solution. A solution of di-t-butyldicarbonate (99.5 mg, 0.456 mmol) in THF (1 mL) was added. After 10 min, the reaction was quenched by the addition of ammonium chloride (saturated, aqueous solution) and was extracted with EtOAc (2×). The organics were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by column chromatography (Biotage 10 g column, 0−12% EtOAc in hexanes gradient), yielding 13.6 mg (17%) of the product. Rf = 0.22 in 10% EtOAc/hexanes. 1H NMR (400 MHz, CDCl3) δ 7.40−7.28 (m, 7 H), 7.28−7.22 (m, 1 H), 7.05−7.00 (m, 2 H), 4.90 (s, 2 H), 2.79 (s, 6 H), 2.34 (s, 3 H), 1.76−1.66 (m, 2 H), 1.56−1.50 (m, 2 H), 0.91 (t, J = 7.56 Hz, 3 H). MS (ESI) m/z 520.38 (M + H).
N-[(4aS,11aR,12aS,13S)-10,13-Bis(dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4,4a,6,11,11a,12,12a,13-octahydro-5-hydroxy-4,6-dioxo-3,7-bis(phenylmethoxy)isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinolin-8-yl]-N-propyl-carbamic Acid, 1,1-Dimethylethyl Ester (23b)
A solution of 22b (24 mg, 0.047 mmol) in THF (0.5 mL) was added dropwise to a −78 °C solution of LDA (0.5 M solution in THF, 0.102 mL, 0.051 mmol) and TMEDA (0.041 mL, 0.27 mmol) in THF (2 mL), resulting in a yellowish−orange colored solution. After 10 min, a solution of 8 (16.4 mg, 0.034 mmol) in THF (0.5 mL) was added dropwise. After complete addition, the reaction mixture was allowed to warm to −20 °C over ∼45 min. The reaction was quenched by the addition of ammonium chloride (saturated, aqueous solution) and was extracted with EtOAc (2×). The combined extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was purified by preparative HPLC (Sunfire Prep C18 column, 90−100% B gradient), yielding 6.1 mg (20%) of the desired product as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 15.56 (s, 1 H), 7.52−7.24 (m, 10 H), 5.36 (s, 2 H), 4.99−4.64 (m, 2 H), 4.02 (d, J = 11.0 Hz, 1 H), 3.10−2.81 (m, 2 H), 2.78 (s, 6 H), 2.62−2.40 (m, 10 H), 2.20−2.14 (m, 1 H), 1.80−1.45 (m, 2 H), 1.44−1.15 (m, 11 H), 1.00−0.70 (m, 12 H), 0.25 (s, 3 H), 0.12 (s, 3 H). MS (ESI) m/z 908.65 (M + H).
6-Demethyl-6-deoxy-7-(dimethylamino)-9-(propylamino)-8-azatetracycline Dihydrochloride (24o)
Aqueous HF (48%, 0.4 mL) was added to a solution of 20o (6.1 mg, 0.007 mmol) in CH3CN (0.6 mL) in a plastic vial. After 18 h, the reaction mixture was poured into a solution of K2HPO4 (4.8 g) in water (15 mL). The mixture was extracted with EtOAc (3×). The combined EtOAc extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was dissolved in MeOH (1 mL) and 1,4-dioxane (1 mL), and palladium on carbon (Degussa, 10 wt %, ∼2 mg) was added. An atmosphere of hydrogen was introduced, and the reaction mixture was stirred for 4 h. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. The material was purified by preparative HPLC (Phenomenex Polymerx, 0−100% B gradient). Fractions with the desired MW were collected and freeze-dried to yield 2.8 mg (70%, 2 steps) of the desired product as a yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 4.19 (s, 1 H), 3.44 (t, J = 7.6 Hz, 2 H), 3.24−3.12 (m, 7 H), 3.12−2.97 (m, 8 H), 2.39−2.26 (m, 2 H), 1.73−1.56 (m, 3 H), 0.98 (t, J = 8.4 Hz, 3 H); MS (ESI) m/z 516.42 (M + H).
