

Abstract
The molybdenum-dependent nitrogenase catalyzes the multi-electron reduction of protons and N2 to yield H2 and 2NH3. It also catalyzes the reduction of a number of non-physiological doubly and triply bonded small molecules (e.g. C2H2, N2O). Carbon monoxide (CO) is not reduced by the wild-type molybdenum nitrogenase but instead inhibits the reduction of all substrates catalyzed by nitrogenase except protons. Here, we report that when the nitrogenase MoFe protein α-Val70 residue is substituted by alanine or glycine, the resulting variant proteins will catalyze the reduction and coupling of CO to form methane (CH4), ethane (C2H6), ethylene (C2H4), propene (C3H6), and propane (C3H8). The rates and ratios of hydrocarbon production from CO can be adjusted by changing the flux of electrons through nitrogenase, by substitution of other amino acids located near the FeMo-cofactor, or by changing the partial pressure of CO. Increasing the partial pressure of CO shifted the product ratio in favor of the longer chain alkanes and alkenes. The implications of these findings in understanding the nitrogenase mechanism and the relationship to Fischer-Tropsch production of hydrocarbons from CO are discussed.
Keywords: Enzymes, Iron-Sulfur Protein, Molybdenum, Nitrogen Metabolism, Nitrogenase
Introduction
Nitrogenase is the bacterial enzyme responsible for the biological reduction of N2 to ammonia, accounting for more than half of the input of fixed nitrogen into the biogeochemical nitrogen cycle (1). Three different nitrogenases have been identified, with each being coded for by unique sets of genes (2, 3). The three nitrogenase systems are composed of a smaller protein called the Fe protein2 and a larger protein called the MoFe protein, VFe protein, or FeFe protein, depending on the metal composition of the corresponding cofactors. The Fe protein serves as a reductant of the larger protein, with electron transfer from the Fe protein being coupled to the hydrolysis of two MgATP molecules for each electron transferred (4). The nitrogenase system containing molybdenum is the most widely occurring and is preferentially expressed if sufficient molybdenum is present in the cell growth medium, with the other nitrogenases being expressed secondarily if molybdenum is deficient (3, 5).
All three nitrogenase systems catalyze the reduction of N2 to ammonia and the reduction of protons to H2. In addition, all three systems catalyze the reduction of a range of other small molecules that contain double and triple bonds, such as acetylene (C2H2) to ethylene (C2H4), with different rates and ratios of products being observed for the three systems (2). Carbon monoxide (CO) is a well established inhibitor of all three nitrogenase systems, where it inhibits the reduction of all known substrates except protons (2, 6, 7). Recently, Lee et al. (8) discovered that the vanadium nitrogenase system can reduce and couple CO at low rates to form the hydrocarbons ethane, ethylene, and propane. The molybdenum nitrogenase was not observed to catalyze any detectable reduction of CO to hydrocarbons.
We have recently shown that the substrate range for the molybdenum nitrogenase can be expanded to larger substrates by substitution of an amino acid residue located near the active site FeMo-cofactor (α-Val70) (9, 10). In light of our ability to change the substrate range of nitrogenase and in light of the observation of CO reduction and coupling by the vanadium nitrogenase, it was of interest to determine whether CO might be reduced and coupled to form hydrocarbons in the molybdenum nitrogenase having amino acid substitutions at the α-Val70 residue.
EXPERIMENTAL PROCEDURES
Reagents and Protein Purification
All reagents were obtained from Sigma-Aldrich or Fisher Scientific and were used without further purification, unless specified otherwise. CO was purchased from Matheson Tri-Gas (Basking Ridge, NJ) with a purity of ≥99.9%. Ethylene (99.9%) was obtained from Praxair Inc. (Danbury, CT). Methane gas was obtained from a household natural gas line containing about 3% ethane as an impurity. Propane gas was obtained from a propane fuel tank with an estimated purity of 86%. All other gases were from Air Liquide (Plumsteadville, PA). Azotobacter vinelandii strains DJ1260 (wild-type, WT, or α-Val70), DJ997 (α-Gln195), DJ1310 (α-Ala70), DJ1316 (α-Ala70/α-Gln195), DJ1313 (α-Gly70), DJ1391 (α-Ala70/α-His96), and DJ1495 (α-Ala70/α-Ala191) were grown, and the corresponding nitrogenase MoFe proteins were expressed and purified as described previously (11). All MoFe proteins in this study contain a seven-His tag addition near the carboxyl-terminal end of the α-subunit. The purification of these proteins was accomplished by following the previously developed zinc affinity purification protocol (11). Protein concentrations were determined by the Biuret assay using bovine serum albumin as standard. The purities of these proteins were >95% based on SDS-PAGE analysis with Coomassie Blue staining. Manipulation of proteins and buffers was done in septum-sealed serum vials under an argon atmosphere or on a Schlenk vacuum line. All gases and liquid transfers used gas-tight syringes.
