

Abstract
Methylmercury (MeHg) is a neurotoxin capable of causing severe damage to the CNS, especially in the developing fetus. Glia in the CNS release a number of cytokines that are important for proper CNS development and function. We reported earlier that MeHg could induce interleukin-6 (IL-6) release in primary mouse glia. This finding is significant considering previous reports indicating that sustained IL-6 exposure could be detrimental to cerebellar granule neurons, one of the major cellular targets of MeHg cytotoxicity. By using pharmacological antagonists against phophatidycholine- and phosphoinositol-specific phospholipase C, the current study indicated that phospholipase C activity was necessary for MeHg-induced IL-6 release. Results from pharmacological antagonists further suggested that the calcium signaling initiated by phospholipase C appeared essential for this event. In contrast, protein kinase C activity did not appear to be important. Even though mitogen-activated protein kinases were important for IL-6 release in some experimental systems, these enzymes did not appear to be required for MeHg-induced IL-6 release in glia. Based on these data and those reported by us and others, there is a possibility that MeHg-induced phospholipase C activation initiates a calcium signaling that causes phospholipase A2 activation. This, in turn, leads to arachidonic acid and lysophosphatidyl choline generation, both of which are potent inducers for IL-6 release.
Keywords: calcium, cytokine, IL-6, methylmercury, phospholipase C
Introduction
Glia in the central nervous system (CNS) release a number of cytokines, which are important for proper CNS development and function [8]. Insults to the CNS can alter the pattern of cytokine release. For example, methylmercury (MeHg) can increase interleukin (IL)-6 release from the glial cells of the CNS. This is observed in several glial cell lines derived from various species including those from human [5], rat [5] and mouse [11] as well as primary cultures of mouse glia [6]. Results from human glioma cells indicate that MeHg specifically induces IL-6 release without causing the release of either tumor necrosis factor (TNF)-α or IL-1β in the same cultures [5]. Interestingly, even though some cytotoxicity is associated with MeHg induced IL-6 release, cytotoxicity itself caused by a heavy metal is not a sufficient cause for IL-6 release. For example, even though CdCl2 (another toxic metal) and HgCl2 (another toxic mercury compound) cause cytotoxicity, they do not cause IL-6 release [6]. Sustained glial IL-6 release can be detrimental to cerebellar granule neurons [14, 22], one of the major cellular targets of MeHg cytotoxicity [4, 9]. This is because pretreatment of cerebellar granule neurons with IL-6 can increase glutamate-induced death in this neuronal population. Thus, IL-6 release caused by MeHg has significant pathological importance.
MeHg is known to activate cytosolic phospholipase A2 (PLA2) activity in a variety of cell types [20, 31] including CNS glial cells [25]. Results from our previous study indicate that PLA2 activation is required for MeHg to induce IL-6 release [6]. This is because inhibition of PLA2 activity by various pharmacological inhibitors prevents MeHg-induced IL-6 release from primary mouse glia. The current study was designed to explore the events upstream of PLA2 activation that lead to MeHg-induced IL-6 release.
There is evidence that phospholipase C (PLC) activation is an upstream event that leads to PLA2 activation [16]. Specifically, MeHg can induce phosphatidylcholine-specific phospholipase C (PC-PLC) activation. This is followed by several events including generation of diacylglycerol (an activator of protein kinase C, PKC), an increase in intracellular calcium levels and PLA2 activation. The pharmacological inhibitor of PC-PLC, D609, cab block all these events subsequent to PC-PLC activation in cells treated with MeHg [16]. In addition to PC-PLC, MeHg can also activate the phosphoinositol-specific PLC (PI-PLC). This is because MeHg can increase intracellular phosphoinositol levels [24], a product of PI-PLC activation.
PLC activation leads to two signaling pathways, i.e., PKC and calcium. PI-PLC activation can generate IP3, which can cause release of intracellular calcium stores. This can subsequently lead to store-activated calcium entry from the extracellular compartment, which serves as a mechanism to replenish the intracellular calcium stores (reviewed in [30]). Even though the precise mechanism is unknown, PC-PLC activation also causes elevation of intracellular calcium levels in several experimental systems [1, 26, 29] including MeHg treated MDCK cells [16]. This PI-PLC and PC-PLC induced calcium signaling is likely responsible for the observation that MeHg causes elevation of intracellular calcium levels, first by release of intracellular stores, followed by calcium entry from the extracellular compartment [12, 13].
