

Abstract
Mid-to-late transition metal complexes that feature terminal, multiply bonded ligands such as oxos, imides, and nitrides have been invoked as intermediates in several catalytic transformations of synthetic and biological significance. Until about ten years ago, isolable examples of such species were virtually unknown. Over the past decade or so, numerous chemically well-defined examples of such species have been discovered. In this context, the presentreview summarizes the development of 4- and 5-coordinate Fe(E) and Co(E) species under local three-fold symmetry.
Keywords: Atom and Group transfer, Metal-to-ligand multiple bond, Oxos, Imides, Nitrides
1. Introduction
Multiply bonded species of the late transition metals (M≡E and M=E, where E = O, N, NR, CR) have been postulated as key intermediates in many synthetic and enzymatic transformations[1-8]. These transformations include, for example, olefin epoxidation and aziridination[9-14], C-H bond oxygenation and amination[15-19], and e-/H+ transfer processes related to nitrogen fixation[20, 21]. The diverse multi-electron reactivity exhibited by such species is in part attributed to their multiple bond character, and there has hence been much interest in understanding the electronic structures and reactivity patterns of such species[22-27].
The molecular orbital (MO) picture of complexes of the type C4v-L5M(E) was developed in the classic study of the d1 vanadyl ion (VO2+) by Ballhausen and Gray (Figure 1)[28]. This MO picture stressed a large splitting of the t2g orbitals (Δπ≈ 13,000 cm-1), arising from the presence of a strongly π donating oxo ligand, and the presence of a V≡O triple bond. As a consequence, complexes of the type LnM(E) should feature bona fide multiple bond character, and hence electronic stability, when M(E) π* orbitals are vacant or only partially filled [1], regardless of local symmetry.
Figure 1.
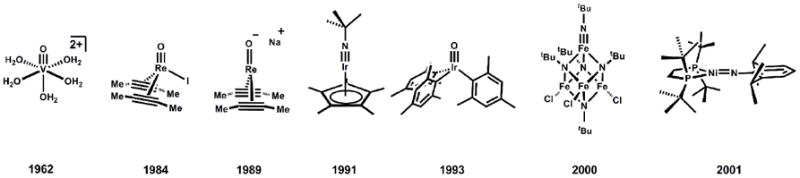
Examples of M≡E and M=E species, with year of publication noted.
With this generalization in mind, there are at present three well-identified pathways to achieve a reasonable degree of electronic stability in an M≡E or M=E interaction (also abbreviated as M(E)). The most straightforward and hence historically most familiar way is to preserve a low d-electron count at the metal. For a prototypical 6-coordinate metal center that features one terminal multiply bonded ligand L5M(E), the σ* and π* bonding interaction results in the destabilization of four orbitals of d-parentage. It is therefore not surprising that high-valent early transition metals that feature d0, d1, or d2 electron counts have historically dominated the literature of terminal L5M(E) complexes[29]. To a first order approximation, the π-bond order decreases from 2 to 3/2, 1, and ½ as the d3, d4, and d5 configurations are respectively populated. Such an approach is of course not limited to complexes of four-fold symmetry. The high stability of complexes such as trigonal bipyramidal (TBP) {(Me3SiNCH2CH2)3N}V(O) [30] and tetrahedral (iPr2N)3Cr(N) [31] drives home this point. A second means of achieving electronic stability in M(E) species constitutes using a combination of ligands that are both σ donating and π-accepting. Synergism in this context can in principle, via orbital mixing, serve to stabilize d-electrons that would otherwise be destabilized via a strong π* interaction. Such a scenario has been used, for example, to offer an explanation for the unanticipated stability of the d6 L5Pt(O) species prepared by Hill and coworkers [32]. A third mode of achieving electronic stabilization for multiply bonded species is to remove donor ligands from the central metal under consideration. This provides a distinct electronic structure that may be able to accommodate a higher number of d-electrons, so long as those electrons do not fill strongly destabilized orbitals. For example, under three-fold symmetry, both 4- and 5-coordinate species can accommodate M(E) species with diverse d-electron configurations (Figure 2)[6, 27, 33-35].
Figure 2.
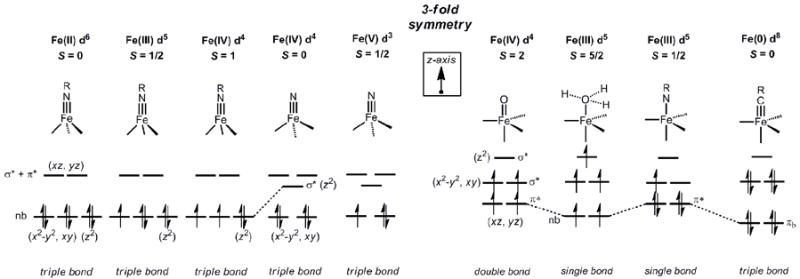
Qualitative d-orbital splitting diagrams for L3-Fe(E) and L’L3-Fe(E) structure types discussed throughout the review. The diagram is meant as a guide to the discussion in the text. The relative ordering of the orbital energies for the lower-lying e set (xy, x2-y2) and the a1 orbital (z2) is not implied for L3Fe(NR) structures, and are arbitrarily set as degenerate, for simplicity.
Synthetic entry to M(E) complexes featuring higher d- electron counts first surfaced about 25 years ago when Mayer and Tulip isolated and characterized an intriguing Re(O)I(MeC≡CMe)2 complex (Figure 1)[33]. This was the first well-characterized terminal M(E) multiple bond linkage for a complex formulated as d4. The Re-O bond distance of 1.697(3) Å in Re(O)I(MeC≡CMe)2 is similar to that of higher valent Re(O) complexes[36], suggesting that the π* orbitals are not populated. To achieve such stabilization, the Re adopts a distorted geometry that can be crudely described as tetrahedral, with approximate three-fold symmetry about the Re-O bond[37]. The two electron reduction of a related complex, Re(O)I(PhC≡CPh)2, furnished an equally interesting d6, 3-coordinate [Re(O)(PhC≡CPh)2][Na] species[35]. In this latter complex, partial population of π* orbitals is presumed to cause elongation of the Re-O bond to 1.756(3) Å, a destabilization that leads to a more reactive metal center (Figure 1). At a similar time, Meyer and coworkers identified the interesting intermediate-spin, d4 ruthenium oxo species (bpy)2(py)Ru(O)2+[38-40]. Again, partial population of π* orbitals was presumed to give rise to a more reactive Ru(O) linkage.
By establishing the viability of high d-count structures featuring M(E) bonds, these studies anticipatedthe possibility that M≡E and M=E complexes of the mid-to-late transition metals might be more generally accessible. Such species would be expected to exhibit the highest degree of kinetic stability for third row ions, and it is hence not surprising that about 15 years ago the groups of Bergman and Wilkinson isolated a d6 iridium(III) imido complex[41], and a d4 iridium(V) oxo complex[42], respectively (Figure 1). Wilkinson’s d4 oxo owes its stability to the fact that itsfour d-electrons reside in largely non-bonding orbitals that are orthogonal to the Ir-O bond vector. As a consequence of its pseudotetrahedral geometry, its higher energy combined σ*/π* orbitals are unfilled.
Until about ten years ago it had been commonly held that, owing to their propensity to populate intermediate and high spin states, first row ions that feature multiply bonded ligands would be highly reactive and hence far more difficult to isolate and thoroughly characterize[1, 43-45]. However, this has turned out not to be the case. For example, the ferrocene-like electronic structure of Bergman’s Cp*Ir(NtBu) imide closely resembles that of the isolobal pseudotetrahedral L3Co(NR) and L3Fe(NR) imides that are reviewed herein (Figure 2). Similarly, while the electronic structure of Wilkinson’s d4 iridium oxo was not described at the time of its synthesis, its relative stability predicted the electronic stability of pseudotetrahedral iron nitrides of the type d4 L3Fe(N) (Figure 2).
Indeed, over the past ten years, there have been several reports of Fe[6, 27, 46-56], Co[57-60], and Ni[61-64] complexes that feature multiply bonded ligands, in both high- and low-spin configurations. In many instances, the coordination number and hence symmetry of the species has been reduced. From these reports a general picture of the electronic structures and reactivity patterns of such complexes is emerging. This article reviews the development of terminal M(E) species of cobalt and iron with a specific emphasis on complexes that reside in local three-fold symmetry. Hillhouse[62-64], Holland[65-68], and Warren[60, 61] have advanced the field of 3-coordinate M(E) species in approximate C2v symmetry, though a discussion of these systems is beyond the scope of this review.