Carbon Monoxide Reduction Assays
Unless stated otherwise, CO reduction assays were conducted in serum vials having a 9.4-ml total volume and containing 1 ml of an assay buffer consisting of 200 mm MOPS, pH 7.0, with MgATP and a MgATP-regenerating system (13.4 mm MgCl2, 10 mm ATP, 60 mm phosphocreatine, 1.3 mg/ml bovine serum albumin, and 0.4 mg/ml creatine phosphokinase). After the solution was made anaerobic, 150 μl of 1 m dithionite solution and the MoFe protein were added. Reactions were initiated by the addition of Fe protein and incubated at 30 °C. Reactions were quenched by the addition of 300 μl of 400 mm EDTA, pH 8.0, solution. All assays were done under 1 atm of CO pressure except for wild-type and α-Gln195 MoFe proteins (0.106 atm of CO for these two cases). Methane (CH4), ethylene (C2H4), and ethane (C2H6) were quantified by gas chromatography by injection of 200 μl of the gas phase of the reaction vial (400 μl for wild-type and α-Ala70/α-Gln195 MoFe samples) into a Shimadzu GC-8A equipped with a flame ionization detector fitted with a 30 × 0.3-cm Porapak N column with nitrogen as the carrier gas. The injection/detection temperature was set to 180 °C, and the column temperature was set to 110 °C. The standard curves with high linearity were created using methane, ethylene, and ethane gases diluted with argon in 9.4-ml serum vials. Propane (C3H8) and propene (C3H6) were quantified by gas chromatography by injection of 100 μl of the gas phase of the reaction vial into a Shimadzu GC-2010 gas chromatograph equipped with a flame ionization detector fitted with a Rt-Alumina BOND/KCl column (30 m, 0.32-mm inner diameter, and 5-μm film thickness) (Restek, Belafonte, PA). Helium was used as the carrier gas set at a linear velocity of 45 cm/s. The injection/detection temperature was set to 200 °C, and the column temperature was set to 60 °C. Identification of three- and four-carbon hydrocarbons was confirmed by mass spectroscopy (GCMS-QP2010S, Shimadzu Scientific). After the addition of 300 μl of EDTA solution, the total liquid volumes in the standard vials were the same as those in CO reduction assay vials.
For the electron flux dependence study, 1 mg of MoFe protein was used for each assay vial. Molar ratios of Fe protein to MoFe protein from 4:1 to 20:1 were tested. All these assays were incubated for 2 h before quenching with EDTA. For time dependence measurements and CO pressure dependence studies, the amount of MoFe protein and the electron flux for different mutants was varied.
RESULTS
Reduction of CO by α-Val70-substituted MoFe Proteins
Consistent with the earlier report (8), we find that the wild-type MoFe protein shows no catalytic production of hydrocarbons by reduction and coupling of CO when analyzed over 90 min under turnover conditions. In contrast, when the MoFe protein residue α-Val70 was substituted by alanine (α-Ala70) or glycine (α-Gly70), the variant MoFe proteins were found to reduce and couple CO to hydrocarbons in a reaction dependent on the presence of Fe protein, MgATP, and reductant (Fig. 1). Four hydrocarbon products were quantified for both the α-Ala70 and the α-Gly70 MoFe proteins: the alkanes ethane (C2H6) and propane (C3H8) and the alkenes ethylene (C2H4) and propene (C3H6). For both proteins, the highest yielding product was ethylene followed by propene, ethane, and propane. The amounts of hydrocarbons produced were ∼7 nmol of ethylene (C2H4)/nmol of MoFe protein, 4 nmol of propene (C3H6)/nmol of MoFe protein, 2 nmol of ethane (C2H6)/nmol of MoFe protein, and 1 nmol of propane (C3H6)/nmol of MoFe protein over 60 min. Three of these products (ethylene, ethane, and propane) were reported for CO reduction by the vanadium nitrogenase system (8), although the relative ratio of products and rates of production was different between the two nitrogenase systems. For the vanadium nitrogenase, product production rates were 140 nmol of ethylene/nmol of VFe protein, 1.5 nmol of propane/nmol of VFe protein, and 5 nmol of ethane/nmol of VFe protein over 60 min. No propene product was reported for the vanadium nitrogenase, whereas this was the second most abundant product in the molybdenum nitrogenase.