Both the PKC pathway and calcium signaling initiated by PLC can activate PLA2 (reviewed by [28]). Based on this, we hypothesized that MeHg-induced activation of PLC and the subsequent PKC and/or calcium signaling were the signaling pathway that led to PLA2 activation and the observed glial IL-6 release. Because IL-6 production in other experimental systems can be inhibited by various mitogen-activated protein (MAP) kinase inhibitors [18, 23], possible involvement of MAP kinases on MeHg-induced IL-6 was also investigated.
Material and Methods
Mixed mouse cerebral glia derived from 1-2 day old C57BL/6 mice were prepared as described previously [7]. As reported earlier, astrocytes constituted the majority of cells in these cultures because more than 90% of the culture surface was covered by cells positive for glial fibrillary acidic protein (GFAP) staining [7]. Even though some microglia were present in these cultures, they had only a minor contribution to the IL-6 release detected in this mixed glia culture system [6]. The PLC inhibitors D609, U73122 and ET-18-OCH3 were purchased from Calbiochem (Gibbstown, NJ, USA). The PKC inhibitors H7 and chelerythine were from Alexis Biochemicals (San Diego, CA, USA). The inhibitor for IP3-induced calcium release and stored-operated calcium entry, 2-APB, as well as the MAP kinase inhibitors PD 98059 (against ERK), SB 203580 (against p38) and SP 600125 (against JNK) were from Cayman Chemical (Ann Arbor, MI, USA). Methylmercury chloride and other general chemicals were from Sigma (St. Louis, MO, USA) unless otherwise stated. The growth medium was composed of Dulbecco’s Modified Eagle’s Medium/F12 (DMEM/F12 medium) supplemented with 5% newborn calf serum and 2.5 mM glutamine. To prepare for experiments, cells were plated into culture plates in this growth medium at 140,000 cells/well in 24-well plates with 700 μl medium per well. A 10X concentration of each testing agent was added to each well two hours after plating to reach the final concentration. The only exception to this was SP 600125. Because of its low solubility, it was difficult to make a 10X solution with this agent. Consequently, the final concentration of SP 600125 used was prepared directly in growth medium, and then was used to replace the initial plating medium 2 hours after plating. Following overnight incubation, the medium was switched to a medium containing MeHg (but without the testing agent) consisting of DMEM/F12 supplemented with 1% newborn calf serum and 2.5 mM glutamine.
For IL-6 measurement, culture medium from each well was collected after overnight (≥18 hrs) MeHg treatment, stored at -70°C for later ELISA analysis. Mouse IL-6 ELISA kits were obtained from eBioscience (San Diego, CA). The assay was set up in duplicates, and a standard curve was run in parallel, per the manufacturer’s instructional manual. Cell viability was determined immediately by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay [10, 11]. The OD of each well was measured by a plate reader (Spectra Max 190, Molecular Devices, Sunnyvale, CA) with a filter setting at 570 nm (reference filter setting was 630 nm). Our previous studies indicated that the MTT viability assay agreed well with results from the trypan blue exclusion viability assay [11].
We have previously reported that overnight (≥18 hrs) treatment of mouse glia with 5 μM MeHg could induce IL-6 release from ~14 (control) to ~185 pg/ml. [6]. Concurrent analyses indicated that this concentration of MeHg reduced cell viability from 100% (control) to ~88%. These results indicated that 5 μM MeHg could cause significant (more than 10-fold) IL-6 release with only mild cytotoxicity. We thus used this MeHg concentration for the current study: The level of IL-6 release caused by 5 μM MeHg was defined as 100% (designated as “MeHg control”), and was used for comparison with the level of IL-6 released in the presence of other testing agents. Concurrent assays were performed to determine cell viability under each experimental condition. The viability of untreated cells was defined as 100%, and was used to determine the cytotoxic effect caused by 5 μM MeHg alone as well as a testing agent plus MeHg. For most compounds tested, the results were derived from 4 independent experiments with 3 replicates in each experiment. For chelerythrine and SB 203580, the results were derived from 5 independent experiments with 3 replicates in each experiment. Data were expressed as Mean ± SEM. Results of IL-6 measurements were presented as solid circles (use the right Y axis); results of viability assay were presented as open circles (use the left Y axis). Statistical analyses were performed using one-way ANOVA. The Bonferroni test was used for post-hoc analysis; level of significance was expressed as: * p< 0.05; ** p< 0.01; *** p< 0.001.
