

A decrease in N-glycan mannose content significantly diminishes in vivo glucosidase II–mediated deglucosylation rates but does not affect in vivo UDP-glucose:glycoprotein glucosyltransferase–mediated glucosylation, thus increasing the possibility of displaying monoglucosylated structures able to interact with calnexin/calreticulin for longer time periods.
Abstract
Glucosidase II (GII) sequentially removes the two innermost glucose residues from the glycan (Glc3Man9GlcNAc2) transferred to proteins. GII also participates in cycles involving the lectin/chaperones calnexin (CNX) and calreticulin (CRT) as it removes the single glucose unit added to folding intermediates and misfolded glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase (UGGT). GII is a heterodimer in which the α subunit (GIIα) bears the active site, and the β subunit (GIIβ) modulates GIIα activity through its C-terminal mannose 6-phosphate receptor homologous (MRH) domain. Here we report that, as already described in cell-free assays, in live Schizosaccharomyces pombe cells a decrease in the number of mannoses in the glycan results in decreased GII activity. Contrary to previously reported cell-free experiments, however, no such effect was observed in vivo for UGGT. We propose that endoplasmic reticulum α-mannosidase–mediated N-glycan demannosylation of misfolded/slow-folding glycoproteins may favor their interaction with the lectin/chaperone CNX present in S. pombe by prolonging the half-lives of the monoglucosylated glycans (S. pombe lacks CRT). Moreover, we show that even N-glycans bearing five mannoses may interact in vivo with the GIIβ MRH domain and that the N-terminal GIIβ G2B domain is involved in the GIIα–GIIβ interaction. Finally, we report that protists that transfer glycans with low mannose content to proteins have nevertheless conserved the possibility of displaying relatively long-lived monoglucosylated glycans by expressing GIIβ MRH domains with a higher specificity for glycans with high mannose content.
INTRODUCTION
Protein N-glycosylation involves the initial transfer of a glycan (Glc3Man9GlcNAc2; Figure 1) from a dolichol (Dol)-P-P derivative to Asn residues in the consensus sequence Asn-X-Ser/Thr in proteins in the endoplasmic reticulum (ER). Transfer is immediately followed by the removal of the external glucose unit (residue n, Figure 1) by glucosidase I (GI) and the removal of the two remaining glucose residues (residues l and m, Figure 1) by glucosidase II (GII). One or more mannose residues may be removed in the ER by ER mannosidase(s). Both GII-mediated cleavages are determining factors in the quality control of glycoprotein folding in the ER. Monoglucosylated glycan-bearing glycoproteins may interact with calnexin (CNX) and/or calreticulin (CRT), two highly homologous ER lectin and chaperones that enhance folding efficiency by preventing aggregation and facilitating correct disulfide bond formation through their interaction with ERp57, a protein disulfide isomerase. Furthermore, the interaction of the folding intermediates and misfolded glycoproteins with the lectin and chaperones prevents exit from the ER to the Golgi. The second GII-mediated cleavage, which generates unglucosylated molecules, abolishes the glycoprotein-lectin and chaperone interaction, thus allowing glycoproteins to pursue their transit through the secretory pathway. If not yet properly folded, however, glycoproteins may be reglucosylated by the UDP-Glc:glycoprotein glucosyltransferase (UGGT), a soluble ER luminal enzyme that specifically glucosylates nonnative conformers and regenerates monoglucosylated glycans. These, in turn, interact again with the lectin and chaperones. Cycles of reglucosylation and deglucosylation catalyzed by the opposing activities of UGGT and GII continue until the glycoproteins acquire their native tertiary structure or, if unable to properly fold, are driven to the cytosol for proteasomal degradation (Caramelo and Parodi, 2007, 2008; D'Alessio et al., 2010; Parodi, 2000). Cell-free assays have shown that UGGT-mediated glucosylation decreases drastically when the N-glycan mannose content is reduced (Sousa et al., 1992).
FIGURE 1:

Glycan structures. The structure depicted is that of the full-length glycan transferred to Asn residues in N‑glycosylation. Lettering (a, b, c…) follows the order of addition of the monosaccharides in the synthesis of the Dol-P-P derivatives. Alg genes involved in the synthetic process are indicated. GI removes residue n, and GII removes residues m and l. UGGT re-adds residue l. Glycans have the following structures: M9, residues a–k; M7, a–i; M6, a–h; and M5, a–g. The respective mono-, di-, and triglucosylated derivatives have, additionally, residues l, l–m, and l–n. Because glycans with nine, seven, or six mannoses were released with Endo H, they lack residue a when separated by paper chromatography or HPLC. In contrast, glycans with five mannoses were released with N-glycanase and therefore have residue a. Arm A comprises residues d, f, g, and l–n, arm B comprises residues h and I, and arm C comprises residues j and k.
GII is an ER-soluble heterodimeric protein composed of catalytic (GIIα) and regulatory (GIIβ) subunits (Trombetta et al., 1996; D'Alessio et al., 1999). The latter subunit displays at its C terminus a domain highly homologous to the lectin domain of the mammalian mannose 6-phosphate (Man6P) receptor (MRH, for Man6P receptor homologous domain), which is involved in the transport of glycoprotein enzymes from the Golgi to lysosomes (Munro, 2001). We and others have shown that point mutations of conserved amino acids in the GIIβ MRH domain that are involved in mannose recognition in the Man6P receptor sharply reduce in vivo deglucosylation rates, thus strongly suggesting that the MRH domain somehow accelerates enzymatic reactions upon recognition of the mannose units in the glycan (Quinn et al., 2009; Stigliano et al., 2009). As has been shown for UGGT, cell-free assays have shown that the removal of mannose residues from the glycan results in a decrease in the deglucosylation rates and in the affinity of the GIIβ MRH domain for glycans (Grinna and Robbins, 1980; Totani et al., 2006; Hu et al., 2009). It was suggested, therefore, that misfolded glycoproteins could exit from futile CNX/CRT cycles if a more pronounced decrease in in vivo reglucosylation than deglucosylation occurred upon ER mannosidase removal of the N-glycan mannose units (Cabral et al., 2001). Interestingly, a lectin (Yos9p in yeasts, OS-9 in humans) that is apparently involved in driving misfolded glycoproteins to proteasomal degradation also displays an MRH-like domain, but, in contrast to the MRH domain of GIIβ, the Yos9p/OS-9 MRH-like domain has a higher affinity for glycans with a lower mannose content (Hosokawa et al., 2009; Quan et al. 2009, Satoh et al., 2010). This finding is consistent with a model in which long ER permanence of misfolded glycoproteins results in the generation of partially demannosylated molecules that can be driven to the cytosol.
The ER is a crowded environment in which the protein content may reach concentrations as high as 200–300 mg/ml. Mimicking crowded conditions by adding different proteins (bovine serum albumin, RNAse A) or polyethylene glycol to enzymatic assays has revealed that the removal by GII of the more internal (residue l, Figure 1) but not the middle glucose (residue m, Figure 1) increases sharply in crowded environments. These experimental conditions also triggered a conformational change in GII. A similar activating effect was found for mannose removal by an α-1,2-mannosidase (Totani et al., 2008). Cell-free assays may thus not always reflect what occurs in vivo in the processing of N-glycans in the ER.
The main purpose of this work was to examine in vivo whether variations in the opposing activities of GII and UGGT triggered by removal of mannoses favored generation of unglucosylated or of monoglucosylated N-glycans; that is, whether demannosylation resulted in the exit or a prolonged permanence of misfolded and slow-folding glycoproteins in the CNX/CRT cycles. The system used for these studies was the fission yeast Schizosaccharomyces pombe because of the of availability of genetic and biochemical tools and because this microorganism displays an ER quality control mechanism of glycoprotein folding similar to that of mammalian cells (D'Alessio et al., 1999). Saccharomyces cerevisiae lacks UGGT activity and therefore also the cycles mentioned above (Fernández et al., 1994). Contrary to most other eukaryotic cells, both yeast species lack CRT but express CNX. Therefore cycles involving unglucosylated and monoglucosylated glycans in S. pombe will be referred to as CNX cycles.
Here we show that, in live S. pombe cells, the removal of mannoses from the B and/or C branches of the transferred glycan (Figure 1) results in a reduced glucose removal by GII but not in a reduced glucose addition by UGGT. We suggest that ER α-mannosidase–mediated glycoprotein demannosylation would prolong the half-lives of monoglucosylated glycans, thus preventing exit to the Golgi of misfolded and slow-folding glycoproteins and increasing their possibility of forming proper native structures.
RESULTS
In vivo N-glycan deglucosylation diminishes as a function of the mannose content
To study whether a reduced mannose content in the protein-linked N-glycan affects GII glucose trimming activity in vivo, we constructed a series of fission yeast S. pombe mutants transferring truncated N-glycans to nascent proteins. Except where otherwise stated, the yeast cells used displayed a Δalg10 mutation. Because Alg10p catalyzes the transfer of the last glucose from Dol-P-Glc to Glc2Man9GlcNAc2-P-P-Dol, the mutants used transferred N-glycans with only two glucoses (Table 1 and Figure 1). The rationale for introducing this mutation was to eliminate any possible effect of the N-glycan mannose content on GI activity, which could affect the relative levels of N-glycans bearing two, one, or no glucose units to be observed.