(4aS,11aR,12aS,13S)-10,13-Bis(dimethylamino)-4a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-11a,12,12a,13-tetrahydro-5-hydroxy-3,7-bis(phenylmethoxy)-8-(2-propen-1-yl)-isoxazolo[5′,4′:6,7]naphth[2,3-g]isoquinoline-4,6(4aH,11H)-dione (25a)
Compound 19m (105 mg, 0.126 mmol), tetrakis(triphenylphosphine)palladium(0) (15 mg, 0.013 mmol), and K3PO4 (80 mg, 0.38 mmol) were weighed into an 8 mL vial. This was sealed with a septum and was evacuated and backflushed with nitrogen (3×). Toluene (1 mL), 1,4-dioxane (1 mL), and water (0.5 mL) were added, followed by allylboronic acid pinacol ester (63.7 mg, 0.379 mmol). The reaction mixture was heated to 90 °C. After 2.5 h, the reaction mixture was cooled to room temperature and was concentrated under reduced pressure. The material was purified by preparative HPLC (Sunfire Prep C18 column, 90−100% B gradient), yielding 28.6 mg (29%) of the title compound as a yellow solid. MS (ESI) m/z 791.64 (M + H).
6-Demethyl-6-deoxy-7-(dimethylamino)-9-propyl-8-azatetracycline Dihydrochloride (26a)
Aqueous HF (48%, 0.4 mL) was added to a solution of 25a (28.6 mg, 0.036 mmol) in 1,4-dioxane (1 mL) in a plastic vial. After 18 h, the reaction mixture was poured into a solution of K2HPO4 (4.8 g) in water. The mixture was extracted with EtOAc, and the combined extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The material was dissolved in MeOH (1 mL), 0.5 M HCl in MeOH (0.5 mL), and 1,4-dioxane (1 mL), and palladium on carbon (Degussa, 10 wt %, ∼5 mg) was added. An atmosphere of hydrogen was introduced, and the reaction mixture was stirred for 1 h. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. The material was purified by preparative HPLC (Phenomenex Polymerx column, 0−70% B gradient). Fractions with the desired MW were collected and freeze-dried to yield 14.3 mg (69%) of the title compound as a yellow solid. 1H NMR (400 MHz, CD3OD with 1 drop DCl) δ 4.20 (s, 1 H), 3.38−3.15 (m, 7 H), 3.14−2.94 (m, 8 H), 2.91−2.82 (m, 2 H), 2.56 (t, J = 13.8 Hz, 1 H), 2.40−2.32 (m, 1 H), 1.86−1.76 (m, 2 H), 1.74−1.62 (m, 1 H), 1.01 (t, J = 7.34 Hz, 3 H). MS (ESI) m/z 501.35 (M + H).
The following compounds were prepared according to methods similar to 26a:
7-Chloro-6-demethyl-6-deoxy-9-propyl-8-azatetracycline Hydrochloride (26b)
Prepared from 19h and allylboronic acid pinacol ester, yellow solid. 1H NMR (400 MHz, CD3OD) δ 4.10 (s, 1 H), 3.22−2.92 (m, 9 H), 2.81−2.74 (m, 2 H), 2.49−2.38 (m, 1 H), 2.26−2.19 (m, 1 H), 1.77 −1.60 (m, 3 H), 0.97 (t, J = 7.3 Hz, 3 H). MS (ESI) m/z 492.27 (M + H).
7-Chloro-6-demethyl-6-deoxy-9-methyl-8-azatetracycline Hydrochloride (26c)
Prepared from 19h and methylboronic acid, yellow solid. 1H NMR (400 MHz, CD3OD) δ 4.09 (s, 1 H), 3.22−2.92 (m, 9 H), 2.85 (s, 3 H), 2.49−2.38 (m, 1 H), 2.26−2.19 (m, 1 H), 1.77 −1.60 (m, 1H). MS (ESI) m/z 464.30 (M + H).