Results
The first set of experiments was performed to test whether D609, a specific PC-PLC inhibitor, could block MeHg-induced IL-6 release from primary mouse glia. Results indicated that D609 at 5, 10 or 20 μM caused concentration-dependent decrease of MeHg-induced IL-6 release such that the IL-6 levels reduced to ~89%, ~67% or ~50% of MeHg control, respectively (Fig. 1A, solid circles, right-side axis). D609 at these concentrations also led to a concentration-dependent increase of cell viability (Fig. 1A, open circles, left-side axis), consistent with the findings of Kang et al. [16].
Fig. 1. PLC inhibitors prevented MeHg-induced IL-6 release.
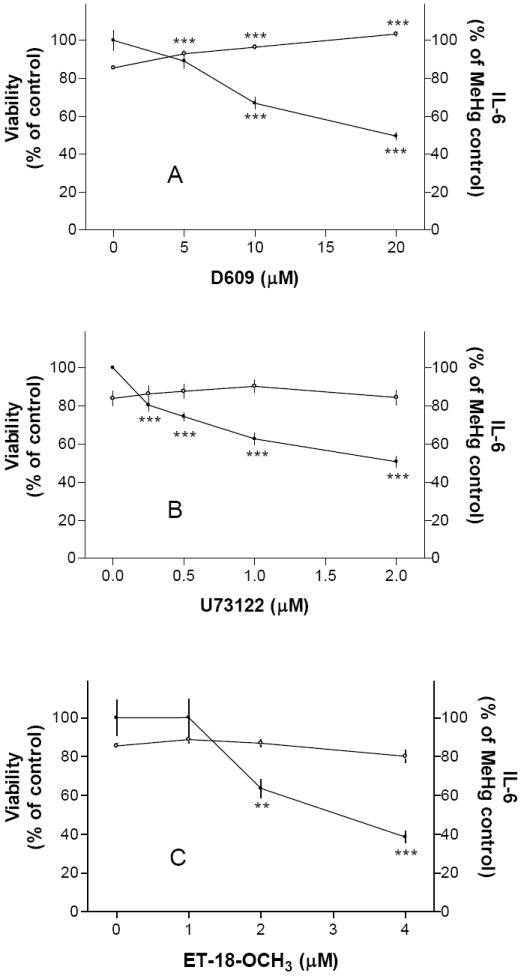
Cells were pretreated with a PC-PLC inhibitor (D609, Fig. 1A) or PI-PLC inhibitors (U73122, Fig. 1B; ET-18-OCH3, Fig. 1C) overnight followed by 5 μM MeHg (without the testing agent) overnight, and then IL-6 production and cell viability were determined. Results indicated that all PLC inhibitors tested could prevent IL-6 release.
Subsequent experiments indicated that U73122, a PI-PLC inhibitor, also prevented MeHg induced IL-6 release in a concentration-dependent manner (Fig. 1B). A Significant inhibition of IL-6 release could be observed at 0.25 μM U73122. Another PI-PLC inhibitor, ET-18-OCH3, also prevented MeHg induced IL-6 release in a concentration-dependent manner (Fig. 1C). This agent at 2 and 4 μM reduced MeHg induced IL-6 release to ~64% and ~39% of MeHg control, respectively. These two agents did not affect MeHg cytotoxicity in the concentration range tested. Together with the previous set of experiments, these results using pharmacological agents suggested that PLC activities were important for MeHg to induce IL-6 release from mouse glia. In addition, prevention of MeHg-induced IL-6 release was not necessarily linked to prevention of MeHg-induced cytotoxicity.