TABLE 1:
Structure of N-glycans transferred to proteins in S. pombe mutants.
| N-Glycans Recognized by GI | N-Glycans Recognized by GII | N-Glycans Recognized by GT | ||||||
| Mutant | Transferred oligosaccharide | Structure | Mutant | Transferred oligosaccharide | Structure | Mutant | Transferred oligosaccharide | Structure |
| Wild type | G3M9 | 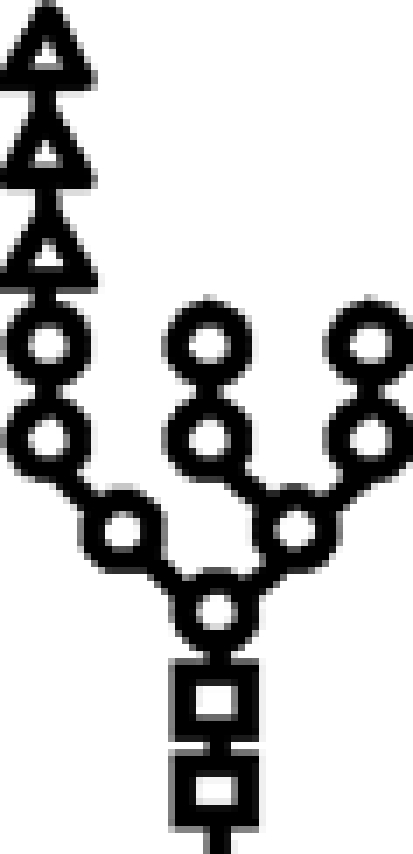 |
Δalg10 | G2M9 |  |
Δalg6 | M9 | 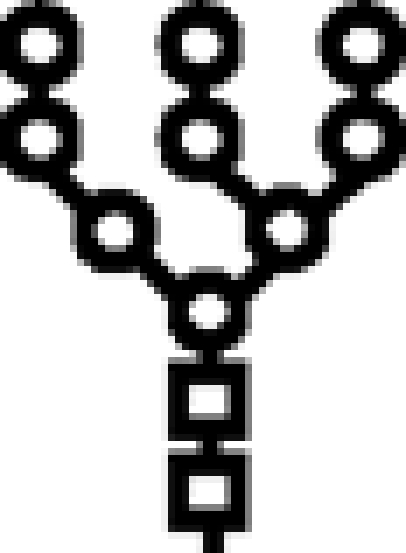 |
| Δalg12 | G3M7 | 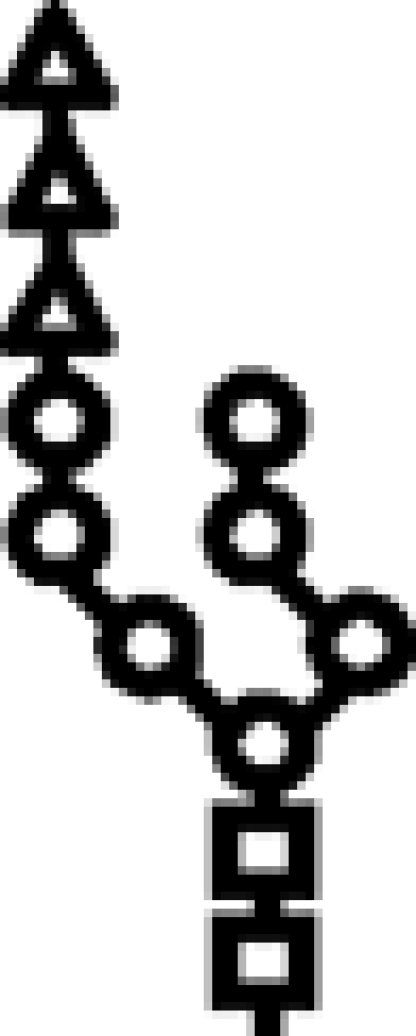 |
Δalg10/Δalg12 | G2M7 |  |
Δalg6/Δalg12 | M7 | 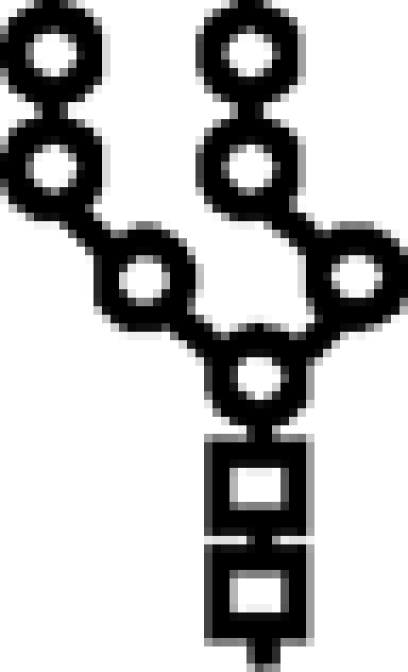 |
| Δalg9 | G3M6 |  |
Δalg10/Δalg9 | G2M6 |  |
Δalg6/Δalg9 | M6 |  |
| Δalg3 | G3M5 | 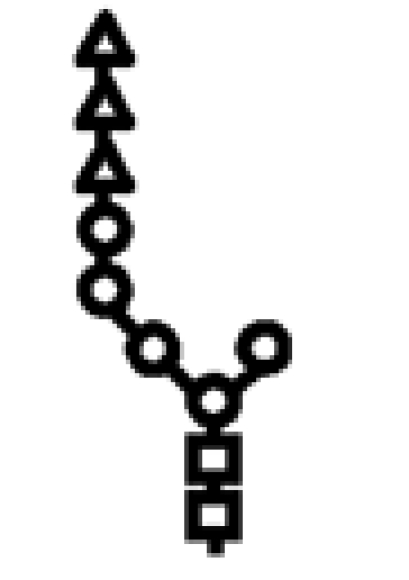 |
Δalg10/Δalg3 | G2M5 |  |
Δalg6/Δalg3 | M5 | 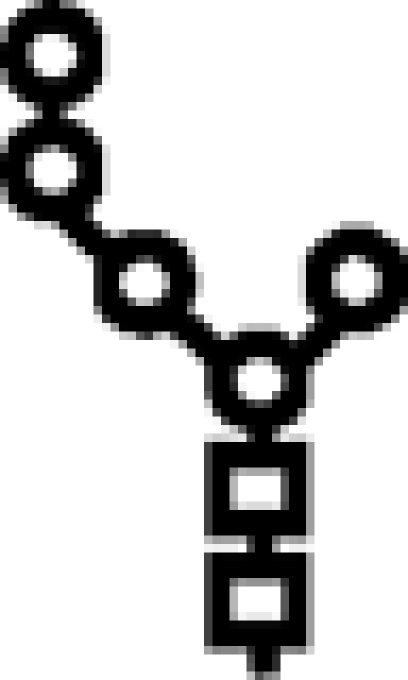 |
In mutants lacking GIIβ gene, the glycan transferred is the same. Triangle: Glc; circle: Man; square: GlcNAc.
Cells were incubated with [14C]Glc for 15 min in the presence of 5 mM dithiothreitol (DTT), and whole-cell N-glycans were isolated and analyzed as described in Materials and Methods. The total incorporation of the label into the N-glycans was linear during the incubation time, and the presence of DTT prevented the passage of glycoproteins to the Golgi, hindering further extension of the recently synthesized N-glycans (Fernández et al., 1998). Addition in the Golgi of many mannose and galactose units would have prevented the measurement of the proportion of N-glycans bearing two, one, or no glucose units. The short incubation time of the cells with 5 mM DTT (a total of 20 min) ensured that the unfolded protein response triggered by the addition of the drug did not affect the levels of GII and UGGT (Pincus et al., 2010).
The N-glycan patterns obtained with Δalg10, Δalg10/Δalg12, Δalg10/Δalg9, and Δalg10/Δalg3 (i.e., transferring Glc2Man9GlcNAc2, Glc2Man7GlcNAc2, Glc2Man6GlcNAc2, and Glc2Man5GlcNAc2, which will be respectively abbreviated as G2M9, G2M7, G2M6, and G2M5; the structures of all glycans mentioned in this article are given in Table 1 and Figure 1) revealed that, whereas deglucosylation of G2M9 was so rapid that no glucose-containing glycans were detected, the amount of glucosylated glycans increased as the N-glycan mannose content decreased (Figure 2, A–E). These results agree with those of previous cell-free assays in which a decrease in the glycan mannose content resulted in a decrease in both GII-catalyzed reaction rates (Grinna and Robbins, 1980; Totani et al., 2006). The relative rates of deglucosylation of the diglucosylated and monoglucosylated glycans cannot be calculated from the results shown in Figure 2, A–E, because of the continuous refeeding of diglucosylated glycans during the 15-min labeling period, but a decrease in mannose content clearly reduces the conversion of glucosylated glycans to unglucosylated glycans (Figure 2E). Mutants that transfer G2M8 could not be obtained because the same mannosyltransferase (Alg9p) is responsible for the addition of the seventh and ninth mannose residues to the Dol-P-P–linked derivative (Figure 1).
FIGURE 2:

Glycan patterns synthesized by mutants transferring diglucosylated glycans containing nine to five mannoses. (A) G2M9 (Δalg10); (B) G2M7 (Δalg10/Δalg12); (C) G2M6 (Δalg10/Δalg9), and (D) G2M5 (Δalg10/Δalg3). The structures of the glycans transferred by each mutant are indicated in the corresponding panels. (E) Quantification of the relative amounts of the di-, mono-, and unglucosylated species from panels A–D. In panel A, the label in M8 was added to that in M9 to account for unglucosylated species.
The effect of N-glycan mannose content on the deglucosylation levels of mutants lacking the GII regulatory subunit (GIIβ)
The N-glycan pattern experiments were repeated with mutant strains that additionally lacked GIIβ (strains Δalg10/ΔGIIβ, Δalg10/Δalg12/ΔGIIβ, Δalg10/Δalg9/ΔGIIβ, and Δalg10/Δalg3/ΔGIIβ). The absence of the regulatory subunit resulted in a decrease in GIIα content as judged by Western blot analysis (Figure 3, lanes 3, 6, 9, and 12), confirming the role of GIIβ in GIIα ER retention ( Stigliano et al., 2009). Similar N-glycan patterns were observed for mutants transferring G2M7, G2M6, and G2M5 and lacking GIIβ, demonstrating that the mannose content did not influence the deglucosylation of those glycans by GIIα in the absence of the regulatory subunit, but the pattern produced by the mutant transferring G2M9 indicated that the glycan containing the full complement of mannoses was deglucosylated at a slower rate because almost no glycans devoid of glucoses were observed (Figure 4, A–E). Mannose residues j and/or k (Figure 1) probably interact with either the GIIα active site or the glucose units, thus reducing the rates of deglucosylation.