2. The L3M-X Structure Type
2.1 Electronic Lessons Learned from Pseudotetrahedral Cobalt(II)
Prior to delving into a discussion of M(E) species of iron and cobalt, it is instructive to first consider the electronic structures of pseudotetrahedral L3CoII-X species, as they provided an important lesson with respect to the stabilization of L3M(E) species. Our group has extensively studied a family of low-spin, high-spin, and spin-crossover complexes of the type L3CoII-X[69-71], in which the L3 donor ligand is the anionic tris(phosphino)borate ligand, [PhBPR3] ([PhBPR3] = [PhB(CH2PR2)3]-; R = Ph, iPr)[72, 73]. The electronic structure model we proposed to account for the magnetic properties of these pseudotetrahedral d7 ions provided a useful guide to further consider the stability (or instability) of other d-electron configurations for related structure types, especially where the terminal ligand would be multiply bonded to the metal center.
Prior to the characterization of low-spin [PhBPPh3]CoII-X complexes, all of the 4-coordinate cobalt(II) systems known to exhibit low-spin ground state configurations were classified as square planar[74]. Ions of approximate tetrahedral geometries, whether of nearly perfect Td symmetry, or species more appropriately described as pseudotetrahedral, distorted tetrahedral, or trigonal bipyramidal had been, without exception, classified as high-spin[75-78]. A similar situation existed for the well-documented cases of iron(II) and nickel(II) ions. These latter systems had been reported to populate high-spin electronic configurations when approximately tetrahedral (S = 2 and S = 1, respectively), and low-spin configurations when square planar[79-81].
The familiar tetrahedral ligand-field for divalent cobalt ions places three degenerate orbitals, the t set, at a significantly higher energy than a nonbonding e set (Figure 3). This splitting is not generally large enough to enforce a low-spin configuration, and hence a high spin S = 3/2 electronic configuration is typically observed. Lowering the symmetry, whether to C3, Cs, or C1, does not typically have a large effect on the relative arrangement of the d-orbitals; the splitting of the degenerate t set is generally small by comparison to the pairing energy that would be required to achieve a low-spin configuration. A strong axial distortion, whereby the angles between three of the ligands decrease to well below 109.5° (as would be enforced by a tripodal ligand), stabilizes the orbital of a1 symmetry (in C3v) from the upper set by decreasing angular overlap between the L donors and dz2[69, 71, 82, 83]. When the ligand-field splitting becomes unusually large, which will be the case for highly covalent, strongly σ–donating and possibly π-accepting L3 donor sets, a low-spin ground state configuration can be attained (Figure 3)[69-71].
Figure 3.
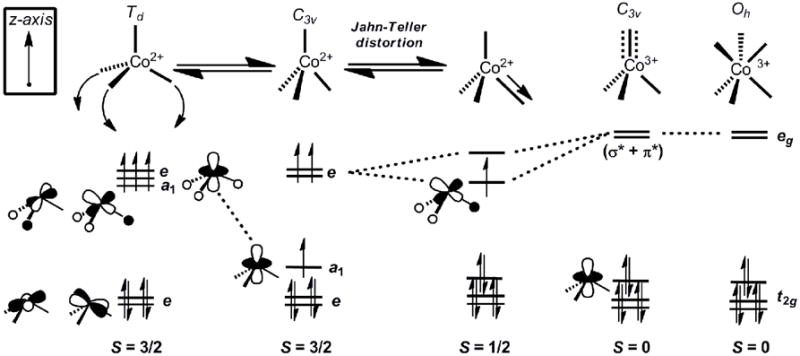
Qualitative d-orbital correlation diagram that summarizes the electronic relationships discussed in the text regarding 4-coordinate L3Co-X species.
The d-orbital splitting diagram achieved under this scenario recalls the “two-over-three” splitting commonly used to describe octahedral coordination complexes. A low-spin configuration for a pseudotetrahedral d7 L3M-X system is thus expected to exhibit a stabilizing Jahn-Teller distortion, away from three-fold symmetry, to split the upper e set. This distortion attenuates a σ antibonding interaction between a phosphine donor and the metal centered SOMO, and is similar to that observed in pseudo-octahedral low-spin d7 ions. Such a distortion is evident in the solid-state structures of numerous low-spin [PhBPR3]Co-X complexes that have been characterized[71], representative examples of which are shown in Figure 4. The generality of this phenomenon has been probed by comparing the structural and magnetic data for a series of [PhBPR3]Co-X complexes. Subtle electronic and steric changes, both within the immediate coordination sphere and well removed from it, have been found to affect the spin-state.
Figure 4.
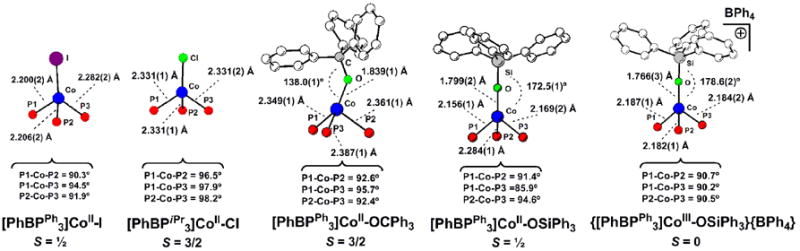
Select structural data for a series of pseudotetrahedral [PhBPR3]Co-X species that feature both high- and low-spin ground-states.
The electronic structure depicted in Figure 3 suggested that removal of a single electron from pseudotetrahedral L3CoII-X systems might give rise to an S = 0 ground state, particularly if significant π donation from the X-type linkage would complement the σ-donor character of the tripodal L3 scaffold. The complex {[PhBPPh3]CoIII-OSiPh3}{BPh4} provided one such case (Figure 4), and clearly illustrated the close electronic structure relationship that exists between the trigonal ligand-field of 4-coordinate L3CoIII-X complexes and the conventional ligand-field of octahedral L6CoIII species.
Around the time that we were developing the MO description of L3CoII-X and L3CoIII-X+ complexes, Lee and coworkers reported a tetranuclear iron cluster that featured an Fe(NR) terminal linkage[84]. The iron that coordinates the terminal imido functionality in this system resides in a pseudotetrahedral geometry. Despite the low synthetic yield of the tetranuclear iron cluster (1-2 %), they were able to characterize it by several spectroscopic techniques, including X-ray crystallography. Also, Hillhouse and coworkers reported the isolation of a structurally distinct, 3-coordinate nickel imide in 2001 (Figure 1)[62]. These two results, combined with our electronic structure description for 4-coordinate L3M-X systems, suggested that a variety of d-electron configurations (i.e. d3, d4, d5, d6) might be electronically stabilized by strong π-bonding at mid-to-late first row transition ions (Figure 2).
2.2 Pseudotetrahedral L3Co(NR) Species
Intrigued by the possibility that L3M(E) species might prove sufficiently stable to isolate, our group began to systematically explore the synthetic feasibility of accessing such complexes. Given our observation that low-spin cobalt(II) systems supported by tris(phosphino)borate ligands were accessible, cobalt(III) seemed like a good place to begin. The soft, polarizable phosphine donors of [PhBPR3] ligands provided straightforward access to monovalent cobalt(I) precursors of the type [PhBPPh3]CoI-L[57]. Subsequent two-electron [NR] group transfer from an organic azide to cobalt proved a reliable means of generating terminal cobalt(III) imides. This reaction manifold moreover exposed for the first time the viability of a CoIII/I two-electron redox couple.