We next determined whether the two PLC-activated pathways, i.e., PKC or calcium, were involved in MeHg-induced IL-6 release. The PKC inhibitor chelerythrine at 2 μM had some inhibitory effect (Fig. 2A). It was toxic to cells at higher concentrations. The other PKC inhibitor H7 did not prevent MeHg-induced IL-6 release (Fig. 2B). In contrast, 2-APB (5-40 μM), an inhibitor of IP3-induced calcium release and store-activated calcium entry [2], prevented MeHg-induced IL-6 release (Fig. 2C). Based on these results, elevation of calcium levels subsequent to PLC activation appeared to be an essential step in MeHg-induced IL-6 release. Even though PLC-dependent IL-6 production in other experimental systems was under the control of MAP kinases [18, 23], the inhibition of ERK by PD 98059 (5-20 μM, Fig. 3A), p38 by SB 203580 (5-20 μM, Fig. 3B) or JNK by SP 600125 (5-20 μM, Fig. 3C) did not prevent MeHg-induced IL-6 release. Thus, these pathways did not appear to be necessary for MeHg-induced IL-6 release.
Fig. 2. Calcium signaling was essential for MeHg-induced IL-6 release.
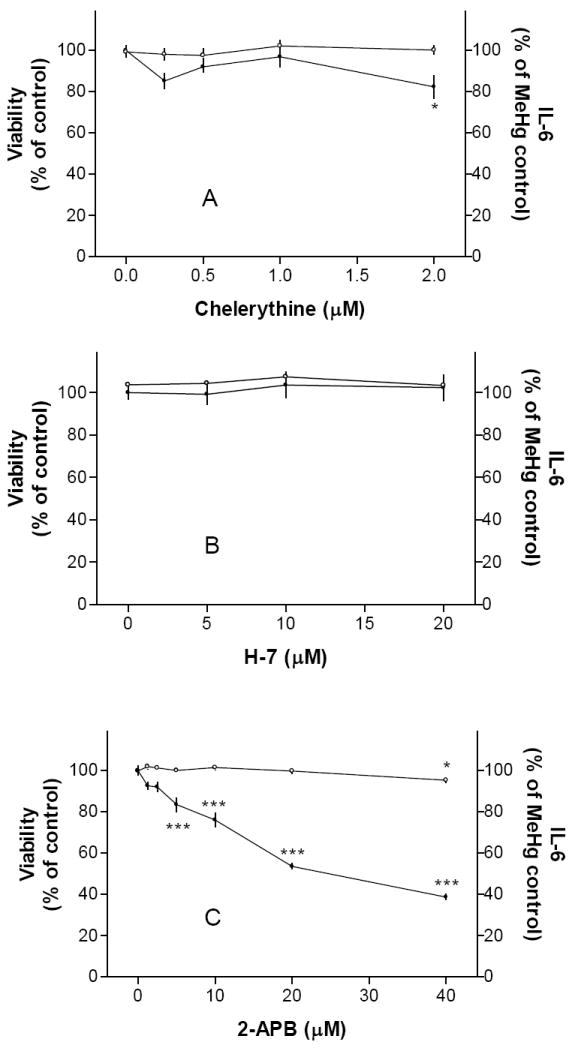
Cells were pre-treated with PKC inhibitors (chelerythrine, Fig. 2A; H-7, Fig. 2B) or a calcium signaling inhibitor (2-APB, Fig. 2C) overnight followed by 5 μM MeHg (without the testing agent) overnight, and then IL-6 production and cell viability were determined. Results indicated that PKC inhibition had little effect on IL-6 release. In contrast, prevention of calcium signaling could significantly reduce IL-6 release. See text for more discussion on this issue.
Fig. 3. MAP kinase inhibitors did not prevent MeHg-induced IL-6 release.