FIGURE 3:

ER GIIα content in S. pombe cells expressing wild-type, mutant, or no GIIβ. Each lane was loaded with 250 μg of microsomal proteins of ΔGIIβ cells or ΔGIIβ cells expressing exogenous GIIβ or GIIβ-MRH* (MRH*). The membrane was blotted using mouse polyclonal anti-GIIα subunit (1:500) and rabbit polyclonal anti-CNX (1:100,000) primary antibodies. Goat HRP anti–mouse or –rabbit IgG (1:5000 and 1:30,000, respectively) were used as secondary antibodies. Reactions were detected by chemiluminescence.
FIGURE 4:

Glycan patterns synthesized by mutants transferring diglucosylated glycans containing nine to five mannoses and lacking GIIβ. (A) G2M9 (Δalg10/ΔGIIβ); (B) G2M7 (Δalg10/Δalg12/ΔGIIβ); (C) G2M6 (Δalg10/Δalg9/ΔGIIβ); (D) G2M5 (Δalg10/Δalg3/ΔGIIβ). The structures of the glycans transferred by each mutant are indicated in the corresponding panels. (E) Quantification of the relative amounts of the di-, mono-, and unglucosylated species from panels A–D.
GIIβ was not essential for the removal of the innermost glucose unit as has been previously suggested (Wilkinson et al., 2006); no such requirement was observed for G2M7, G2M6, or G2M5 (Figure 4, B–D). Furthermore, as already reported, a longer incubation of the ΔGIIβ cells transferring G3M9 led to the production of M9 (Stigliano et al., 2009). The interaction of mannoses j and/or k (Figure 1) with the GIIβ MRH domain likely results not only in the presentation of the glycan to the GIIα active site as previously speculated (Stigliano et al., 2009) but also in the elimination of the interaction of those mannose residues with either the active site or the glucoses as suggested earlier in text.
G2M9 to G2M5 N-glycans are recognized in vivo by the GIIβ MRH domain
We have previously reported that the poor deglucosylation levels observed in mutants lacking the regulatory subunit and transferring G3M9 could be restored to wild-type levels by complementation with exogenous GIIβ, whereas the expression of GIIβ with mutations of residues in the MRH domain that have been shown to interact with mannoses in the Man6P receptor failed to correct the deficiency, even though wild-type GIIα levels in the ER were totally restored (Stigliano et al., 2009). In this work we performed the same experiment but with mutants transferring G2M7, G2M6, and G2M5 (Δalg10/Δalg12/ΔGIIβ, Δalg10/Δalg9/ΔGIIβ, and Δalg10/Δalg3/ΔGIIβ mutants). Expression of wild-type GIIβ restored the N-glycan patterns observed in cells expressing endogenous GIIβ (compare Figure 5, A–D, with Figure 2, B–E). In contrast, expression of GIIβ with mutations Y462F/E456Q in the MRH domain (hence referred to as GIIβ-MRH*) failed to fully restore the original patterns, although the difference between mutants expressing wild type and mutant GIIβ was much higher for mutants transferring N-glycans with a higher mannose content (compare Figure 5, E–H, with Figure 5, A–D). Western blot analysis showed that the expression of either GIIβ or GIIβ-MRH* restored GIIα to similar levels (Figure 3, lanes 4 and 5, 7 and 8, 11 and 12, and 13 and 14), and the results shown in Figure 5, as well as those previously reported, indicate that the MRH domain recognizes N-glycans bearing from nine to five mannose units in vivo, albeit with different affinities.
FIGURE 5:

Glycan patterns synthesized by mutants transferring diglucosylated glycans containing seven to five mannoses and lacking GIIβ but expressing exogenous wild-type GIIβ (A–D) or exogenous GIIβ with a mutated MRH domain (E–H). (A) G2M7 (Δalg10/Δalg12/ΔGIIβ + pGIIβ); (B) G2M6 (Δalg10/Δalg9/ΔGIIβ + pGIIβ); (C) G2M5 (Δalg10/Δalg3/ΔGIIβ + pGIIβ); (E) G2M7 (Δalg10/Δalg12/ΔGIIβ + pGIIβ-MRH*); (F) G2M6 (Δalg10/Δalg9/ΔGIIβ + pGIIβ-MRH*); (G) G2M5 (Δalg10/Δalg3/ΔGIIβ + pGIIβ-MRH*). The structures of the glycans transferred by each mutant are indicated in the corresponding panels. (D and H) Quantification of the relative amounts of the glycans shown in panels A–C (D) and E–G (H).
The G2B domain is involved in the GIIα–GIIβ interaction in S. pombe
In addition to the C-terminal MRH domain, a domain near the N terminus that is conserved among GIIβ subunits of different species (G2B, Supplemental Figure S1) has been reported to be involved in the interaction of both GII subunits in mammalian cells and in the removal of the middle glucose in S. cerevisiae (Arendt and Ostergaard, 2000; Quinn et al., 2009). A series of mutations were introduced in the S. cerevisiae GIIβ G2B domain. Mutation E132A did not prevent the GIIα–GIIβ interaction but did result in reduced G1M9 production in vivo. We mutated the corresponding amino acid (E114A) and amino acid E73A in the S. pombe GIIβ G2B domain (Supplemental Figure S1). Microsomes of S. pombe mutants lacking both GIIα and GIIβ (ΔGIIαβ) but expressing GIIβ-E73A or GIIβ-E114A (source of GIIβ) failed to complement the trimming of G1M9 by microsomes of S. pombe cells expressing only GIIα in the ER (ΔGIIαβ + pGIIα.VDEL) (Figure 6A). Similar results were obtained with the same mutants expressing the mutated GIIβ MRH domain (ΔGIIαβ + pGIIβ-MRH*). In contrast, microsomes of S. pombe mutants expressing only wild-type GIIβ restored the ability of microsomes expressing only GIIα to hydrolyze G1M9 (Figure 6A).
FIGURE 6:

Mutations in the GIIβ G2B domain affect the GIIα–GIIβ interaction. (A) Complementation assay between GIIα and GIIβ. Microsomes from ΔGIIαβ S. pombe cells transformed with pGIIαVDEL (the source of the GIIα subunit) were preincubated in the presence of 1% Triton X-100 with microsomes from ΔGIIαβ cells expressing wild-type GIIβ (pGIIβ), GIIβ with a mutated MRH domain (pGIIβ-MRH*), or mutated G2B domain (pGIIβ-E73A or pGIIβ-E114A) (sources of the GIIβ subunit). GII activity assays with [14C-Glc]G1M9 were performed as described in Materials and Methods. (B) GII activity in microsomal fractions of S. pombe wild-type, ΔGIIβ mutant cells or the same mutants expressing exogenous wild-type GIIβ or GIIβ with mutated MRH or G2B domains. Assays were performed with pNPG or G1M9 as substrates. In both cases, the activity was stated relative to that of the wild-type strain (100%). (C) Immunodetection of GIIα and GIIβ in microsomal fractions of S. pombe. Each lane was loaded with 250 μg of microsomal proteins from wild type, ΔGIIα or ΔGIIβ transformed with vector alone (–), wild-type GIIβ (GIIβ), or GIIβ bearing mutations in the MRH (MRH*) or G2B domains (E73A and E113A). The membrane was blotted using mouse polyclonal anti-GIIα subunit (1:500) or -GIIβ subunit (1:1000). Goat HRP anti-mouse 1:5000 was used as the secondary antibody. Reactions were detected by chemiluminescence.
We have previously reported that measurements of microsomal GII activity with the small molecule pNPG (p-nitrophenyl α-d-glucopyranoside) reflect ER GIIα content. We have also previously shown that GIIβ is involved in GIIα ER localization (Stigliano et al., 2009). Mutations in G2B but not in the MRH domains affected microsomal GII activity when measured with pNPG as the substrate, indicating that the former, but not the latter, mutations influence ER GIIα retention. In contrast, mutations in both domains influenced GII activity when G1M9 was the substrate: Microsomes of S. pombe mutants lacking GIIβ but expressing GIIβ with mutations in the G2B domain (ΔGIIβ + pGIIβ-E114A and ΔGIIβ + pGIIβ-E73A) were inactive toward G1M9 as a substrate, as were the same mutants expressing GIIβ-MRH* (Figure 6B). As expected, Western blot analysis revealed that the E114A and E73A mutations in GIIβ resulted in reduced ER GIIα levels that were similar to those present in ΔGIIβ mutants, despite the fact that the G2B mutation only slightly affected GIIβ levels (Figure 6C). In contrast, the expression of wild-type GIIβ or GIIβ with mutations in the MRH domain fully restored GIIα levels. A lower band that reacted with the GIIβ antibodies appeared in cells expressing GIIβ with mutations in the G2B domain. We have not yet identified this faster migrating protein, but it likely resulted from a proteolytic cleavage of perhaps a less conformationally stable mutant subunit.
The N-glycan patterns obtained by in vivo labeling of cells transferring G2M9 or G2M6 and lacking endogenous GIIβ but expressing GIIβ with a mutation in the G2B domain (Δalg10/ΔGIIβ + pGIIβ-E114A and Δalg10/Δalg9/ΔGIIβ + pGIIβ-E114A) were similar to those produced by mutant cells transferring the same glycans and lacking the regulatory subunit (Δalg10/ΔGIIβ and Δalg10/Δalg9/ΔGIIβ) (Supplemental Figure S2; compare Figures S2A with 4A, S2B with 4C, and S2C with 4E). Similar results were obtained for the E73A mutation (unpublished results).