Access to [PhBPR3]CoI-L precursors could be accomplished in a two-step sequence starting from a [PhBPR3]CoII-X precursor via addition of the donor L ligand followed by reduction (Scheme 1)[57, 73]. In our initial report, treatment of green [PhBPPh3]CoII-I with PMe3 afforded the red 5-coordinate cobalt(II) complex, [PhBPPh3]CoII(I)(PMe3). Reduction by sodium/mercury amalgam then provided the desired bright green cobalt(I) precursor, [PhBPPh3]CoI-PMe3, as a d8S = 1 species (the two unpaired electrons populate nearly degenerate σ* orbitals of dxz/dyzparentage). Treatment of [PhBPPh3]CoI-PMe3 with two equivalents of N3(p-tolyl) resulted in N2 extrusion and successful formation of the red and diamagnetic terminal imide [PhBPPh3]CoIII(N-p-tolyl) (Scheme 1). Two equivalents of N3(p-tolyl) are required in this reaction because one equivalent serves to oxidize PMe3 to PMe3=N(p-tolyl).
Scheme 1.
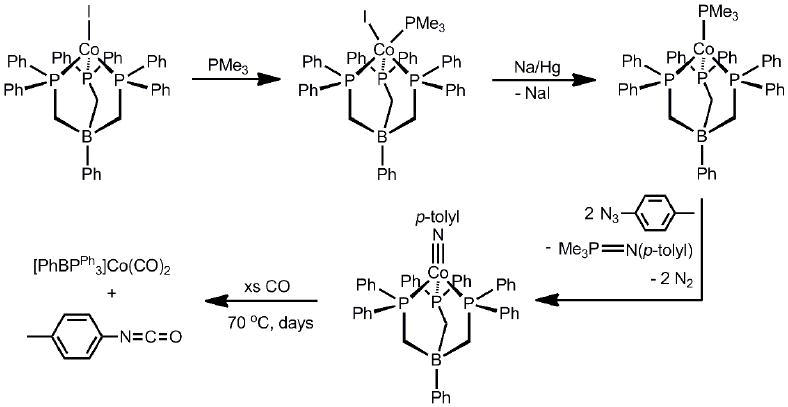
The solid-state structure of [PhBPPh3]CoIII(N-p-tolyl)revealed a very short Co-N bond distance of 1.658(2) Å, establishing the presence of significant multiple bond character at the Co-NR linkage[57]. The relatively linear Co-N-C angle of 169.51(2)° indicated sp-hybridization at the imide N-atom, establishing two π-bonds between the cobalt and nitrogen atoms (Table 1). Such a [PhBPPh3]CoIII≡N(p-tolyl) bonding description is consistent with the qualitative d-orbital splitting scenario described above, and was corroborated by DFT calculations[88]. Hence, three low-lying orbitals of dz2, dxy, and dx2-y2 parentage (a1 + e) accommodate six electrons, whereas the σ and π antibonding orbitals of dxy and dyz parentage lie at higher energies (Figure 3).
Table 1.
Select metrical parameters for pseudotetrahedral L3CoIII(NR) complexes.
| Complex | Co-N bond length (Å) | N-C bond length (Å) | Co-N-C angle (°) | Displacement of Co from L3 plane (Å) | Ref |
|---|---|---|---|---|---|
| [PhBPPh3]Co(Np-tolyl) | 1.658(2) | 1.367(2) | 169.5(1) | 1.23 | [57] |
| [PhBPPh3]Co(NtBu) | 1.633(2) | 1.442(2) | 176.7(1) | 1.24 | [85] |
| [PhBPiPr3]Co(Np-tolyl) | 1.667(2) | 1.315(3) | 173.2(2) | 1.21 | [73] |
| [(TIMENmes)Co(Np-OMeC6H4)][BPh4] | 1.675(2) | 1.386(4) | 168.7(2) | 0.90 | [58] |
| (TptBu,Me)Co(NtBu) | 1.660(3) | 1.449(5) | 179.4(3) | 1.20 | [86] |
| (TptBu,Me)Co(NAd) | 1.655(2) | 1.441(3) | 178.3(2) | 1.196 | [87] |
| [PhB(tBuIm)3]Co(NtBu) | 1.659(3) | 1.463(5) | 179.7(3) | 1.17 | [59] |
Of interest with respect to chemical reactivity was our observation that, despite the high kinetic stability of [PhBPPh3]Co(N-p-tolyl), it was possible to release the imide functionality from cobalt to a nitrene acceptor, thereby regenerating cobalt(I)[57]. For example, addition of CO to of [PhBPPh3]CoIII≡N(p-tolyl) released the free isocyanate O=C=N(p-tolyl), whilst generating the 5-coordinate dicarbonyl, [PhBPPh3]CoI(CO)2(Scheme 1). This reaction, and the analogous release of carbodiimides by addition of C≡NR, have since become a useful diagnostic probe for the presence of imides coordinated to late transition metals[53, 67, 89, 90]. Additionally, it suggested the viability of a catalytic MIII/I redox loop (vide infra).
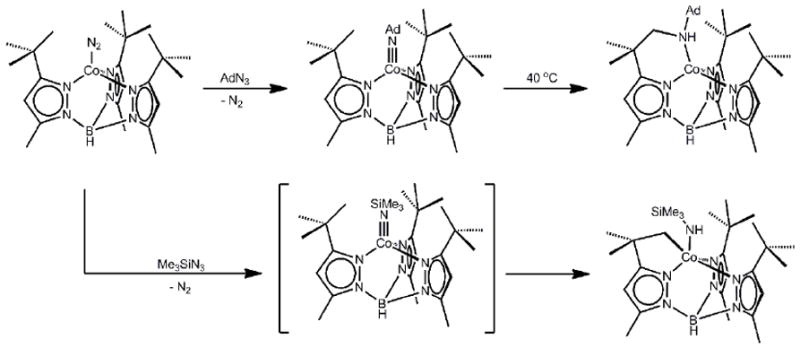
Around the time that our studies of [PhBPR3]Co(NR)species were underway, Theopold and coworkers were exploring conceptually related transformations at substituted tris(pyrazolyl)borate (Tp’)ligated cobalt(I) precursors in the hope of gaining evidence for terminally bonded cobalt imide and oxo functionalities[87, 91, 92]. Whereas they ultimately established the fidelity of the L3CoIII(NR) framework for certain (Tp’) and NR functionalities, early studies exposed radical degradation pathways. For example, addition of Me3SiN3 to (TptBu,Me)Co(N2) ((TpR,R’)- = hydrotris(3-R,5-R’-pyrazolyl)borate) resulted in formation of cylometallated, 5-coordinate (HB(pz)2(κ2-pz’))Co(NHSiMe3) (Scheme 2)[92]. This product in all likelihood formed via H-atom transfer (HAT) from a ligand C-H bond to in situ generated (TptBu,Me)CoIII(NSiMe3). This idea found merit in the reactivity patterns of isolable (Tp’)Co(NR) species (vide infra). An example of such a species is (TptBu,Me)CoIII(NAd), prepared by treatment of (TptBu,Me)Co(N2) with AdN3 (Scheme 2)[87, 91]. The metrical parameters of the imide functionality in (TptBu,Me)CoIII(NAd)(Co-N = 1.655(2) Å, Co-N-C = 178.3(2)°) are highly similar to those of [PhBPR3]CoIII(NR) species, establishing their closely related electronic structures (Table 1). (TptBu,Me)CoIII(NAd) exhibits a low-spin d6 electronic configuration, and hence is diamagnetic even at room temperature in solution. For example, (TptBu,Me)CoIII(NAd) has a well resolved 13CNMR spectrum. But (Tp’) is a weaker field ligand than [PhBPR3], which may afford thermal access to open-shell states that would in part explain the propensity of (Tp’)CoIII(NR) derivatives to display H-atom abstraction behavior. For example, gentle heating of solutions of (TptBu,Me)CoIII(NAd) resulted in partial insertion of the nitrene into a ligand C-H bond (Scheme 2).
Scheme 2.