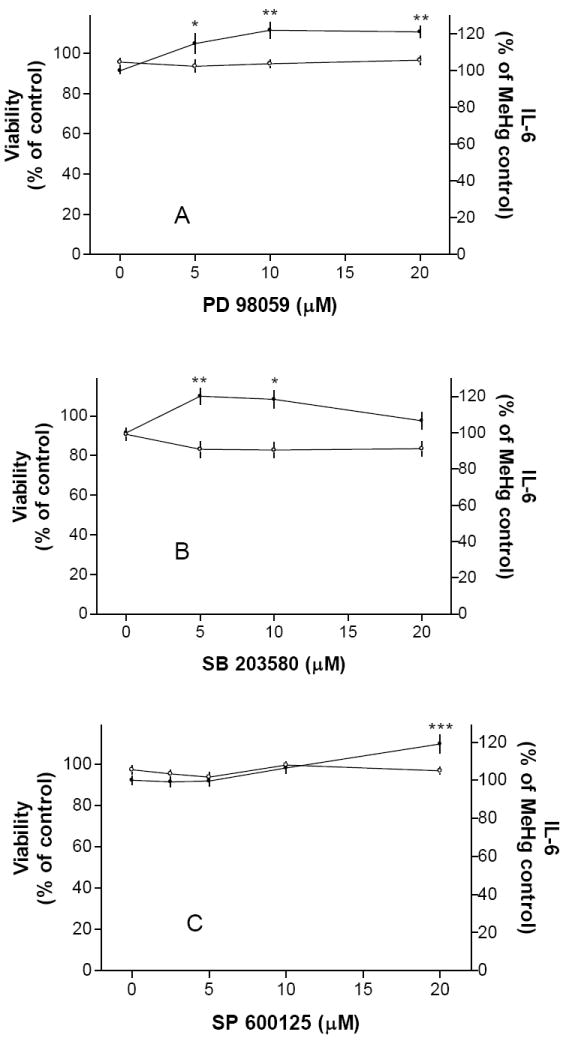
Cells were pre-treated with MAP kinase inhibitors against ERK (PD 98059, Fig. 3A), P38 (SB 203580, Fig. 3B) or JNK (SP 600125, Fig. 3C) overnight followed by 5 μM MeHg (without the testing agent) overnight, and then IL-6 production and cell viability were determined. Results indicated that inhibition of these MAP kinases did not prevent IL-6 release. Thus, MAP kinases were not required for MeHg-induced IL-6 release. These MAP kinase inhibitors alone (without MeHg) did not increase IL-6 release.
Discussion
Results from the current study suggested that MeHg activation of PLC (PC-PLC and PI-PLC) was an important step that led to IL-6 release. The role of PC-PLC in IL-6 release was investigated in other studies. For example, Monick et al. reported that PC-PLC was activated in bacterial lipopolysaccharide (LPS)-treated macrophages [21]. This led to a series of events, such as release of diacylglycerol, PKC activation, MAP kinase (ERK) activation and IL-6 generation. All these events were blocked by the PC-PLC inhibitor, D609. The IL-6 release in that study was reduced to ~60% by 10 μM D609, a result similar to the current study (Fig. 1A). These authors concluded that PC-PLC activation was the first step that led to IL-6 generation in this experimental system. Other than PC-PLC, inhibition of PI-PLC also prevented MeHg induced IL-6 release (Fig. 1 B, U73122; Fig. 1C, ET-18-OCH3). Similar inhibition of IL-6 release by U73122 was reported before by Rees et al. [23]. These investigator reported that adenosine-induced IL-6 release in pituitary cells was depend on PI-PLC. Furthermore, PKC and p38 activation were required for IL-6 release [23].
Those papers cited above suggested PKC and MAP kinases were critical downstream events required for PLC-dependent IL-6 production. We thus explored whether PKC and MAP kinases were important for MeHg-induced IL-6 release. Results from this study indicated that the PKC inhibitors tested had slight (chelerythrine, Fig. 2A) or no (H7, Fig. 2B) inhibitory effects. These results did not support the notion that PKC was critically important in MeHg-induced IL-6 release. Further experiments indicated that inhibition of MAP kinases (ERK, Fig. 3A; p38, Fig. 3B and JNK, Fig. 3C) did not prevent MeHg-induced IL-6 release. In fact, the inhibition of these enzymes led to 10-20% increase of MeHg-induced IL-6 release. We thus concluded that activities of MAP kinases were not required for MeHg-induced IL-6 release.
In addition to PKC, PLC activation is known to initiate the calcium signaling system. We thus explored whether PLC-induced calcium elevation was involved in MeHg-induced IL-6 production. 2-APB is known to inhibit both IP3-induced and store-operated calcium entry [2]. Results (Fig. 2C) indicated that 2-APB could prevent MeHg-induced IL-6 release. This observation was consistent with earlier reports that showed 2-APB could inhibit calcium signaling-induced IL-6 mRNA synthesis [17] and IL-6 protein production [15]. There is a possibility that calcium signaling was required for MeHg-induced IL-6 release in this experimental system.