These results show that the G2B domain is indeed involved in the GIIα–GIIβ interaction in S. pombe. We did not obtain evidence that it also participates in the removal of the middle glucose in this yeast as cells expressing the E114A mutant of the regulatory subunit were able to deglucosylate G2M9 to G1M9 and G2M6 to G1M6 (Supplemental Figure S2).
Does the N-glycan mannose content influence GI-mediated deglucosylation?
The transfer of glycans to proteins containing three glucoses but fewer than nine mannoses occurs in certain protists, such as Tetrahymena pyriformis, in which the glycan involved in protein glycosylation has the composition Glc3Man5GlcNAc2 (G3M5, residues a-g, l-n, Figure 1) (Yagodnik et al., 1987). There are conflicting reports on the in vitro effect of N-glycan mannose content on GI-mediated deglucosylation. Similar to GII, deglucosylation rates for the mammalian enzyme have been reported to decrease as the number of mannoses decreases from nine to five. It has also been reported that the rate for the N-glycan bearing five mannose units is similar to that of the compound bearing the full complement of mannose units, whereas glycans with six or seven mannoses are deglucosylated at lower rates (Grinna and Robbins, 1980; Schweden et al., 1986). In contrast, no difference in the deglucosylation rates of glycans displaying from nine to five mannoses was observed in the case of a plant GI (Zeng and Elbein, 1998).
No difference was observed between the N-glycan patterns of live S. pombe cells transferring G3M7, G3M6, and G3M5 (Δalg12, Δalg9, and Δalg3 mutants; see Table 1 and Supplemental Figure S3) and those of cells transferring the corresponding diglucosylated compounds (Δalg10/Δalg12, Δalg10/Δalg9, and Δalg10/Δalg3 mutants; Figure 2). In S. pombe, one of the following situations applies: The mannose content does not influence removal of the external glucose by GI, the effect of mannose content is lower than for GII-mediated deglucosylation, or the higher activity of GI relative to GII compensates for all possible effects.
The effect of N-glycan mannose content on in vivo UGGT-mediated N-glycan glucosylation
As mentioned above, UGGT-mediated glucosylation rates decrease in cell-free assays as the number of mannoses in the acceptor glycans decreases (Sousa et al., 1992). To determine whether a similar effect occurs in vivo, we preincubated S. pombe mutants transferring M9, M7, M6, and M5 (Δalg6, Δalg6/Δalg12, Δalg6/Δalg9, and Δalg6/Δalg3 mutants; see Table 1) with 5 mM N-methyl 1-deoxynojirimycin (NMDNJ) for 60 min before incubating the cells with [14C]glucose for 15 min in the presence of 5 mM DTT. NMDNJ is a cell-permeable compound with an half maximal inhibitory concentration for GII inhibition of 3–5 μM. Alg6p catalyzes the transfer of the first glucose from Dol-P-Glc to the Dol-P-P-derivative (Figure 1). Similar ratios of monoglucosylated and nonglucosylated glycans were obtained for all of the mutants (Figure 7, A–D and I), demonstrating that the mannose content did not influence the in vivo UGGT activity. To confirm this assertion, we performed the same experiments in the absence of NMDNJ. No G1M9 formation was observed for the Δalg6 mutant, but detectable amounts of the respective monoglucosylated N-glycans were observed for the other mutants as their mannose contents decreased (Figure 7, E–H and J). Because a decrease in N-glycan mannose content significantly diminished in vivo GII-mediated deglucosylation, the results of these experiments confirm that the mannose content either does not affect in vivo UGGT-mediated glucosylation or that its effect is much lower than that on GII activity, and suggest that GII may be an in vivo regulator of misfolded and slow-folding glycoprotein permanence in the ER as these glycoproteins are characterized by a detectable ER mannosidase(s)-catalyzed N-glycan demannosylation and, therefore, an increased possibility of displaying monoglucosylated structures for longer time periods.
FIGURE 7:

Glycan patterns synthesized by mutants transferring unglucosylated glycans containing nine to five mannoses with inhibition of deglucosylation (plus 5 mM NMDNJ) (A–D) or without inhibition of deglucosylation (minus NMDNJ) (E–H). (A and E) M9 (Δalg6); (B and F) M7 (Δalg6/Δalg12); (C and G) M6 (Δalg6/Δalg9); (D and H) M5 (Δalg6/Δalg3). The structures of the glycans transferred by each mutant are indicated in the corresponding panels. (I and J) Quantification of the relative amounts of the glucosylated and nonglucosylated labeled glycans from panels A–D (I) and E–H (J). In panel E, the label in M8 was added to that of M9. The label in the unidentified peak from panel F was omitted for quantification.
The curious case of Leishmania mexicana
Is the prolonged existence of monoglucosylated glycans due to an ER demannosylation-triggered loss of GII activity relevant for preventing the exit of misfolded species and/or enhancing the probability that glycoproteins with a long and difficult folding process will properly fold? To attempt to answer this question, we first studied the N-glycan specificity of L. mexicana GII. Many years ago, we described the transfer of unglucosylated glycans to proteins by trypanosomatid protozoa (M9, M7, or M6, depending on the species) (Parodi, 1993). These protozoa lack the Dol-P-Glc–synthesizing enzyme and, depending on the species, one or more Dol-P-Man–dependent mannosyltransferases (de la Canal and Parodi, 1987). Furthermore, Samuelson et al. (2005) demonstrated that the glycan-related enzymatic pattern displayed by trypanosomatids represented a secondary loss of glycosyltransferases from an ancestor that displayed all of them. For L. mexicana, we proposed the following ER N-glycosylation–N-glycan processing reactions (Parodi et al., 1984a):
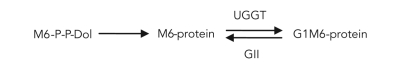 |
No N-glycan demannosylation was detected. Together with the low mannose content of the glycan, this result indicated that ER-demannosylation–triggered loss of GII activity did not occur in this protozoon. The L. mexicana genome has putative GIIα and GIIβ subunit–encoding genes, and the latter also has G2B and MRH domains (Supplemental Figure S1). Has the L. mexicana GIIβ MRH domain evolved to a structure with a higher affinity for smaller glycans, or has it conserved the higher affinity for M9 of the ancestor microorganism? Proteins with MRH domains with higher affinities for glycans shorter than M9 do exist. One example is Yos9p/OS-9, which displays MRH domains with a higher affinity for shorter glycans, although the glycan structure is not identical to the M6 involved in protozoon N-glycosylation. If the L. mexicana GIIβ MRH domain had a higher affinity for smaller glycans, this would eliminate the possibility of preventing Golgi exit of misfolded and slow-folding glycoproteins by extending the existence of monoglucosylated glycans. In contrast, maintenance of the higher affinity for larger glycans would result in a slow deglucosylation of all glycoproteins, including misfolded and slow-folding species.
A comparison of the deglucosylation of G1M9 and G1M6 having the same specific activities by an L. mexicana microsomal soluble fraction revealed that the parasite GIIβ MRH has maintained the original N-glycan specificity (Figure 8). Because these glycans were labeled at both the glucose and mannose residues, we confirmed that we were measuring deglucosylation and not demannosylation by adding 1-deoxymannojirimycin (an inhibitor of ER mannosidase) alone or in combination with NMDNJ to the incubation mixtures. Substrate deglycosylation was completely inhibited only when 1-deoxymannojirimycin was added in combination with NMDNJ (Figure 8). Our results suggest that the GII-mediated regulation of the existence of monoglucosylated glycans is an important factor in ER glycoprotein folding quality control.
FIGURE 8:

Glucose release from either G1M9 or G1M6 by L. mexicana GII. In all cases, the incubation mixtures contained 1 mM 1-deoxymannojirimycin and, where indicated, the same concentration of NMDNJ. The percentage of the total glucose liberated was calculated from each glycan [14C]glucose content (see Materials and Methods). The value for G1M9 was taken as 100%.
DISCUSSION
The glycans with the highest in vivo deglucosylation levels in the fission yeast S. pombe were G2M9 and G1M9; no traces of these glycans were detected after the 15-min labeling period (Figure 2A). The deglucosylation progressively decreased in glycans displaying seven, six, or five mannoses; increasing amounts of their di- and monoglucosylated derivatives were observed as the mannose content decreased (Figure 2, B–E). Nevertheless, even the smallest glycan was recognized in vivo by the MRH domain because the expression of wild-type GIIβ, but not of GIIβ with mutations in the MRH domain, in ΔGIIβ cells fully restored the glycan patterns to that of cells expressing endogenous wild-type GIIβ (Figure 5). This result suggests that, although the affinity of the MRH domain for G1M9 is approximately seven times higher than that for G1M5, MRH recognition of residue e in addition to residues i and k (see Figure 1 and below; Hu et al., 2009) may influence GII catalytic rates when it is exposed in a glycan, such as in G1M5. A similar influence may exist for mannose i in the case of G2M7 and G1M7 and mannose h for G2M6 and G1M6 (Figure 1).
Unexpectedly, although the in vivo deglucosylation of G2M7, G2M6, and G2M5 was similar for strains expressing GIIα but not GIIβ, that of G2M9 was somewhat lower (Figure 4). This result points to an interaction of residues j and/or k with either the catalytic site or with the glucoses that slow deglucosylation. In an operative holoenzyme, the interaction between the MRH domain and the complete glycan may not only present the latter to the catalytic site, thus somehow strongly accelerating deglucosylation, but also annul the inhibitory effects of residues j and/or k.
G2B, a domain located close to the GIIβ N terminus, has been proposed to be involved in the GIIα–GIIβ interaction or, additionally, in G2M9 deglucosylation (Arendt and Ostergaard, 2000; Quinn et al., 2009). Our results support the former role because microsomes of cells lacking endogenous GIIβ but expressing GIIβ with a mutant G2B domain failed to correct the poor ability of GIIα to trim glucose residues from G1M9 in the absence of GIIβ. This result was most likely due to a reduced GIIα content as judged by Western blot, supporting a role for GIIβ in which it is partially responsible for GIIα ER retention (Figure 6C). Moreover, the glycan patterns formed upon transfer of either G2M9 or G2M6 in live cells lacking wild-type GIIβ but expressing GIIβ with a G2B mutation were similar to those of cells lacking GIIβ (Supplemental Figure S2). Our interpretation is that the absence of the GIIα–GIIβ interaction results in a failure to both allow MRH-mediated deglucosylation enhancement and relieve the inhibition of deglucosylation mediated by residues j and/or k. Our results show that the G2B domain is not directly involved in G2M9 or G2M6 deglucosylation in S. pombe.