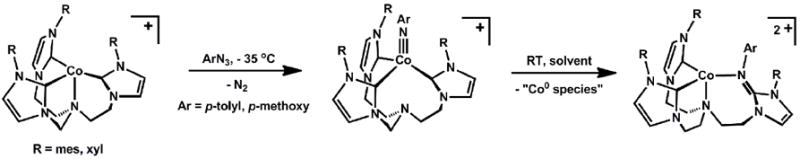
Meyer and Hu extended the generality of the L3CoIII≡NR motif and underscored its stability in preference to a 5-coordinate, trigonal bipyramidal L’L3CoIII(NR) structure type using a flexible tris(carbene)amine scaffold, which features a hemi-labile amine donor at the axial position[58]. Once again, oxidative group transfer to cobalt(I) proved the synthetic method of choice. For example, addition of p-tolylazide to trigonal pyramidal [(TIMENR)Co][Cl] led to the terminal imide complex[(TIMENR)Co(N-p-tolyl)][Cl] (TIMENR = tris-[2-(3-R-imidazol-2-ylidene)ethyl]amine; xyl = 2,6-dimethylphenyl; mes = 2,4,6-trimethylphenyl) (Scheme 3). The solid-state structure of the imide complex clearly established the absence of an interaction between the d6 cobalt centerand the amine N-donor of the (TIMENmes) ligand (the Co-Nax distance is 4.01 Å). Accordingly,complexes of the type [(TIMENR)Co(NArR’)][Cl] are low-spin, and feature similar metrical parameters to the cobalt imides described above. Were there an appreciable interaction between the apical N-donor and the cobalt center, a triplet ground state would instead be expected (see discussion below for 5-coordinate d6L’L3M(E) species). Despite the clear preference for a low-spin ground state, these [(TIMENR)CoIII(NArR’)][Cl]imide complexes are thermally unstable and undergo nitrene insertion into one Co-carbene bond at room temperature (Scheme 3). This transformation may be facilitated by the flexible ligand scaffold, as nitrene transfer is accompanied by coordination of the axial amine. Hence, it is cautioned that the reactivity patterns in such systems are not easily correlated with observed ground spin states.
Scheme 3.
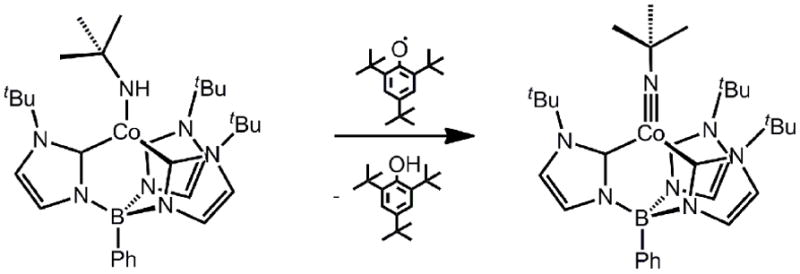
Another cobalt(III) imide system worth noting in the present context comes from Smith and coworkers[59]. Using sterically encumbering tris(carbene)borate ligands that are isomers of the ubiquitous tris(pyrazolyl)borates, they have prepared structurally related L3CoIII(NR) species such as[PhB(tBuIm)3]Co(NtBu) ([PhB(RIm)3]= [PhB(1-R-2-ylidene)3]-). As for tris(pyrazolyl)borate derivatives, vertical bulk is readily accomplished with these ligands, yet they differ by virtue of a stronger ligand field conferred by the carbene donors[93]. The cobalt(III) imides derived from these platforms are electronically similar to those already described (Table 1). Their method of synthesis provided a noteworthy distinction, however (Scheme 4). H-atom abstraction from the cobalt(II) amide precursor, [PhB(tBuIm)3]Co(NHtBu), was accomplished by addition of 2,4,6-tri(tert-butyl)phenoxy radical to furnish the d6 cobalt(III) imide, [PhB(tBuIm)3]CoIII(NtBu). This strategy is related to the stepwise oxidation/deprotonation protocol first employed by Hillhouse and Mindiolato prepare a terminal nickel(II)imide[62]. Worth noting is that in all instances, these cobalt(III) imides lack reversible redox couples. Though this is perhaps intuitive for a reduction event, as a d7 cobalt(II) species would populate a high-lying orbital, the reason why oxidation to a d5 cobalt(IV) species is not accessible is less obvious (Figure 3).
Scheme 4.
2.3 Pseudotetrahedral L3Fe(NR) Species
With the aforementioned ‘[PhBPR3]Co’ chemistry as a backdrop, our group was eager to explore whether related L3Fe(NR) species could be generated and thoroughly characterized. Whereas terminal imido complexes of ruthenium and osmium had been known for many years[94-97], terminally bonded iron imido complexes were essentially unknown. As noted already, Lee and coworkers had provided the singular exception to this statement in 2000 with the low-yielding synthesis of a tetranuclear iron cluster, {ClFe}3{NtBuFe}(μ3-NtBu)4, that featured a terminal Fe-NtBu linkage (Fe-N = 1.635(4) Å, Fe-N-C = 178°)[84].
We were fortunate to find that, akin to the mononuclear L3Co(NR) systems, iron(III) imides of the type [PhBPR3]Fe(NR) were synthetically accessible via two-electron oxidative group transfer to iron(I) precursors[53, 73, 85, 98]. Again, access to well-defined iron(I) synthons was therefore necessary. Despite the paucity of iron(I) complexes at the outset of our studies[99, 100], such species proved readily accessible by reduction of [PhBPR3]FeII-X precursors in the presence of a coordinating L ligand (e.g., L = PR3 or N2). For example, we initially reported that the sodium/mercury amalgam reduction of the yellow and high-spin (S = 2) complex [PhBPPh3]FeCl in the presence of PPh3 afforded monovalent, orange and high-spin (S = 3/2) [PhBPPh3]Fe(PPh3)[53]. As for the related cobalt system, addition of two equivalents of p-tolylazide generated the desired terminal d5 imide species [PhBPPh3]Fe(N-p-tolyl), in addition to Ph3P=N(p-tolyl) (Scheme 5). Of particular interest to us was the low-spin ground state of [PhBPPh3]Fe(N-p-tolyl), further buttressing the electronic structure picture that we had forwarded to account for the low-spin d7L3CoII-X and d6 and L3CoIII(NR) systems reviewed above.
Scheme 5.
As for the cobalt(III) imides, iron(III) imides containing a variety of ancillary organic groups have proven accessible using this methodology (Table 2). These iron(III) imides are generally highly colored and exhibit intense low-energy absorptions that can be assigned as imide-to-iron(III) charge-transfer bands[85]. As would therefore be expected, substitution of an alkylimide for an arylimide greatly reduces the intensity of these transitions, while also increasing the energy at which they are observed. Consistent with this observed blue-shift, the vibrations that are predominantly associated with Fe-N-R stretching character are observed around 1100 cm-1 for alkylimides and near 960 cm-1 for arylimides. While the alkylimides Fe-N-R stretching mode can be very crudely approximated as a harmonic oscillator, the latter arylimides feature strongly vibrationally coupled Fe-N-Cipso and Fe-N-Cipso modes that cannot be decoupled. The d-orbital electronic structures of these iron(III) imides are well described using arguments originally developed for Cp2Fe+ by Gray and coworkers[101, 102].
Table 2.
Select metrical parameters for pseudotetrahedral L3Fe(NR) complexes.