Considering the fact that this project relied exclusively on the use of pharmacological reagents to explore the mechanism by which MeHg caused IL-6 release in glia, there were some limitations of this report that must be mentioned. First, the actual enzymatic activities of PLC, PKC and MAP kinases were not determined. Even though we used three inhibitors for PLC and two inhibitors for PKC to examine the involvement of these two enzymes, we realized that the results obtained were good, but indirect, evidence of the conclusions we made. Similarly, the results of the MAP kinases suffered the same limitation. Second, even though results of 2-APB suggested the involvement of calcium signaling, we did not measure the calcium levels directly. We did not have enough evidence to show whether the IP3-induced calcium release or the store-operated calcium entry was responsible for MeHg induced IL-6 release in our experimental system. This will be addressed in future reports. Third, there is a possibility that the pharmacological agents used in this study could alter glial IL-6 release in the absence of MeHg. Regarding this, we were especially concerned with results from MAP kinases (Fig. 3) because these agents slightly enhanced MeHg-induced IL-6 release. Subsequent experiments demonstrated that these agents did not cause IL-6 release above basal levels in the absence of MeHg, as indicated in Fig. 3 figure legends. Those agents used in Fig. 1 (D609, U73122, ET-18-OCH3), Fig. 2A (chelerythrine) and Fig. 2C (2-APB) reduced MeHg-induced IL-6 release while that used in Fig. 2B (H-7) did not affect MeHg-induced IL-6 release. While we had no reason to believe they could stimulate IL-6 release by themselves because they were mostly inhibitory in nature (except H-7), we did not determine whether these agents could reduce basal IL-6 release. This was because, as indicated in the Material and Methods, the basal IL-6 release was very low (~14 pg/ml), which was at the low end of the ELISA standard curve. Even if these agents generated a set of readings slightly below the basal level, we would not consider them reliable and/or biologically significant. However, we do recognize this is indeed a limitation of this report.
In conclusion, the current study was guided by our previous findings that PLA2 activation is required for MeHg-induced IL-6 release in mouse glia [6], and the report that MeHg-induced PLC activation is an upstream event of PLA2 activation [16]. Results from this study using pharmacological antagonists indicated that PLC activation was critical for MeHg-induced IL-6 release. Results derived from pharmacological inhibitors also suggested that calcium, but not PKC, was necessary for IL-6 induction. There is a possibility that PLC-activated calcium signaling could cause PLA2 activation, as reported by other investigators [19]. Based on others’ reports, PLA2 activation can cause generation of arachidonic acid and lysophosphatidyl choline, both of which are potent inducers of IL-6 production [3, 27]. We propose that this is likely the pathway by which MeHg causes IL-6 release in glia.
Acknowledgments
This work was partially supported by funds from Research to Prevent Blindness. Technical support of Ms. Gail Wagner provided by a P30 facility funded by the NIH (NS047546) was greatly appreciated.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Andrei C, Margiocco P, Poggi A, Lotti LV, Torrisi MR, Rubartelli A. Phospholipases C and A2 control lysosome-mediated IL-1 beta secretion: Implications for inflammatory processes. Proc Natl Acad Sci USA. 2004;101:9745–9750. doi: 10.1073/pnas.0308558101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bootman MD, Collins TJ, Mackenzie L, Roderick HL, Berridge MJ, Peppiatt CM. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 2002;16:1145–1150. doi: 10.1096/fj.02-0037rev. [DOI] [PubMed] [Google Scholar]