If the mannose content affects GI-mediated removal of the external glucose (residue n, Figure 1) as has been claimed from cell-free assays of the mammalian but not of a plant enzyme (Grinna and Robbins, 1980; Schweden et al., 1986; Zeng and Elbein, 1998), there is either no such effect in S. pombe, or the effect is much lower than that on GII activity. Alternatively, GI activity levels are much higher than those of GII because the glycan patterns of cells transferring either three or two glucoses and seven, six, or five mannoses were similar (Supplemental Figure S3).
Contrary to what we reported previously when assaying UGGT activity using glycans linked to a single amino acid (Asn) and not denatured glycoproteins as acceptors (Sousa et al., 1992), no influence of the mannose content on in vivo UGGT enzymatic activity was observed (Figure 7). Because the mannose content influenced GII activity but not UGGT activity, no monoglucosylated UGGT-generated glycans were observed in mutants transferring M9, but increased amounts of those compounds were observed in cells transferring M7 to M5. We can speculate that, as the demannosylation of misfolded and slow-folding glycoproteins proceeds in the ER, the content of monoglucosylated glycans increases. This increase prevents surreptitious exit to the Golgi of misfolded and slow-folding glycoproteins and enhances their probability of folding properly by interacting with CNX (Figure 9). The decrease in affinity of CRT (and probably that of CNX) for N-glycans upon mannose removal (that for G1M5 is 65% of that for G1M9) is apparently more than compensated for by the reduction of the GII-mediated rate of monoglucosylated N-glycan deglucosylation (that of G1M5 is approximately 3% of that of G1M9 in cell-free assays) (Grinna and Robbins, 1980; Spiro et al., 1996). The presence of the MRH domain in GIIβ therefore confers upon the enzyme a major role in ER glycoprotein folding quality control. Up to four mannoses may be removed from misfolded glycoproteins in the mammalian ER. The exit of those species from futile CNX cycles most likely occurs upon removal of mannose unit g in Arm A (Figure 1), the last or one of the last demannosylation events that yields glycans unable to be reglucosylated by UGGT (Frenkel et al., 2003). In vivo formation of protein-linked G1M9, G1M8, G1M7, and G1M6 has been detected in the rat liver (Parodi et al., 1984b).
FIGURE 9:

Model proposed for GII as an in vivo regulator of misfolded/slow-folding glycoprotein ER permanence. Misfolded/slow-folding species are characterized by an ER mannosidase(s)-catalyzed N-glycan demannosylation. A decrease in N-glycan mannose content significantly diminishes in vivo GII-mediated deglucosylation rates but does not affect in vivo UGGT-mediated glucosylation, thus increasing the possibility of displaying monoglucosylated structures able to interact with CNX/CRT for longer time periods. The exit of irreversibly misfolded glycoproteins from futile CNX cycles most likely will occur, at least in mammalian cells, upon removal of mannose unit g in Arm A (see Figure 1).
The order in which mannose units are removed in the ER is not the reverse of that of mannose addition in Glc3Man9GlcNAc2-P-P-Dol synthesis (outlined in Figure 1). In removal, a preferential excision of the first residue, i, followed by that of residue k has been documented in S. pombe as well as in other cell types (Movsichoff et al., 2005). There is evidence indicating that the glycans studied in the present work (i.e., bearing the structures of the biosynthetic intermediates) as well as those produced by ER demannosylation of the transferred compound will be deglucosylated in vivo, after they are glucosylated by UGGT, at diminishing rates as their mannose content decreases. First, as mentioned earlier in text, cell-free assays have shown that GII activity decreases as the mannose content of the glycans produced by ER processing decreases (Grinna and Robbins, 1980; Totani et al., 2006). The known affinities of the MRH domain for glycans produced by ER processing also support this hypothesis (Hu et al., 2009). For example, the affinities of the MRH domain for G1M8 lacking either residue i or k are 36 and 22%, respectively, of that for G1M9, and the affinity for G1M7 lacking residues i and k is approximately 7% of that for G1M9. These results indicate that the MRH domain primarily recognizes residue k, followed by residue i. From these data, it may be concluded that the G1M8 normally produced in cells by glycan processing (i.e., lacking residue i, Figure 1) will be deglucosylated at a lower rate than G1M9. The next glycan produced in vivo (G1M7 lacking residues i and k, Figures 1 and 9) will be deglucosylated at an even slower rate because it lacks both residues that are primarily recognized by the MRH domain. The G1M7 produced by ER processing (lacking residues i and k) may be deglucosylated at a slower rate than the G1M7 studied in the present work (lacking residues j and k) because the former, but not the latter, lacks residue i, which is the second most important residue recognized by the MRH domain. Additional ER removal of α1,2-linked mannose units is expected to further reduce the affinity of the MRH domain for glycans. Point mutations in the MRH domain resulted in a reduction of both its affinity for glycans produced by ER processing and in the deglucosylation rates of those glycans in cell-free assays (Hu et al., 2009). Those same point mutations also resulted in reduced in vivo glycan deglucosylation (Stigliano et al., 2009). Therefore a decrease in the affinity of MRH for glycans, such as that mentioned earlier in text for ER-produced G1M8 and G1M7, will result in reduced in vivo deglucosylation. With respect to UGGT, our results indicate that all glycans bearing residue g will be equally glucosylated, irrespective of their mannose content or structure (Figure 7).
The relevance of displaying a slow deglucosylation mechanism to increase the time frame in which a particular glycan has a monoglucosylated epitope is supported by the fact that L. mexicana, a protist transferring M6 and lacking ER mannosidase activity that has retained an ancestor-derived GIIβ with an MRH domain with higher affinity for high mannose content glycans (Figure 8). Proteins with MRH domains with higher affinity for glycans shorter than M9 do exist, such as Yos9p/OS-9, which displays MRH domains with a higher affinity for shorter glycans; however, those glycans do not have the same structure as the M6 involved in the protozoon N-glycosylation. Our results provide new insight into the elements that regulate the quality control of the glycoprotein folding cycle.
The present studies were conducted in S. pombe, but the conclusions drawn are assumed to also be applicable to mammalian cells. No significant differences in the specificity, kinetics, and regulatory properties of UGGT and GII from either species have been reported (Trombetta and Parodi, 1992; Fernández et al., 1994; Stigliano et al., 2009).
MATERIALS AND METHODS
Materials
Yeast extract, Bacto-Peptone, and yeast nitrogen base were obtained from Difco (Detroit, MI). Malt extract was obtained from Britania (Buenos Aires, Argentina). Endo-β-N‑acetylglucosaminidase H (Endo H), N-glycanase (PNGase F), porcine trypsin, protease inhibitors, pNPG, DTT, amino acids, and supplements for culture media were obtained from Sigma (St. Louis, MO). [14C]glucose (301 Ci/mol) was obtained from PerkinElmer Life Sciences (Waltham, MA). NMDNJ and 1-deoxymannojirimycin were obtained from Toronto Biochemicals. Geneticin (G418) was obtained from Invitrogen (Carlsbad, CA), and nourseothricin was from WERNER BioAgents (Jena, Germany).
Strains and media
Escherichia coli DH5α was used for cloning purposes, and recombinant protein expression was performed in BL26 cells. Bacteria were grown at 37ºC in LB medium (0.5 % NaCl, 1% tryptone, 0.5% yeast extract) supplemented with 200 μg/ml ampicillin or 50 μg/ml kanamycin as needed. S. pombe cells were grown at 28ºC in rich YES medium (0.5% yeast extract, 3% glucose, and 75 μg/l adenine) or EMM minimal medium (Moreno et al., 1991; Alfa et al., 1993), supplemented with adenine (75 μg/l), uracil (75 μg/l), and/or leucine (250 μg/l) for selective growth. Malt extract medium (3% Bacto Malt Extract pH 5.5 supplemented with adenine, uracil, and leucine) was used for matings. Geneticin was added to the medium at 200 μg/ml for KanMX6 selection, and nourseothricin was added to the medium at 100 μg/ml for NatMX6 marker selection in rich medium. When double selection for geneticin and auxotrophic markers was needed, NH4Cl was replaced by 0.37% monosodium l-glutamate as the nitrogen source in EMM. L. mexicana cells were grown as previously described (Parodi et al., 1984a). The S. pombe strains used are summarized in Table 2.