| Complex | Oxidation State | Fe-N bond length (Å) | N-C bond length (Å) | Fe-N-Cangle(°) | Displacement of Fe from L3 plane (Å) | Ref |
|---|---|---|---|---|---|---|
| {ClFe}3{NtBuFe}(μ3-NtBu)4 | 4+ | 1.635(4) | -- | 178.6(3) | -- | [84] |
| {[PhBPPh3]Fe(NAd)}{NnBu4} | 2+ | 1.651(2) | 1.434(3) | 178.6(1) | 1.24 | [54] |
| [PhBPPh3]Fe(NAd) | 3+ | 1.641(2) | 1.428(3) | 176.3(2) | 1.32 | [54] |
| [PhBPPh3]Fe(Np-tolyl) | 3+ | 1.658(3) | 1.383(3) | 170.0(2) | 1.31 | [53] |
| 1.661(3) | 1.374(3) | 167.3(2) | 1.29 | |||
| [PhBPPh3]Fe(NtBu) | 3+ | 1.635(1) | 1.442(2) | 179.2(1) | 1.29 | [85] |
| [PhBPiPr3]Fe(NAd) | 3+ | 1.638(2) | 1.438(3) | 176.0(2) | 1.27 | [73] |
| [PhBPCH2Cy3]Fe(NAd) | 3+ | 1.622(4) | 1.433(6) | 176.3(3) | 1.24 | [98] |
| [PhB(mesIm)3]Fe(NCPh3) | 3+ | 1.654(3) | 1.473(4) | 177.7(3) | 1.20 | [48] |
| [PhB(PtBu)2(pz)]Fe(NAd) | 3+ | 1.626(8) | 1.33(3) | 176(1) | 1.27 | [55] |
| 1.634(8) | 1.59(3) | 170(1) | 1.26 | |||
| {[PhB(PtBu)2(pz)]Fe(NAd)}{BArF24} | 4+ | 1.61(1) | 1.44(2) | 174(1) | 1.13 | [55] |
| [PhB(PtBu)2(pzMe,Me)]Fe(NAd) | 3+ | 1.647(8) | 1.45(1) | 172.2(6) | 1.22 | [55] |
| 1.652(8) | 1.44(1) | 172.4(6) | 1.23 | |||
| {[PhB(PtBu)2(pzMe,Me)]Fe(NAd)}{BArF24} | 4+ | 1.634(4) | 1.456(6) | 176.3(3) | 1.11 | [55] |
| [PhB(mesIm)3]Fe(NAd) | 3+ | 1.625(5) | 1.441(7) | 177.0(3) | 1.13 | [50] |
| {[PhB(mesIm)3]Fe(NAd)}{BPh4} | 4+ | 1.618(3) | 1.439(5) | 176.8(3) | 1.15 | [50] |
Akin to the cobalt(III) imides, nitrene transfer to CO also proved to be facile for the iron(III) [PhBPR3]Fe(NR) imides[53]. For instance, addition of one atmosphere of CO to [PhBPPh3]FeIII(N-p-tolyl) resulted in formation of O=C=N-p-tolyl and [PhBPPh3]FeI(CO)2. Alternatively, addition of C≡NtBu released the carbodiimide tBuN=C=N(p-tolyl). We were moreover able to show that the addition of N3(p-tolyl) to [PhBPPh3]FeI(CO)2 regenerated [PhBPPh3]Fe(N-p-tolyl), forming the basis for a catalytic nitrene transfer cycle[67].
In addition, the iron imide functionality proved susceptible to hydrogenolysis, a reactivity pattern that Chirik has since reported for a geometrically and electronically distinct iron(III) imide[47]. Treatment of [PhBPPh3]FeIII(N-p-tolyl) with one atmosphere of H2 resulted in complete scission of the Fe-N triple bond, liberating p-tolyl-aniline over the course of days at room temperature (Scheme 5)[103]. The high-spin S = 2 anilide species, [PhBPPh3]FeII(HN-p-tolyl), was observed as a long-lived intermediate and could be isolated. Further exposure to H2 released H2N-p-tolyl and ‘[PhBPPh3]Fe(H)’. The latter hydride species [104] was not observed but inferred from a subsequent benzene insertion reaction to generate [PhBPPh3]Fe(η5-C6H7). The solid-state structure of [PhBPPh3]FeII(HN-p-tolyl) showed an Fe-N-C angle of 127.4(2)° and a Fe-N bond distance of 1.913(2) Å, compared with the respective parameters of 1.658(3) Å and 170.0(2)° for [PhBPPh3]FeIII(N-p-tolyl). These distinct metrical parameters establish sp2-hybridization at nitrogen, which disrupts the degenerate π bonding manifold of the terminally bonded imide precursor. This disruption in turn leads to a preference for a high-spin configuration, owing to a weakened ligand field.
According to the qualitative d-orbital electronic structure picture we had advanced for these pseudotetrahedral L3MIII(NR) imides (Figures 2 and 3), we anticipated that iron(II) imides should also be accessible. We were gratified to observe that treatment of [PhBPPh3]Fe(NAd) with one equivalent of sodium/amalgam, followed by addition of [nBu4N][Br], resulted in the clean generation of {[PhBPPh3]FeII(NAd)}{nBu4N}[54]. The metrical parameters of the imide ligand in this d5 and d6 redox pair are similar, preserving short Fe-N bonds (approximately 1.65 Å) and near linear Fe-N-C bond angles (Table 2). These observations are consistent with a high degree of Fe-N π-bonding. The presence of two strong π-bonds from the imide linkage, along with the strong field tris(phosphino)borate ligands, once again confers the low spin configuration for the iron(II) system. The electronic structure of {[PhBPPh3]Fe(NAd)} {nBu4N} was probed by DFT methods, and is best described by a splitting diagram in which an orbital of 3dz2 parentage lies at low energy, close to 3dxy and 3dx2-y2 orbitals that are orthogonal to the Fe-N bond vector. The electronic structure pictures of low-spin L3FeII(NR)-, and isoelectronic L3CoIII(NR), are hence closely related to that which was originally elucidated for ferrocene in the classic study by Gray and coworkers nearly four decades ago[102, 105].
Figure 5a plots the visible absorption spectra of select L3FeII(NR)- and L3CoIII(NR) species. If one uses the spin-allowed optical transition assignments (two 1A1g→1E1g and one 1A1g→1E2g) for Cp2Fe and Cp2Co+ as a basis to assign the optical transition assignments for d6L3Fe(NR)- and L3Co(NR), then one derives a splitting Δ1 of ca. 1 eV between the lowest nonbonding xy/x2-y2 e-set and the z2a1 orbital (Figure 5b). This splitting is substantially smaller than the splitting Δ2 of ca. 2 eV between the a1 orbital and the highest-lying xz/yz e-set that is strongly σ* and π* in character. The splitting Δ1 is not anticipated by ground state DFT calculations of d6L3M(NR) imides, which place the a1 orbital nearly degenerate with the nonbonding e-set. It would be of obvious interest to theoretically calculate the optical transitions of d6L3M(NR) imides to further explore this issue.
Figure 5.
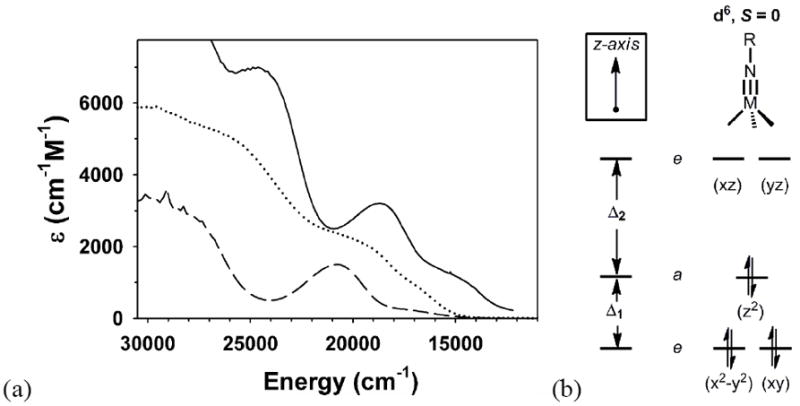
(a) Visible absorption spectra of d6 pseudotetrahedral metal imides. All spectra were recorded at room temperature. (A) [PhBPPh3]Co(NPh) (—); (B) {[PhBPPh3]Fe(NAd)}- (⋯) in THF; and (C) [PhBPPh3]Co(NtBu) (—). (b) d-orbital splitting diagram defining splittings Δ1 and Δ2.
An obvious progression was to next begin exploring whether the d4 iron(IV) configuration might also be compatible within the L3Fe(NR) framework. In parallel with our own studies of L3FeIII(NR) systems, terminally bonded 6-coordinate iron(IV) oxos had begun to emerge as rigorously isolable species, for example by the groups of Que and Nam[106-109]. Our initial efforts towards L3FeIV(NR) species focused on the one-electron chemical oxidation of [PhBPR3]FeIII(NR) precursors, but we were unsuccessful: if an Fe(IV) species was generated in such reaction mixtures, it was not sufficiently long-lived to be detected by the routine characterization methods we adopted. Interrogation of [PhBPR3]FeIII(NR) complexes by cyclic voltammetry revealed a chemically irreversible Fe(IV/III) redox couple, contrasting the reversible Fe(III/II) couple that was present at more negative potentials. This observation enforced the notion that [PhBPR3]FeIV(NR)+ species are kinetically unstable, and we thus began to explore related 3-coordinate ligand scaffolds in the hopes of lending a higher degree of stability to the iron(IV) state. One path of exploration that ultimately proved efficacious was using hybrid scaffolds, whereby one of the phosphine donor ligands was replaced by a pyrazolyl group. To access such ligands, it proved synthetically necessary to decorate the two phosphine donor arms with tert-butyl groups, as the intermediate bis(phosphino)boranes, PhB(CH2PR2)2, were unstable to dimerization when less encumbering groups were employed[110]. Addition of pyrazole anions to PhB(CH2PtBu2)2 afforded bis(phosphino)pyrazolylborate ligands, abbreviated as [PhB(PtBu)2(pzR,R’)] (pzR,R’ = pyrazole substituted at the 3 and 5 positions, respectively).