FIGURE 1.

Time course of hydrocarbon production by the α-Ala70- and α-Gly70-substituted MoFe proteins. The quantity of (●) ethylene (C2H4), (■) ethane (C2H6), (▾) propene (C3H6), and (▴) propane (C3H8) produced per MoFe protein (nmol of product per nmol of MoFe protein) as a function of time is shown for the α-Ala70 (A) and α-Gly70 (B) MoFe nitrogenases. Assay conditions are described under “Experimental Procedures.”
Effects of Additional Amino Acid Substitutions
In addition to the pivotal role of α-Val70 in defining substrate binding to the FeMo-cofactor, we have earlier shown that two additional amino acids located near the FeMo-cofactor, α-Arg96 and α-Gln191, can affect substrate binding (12, 13). Substituting these amino acids by histidine or alanine, respectively, has been shown to further expand the substrate range of nitrogenase to include even longer chain alkynes. We therefore examined the doubly substituted α-Ala70/α-His96 and α-Ala70/α-Ala191 MoFe proteins to test whether they might reduce and couple CO. Both of the doubly substituted MoFe proteins showed an altered product profile for hydrocarbon production, with the yield of the longer chain products propane and propene rising significantly (Fig. 2). For the α-Ala70/α-Ala191 MoFe protein, 8 nmol of propene were accumulated per nmol of MoFe protein over 60 min. A trace of methane (CH4) was also detected as a product of CO reduction for both doubly substituted MoFe proteins, whereas methane was not detected for any of the singly substituted MoFe protein variants. The doubly substituted MoFe proteins demonstrated a non-linear production of products, suggesting possible inactivation of these enzymes during the course of the assay.
FIGURE 2.

Time course of hydrocarbon production by the α-Ala70/α-His96- and α-Ala70/α-Ala191-substituted MoFe proteins. The quantity of (♦) methane (CH4), (●) ethylene (C2H4), (■) ethane (C2H6), (▾) propene (C3H6), and (▴) propane (C3H8) produced per MoFe protein (nmol of product per nmol of MoFe protein) as a function of time is shown for the α-Ala70/α-His96 (A) and α-Ala70/α-Ala191 (B) MoFe nitrogenases.
The amino acid α-His195 is located near the FeMo-cofactor and has been implicated in the delivery of protons for the reduction of nitrogenous substrates (14, 15). We find that when α-His195 is substituted by glutamine in combination with the α-Ala70 substitution, the rates of product formation for CO reduction and coupling are greatly decreased (Fig. 3), consistent with the possibility that α-His195 functions to deliver protons for the reduction of CO.
FIGURE 3.