TABLE 2:
Yeast strains used in this study.
| Strains (nickname) | Genotype | Source |
|---|---|---|
| Sp61 (WT) | h−, leu1–32, ade6-M210, ura4-D18, ade1 | Our stock ( D'Alessio et al., 1999) |
| ADm (WT) | h−, leu1–32, ade6-M210, ura4-D18 | Our stock ( D'Alessio et al., 1999) |
| ADp (WT) | h+, leu1–32, ade6-M216, ura4-D18 | Our stock ( D'Alessio et al., 1999) |
| Sp61IIα (ΔGIIα) | h−, leu1–32, ade6-M210, ura4-D18, ade1, Δgls2α::ura4+ | Our stock ( Soussilane et al., 2009) |
| ADmIIβ (ΔGIIβ) | h−, leu1–32, ade6-M210, ura4-D18, Δgls2β::ura4+ | Our stock ( D'Alessio et al., 1999) |
| SpADIIαβ (ΔGIIαβ) | h−, leu1–32, ade6-M216, ura4-D18 Δgls2α::ura4+, Δgls2β::ura4+ | Our stock ( Soussilane et al., 2009) |
| SPAC56F8.06c (Δalg10) | h+, leu1–32, ade6-M216, ura4-D18, Δalg10::KanMX4 | Bioneer |
| SpA10–4AK (Δalg10-K) | h−, leu1–32, ade6-M210, ura4-D18, Δalg10::KanMX4 | This study |
| SpA10–4AN (Δalg10-N) | h−, leu1–32, ade6-M210, ura4-D18, Δalg10::NatMX4 | This study |
| SPBC1734.12C (Δalg12) | h+, leu1–32, ade6-M216, ura4-D18, Δalg12::KanMX4 | Bioneer |
| SPAC1834.05 (Δalg9) | h+, leu1–32, ade6-M216, ura4-D18, Δalg9::KanMX4 | Bioneer |
| ADpA3 (Δalg3) | h+, leu1–32, ade6-M216, ura4-D18, Δalg3::KanMX6 | This study |
| SpA10A12–2B (Δalg10/Δalg12) | h−, leu1–32, ade6-M210, ura4-D18, Δalg10::KanMX4, Δalg12::KanMX4 | This study |
| SpA10A9–7C (Δalg10/Δalg9) | h−, leu1–32, ade6-M216, ura4-D18, Δalg10::NatMX4, Δalg9::KanMX4 | This study |
| SpA10A3–7A (Δalg10/Δalg3) | h+, leu1–32, ade6-M216, ura4-D18, Δalg10::NatMX4, Δalg3::KanMX6 | This study |
| SpA10GIIβ-1C (Δalg10/ΔGIIβ) | h−, leu1–32, ade6-M210, ura4-D18, Δalg10::KanMX4, Δgls2β::ura4+ | This study |
| SpA10A12GIIβ-10B (Δalg10/Δalg12/ΔGIIβ) | h+, leu1–32, ade6-M210, ura4-D18, Δalg10::KanMX4, Δalg12::KanMX4, Δgls2β::ura4+ | This study |
| SpA10A9GIIβ-12D (Δalg10/Δalg9/ΔGIIβ) | h−, leu1–32, ade6-M216, ura4-D18, Δalg10::NatMX4, Δalg9::KanMX4, Δgls2β::ura4+ | This study |
| SpA10A3GIIβ-14A (Δalg10/Δalg3/ΔGIIβ) | h−, leu1–32, ade6-M216, ura4-D18, Δalg10::NatMX4, Δalg3::KanMX6, Δgls2β::ura4+ | This study |
| Sp61A (Δalg6) | h−, leu1–32, ade6-M210, ura4-D18, ade1, Δalg6::ura4+ | This study |
| SpA6A12–3D (Δalg6/Δalg12) | h−, leu1–32, ade6-M210, ura4-D18, Δalg6:: ura4+, Δalg12::KanMX4 | This study |
| SpA6A9–5C (Δalg6/Δalg9) | h−, leu1–32, ade6-M216, ura4-D18, Δalg6:: ura4+, Δalg9::KanMX4 | This study |
| SpA6A3–8C (Δalg6/Δalg3) | h−, leu1–32, ade6-M210, ura4-D18, Δalg6:: ura4+, Δalg3::KanMX6 | This study |
WT: wild type.
Genetic and DNA procedures
DNA procedures were as described previously (Sambrook and Russell, 2001). Yeast DNA extraction was performed as described previously (Hoffman and Winston, 1987). S. pombe transformations were performed by electroporation with 0.5 μg of plasmid DNA or 1 μg of linear DNA as described in Stigliano et al. (2009). Cells were recovered in 0.5 M sorbitol in YES medium for 1 h at 28ºC and plated on the appropriate selective medium.
S. pombe Δalg10, Δalg12, and Δalg9 strains were purchased from Bioneer (Daejeon, Korea). S. pombe Δalg6 mutants were obtained as in Fanchiotti et al. (1998), but in a Sp61 strain genetic background. S. pombe Δalg3 mutants were constructed as follows: a disruption cassette containing the sequence of the KanMX6 selective marker flanked by 307 and 266 base pairs of the 5′ and 3′ portions of the S. pombe alg3+ gene (SPAC7D4.06c) was obtained in two rounds of PCR as described by Krawchuk and Wahls (1999). In the first round of PCR, two long primers containing coding regions of alg3+ gene and 26–27 bases homologous to the KanMX6 geneticin resistance marker cassette were obtained with the primers W-alg3s: 5′-TCACAGTAATGATTACGCTCCG-3’ and X-KanMX-alg3a: 5′-GGGGATCCGTCGACCTGCAGCGTACGAGTAACCAGCCATAGGAACCAAG-3′ and Y-KanMX-alg3s: 5′-GTTTAAACGAGCTCGAATTCATCGATCACATCCGACTACAGAAAACCC-3′ and Z-alg3a: 5′-ATCAATCTGTCAATCCCCTGAG-3′ using genomic S. pombe DNA as the template (the region homologous to KanMX6 is underlined). In the second round of PCR, both synthesized long primers were used with an excess of primers W-alg3s and Z-alg3a to amplify the 2053 base pair disruption cassette using the plasmid pFA6A-KanMX6 (Bähler et al., 1998) as the template. The resulting DNA was electroporated into competent S. pombe ADp cells, and homologous recombination in geneticin-resistant colonies was confirmed by PCR with the primers alg3–5′NCs: 5′-TACGACTTTTTGGTTAGGCG-3′ and KanMXrev: 5′-AAACAACTCTGGCGCATCG-3′ or alg3–5′NCs and Z-alg3a. The resulting strain (Δalg3) was called ADpA3 (Table 2).
S. pombe double or triple mutants were obtained using standard techniques for mating, sporulation, tetrad dissection, and analysis as previously described (Alfa et al., 1993; D'Alessio et al., 2003). Spore micromanipulations were carried out with a Singer Manual Micromanipulator (Somerset, UK). Relevant genotypes were determined by antibiotic resistance or auxotrophic growth in the appropriate medium and also by colony PCR with the primers described later in text.
S. pombe Δalg10 was mated with ADm to obtain Δalg10-K. The exchange of the geneticin marker for the nourseothricin selective marker in this strain to obtain the Δalg10-N mutants was performed according to Sato et al. (2005), as follows: primers MD1 and MD2 were used to obtain NatMX6 (nourseothricin-resistant marker) flanked by PTEF and TTEF by PCR from the pCR2.1-nat plasmid. This DNA fragment was gel-purified and used to transform electrocompetent Δalg10-K mutants. Nourseothricin-resistant colonies were tested for sensitivity to geneticin, and homologous recombination was verified by PCR.
The following primers were used to verify the genotypes of the strains: for Δalg10::KanMX4, SpAlg10s 5′NC: 5′-CCAAACTTCCTGCCAACAAC-3′ and KanMXrev; for Δalg10::NatMX4, ClonNat Forward: 5′-CTTCGTGGTCGTCTCGTAC-3′ and SpAlg10 3′NCa: 5′-ACCAAAAATCAACGAGCACC-3′; for Δalg12::kanMX4, Walg12s: 5′-GATGAAGCGGGATGACTTCT-3′ and KanMX reverse; for Δalg9::kanMX4, alg9s 5′NC and KanMX reverse; for Δalg6::ura4+, the primers were described in Fanchiotti et al. (1998), and for ΔGIIβ::ura4+, the primers were described in Stigliano et al. (2009).
GIIβ mutagenesis
The gateway pDONR201 plasmid containing clone 26/D11 (S. pombe GIIβ subunit, which was obtained from RIKEN DNA Bank; Matsuyama et al., 2006) was used as the template for single amino acid PCR mutagenesis of the GIIβ GIIB domain (E73A or E114A). The amplified mutant DNA containing both GIIβ and vector was phosphorylated, religated, and electroporated into DH5α cells. The primers used were the following (mutagenic codons are underlined): E73A forward mutagenic primer 5′-GATGCACCAGGTAATAAGC-3′, reverse primer 5′-TGACCCGTCAGGGCAATCAC-3′; E114A forward mutagenic primer 5′-GATGCAAGTCTTATTAAATGC-3′, reverse primer 5′-AGAACCGTCACAGCAATC-3′. Wild type and mutant GIIβ DNA clones were transferred to the pREP1-ccdb2 Gateway-compatible S. pombe destination expression vector (RIKEN DNA Bank) by the LR recombination reaction (Invitrogen). S. pombe competent ΔGIIβ cells were electroporated with the pREP1-GIIβ episomal constructs. The MRH domain double mutant construct used in this study (Y462F/E456Q) was described in Stigliano et al., (2009) (by mistake, in that reference the numbering of the amino acids was Y463F/E457Q).
Microsomal fraction preparations
S. pombe microsomes were prepared from 250 ml of cultures at A600 = 2. Cells were harvested, washed with 5 mM NaN3, and broken by 10 repetitive cycles of 1-min vortexing on ice with glass beads in 0.25 M sucrose, 20 mM imidazole, and 5 mM EDTA with protease inhibitors (100 μM phenylmethylsulfonyl fluoride, 10 μM l-1-tosylamido-2-phenylethyl chloromethyl ketone, 10 μM Nα-p-tosyl-l-lysine chloromethyl ketone, 10 μM leupeptin, 10 μM pepstatin, and 10 μM E64), and the microsomal fraction was obtained as described (D'Alessio et al., 1999). For L. mexicana, the ER soluble content was prepared by freeze-thawing the parasites as previously described for Trypanosoma cruzi (Labriola et al., 1995). Protein concentrations were determined by the Bio-Rad Protein Assay as described by the manufacturer (Hercules, CA).