6-Demethyl-6-deoxy-9-methyl-8-azatetracycline Dihydrochloride (26d)
Prepared from 19h as an over-reduced side product in the synthesis of 26c. 1H NMR (400 MHz, CD3OD) δ 7.78 (s, 1 H), 4.12 (s, 1 H), 3.22−2.91 (m, 9 H), 2.78 (s, 3 H), 2.49−2.21 (m, 1 H), 1.77−1.62 (m, 1H). MS (ESI) m/z 430.46 (M + H).
7-Chloro-6-demethyl-6-deoxy-9-phenyl-8-azatetracycline Hydrochloride (26e)
Prepared from 19h and phenylboronic acid. 1H NMR (400 MHz, CD3OD) δ 8.04−7.98 (m, 2 H), 7.47−7.39 (m, 3 H), 4.08 (s, 1 H), 3.14−2.92 (m, 9 H), 2.57−2.47 (m, 1 H), 2.28−2.07 (m, 1 H), 1.73−1.65 (m, 1H). MS (ESI) m/z 526.26 (M + H).
6-Demethyl-6-deoxy-9-phenyl-8-azatetracycline Dihydrochloride (26f)
Prepared from 19h as an over-reduced side product in the synthesis of 26e. 1H NMR (400 MHz, CD3OD) δ 8.04−7.98 (m, 2 H), 7.47−7.39 (m, 3 H), 4.08 (s, 1 H), 3.14−2.92 (m, 9 H), 2.57−2.47 (m, 1 H), 2.28−2.07 (m, 1 H), 1.73−1.65 (m, 1H). MS (ESI) m/z 526.26 (M + H).
Susceptibility Testing
Compound stocks were prepared in sterile deionized water. Tetracycline-susceptible isolates SA100 (S. aureus ATCC 13709, Smith), SA101 (S. aureus ATCC 29213), SP106 (S. pneumoniae ATCC 49619), EF103 (E. faecalis ATCC 29212), EC107 (E. coli ATCC 25922), KP109 (K. pneumoniae ATCC 13883), AB110 (Acinetobacter baumannii ATCC 19606), EC108 (Enterobacter cloacae ATCC 13047), and PA111 (Pseudomonas aeruginosa ATCC 27853) were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Tetracycline-resistant isolates S. aureus SA158 (tet(K)), S. pneumoniae SP160 (tet(M)), E. faecalis EF159 (tet(M)), E. coli EC155 (tet(A)), K. pneumoniae KP153 (tet(A)) were obtained from Marilyn Roberts’ lab at the University of Washington. S. aureus SA161 (tet(M)) was obtained from Micromyx (Kalamazoo, MI). Minimal inhibitory concentration (MIC) determinations were performed in liquid medium in 96-well microtiter plates according to the methods described by the Clinical and Laboratory Standards Institute (CLSI).(26) Cation-adjusted Mueller Hinton broth was obtained from BBL (cat. no. 212322, Becton Dickinson, Sparks, MD), prepared fresh and kept at 4 °C prior to testing. Defibrinated horse blood (cat. no. A0432, PML Microbiologicals, Wilsonville, OR) was used to supplement medium, as appropriate. All test methods met acceptable standards based on recommended quality control ranges for all comparator antibiotics and the appropriate ATCC quality control strains.
In vitro Transcription/Translation Assay
Compound stocks prepared and diluted in sterile deionized water were assayed for inhibition of coupled in vitro transcription/translation using an E. coli S30 extract system with a firefly luciferase readout from Promega (cat. no. L1020, Madison, WI). Briefly, compounds were diluted into water and added to reaction mix aliquoted to black-walled 96-well microtiter plates (cat. no. 3650, Costar, Corning, NY). An appropriate three-point titration was used for each compound, and reactions were run in duplicate. The final total reaction volume was 20 μL. Plates were incubated at 37 °C for one hour and then placed on ice for 5 min to arrest transcription/translation. Luciferase substrate (25 μL, Promega cat. no. E1500) was added to each well and luminescence was detected on a BMG LabTech LUMIstar-OPTIMA instrument. Positive assay control values, from reactions without inhibitor, were averaged per plate to determine percent inhibition of luciferase production. Results were plotted using Microsoft Excel and 50% inhibition values (IC50) were determined.