Using a hybrid scaffold of this type, we found that low-spin terminal iron(III) imides could be readily generated, as for the complexes [PhB(PtBu)2(pz)]Fe(NAd) and [PhB(PtBu)2(pzMe,Me)]Fe(NAd)[55]. The cyclic voltammograms of these species were distinct from those oftris(phosphino)borate congeners, in that now, the Fe(III/II) couple was irreversible, but the Fe(III/IV) couple was quasi-reversible or reversible at room temperature depending on the scan-rate. Chemical oxidation of these iron(III) imides with [Cp2Fe][BArF24] (BArF24= tetra(3,5-di(trifluoromethyl)phenyl)borate) afforded the bright green iron(IV) imides{[PhB(PtBu)2(pz)]FeIV(NAd)}{BArF24} and {[PhB(PtBu)2(pzMe,Me)]FeIV(NAd)}{BArF24}, the former of which was very thermally unstable (Scheme 6). The Fe-N bond distance in {[PhB(PtBu)2(pzMe,Me)]FeIV(NAd)}{BArF24} is 1.634(4) Å and the Fe-N-C angle is 176.2(3)°, similar to that of all of the pseudotetrahedral iron(III) imides we had characterized. This is consistent with removal of an electron from an orbital that is orthogonal to the Fe-NAd bond vector, and a (dz2)2(dxy)1(dx2-y2)1(dxz)0(dyz)0 electronic configuration (Figure 2). Solution Evans method and DFT data were consistent with this S = 1 electronic structure picture. Curiously, upon oxidation, the distance between the iron center and the plane defined by the donor atoms of the tripodal ligand (N-P-P) decreased by ca. 0.11 Å to 1.11 Å. This decrease results in slightly longer Fe-Pave bond distances (ca. 0.03 Å) in the iron(IV) imide compared tothe iron(III) congener, which might result in a net decrease of π backbonding from iron into ligand σ* orbitals of appropriate symmetry. This hypothesis is consistent with the observation that the average Fe-P distance decreases by ca. 0.1 Å when neutral [PhBPPh3]FeIII(NAd) is reduced by one electron to {[PhBPPh3]FeII(NAd)}{nBu4N}, which presumably leads to a net increase in π backbonding into the phosphine ligand σ* orbitals [54].
Scheme 6.
Smith and coworkers[50] have recently extended the number of well-characterized iron(IV) imides of the pseudotetrahedral L3Fe(NR) structure type via the isolation of an intermediate-spin iron(IV) imide, {[PhB(mesIm)3]FeIV(NAd)}{OTf}, whose electronic structure is similar to that of {[PhB(PtBu)2(pzMe,Me)]FeIV(NAd)}{BArF24}. This species was generated by oxidation of the precursor iron(III) imide with [Cp2Fe][OTf] (Scheme 6). The metrical parameters for the core atoms of both the iron(III) and iron(IV) imide congeners are quite similar. For example, the Fe-Nimide distances (1.625(4) Å for Fe(III) and 1.618(3) Å for Fe(IV)), the Fe-N-C angle (177.0(3)° for Fe(III) and 176.8(3)° Fe(IV)), and the distance between the iron and the plane defined by the tris(carbene) C-donor atoms (1.13 Å for Fe(III) and 1.15 Å for Fe(IV)) change very little. However, the average Fe-C distance increased by 0.04 Å upon oxidation. Again, diminished π backbonding, in this case into σ* orbitals of the tris(carbene) ligand might offer a plausible explanation.
2.4 Pseudotetrahedral L3Fe(N) Species
Well-defined and terminally bound nitrides of iron remain relatively rare. Bridging species are instead often obtained[6, 111-114]. Nakamoto and coworkers were first to describe the generation of a terminally bonded iron nitride species using an octaethylporphyrin (OEP) supporting ligand framework[115, 116]. They employed a very elegant vibrational analysis to support their assignment. However, the high thermal instability of the (OEP)FeV(N) species precluded its further characterization. Wieghardt and coworkers have since characterized low-spin 6-coordinate iron(V) [117, 118] and iron(VI) [119] nitrides using cyclam and cyclam-acetate supporting ligands, respectively. These complexes also exhibit limited thermal stability but have been characterized by a suite of spectroscopic techniques, in addition to having been analyzed theoretically[120]. They populate low-spin ground states. The S = ½ iron(V) d3 species appears less stable than the diamagnetic iron(VI) species. The latter d2 species finds close structural and electronic analogy to the well-established and studied Mn(V) nitride systems[12, 121, 122].
Our own group’s interest in this area focused instead on exploring the feasibility of terminally bonded nitrides under local three-fold symmetry of the type L3Fe(N). Theoretical analysis suggested to us that replacement of the terminal NR2- ligand by an N3- ligand in systems of this type would strongly destabilize the a1 orbital of dz2 parentage that is low-lying in L3Fen(NR) (n = II, III, IV) species (Figure 2)[52]. Such destabilization would thereby render d5 and d6 configurations comparatively less favorable due to population of a strongly antibonding σ* orbital. By contrast, d4 and perhaps even lower d-count species might be electronically favored, as there are only two energetically low-lying d-orbitals (dx2-y2/dxy). For an iron(IV) species, the predicted electronic configuration is therefore (xy)2(x2-y2)2(z2)0(xz)0(yz)0 (Figure 6). Such a configuration is similar to Wilkinson’s d4Mes3Ir(O) complex. The destabilization of dz2 relative to the structurally related imides is a consequence of better overlap with the nitrogen sp-hybrid orbital, and a distortion that renders the geometry about the iron center more tetrahedral. The calculated S = 0 [PhBPiPr3]Fe(N) structure features P-Fe-P angles between 99-101°, whereas these calculated angles for the corresponding imide, S = 0 {[PhBPiPr3]Fe(NtBu)}-, are between 90-95°, in accord with a wealth of experimental data. It was therefore incumbent upon us to select an appropriate N-atom transfer agent to test whether an S = 0 nitride would be accessible using the tris(phosphino)borate iron frameworks.
Figure 6.
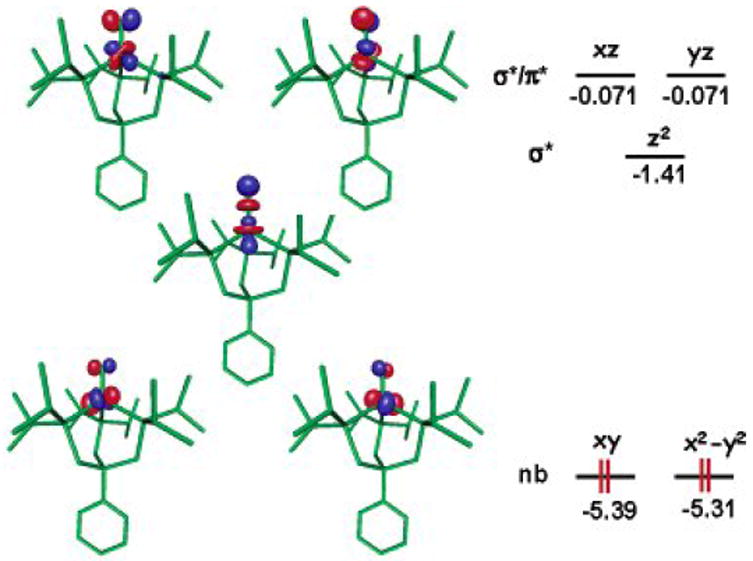
Theoretically predicted geometry and electronic structure (DFT,JAGUAR 5.0, B3LYP/LACVP**) for S = 0 [PhBPiPr3]Fe≡N. Lobal representations correspond to the frontier orbitals (energies in eV). Structural parameters: Fe-P = 2.28, 2.28, 2.29 Å; N-P-Fe = 117, 117, 119°; P-Fe-P = 99, 101, 101°; Fe-N = 1.490 Å. Reprinted with permission from ref[52]. Copyright 2004 American Chemical Society.