- 3.Bordin L, Priante G, Musacchio E, Giunco S, Tibaldi E, Clari G, Baggio B. Arachidonic acid-induced IL-6 expression is mediated by PKC alpha activation in osteoblastic cells. Biochemistry. 2003;42:4485–4491. doi: 10.1021/bi026842n. [DOI] [PubMed] [Google Scholar]
- 4.Castoldi AF, Barni S, Turin I, Gandini C, Manzo L. Early acute necrosis, delayed apoptosis and cytoskeletal breakdown in cultured cerebellar granule neurons exposed to methylmercury. J Neurosci Res. 2000;59:775–787. doi: 10.1002/(SICI)1097-4547(20000315)59:6<775::AID-JNR10>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 5.Chang JY. Methylmercury causes glial IL-6 release. Neurosci Lett. 2007;416:217–220. doi: 10.1016/j.neulet.2007.01.076. [DOI] [PubMed] [Google Scholar]
- 6.Chang JY, Tsai PF. IL-6 release from mouse glia caused by MeHg requires cytosolic phospholipase A(2) activatio. Neurosci Lett. 2009;2009:85–89. doi: 10.1016/j.neulet.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang JY, Tsai PF. Prevention of methylmercury-induced mitochondrial depolarization, glutathione depletion and cell death by 15-deoxy-delta-12,14-prostaglandin J(2) Neurotoxicology. 2008;29:1054–1061. doi: 10.1016/j.neuro.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong Y, Benveniste EN. Immune function of astrocyte. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- 9.Edwards JR, Marty MS, Atchison WD. Comparative sensitivity of rat cerebellar neurons to dysregulation of divalent cation homeostasis and cytotoxicity caused by methylmercur. Toxicol Appl Pharmacol. 2005;208:222–232. doi: 10.1016/j.taap.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 10.Garg TK, Chang JY. 15-deoxy-delta 12, 14-Prostaglandin J2 prevents reactive oxygen species generation and mitochondrial membrane depolarization induced by oxidative stress. BMC Pharmacol. 2004;4:6. doi: 10.1186/1471-2210-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garg TK, Chang JY. Methylmercury causes oxidative stress and cytotoxicity in microglia: Attenuation by 15-deoxy-delta 12, 14-Prostaglandin J2. J Neuroimmunol. 2006;171:17–28. doi: 10.1016/j.jneuroim.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 12.Hare MF, Atchison WD. Methylmercury mobilizes Ca++ from intracellular stores sensitive to inositol 1,4,5-trisphosphate in NG108-15 cells. J Pharmacol Exp Ther. 1995;272:1016–1023. [PubMed] [Google Scholar]
- 13.Hare MF, McGinnis KM, Atchison WD. Methylmercury increases intracellular concentrations of Ca++ and heavy metals in NG108-15 cells. J Pharmacol Exp Ther. 1993;266:1626–1635. [PubMed] [Google Scholar]
- 14.Holliday J, Parsons K, Curry J, Lee SY, Gruol DL. Cerebellar granule neurons develop elevated calcium responses when treated with interleukin-6 in culture. Brain Res. 1995;673:141–148. doi: 10.1016/0006-8993(94)01417-g. [DOI] [PubMed] [Google Scholar]
- 15.Ihara H, Hirukawa K, Goto S, Togari A. ATP-stimulated interleukin-6 synthesis through P2Y receptors on human osteoblasts. Biochem Biophys Res Commun. 2005;326:329–334. doi: 10.1016/j.bbrc.2004.11.037. [DOI] [PubMed] [Google Scholar]
- 16.Kang MS, Jeong JY, Seo JH, Jeon HJ, Jung KM, Chin MR, Moon CK, Bonventre JV, Jung SY, Kim DK. Methylmercury-induced toxicity is mediated by enhanced intracellular calcium through activation of phosphatidylcholine-specific phospholipase C. Toxicol Appl Pharmacol. 2006;216:206–215. doi: 10.1016/j.taap.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi D, Ohkubo S, Nakahata N. Contribution of extracellular signal-regulated kinase to UTP-induced interleukin-6 biosynthesis in HaCaT keratinocytes. J Pharmacol Sci. 2006;102:368–376. doi: 10.1254/jphs.fp0060669. [DOI] [PubMed] [Google Scholar]