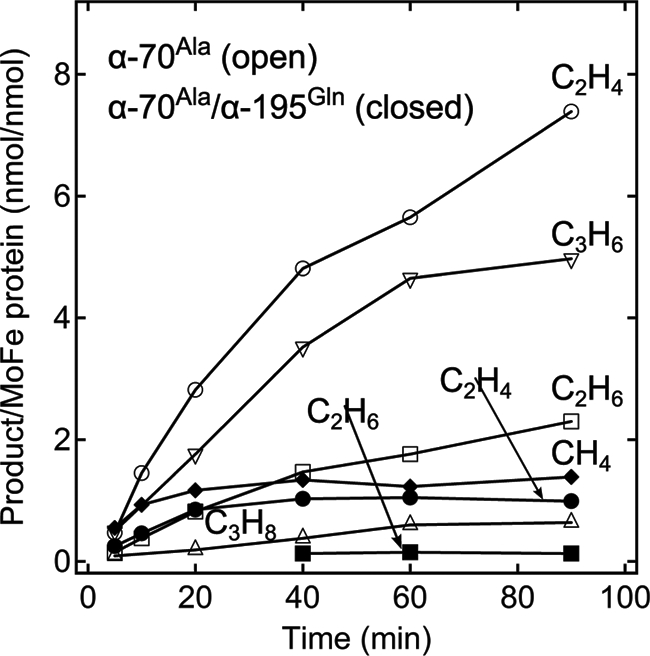
Time course of hydrocarbon production by the α-Ala70- and α-Ala70/α-Gln195-substituted MoFe proteins. The quantity of (circles) ethylene (C2H4), (squares) ethane (C2H6), (inverted triangles) propene (C3H6), and (triangles) propane (C3H8) produced per MoFe protein (nmol of product per nmol of MoFe protein) as a function of time is shown for the α-Ala70 (open symbols) and α-Ala70/α-Gln195 MoFe proteins (closed symbols).
Influence of Electron Flux on Product Ratio
Electron flux in nitrogenase is defined as the rate of total electrons flowing through the enzyme going to substrates. The flux can be controlled by varying the ratio of Fe protein to MoFe protein, with a low ratio corresponding to low flux and a higher ratio corresponding to higher flux (2). Increasing electron flux affected the product profile for the α-Ala70, α-Gly70, and α-Ala70/α-His96 MoFe proteins (Fig. 4). In each case, however, ethylene remained the most abundant product over the range tested.
FIGURE 4.

Electron flux dependence on CO reduction product profile. The quantity of (♦) methane (CH4), (●) ethylene (C2H4), (■) ethane (C2H6), (▾) propene (C3H6), and (▴) propane (C3H8) produced per MoFe protein (nmol of product per nmol of MoFe protein) is shown as a function of the molar ratio of Fe protein to MoFe protein for the α-Ala70 (A), α-Gly70 (B), and α-Ala70/α-His96 (C) MoFe proteins. The time of the assay was 120 min.
Influence of CO Partial Pressure on Product Profile
Varying the partial pressure of CO has been shown to result in different numbers of CO bound to the FeMo-cofactor (6, 16). Under low concentrations of CO (∼0.08 atm partial pressure), a single CO species binds to the FeMo-cofactor, referred to as the low CO state. At partial pressures of CO above 0.4 atm, two CO molecules bind to the FeMo-cofactor, referred to the high CO state (17). It was therefore of interest to determine whether varying the partial pressure of CO might also alter the product profile for CO reduction and coupling. As can be seen in Fig. 5, the partial pressure of CO significantly changes the product profile for the four MoFe protein variants examined (α-Ala70, α-Gly70, α-Ala70/α-His96, α-Ala70/α-Ala191 MoFe proteins). At lower CO partial pressure (0.08 atm), the shorter chain C2 products are favored. At this low CO concentration, the α-Ala70/α-His96 MoFe protein also showed a significant methane production rate. At high CO partial pressure (1.0 atm), the product profile shifted in favor of the longer chain C3 hydrocarbons.
FIGURE 5.

CO pressure dependence on CO reduction product profile. Shown are the methane (CH4), ethylene (C2H4), ethane (C2H6), propene (C3H6), and propane (C3H8) production per MoFe protein (nmol of product per nmol of MoFe protein) under either low CO (0.08 atm) or high CO (1.0 atm) for the α-Ala70 (A), α-Gly70 (B), α-Ala70/α-His96 (C), and α-Ala70/α-Ala191 (D) MoFe proteins. Assay time is 60 min. PCO, Partial pressure of CO.
DISCUSSION
Earlier spectroscopic studies of CO bound to nitrogenase indicate that there are two CO binding sites on the FeMo-cofactor (6). At low CO concentrations, only one CO is bound, whereas at higher CO concentrations, two CO molecules are bound (17–19). The observation that CO can be reduced to two- and three-carbon containing hydrocarbons demands that the two CO binding sites on the FeMo-cofactor are close enough to allow a coupling reaction that would result in the formation of C2 and C3 hydrocarbon products. This fact, coupled with the parallels between this reaction and the Fischer-Tropsch reaction (20–23), suggests a possible mechanism for CO reduction and coupling by nitrogenase (Fig. 6).