Nitride transfer reactions often occur as two-electron processes that deliver an N-atom while liberating a thermodynamically stable molecule. Azide, N3-, is often the reagent of choice owing to its synthetic simplicity and the release of N2 as a byproduct[49, 51, 115, 117, 123]. N2 extrusion can occur spontaneously or it can be effected by heating or photolytic means, the latter often being necessary when the resultant nitride is thermally unstable. We found that such protocols were in effective for generating terminal nitrides with the ‘[PhBPiPr3]Fe’ and ‘[PhBPPh3]Fe’ frameworks. For example, {[PhBPPh3]Fe(N3)}2 can be prepared and is a thermally stable species. While N2 extrusion might have been effected by its one-electron reduction to release the d5 {[PhBPPh3]Fe(N)}- and N2, any such species appears to be rapidly trapped by “[PhBPPh3]FeI” in solution (iron(I) is generated by kinetically competitive loss of N3- upon reduction) to afford the stable diiron(II) μ-nitride complex {[PhBPPh3]Fe}2(μ-N)-, as shown in Scheme 7[54].
Scheme 7.
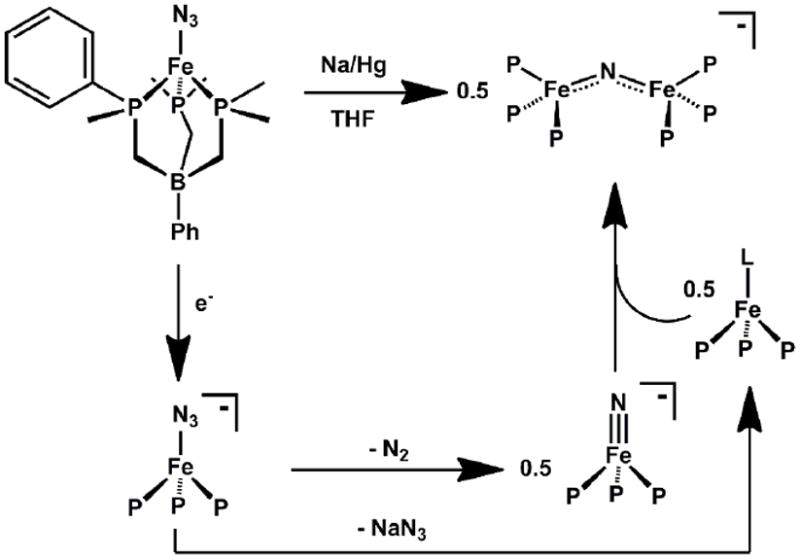
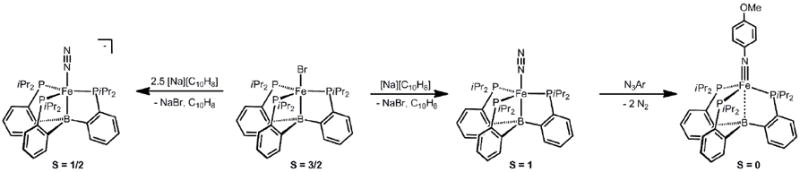
An alternative approach that proved viable involved use of the N-atom transfer agentLi(dbabh) (dbabh = 1,2:5,6-dibenzo-7-aza bicycle[2.2.1]hepta-2,5-diene). This lithium amide was first used by Cummins and Mindiola to prepare a high-valent chromium nitride[124]. Its utility involves initial formation of ametal amide, Mn-(dbabh), which can thermally release Mn+2(N) and one equivalent of anthracene. The utility of this reagent appears to be limited; for example, we have explored its use to install nitride groups at ‘[PhBPR3]Mn’ and ‘[PhBPR3]Ni’ systems and in doing so have uncovered alternative reaction manifolds[125, 126]. But to our satisfaction (and relief!), the synthon worked well for ‘[PhBPiPr3]Fe’.
Using Li(dbabh), the iron(IV) nitride species, [PhBPiPr3]FeIV(N), can be generated and thoroughly characterized (Scheme 8)[52]. Treatment of [PhBPiPr3]FeCl with Li(dbabh) at -35 °C generated the red and thermally unstable iron(II) amide intermediate, [PhBPiPr3]FeII(dbabh). Upon warming to 0 °C, this intermediate underwent clean first order decay to release one equivalent of anthracene and the desired diamagnetic product, [PhBPiPr3]Fe(N). Owing to its many spin active nuclei and its diamagnetism, solution NMR characterization of [PhBPiPr3]Fe(N) was straightforward. For example, a diagnostic 15N NMR chemical shift of 952 ppm was observed for the nitride (Table 3), in accordance with the chemical shift noted for terminal M(N) species[31, 127-129]. The solution IR spectrum showed an Fe-N stretch at 1034 cm-1 which is consistent with that of other M(N) species [130]. Upon15N-labelling, the stretch shifts to 1007 cm-1, in agreement with that calculated (27 cm-1) assuming an Fe-N harmonic oscillator. The Mössbauer spectrum of [PhBPiPr3]Fe(N) was also obtained, and established an isomer shift of δ - 0.34 mm/s, and an unusually large quadrupole-splitting parameter, ΔEQ= 6.01 mms-1[131]. This large quadrupole splitting results from the highly anisotropic electric field gradient that arises from electronic population of dx2-y2 and dxy orbitals orthogonal to the B-Fe-N vector, and has proven diagnostic of L3FeIV(N) species (vide infra). Spontaneous nitride coupling to generate the formal diiron(I)product, {[PhBPiPr3]Fe}2(μ-N2), occured upon concentration or attempts to crystallize [PhBPiPr3]Fe≡N[52]. We canvassed one other scaffold, ‘[PhBPCH2Cy3]Fe’ (CH2Cy = cyclohexylmethyl) and successfully generated and characterized [PhBPCH2Cy3]Fe(N) at low temperature[131]. Though nitride coupling does not occur for this latter system ({[PhBPCH2Cy3]Fe}2(μ-N2) is not an accessible species[98]), [PhBPCH2Cy3]Fe(N) proved to be unstable above -50 °C, and poorly defined degradation pathways frustrated our ability to crystallize this species. Additionally, a short Fe-N bond distance of 1.53(2) Å was obtained for these species by EXAFS [132].
Scheme 8.
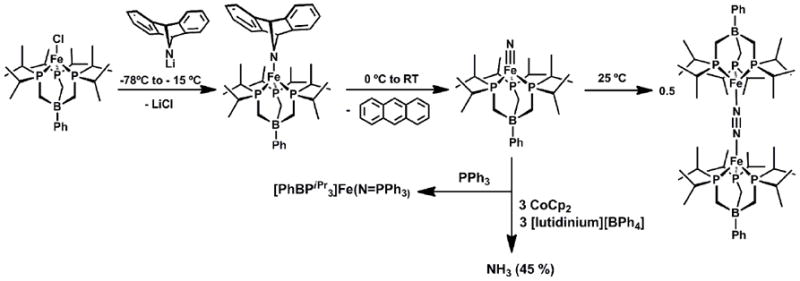
Table 3.
Select spectroscopic and structural parameters of pseudotetrahedral L3FeIV(N) complexes.
| Complex | Fe-N stretch14N/15 N (cm-1) | Fe-N distance (Å) | Displacement of Fe from L3 plane (Å) | 15N NMR chemical shiftaδ | MB δ (mms-1) | MB ΔEQδ (m ms -1) | Ref |
|---|---|---|---|---|---|---|---|
| [PhBPiPr3]FeN | 1034/ 1007 | 1.51-1.54b | -- | 952 | -0.34(1) | 6.01(1) | [52, 131, 132] |
| [PhBPCH2Cy3]FeN | -- | 1.55b | -- | 929 | -0.34(1) | 6.01(1) | [131, 132] |
| [(TIMENmes)FeN][BPh4] | 1008/982 | 1.526(2) | 0.43 | 1121 | -0.27(1) | 6.04(1) | [51] |
| [(TIMENxyl)FeN][BPh4] | -- | 1.527(3) | 0.38 | -- | -- | -- | [51] |
| [PhB(tBuIm)3]FeN | 1028/ 999 | 1.512(1) | 0.98 | 1019 | -- | -- | [49] |
| [PhB(mesIm)3]FeN | -- | 1.499(5) | 1.01 | 1004 | -- | -- | [48] |
Referenced to liquid NH3 at 0 ppm.