Analysis of glycans synthesized in vivo
To assess ER N-glycan composition, S. pombe cells in the exponential growth phase were harvested, extensively washed with 1% YNB medium without glucose, and resuspended in two volumes (vol/wt) of the same medium. Cells were then preincubated for 5 min in 5 mM DTT and pulsed for 15 min in 5 mM glucose with 300 μCi/ml of [14C]glucose. Further details on the labeling procedure and the preparation of whole-cell Endo H–sensitive N-glycans have been described previously (Fernández et al., 1994). For strains carrying the Δalg3 mutation that transfer a pentamannosyl glycan, whole-cell proteins were degraded with porcine trypsin instead of pronase, and the removal of N-glycans was performed with N-glycanase (glycans synthesized by strains harboring the Δalg3 mutation are resistant to Endo H, and the N-glycanase requires both the Asn amino and carboxyl groups to be substituted by amino acids for activity). For strains carrying the Δalg6 mutation, cells were preincubated for 60 min in the presence or absence of 5 mM NMDNJ. Glycans were separated by paper chromatography using Whatman 1 papers and 1-propanol and nitromethane/H2O (5:2:4) as the solvent, and the peaks were identified by standards run in parallel. To improve the resolution, the identified glycans were eluted from the papers and resolved by high performance liquid chromatography (HPLC) using a TSK-GEL Amide-80 column (4.6 mmϕ × 25 cm; Tosoh Bioscience, Tokyo, Japan) with a mobile phase of H2O/CH3CN in a linear gradient from 35:65 to 55:45 over 67 min and a flow rate of 0.75 ml/min at room temperature. Due to slight variations in retention times among runs, the positions of the peaks in paper chromatography and not the retention times from HPLC were used to identify the glycans.
Synthesis of labeled N-glycan substrates
Glucose- and mannose-labeled [14C]Glc1Man9GlcNAc and [14C]Glc1Man6GlcNAc were obtained by in vivo labeling and N-glycan purification of S. pombe cells carrying the Δalg6 or the Δalg6/Δalg9 mutations, respectively. Cells were preincubated with 5 mM NMDNJ for 60 min and with 5 mM DTT for 5 min and were then incubated for 45 min in 5 mM glucose containing 500 μCi [14C]glucose. The Endo H–sensitive monoglucosylated glycans were separated from the unglucosylated compounds by two successive paper chromatographies in n-propanol and nitromethane/H2O (5:2:4). The label in glucose residues was 27 and 35% of the total for [14C]Glc1Man9GlcNAc and [14C]Glc1Man6GlcNAc, respectively, as determined by total acid hydrolysis of glycans followed by paper chromatography in n-butanol and pyridine/H2O (10:3:3). [14C-glucose]Glc1Man9GlcNAc was obtained by glucosylation of denatured bovine thyroglobulin in the presence of UDP-[14C]Glc and rat liver microsomes, followed by glycan purification as described previously (Trombetta et al., 1989).
GII activity assays
GII activity using labeled glycans as substrates was assayed in S. pombe or L. mexicana microsomal fractions as described in Stigliano et al. (2009). Where indicated, 1 mM 1-deoxymannojirimycin was added with or without 1 mM NMDNJ. GII activity in ΔGIIβ mutants transformed with wild type or mutant regulatory subunits was assayed using 5 mM pNPG as the substrate in 0.1 M HEPES buffer, pH 7.2. Incubations lasted for 20 min at 37ºC as described in Stigliano et al. (2009). Complementation assays between GIIα and GIIβ were performed using a mixture of 125 μg of proteins from microsomes containing GIIα but not GIIβ (ΔGIIαβ cells transformed with pREP3x-GIIαVDEL, as described in Stigliano et al., 2009) with 125 μg of proteins from microsomes containing GIIβ but not GIIα (ΔGIIαβ mutant cells expressing either wild-type GIIβ or GIIβ mutated in either the MRH or G2B domains). The mixtures were preincubated for 30 min at 4ºC in the presence of 1% Triton X-100 and then assayed for GII activity with [14C-glucose]Glc1Man9GlcNAc in 40 mM sodium phosphate buffer, pH 7.2, for 15 min at 30ºC to test the ability of GIIβ to partially restore GII N-glycan trimming ability. The liberated glucose was separated from the remaining substrate by ascending paper chromatography using 2-propanol and acetic acid/H2O (25:4:9), and the activity was determined as the percentage of total glucose released.
Antibodies and immunodetection
Microsomal S. pombe proteins (250 μg) were resolved by 9% SDS–PAGE, electroblotted to Immobilon-P membranes (Millipore, Billerica, MA) and incubated with mouse anti–S. pombe GIIα, mouse anti–S. pombe GIIβ, or rabbit anti–S. pombe CNX antibodies. Immunodetection was carried out using enhanced chemiluminescence (West Pico SuperSignal Chemiluminescent Substrate, Thermo Fisher Scientific, Waltham, MA) with horseradish peroxidase (HRP)-conjugated immunoglobulin Gs (IgGs; Sigma).
Mouse polyclonal serum raised against the S. pombe GIIβ protein was obtained as follows: a 447 base pair GIIβ DNA fragment was PCR amplified from a pDONR201 plasmid containing clone 26/D11 using the primers GIIβ-Nde1 forward 5′- GGAATTCCATATGCGAGAATACTTGGCAACATTAG 3′ and GIIβ-Xho1 reverse 5′ CCGCTCGAGATAGTTATGAACATCATCGG 3′, cloned in pET22b+ (Invitrogen) and expressed as a C-terminal His6 fusion protein in E. coli BL26 cells. After a 4-h induction with 1 mM isopropyl-β-d-thiogalactoside, the protein was purified from inclusion bodies by immobilized metal ion affinity chromatography using chelating Sepharose (Amersham, Little Chalfont, UK) in the presence of 6 M urea as described by the manufacturer. Protein (20 μg) was injected intradermally to BALb/C mice, and two boosters of 10 μg were given after 15 and 30 d. The serum was used at a 1:1000 dilution as a primary antibody for S. pombe GIIβ immunodetection. Mouse polyclonal anti-GIIα and rabbit polyclonal anti-CNX sera were obtained as described elsewhere (Stigliano et al., 2009) and were used at 1:500 and 1:100,000 dilutions, respectively.
Supplementary Material
Acknowledgments
Plasmids pFA6a-KanMX6 and pCR2.1-nat were provided by J. Bähler (University College, London, UK) and T. Toda (London Research Institute, Lincoln's Inn Fields Laboratories, London, UK), respectively. We thank M. Rogers and the pathogen sequencing unit at the Wellcome Trust Sanger Institute for their kind help with the L. mexicana GIIβ sequence and Mariela Loschi for her help with Figure 9. UDP-[14C]Glc was efficiently synthesized by Susana Raffo. I.D.S. is a Doctoral Fellow and C.A.L., A.J.P., and C.D. are Career Investigators of the National Research Council (Argentina). Financial support by the Agencia Nacional de Promoción Científica y Tecnológica (ANPCYT), the University of Buenos Aires, the Howard Hughes Medical Institute, and the National Institutes of Health (Grant GM44500) is gratefully acknowledged.
Abbreviations used:
- CNX
calnexin
- CRT
calreticulin
- Dol
dolichol
- DTT
dithiothreitol
- Endo H
endo-β-N-acetylglucosaminidase H
- ER
endoplasmic reticulum
- GI
glucosidase I
- GII
glucosidase II
- HPLC
high performance liquid chromatography
- HRP
horseradish peroxidase
- Man6P
mannose 6-phosphate
- MRH
Man6P receptor homologous
- NMDNJ
N-methyl 1-deoxynojirimycin
- pNPG
p-nitrophenyl α-d-glucopyranoside
- UGGT
UDP-Glc:glycoprotein glucosyltransferase
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-01-0019) on April 6, 2011.