FIGURE 6.

Schematic representation of a possible CO-derived hydrocarbon formation at the nitrogenase active site. Black circles represent CO binding sites, presumably Fe atoms, upon which two CO can bind. Following reduction to semireduced states (-CHx and -CHy), coupling can occur, resulting in formation of a CHx–CHy species. The binding of additional CO molecules can lead to further chain elongation.
Initially, two CO molecules would chemisorb to two adjacent metal (Fig. 6, black circles) atoms of the FeMo-cofactor. Available spectroscopic studies on CO bound to FeMo-cofactor indicate that the CO molecules are bound to iron atoms located in the waist region of the FeMo-cofactor (19, 24, 25). Reduction of the two metal bound CO molecules by protons and electrons (hydrogen) with loss of water would result in the formation of metal-bound -CHx and -CHy groups. Analogous intermediates have been proposed in a mechanism for the Fischer-Tropsch reaction (22). Either the two metal-bound -CHx groups could be further reduced/protonated, resulting in release of methane, or a coupling reaction could result in the formation of a CHx–CHy species bound to one metal. This species could yield ethylene or ethane, or a third CO could bind to and be reduced at the open metal site, leading to the formation of C3 or longer hydrocarbons. Traces of longer chain (C4) hydrocarbon products were detected from molybdenum nitrogenase reduction and from coupling of CO molecules that were tentatively assigned by mass spectroscopy as isobutene and n-butane (data not shown).
In this mechanism, it is expected that higher CO concentrations would favor two bound CO molecules, which in turn would favor formation of longer chain hydrocarbons (e.g. propene and propane) consistent with the observations here. Nevertheless, even under low CO concentrations that have been assigned to a single CO bound to the FeMo-cofactor, we still observe C2 and C3 products, consistent with CO binding at both sites even at the lower CO concentrations.
Essential for the CO reduction reactions is the delivery of protons. For the Fischer-Tropsch reduction of CO, hydrogen atoms come from H2. For the nitrogenase-catalyzed reduction, protons appear to come from multiple sites, although the α-His195 residue located near the FeMo-cofactor has been shown to play a critical role in proton delivery for reduction of nitrogenous substrates (14, 15). Substitution of this residue with the amino acid glutamine effectively eliminates N2 reduction, although N2 still binds to the FeMo-cofactor in the substituted MoFe protein (14, 15). Such an inability to reduce N2 is likely due to a disruption in the proton delivery needed for substrate reduction. To test whether α-His195 might also be responsible for delivery of protons for CO reduction, the α-Gln195 substitution was introduced into the α-Ala70 MoFe protein. The resulting doubly substituted MoFe protein showed significantly lowered CO reduction rates for all products, consistent with the α-His195 being responsible for much, but not all, of the proton delivery for CO reduction in the MoFe protein.
One of the goals for Fischer-Tropsch chemistry is finding catalysts and conditions that favor the formation of the higher value, longer chain hydrocarbons such as propene and propane (23). Here, we report that the introduction of additional amino acid substitutions near the FeMo-cofactor and increasing the CO concentration can significantly shift the product profile of nitrogenase CO reduction and coupling in favor of production of these longer chain hydrocarbons. Thus, the molybdenum nitrogenase offers an experimentally tractable system for examining mechanistic features that favor the production of longer chain hydrocarbons from CO that might be translated to the development of small molecule metal complexes that catalyze such reactions.
In summary, we report that when the active site of the molybdenum nitrogenase is exposed by substitution of α-Val70, CO can be reduced and coupled to yield the hydrocarbons methane, ethylene, ethane, propene, and propane. Further, it is found that the relative ratio of products can be manipulated by altering other amino acids, electron flux, or the CO concentration.
Acknowledgment
We thank Dr. Brett Barney, University of Minnesota, for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant GM-59087 (to L. C. S. and D. R. D.).
This article was selected as a Paper of the Week.
- Fe protein
- iron protein
- MoFe protein
- molybdenum-iron protein
- VFe protein
- vanadium-iron protein
- FeFe protein
- iron-iron protein.