Acknowledgments
We thank members of the Tetraphase Process Chemistry Group for preparing the enone 8 as well as Professor Andrew Myers and Bob Zahler for valuable discussions over the course of this study.
Glossary
Abbreviations Used
- ATCC
American Type Culture Collection
- AUC
area under the curve
- Boc
t-butoxycarbonyl
- Cbz
benzyloxycarbonyl
- Cl
clearance
- Cy
cyclohexyl
- dba
dibenzylideneacetone
- DMAP
4-(dimethylamino)pyridine
- dppf
(diphenylphosphino)ferrocene
- %F
percent oral bioavailability
- IV
intravenous
- LDA
lithium diisopropylamide
- LHMDS
lithium bis(trimethylsilyl)amide
- mCPBA
3-chloroperbenzoic acid
- MIC
minimum inhibitory concentration
- MIC50
minimum inhibitory concentration required to inhibit growth of 50% of organisms
- MIC90
minimum inhibitory concentration required to inhibit growth of 90% of organisms
- MRSA
methicillin-resistant Staphylococcus aureus
- NMP
2-methylpyrrolidinone
- nOe
nuclear Overhouser effect
- PD50
dose at which 50% protection is observed
- PO
oral
- TMEDA
N,N,N′,N′-tetramethylethylenediamine
- Vz
volume of distribution
- Xantphos
9,9-dimethyl-4,5-bis-(diphenylphosphino)xanthenes
Supporting Information Available
MIC data for the complete panel of bacteria for all compounds as well as individual MIC data for the MIC90 calculations for compounds 20f, 24l, tigecycline, and tetracycline. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- a Hlavka J. J.; Ellestad G. A.; Chopra I.. Tetracyclines. In Kirk−Othmer Encyclopedia of Chemical Technology, 4th ed.; John Wiley & Sons, Inc.: New York, 1992; Vol. 3, pp 331−346. [Google Scholar]; b Chopra I.; Hawkey R. M.; Hinton M. Tetracyclines, molecular and clinical aspects. J. Antimicrob. Chemother. 1992, 29, 245–277. [DOI] [PubMed] [Google Scholar]
- a Chopra I.Mode of action of the tetracyclines and the nature of bacterial resistance to them. In The Tetracyclines, Handbook of Experimental Pharmacology; Hlavka J. J., Boothe J. H., Eds.; Springer-Verlag: Berlin, 1985; Vol. 78, pp 317−392. [Google Scholar]; b Brodersen D. E.; Clemons W. M. Jr.; Carter A. P.; Morgan-Warren R. J.; Wimberly B. T.; Ramakrishnan V. The structural basis for the action of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit. Cell 2000, 103, 1143–1154. [DOI] [PubMed] [Google Scholar]; c Pioletti M.; Schlunzen F.; Harms J.; Zarivach R.; Gluhmann M.; Avila H.; Bashan A.; Bartels H.; Auerbach T.; Jacobi C.; Hartsch T.; Yonath A.; Franceschi F. Crystal structures of complexes of the small ribosomal subunit with tetracycline, edeine and IF3. EMBO J. 2001, 20, 1829–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggar B. M. Aureomycin: A Product of the Continuing Search for New Antibiotics. Ann. N. Y. Acad. Sci. 1948, 51, 177–181. [DOI] [PubMed] [Google Scholar]
- a Booth J. H.; Morton J.; Petisi J. P.; Wilkinson R. G.; Williams J. H. Tetracycline. J. Am. Chem. Soc. 1953, 75, 4621. [Google Scholar]; b Conover L. H.; Moreland W. T.; English A. R.; Stephens C. R.; Pilgrim F. J.; Terramycin X. I. Tetracycline. J. Am. Chem. Soc. 1953, 75, 4622–4623. [Google Scholar]; c Minieri P. P.; Sokol H.; Firman M. C. Process for the Preparation of Tetracycline and Chlorotetracycline. U.S. Patent 2,734,018, Feb7, 1956.