- 18.Kondo A, Koshihara Y, Togari A. Signal transduction system for interleukin-6 synthesis stimulated by lipopolysaccharide in human osteoblasts. J Interferon Cytokine Res. 2001;21:943–950. doi: 10.1089/107999001753289550. [DOI] [PubMed] [Google Scholar]
- 19.Leslie CC. Properties and regulation of cytosolic phospholipase A2. J Biol Chem. 1997;272:16709–16712. doi: 10.1074/jbc.272.27.16709. [DOI] [PubMed] [Google Scholar]
- 20.Mazerik JN, Hagele T, Sherwani S, Ciapala V, Butler S, Kuppusamy ML, Hunter M, Kuppusamy P, Marsh CB, Parinandi NL. Phospholipase A2 activation regulates cytotoxicity of methylmercury in vascular endothelial cells. Int J Toxicol. 2007;26:553–569. doi: 10.1080/10915810701707759. [DOI] [PubMed] [Google Scholar]
- 21.Monick MM, Carter AB, Gudmundsson G, Mallampalli R, Powers LS, Hunninghake GW. A phosphatidylcholine-specific phospholipase C regulates activation of p42/44 mitogen-activated protein kinases in lipopolysaccharide-stimulated human alveolar macrophages. J Immunol. 1999;162:3005–3012. [PubMed] [Google Scholar]
- 22.Qiu Z, Sweeney DD, Netzeband JG, Gruol DL. Chronic interleukin-6 alters NMDA receptor-mediated membrane responses and enhances neurotoxicity in developing CNS neurons. J Neurosci. 1998;18:10445–10456. doi: 10.1523/JNEUROSCI.18-24-10445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rees DA, Lewis BM, Lewis MD, Francis K, Scanlon MF, Ham J. Adenosine-induced IL-6 expression in pituitary folliculostellate cells is mediated via A2b adenosine receptors coupled to PKC and p38 MAPK. Br J Pharmacol. 2003;140:764–772. doi: 10.1038/sj.bjp.0705488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarafian TA. Methyl mercury increases intracellular Ca2+ and inositol phosphate levels in cultured cerebellar granule neurons. J Neurochem. 1993;61:648–657. doi: 10.1111/j.1471-4159.1993.tb02169.x. [DOI] [PubMed] [Google Scholar]
- 25.Shanker G, Mutkus LA, Walker SJ, Aschner M. Methylmercury enhances arachidonic acid release and cytosolic phospholipase A2 expression in primary cultures of neonatal astrocytes. Brain Res Mol Brain Res. 2002;106:1–11. doi: 10.1016/s0169-328x(02)00403-5. [DOI] [PubMed] [Google Scholar]
- 26.Snetkov VA, Hapgood KJ, McVicker CG, Lee TH, Ward JP. Mechanisms of leukotriene D4-induced constriction in human small bronchioles. Br J Pharmacol. 2001;133:243–252. doi: 10.1038/sj.bjp.0704076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spangelo BL, Jarvis WD. Lysophosphatidylcholine stimulates interleukin-6 release from rat anterior pituitary cells in vitro. Endocrinology. 1996;137:4419–4426. doi: 10.1210/endo.137.10.8828503. [DOI] [PubMed] [Google Scholar]
- 28.Sun GY, Xu J, Jensen MD, Yu S, Wood WG, Gonzalez FA, Simonyi A, Sun AY, Weisman GA. Phospholipase A2 in astrocytes: responses to oxidative stress, inflammation, and G protein-coupled receptor agonists. Mol Neurobiol. 2005;31:27–41. doi: 10.1385/MN:31:1-3:027. [DOI] [PubMed] [Google Scholar]
- 29.Tao FC, Shah S, Pradhan AA, Tolloczko B, Martin JG. Enhanced calcium signaling to bradykinin in airway smooth muscle from hyperresponsive inbred rats. Am J Physiol Lung Cell Mol Physiol. 2003;284:L90–09. doi: 10.1152/ajplung.00023.2002. [DOI] [PubMed] [Google Scholar]
- 30.Venkatachalam K, van Rossum DB, Patterson RL, Ma HT, Gill DL. The cellular and molecular basis of store-operated calcium entry. Nat Cell Biol. 2002;4:E263–272. doi: 10.1038/ncb1102-e263. [DOI] [PubMed] [Google Scholar]
- 31.Verity MA, Sarafian T, Pacifici EH, Sevanian A. Phospholipase A2 stimulation by methyl mercury in neuron culture. J Neurochem. 1994;62:705–714. doi: 10.1046/j.1471-4159.1994.62020705.x. [DOI] [PubMed] [Google Scholar]