REFERENCES
- Alfa C, Fantes P, Hyams J, McLeod M, Wabrik E. Experiments with Fission Yeast: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. [Google Scholar]
- Arendt CW, Ostergaard HL. Two distinct domains of the β subunit of glucosidase II interact with the catalytic α-subunit. Glycobiology. 2000;10:487–492. doi: 10.1093/glycob/10.5.487. [DOI] [PubMed] [Google Scholar]
- Bähler J, Wu JQ, Longtine MS, Shah NG, McKenzie A 3rd, Steever AB, Wach A, Philippsen P, Pringle JR. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Cabral CM, Liu Y, Sifers RN. Dissecting glycoprotein quality control in the secretory pathway. Trends Biochem Sci. 2001;26:619–624. doi: 10.1016/s0968-0004(01)01942-9. [DOI] [PubMed] [Google Scholar]
- Caramelo JJ, Parodi AJ. How sugars convey information on protein conformation in the endoplasmic reticulum. Sem Cell Dev Biol. 2007;18:10221–10225. doi: 10.1016/j.semcdb.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramelo JJ, Parodi AJ. Getting in and out of calnexin and calreticulin cycles. J Biol Chem. 2008;283:10221–10225. doi: 10.1074/jbc.R700048200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Alessio C, Caramelo JJ, Parodi AJ. UDP-Glc:glycoprotein glucosyltransferase-glucosidase II, the ying-yang of the ER quality control. Semin Cell Dev Biol. 2010;21:491–499. doi: 10.1016/j.semcdb.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Alessio C, Fernández F, Trombetta ES, Parodi AJ. Genetic evidence for the heterodimeric structure of glucosidase II. The effect of disrupting the subunit-encoding genes on glycoprotein folding. J Biol Chem. 1999;274:25899–25905. doi: 10.1074/jbc.274.36.25899. [DOI] [PubMed] [Google Scholar]
- D'Alessio C, Trombetta ES, Parodi AJ. Nucleoside diphosphatase and glycosyltransferase activities can localize to different subcellular compartments in Schizosaccharomyces pombe. J Biol Chem. 2003;278:22379–22387. doi: 10.1074/jbc.M300892200. [DOI] [PubMed] [Google Scholar]
- De la Canal L, Parodi AJ. Synthesis of dolichol derivatives in trypanosomatids. Characterization of enzymatic patterns. J Biol Chem. 1987;262:11128–11133. [PubMed] [Google Scholar]
- Fanchiotti S, Fernández F, D'Alessio C, Parodi AJ. The UDP-Glc:glycoprotein glucosyltransferase is essential for Schizosaccharomyces pombe viability under conditions of extreme endoplasmic reticulum stress. J Cell Biol. 1998;143:625–635. doi: 10.1083/jcb.143.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández F, D'Alessio C, Fanchiotti S, Parodi AJ. A misfolded protein conformation is not a sufficient condition for in vivo glucosylation by the UDP-Glc:glycoprotein glucosyltransferase. EMBO J. 1998;17:5877–5886. doi: 10.1093/emboj/17.20.5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández FS, Trombetta SE, Hellman U, Parodi AJ. Purification to homogeneity of UDP-glucose:glycoprotein glucosyltransferase from Schizosaccharomyces pombe and apparent absence of the enzyme from Saccharomyces cerevisiae. J Biol Chem. 1994;269:30701–30706. [PubMed] [Google Scholar]
- Frenkel Z, Gregory W, Kornfeld S, Lederkremer GZ. Endoplasmic reticulum-associated degradation of mammalian glycoproteins involves sugar chain trimming to Man6‑5GlcNAc2. J Biol Chem. 2003;278:34119–34128. doi: 10.1074/jbc.M305929200. [DOI] [PubMed] [Google Scholar]
- Grinna LS, Robbins PW. Substrate specificities of rat liver microsomal glucosidases which process glycoproteins. J Biol Chem. 1980;255:2255–2258. [PubMed] [Google Scholar]
- Hoffman CS, Winston F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene. 1987;57:267–272. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Kamiya Y, Kamiya D, Kato K, Nagata K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J Biol Chem. 2009;284:17061–17068. doi: 10.1074/jbc.M809725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Kamiya Y, Totani K, Kamiya D, Kawasaki N, Yamaguchi D, Matsuo N, Ito Y, Kato K, Yamamoto K. Sugar-binding activity of the MRH domain in the ERα-glucosidase GIIβ subunit is important for efficient glucose trimming. Glycobiology. 2009;19:1127–1135. doi: 10.1093/glycob/cwp104. [DOI] [PubMed] [Google Scholar]
- Krawchuk MD, Wahls WP. High-efficiency gene targeting in Schizosaccharomyces pombe using a modular, PCR-based approach with long tracts of flanking homology. Yeast. 1999;15:1419–1427. doi: 10.1002/(SICI)1097-0061(19990930)15:13<1419::AID-YEA466>3.0.CO;2-Q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labriola CA, Cazzulo JJ, Parodi AJ. Retention of the glucose unit added by the UDP-Glc:glycoprotein glucosyltransferase delays exit of glycoproteins from the endoplasmic reticulum. J Cell Biol. 1995;130:771–779. doi: 10.1083/jcb.130.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama A, et al. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol. 2006;24:841–847.. doi: 10.1038/nbt1222. Erratum in: Nat Biotechnol, 24, 1033. [DOI] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- Movsichoff F, Castro OA, Parodi AJ. Characterization of Schizosaccharomyces pombe ER α-mannosidase. A re-evaluation of the role of the enzyme on ER-associated degradation. Mol Biol Cell. 2005;16:4714–4724. doi: 10.1091/mbc.E05-03-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S. The MRH domain suggests a shared ancestry for the mannose 6-phosphate receptors and other N-glycan-recognising proteins. Curr Biol. 2001;11:499–501. doi: 10.1016/s0960-9822(01)00302-5. [DOI] [PubMed] [Google Scholar]
- Parodi AJ. N-glycosylation in trypanosomatid protozoa. Glycobiology. 1993;3:193–199. doi: 10.1093/glycob/3.3.193. [DOI] [PubMed] [Google Scholar]
- Parodi AJ. Protein glucosylation and its role in protein folding. Annu Rev Biochem. 2000;69:69–95. doi: 10.1146/annurev.biochem.69.1.69. [DOI] [PubMed] [Google Scholar]
- Parodi AJ, Martín-Barrientos J, Engel JC. Glycoprotein assembly in Leishmania mexicana. Biochem Biophys Res Commun. 1984a;118:1–7. doi: 10.1016/0006-291x(84)91058-1. [DOI] [PubMed] [Google Scholar]
- Parodi AJ, Mendelzon DH, Lederkremer GZ, Martín-Barrientos J. Evidence that transient glucosylation of protein-linked Man9GlcNAc2, Man8GlcNAc2, and Man7GlcNAc2 occurs in rat liver and Phaseolus vulgaris cells. J Biol Chem. 1984b;259:6351–6357. [PubMed] [Google Scholar]
- Pincus D, Chevalier MW, Aragón T, van Anken E, Vidal SE, El-Samad H, Walter P. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol. 2010;8:e1000415. doi: 10.1371/journal.pbio.1000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan EM, Kamiya Y, Kamiya D, Denic V, Weibezahn J, Kato K, Weissman JS. Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol Cell. 2009;32:870–877. doi: 10.1016/j.molcel.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn RP, Mahoney SJ, Wilkinson BM, Thornton DJ, Stirling CJ. A novel role for Gtb1p in glucose trimming of N-linked glycans. Glycobiology. 2009;19:1408–1416. doi: 10.1093/glycob/cwp087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY:: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Samuelson J, Banerjee S, Magnelli P, Cui J, Kelleher DJ, Gilmore R, Robbins PW. The diversity of dolichol-linked precursors to Asn-linked glycans likely results from secondary loss of sets of glycosyltransferases. Proc Natl Acad Sci USA. 2005;102:1548–1553. doi: 10.1073/pnas.0409460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Dhut S, Toda T. New drug-resistant cassettes for gene disruption and epitope tagging in Schizosaccharomyces pombe. Yeast. 2005;22:583–591. doi: 10.1002/yea.1233. [DOI] [PubMed] [Google Scholar]
- Satoh T, Chen Y, Hu D, Hanashima S, Yamamoto K, Yamaguchi Y. Structural basis for oligosaccharide recognition of misfolded glycoproteins by OS-9 in ER-associated degradation. Mol Cell. 2010;22:905–916. doi: 10.1016/j.molcel.2010.11.017. [DOI] [PubMed] [Google Scholar]
- Schweden J, Borgmann C, Legler G, Bause E. Characterization of calf liver glucosidase I and its inhibition by basic sugar analogs. Arch Biochem Biophys. 1986;248:335–340. doi: 10.1016/0003-9861(86)90429-7. [DOI] [PubMed] [Google Scholar]
- Sousa M, Ferrero-García MA, Parodi AJ. Recognition of the oligosaccharide and protein moieties of glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. Biochemistry. 1992;31:97–105. doi: 10.1021/bi00116a015. [DOI] [PubMed] [Google Scholar]
- Soussilane P, D'Alessio C, Paccalet T, Fitchette AC, Parodi AJ, Williamson R, Plasson C, Faye L, Gomord V. N-glycan trimming by glucosidase II is essential for Arabidopsis development. Glycoconj J. 2009;26:598–607. doi: 10.1007/s10719-008-9201-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro RG, Zhu Q, Bhoyroo V, Soling H-D. Definition of the lectin-like properties of the molecular chaperone, calreticulin, and demonstration of its copurification with endomannosidase from rat liver Golgi. J Biol Chem. 1996;271:11588–11594. doi: 10.1074/jbc.271.19.11588. [DOI] [PubMed] [Google Scholar]
- Stigliano ID, Caramelo JJ, Labriola CA, Parodi AJ, D'Alessio C. Glucosidase II β subunit modulates N-glycan trimming in fission yeasts and mammals. Mol Biol Cell. 2009;20:3974–3984. doi: 10.1091/mbc.E09-04-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totani K, Ihara Y, Matsuo I, Ito Y. Substrate specificity analysis of endoplasmic reticulum glucosidase II using synthetic high mannose-type glycans. J Biol Chem. 2006;281:31502–31508. doi: 10.1074/jbc.M605457200. [DOI] [PubMed] [Google Scholar]
- Totani K, Ihara Y, Matsuo I, Ito Y. Effects of macromolecular crowding on glycoprotein processing enzymes. J Am Chem Soc. 2008;130:2101–2107. doi: 10.1021/ja077570k. [DOI] [PubMed] [Google Scholar]
- Trombetta ES, Bosch M, Parodi AJ. Glucosylation of glycoproteins by mammalian, plant, fungal and trypanosomatid protozoa microsomal membranes. Biochemistry. 1989;28:8108–8116. doi: 10.1021/bi00446a022. [DOI] [PubMed] [Google Scholar]
- Trombetta ES, Simons JF, Helenius A. Endoplasmic reticulum glucosidase II is composed of a catalytic subunit, conserved from yeast to mammals, and a tightly bound noncatalytic HDEL-containing subunit. J Biol Chem. 1996;271:27509–27516. doi: 10.1074/jbc.271.44.27509. [DOI] [PubMed] [Google Scholar]
- Trombetta SE, Parodi AJ. Purification to apparent homogeneity and partial characterization of rat liver UDP-Glc:glycoprotein glucosyltransferase. J Biol Chem. 1992;267:9236–9240. [PubMed] [Google Scholar]
- Wilkinson BM, Purswani J, Stirling CJ. Yeast GTB1 encodes a subunit of glucosidase II required for glycoprotein processing in the endoplasmic reticulum. J Biol Chem. 2006;281:6325–6333. doi: 10.1074/jbc.M510455200. [DOI] [PubMed] [Google Scholar]
- Yagodnik C, de la Canal L, Parodi AJ. Tetrahymena pyriformis cells are deficient in all mannose-P-dolichol-dependent mannosyltransferases but not in mannose-P-dolichol synthesis. Biochemistry. 1987;26:5937–5943. [Google Scholar]
- Zeng Y-C, Elbein AD. Purification to homogeneity and properties of plant glucosidase I. Arch Biochem Biophys. 1998;355:26–34. doi: 10.1006/abbi.1998.0717. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.