- a Spencer J. L.; Hlavka J. J.; Petisi J.; Krazinski H. M.; Boothe J. H. 6-Deoxytetracyclines. V. 7,9-Disubstituted Products. J. Med. Chem. 1963, 6, 405–407. [DOI] [PubMed] [Google Scholar]; b Stephens C. R.; Beereboom J. J.; Rennhard H. H.; Gordon P. N.; Murai K.; Blackwood R. K.; Wittenau M. S. 6-Deoxytetracyclines. IV. Preparation, C-6 Stereochemistry, and Reactions. J. Am. Chem. Soc. 1963, 85, 2643–2652. [Google Scholar]
- a Martell M. J.; Boothe J. H. The 6-Deoxytetracyclines. VII. Alkylated Aminotetracyclines Possessing Unique Antibacterial Activity. J. Med. Chem. 1967, 10, 44–46. [DOI] [PubMed] [Google Scholar]; b Church R. F. R.; Schaub R. E.; Weiss M. J. Synthesis of 7-Dimethylamino-6-demethyl-6-deoxytetracycline (Minocycline) via 9-Nitro-6-demethyl-6-deoxytetracycline. J. Org. Chem. 1971, 36, 723–725. [DOI] [PubMed] [Google Scholar]
- Witkowski J. A.; Parish L. C. Antimicrobials in the treatment of acne and rosacea. SKINmed 2003, 2, 202. [DOI] [PubMed] [Google Scholar]
- For a general reviews of antibacterial agents and bacterial resistance, see:; a Wilson D. N. The A−Z of bacterial translation inhibitors. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 393–433. [DOI] [PubMed] [Google Scholar]; b Chu D. T. W.; Plattner J. J.; Katz L. New Directions in Antibacterial Research. J. Med. Chem. 1996, 39, 3853–3874. [DOI] [PubMed] [Google Scholar]; c Neu H. C. The Crisis in Antibiotic Resistance. Science 1992, 257, 1064–1078. [DOI] [PubMed] [Google Scholar]
- For reviews of tetracycline resistance mechanisms, see:; a Chopra I; Roberts M. Tetracycline Antibiotics: Mode of Action, Applications, Molecular Biology, and Epidemiology of Bacterial Resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Speer B. S.; Shoemaker N. B.; Salyers A. A. Bacterial resistance to tetracycline: mechanism, transfer, and clinical significance. Clin. Microbiol. Rev. 1992, 5, 387–399. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Levy S. B. Evolution and Spread of Tetracycline Resistance Determinants. J. Antimicrob. Chemother. 1989, 24, 1–3. [DOI] [PubMed] [Google Scholar]
- a Levy S. B.; McMurry L. Detection of an inductive membrane protein associated with R-factor mediated tetracycline resistance. Biochem. Biophys. Res. Commun. 1974, 56, 1080–1088. [DOI] [PubMed] [Google Scholar]; b McMurry L.; Petrucci R. E. Jr.; Levy S. B. Active efflux of tetracycline encoded by four genetically different tetracycline resistant determinants in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1980, 77, 3974–3977. [DOI] [PMC free article] [PubMed] [Google Scholar]; c McMurry L.; Park B. H.; Burdett V.; Levy S. B. Energy-Dependent Efflux Mediated by Class L (tet L) Tetracycline Resistance Determinant from Streptococci. Antimicrob. Agents Chemother. 1987, 31, 1648–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Burdett V. Streptococcal tetracycline resistance mediated at the level of protein synthesis. J. Bacteriol. 1986, 165, 564–569. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sanchez-Pescador R.; Brown J. T.; Roberts M.; Ureda M. S. Homology of TetM with translational elongation factors: implications for potential modes of tetM conferred tetracycline resistance. Nucleic Acids Res. 1988, 16, 1218. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Connell S. R.; Tracz D. M.; Nierhaus K. H.; Taylor D. E. Ribosomal protection proteins and their mechanism of tetracycline resistance. Antimicrob. Agents Chemother. 2003, 47, 3675–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a summary of tetracycline resistance mechanisms identified to date, see: Marilyn C. Roberts, Ph.D.;http://faculty.washington.edu/marilynr; accessed 11/29/2010.