REFERENCES
- 1. Smil V. (2004) Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production, The MIT Press, Cambridge, MA [Google Scholar]
- 2. Burgess B. K., Lowe D. J. (1996) Chem. Rev. 96, 2983–3012 [DOI] [PubMed] [Google Scholar]
- 3. Eady R. R. (1996) Chem. Rev. 96, 3013–3030 [DOI] [PubMed] [Google Scholar]
- 4. Howard J. B., Rees D. C. (1994) Annu. Rev. Biochem. 63, 235–264 [DOI] [PubMed] [Google Scholar]
- 5. Joerger R. D., Bishop P. E. (1988) Crit. Rev. Microbiol 16, 1–14 [DOI] [PubMed] [Google Scholar]
- 6. Davis L. C., Henzl M. T., Burris R. H., Orme-Johnson W. H. (1979) Biochemistry 18, 4860–4869 [DOI] [PubMed] [Google Scholar]
- 7. Rivera-Ortiz J. M., Burris R. H. (1975) J. Bacteriol. 123, 537–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee C. C., Hu Y., Ribbe M. W. (2010) Science 329, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dos Santos P. C., Igarashi R. Y., Lee H. I., Hoffman B. M., Seefeldt L. C., Dean D. R. (2005) Acc. Chem. Res 38, 208–214 [DOI] [PubMed] [Google Scholar]
- 10. Seefeldt L. C., Hoffman B. M., Dean D. R. (2009) Annu. Rev. Biochem. 78, 701–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Christiansen J., Goodwin P. J., Lanzilotta W. N., Seefeldt L. C., Dean D. R. (1998) Biochemistry 37, 12611–12623 [DOI] [PubMed] [Google Scholar]
- 12. Dos Santos P. C., Mayer S. M., Barney B. M., Seefeldt L. C., Dean D. R. (2007) J. Inorg. Biochem. 101, 1642–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Benton P. M., Mayer S. M., Shao J., Hoffman B. M., Dean D. R., Seefeldt L. C. (2001) Biochemistry 40, 13816–13825 [DOI] [PubMed] [Google Scholar]
- 14. Kim C. H., Newton W. E., Dean D. R. (1995) Biochemistry 34, 2798–2808 [DOI] [PubMed] [Google Scholar]
- 15. Fisher K., Dilworth M. J., Newton W. E. (2000) Biochemistry 39, 15570–15577 [DOI] [PubMed] [Google Scholar]
- 16. Maskos Z., Fisher K., Sørlie M., Newton W. E., Hales B. J. (2005) J. Biol. Inorg. Chem 10, 394–406 [DOI] [PubMed] [Google Scholar]
- 17. Lee H., Cameron L. M., Hales B. J., Hoffman B. M. (1997) J. Am. Chem. Soc. 119, 10121–10126 [Google Scholar]
- 18. Lee H. I., Sørlie M., Christiansen J., Yang T. C., Shao J., Dean D. R., Hales B. J., Hoffman B. M. (2005) J. Am. Chem. Soc 127, 15880–15890 [DOI] [PubMed] [Google Scholar]
- 19. Christie P. D., Lee H., Cameron L. M., Hales B. J., Orme-Johnson W. H., Hoffman B. M. (1996) J. Am. Chem. Soc. 118, 8707–8709 [Google Scholar]
- 20. Rofer-DePoorter C. K. (1981) Chem. Rev. 81, 447–474 [Google Scholar]
- 21. Zhang C., Zhao G., Liu K., Yang Y., Xiang H., Li Y. (2010) J. Mol. Catal. A Chem. 328, 35–43 [Google Scholar]
- 22. Davis B. H. (2009) Catal. Today 141, 25–33 [Google Scholar]
- 23. Khodakov A. Y., Chu W., Fongarland P. (2007) Chem. Rev. 107, 1692–1744 [DOI] [PubMed] [Google Scholar]
- 24. Pollock R. C., Lee H., Cameron L. M., DeRose V. J., Hales B. J., Orme-Johnson W. H., Hoffman B. M. (1995) J. Am. Chem. Soc. 117, 8686–8687 [Google Scholar]
- 25. Yang Z. Y., Seefeldt L. C., Dean D. R., Cramer S. P., George S. J. (2011) Angew. Chem. Int. Ed. Engl. 50, 272–275 [DOI] [PMC free article] [PubMed] [Google Scholar]