- a Sum P.-E.; Lee V. J.; Testa R. T.; Hlavka J. J.; Ellestad G. A.; Bloom J. D.; Gluzman Y.; Tally F. P. Glycylcyclines. 1. A New Generation of Potent Antibacterial Agents through Modification of 9-Aminotetracyclines. J. Med. Chem. 1994, 37, 184–188. [DOI] [PubMed] [Google Scholar]; b Jones C. H.; Petersen P. Tigecycline: a review of preclinical and clinical studies of the first-in-class glycylcycline antibiotic. Drugs Today 2005, 41, 637–659. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Castaner R.; Bolos J.; Estivill C. Amadacycline: tetracycline antibiotic. Drugs Future 2009, 34, 11–15. [Google Scholar]
- Charest M. G.; Lerner C. D.; Brubaker J. D.; Siegel D. R.; Myers A. G. A Convergent Enantioselective Route to Structurally Diverse 6-Deoxytetracycline Antibiotics. Science 2005, 308, 395–398. [DOI] [PubMed] [Google Scholar]
- Brubaker J. D.; Myers A. G. A Practical, Enantioselective Synthetic Route to Key Precursor to the Tetracycline Antibiotics. Org. Lett. 2007, 9, 3523–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C.; Wang Q.; Brubaker J. D.; Wright P. M.; Lerner C. D.; Noson K.; Charest M.; Siegel D. R.; Wang Y.-M.; Myers A. G. A Robust Platform for the Synthesis of New Tetracycline Antibiotics. J. Am. Chem. Soc. 2008, 130, 17913–17927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent reviews, see:; a Suzuki A. Cross-coupling reactions via organoboranes. J. Organomet. Chem. 2002, 653, 83–90. [Google Scholar]; b Miyaura N.; Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar]
- Reddy N. P.; Tanaka M. Palladium-catalyzed amination of aryl chlorides. Tetrahedron Lett. 1997, 38, 4807–4810. [Google Scholar]
- For recent reviews, see:; a Schlummer B.; Scholz U. Palladium-Catalyzed C−N and C−O Coupling—A Practical Guide from an Industrial Vantage Point. Adv. Synth. Catal. 2004, 346, 1599–1626. [Google Scholar]; b Yang B. H.; Buchwald S. L. Palladium-catalyzed amination of aryl halides and sulfonates. J. Organomet. Chem. 1999, 576, 125–146. [Google Scholar]
- Fukuhara T.; Yoneda N.; Suzuki A. A facile preparation of fluoropyridines from aminopyridines via diazotization and fluorodediazoniation in hydrogen fluoride or hydrogen fluoride-pyridine solutions. J. Fluorine Chem. 1988, 38, 435–438. [Google Scholar]
- Shafir A.; Buchwald S. L. Highly Selective Room-Temperature Copper-Catalyzed C−N Coupling Reactions. J. Am. Chem. Soc. 2006, 128, 8742–8743. [DOI] [PubMed] [Google Scholar]
- O’Shea R.; Moser H. E. Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery. J. Med. Chem. 2008, 51, 2871–2878. [DOI] [PubMed] [Google Scholar]
- Cook H. J.; Mundo C. R.; Fonseca L.; Gasque L.; Moreno-Esparza R. Influence of the diet on bioavailability of tetracycline. Biopharm. Drug Dispos. 1993, 14, 549–53. [DOI] [PubMed] [Google Scholar]
- Assuming the relative PK parameters are similar between rat (PK species) and mouse (efficacy species).
- Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, 8th ed.; Approved Standard M07-A8; Clinical and Laboratory Standards Institute: Wayne, PA, 2009; Vol. 29, no. 2. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.