Obtained by EXAFS.
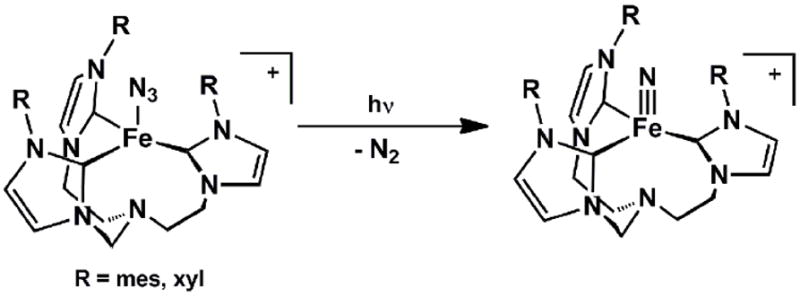
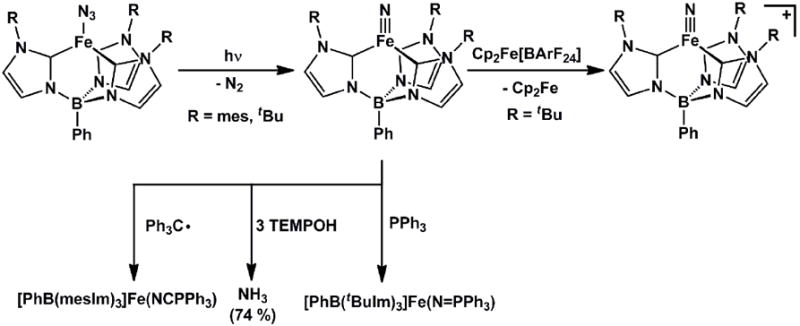
The respective groups of Meyer and Smith have also targeted terminally bonded iron(IV) nitrides in recent years using the carbene ligand frameworks introduced above. Meyer’s 4-coordinate nitrides, [(TIMENR)Fe(N)][BPh4] (R = mes, xyl), formed via photolysis of 4-coordinate azide precursors, [(TIMENR)Fe(N3)][BPh4], proved thermally stable and hence amenable to single crystal XRD characterization (Scheme 9) [51]. Such data revealed an Fe-Nnitride bond distance of 1.526(2) Å for [(TIMENmes)Fe(N)][BPh4](Table 3). This complex represented the first reported terminal iron nitride species to be characterized by X-ray crystallography. Smith’s X-ray structure of [PhB(tBuIm)3]Fe(N) followed soon thereafter[49]. Meyer’s system provided experimental validation that a 4-coordinate L3FeIV(N) system is energetically preferred relative to a corresponding 5-coordinate L’L3FeIV(N) under local three-fold symmetry, since the apical amine donor does not bind the iron center. The geometry about the iron centers in [(TIMENR)Fe(N)][PPh4] are close to trigonal pyramidal, with the iron centers ca. 0.54 Å above the plane defined by the three carbene donors. Therelated [PhB(RIm)3]Fe(N) system reported by Smith and coworkers was likewise generated via photolysis of terminal azide precursors. As noted for the calculated structure of [PhBPiPr3]Fe(N), [PhB(tBuIm)3]Fe(N) is somewhat more tetrahedral in structure compared to the corresponding iron(IV) imides (i.e.,{[PhB(tBuIm)3]Fe(NAd)}{OTf}), as the average C-Fe-Cangle increases from ca. 90° to ca. 96°. DFT calculations on the iron(IV) nitrides supported by the three different ligand scaffolds indicate similar electronic structures[49, 51, 52]. Hence, inall three scaffolds the tridentate L3 donor adopts a geometry such that the a1 orbital is destabilized to accommodate a favorable d4S = 0 (xy)2(x2-y2)2(z2)0(xz)0(yz)0 electronic configuration. Note that this electronic structure is distinct from the iron(IV) imides, which adopt an S = 1 ground state due to the fact that the a1 orbital of dz2 parentage lies much closer in energy to the nonbonding dxy, dx2-y2 e set of orbitals (Figure 2).
Scheme 9.
While the reactivity patterns of these iron(IV) complexes have not been reported in great detail, there are a few transformations worthy of note. Treatment of both [PhBPiPr3]Fe(N) and [PhB(tBuIm)3]Fe(N) with triphenylphosphine effected reductive nucleophilic attack and afforded the corresponding high spin iron(II) phosphiniminato complexes, [PhBPiPr3]Fe(N=PPh3) and [PhB(tBuIm)3]Fe(N=PPh3), respectively[49, 52]. Our own group was interested in determining whether the terminal nitride of [PhBPiPr3]Fe(N) could liberate NH3 on treatment with an e-/H+ source, a transformation motivated by a hypothetical Fe-mediated distal scheme for N2 reduction. It was found that NH3 could be liberated upon addition of [lutidinium][BPh4] in the presence of CoCp2, albeit in moderate yield (ca. 45%)[52]. Smith’s nitride, [PhB(mesIm)3]Fe(N), was treated with a well-defined H-atom transfer equivalent, TEMPOH, and gave rise to an appreciably higher yield of NH3 (74%)(TEMPOH = 1-hydroxy-2,2,6,6-tetramethylpiperidine) [48]. The first step of this transformation is thought to occur via H-atom transfer (HAT) from TEMPOH to the nitride, though a protonation/reduction could not be ruled out from the measured free energy of activation. Smith’s nitride was also exposed to trityl radical to generate the corresponding iron(III) imide, [PhB(tBuIm)3]Fe(NCPh3). This transformation thereby established another synthetic route to iron imide species.
Perhaps the most interesting reaction to note concerns oxidation of [PhB(tBuIm)3]Fe(N). According to the d-orbital configuration proposed for L3FeIV(N) species, d-electron configurations less than d4 should in principle also show electronic stability. Removal of one electron to generate a d3L3FeV(N) species is therefore of paramount interest. Such a reaction has very recently been demonstrated using the [PhB(tBuIm)3]Fe(N) precursor. Low temperature oxidation by [Cp2Fe][BArF24] was used to generate the d3 iron(V) nitride {[PhB(tBuIm)3]Fe(N)}{BArF24}, a low-spin species that proved sufficiently stable to isolate and characterize by X-ray crystallography[133]. As should be expected for an (xy)2(x2-y2)1(z2)0(yz)0 (xz)0 electronic configuration, the Fe-N distance remains virtually unchanged (Fe-N = 1.506(2) Å) relative to its d4 precursor, and the iron center remains ca. 1 Å above the L3 donor plane. Also, {[PhB(tBuIm)3]Fe(N)}{BArF24} likewise affords a diagnostic large quadrapole splitting parameter ΔEQ= 4.25 mms-1 in the Mössbauer spectrum.
3. Trigonal Bipyramidal (TBP) L’L3-M(E) Structure Types
3.1 Orbital Considerations
An interesting question that follows the preceeding discussion of L3M(NR) and L3M(N) species concerns how the electronic structure rules will vary upon coordination of a fifth ligand in the site trans to the multiply bonded ligand E. Such coordination preserves local three-fold symmetry and leads to an L’L3M(E) structure type, where the L3 donor set moves into the equatorial plane and L’ and E occupy axial sites. Qualitative d-orbital splitting diagrams that provide a starting point for discussing L’L3M(E) species are shown in Figure 7. This electronic picture was introduced in a brief in a review article by Miskowski, Hopkins, and Gray [134]. Assuming a σ-only bonding picture, the dxz/yz e set is stabilized by the absence of any σ* interactions with the L’L3 donor set, which contrasts the strongly destabilized σ* dxz/yz e set in pseudotetrahedral L3M(E) structures. As the E ligand px and py orbitals are of π symmetry, they can therefore strongly overlap with the dxz/yz e set. If these p-orbitals and the dxz and dyz orbitals are each filled with electron pairs, as they would be in a hypothetical species such as S = 0 L’L3FeIV(N), then a destabilizing π* interaction is present that removes net π bonding at the Fe-N linkage. By contrast, if the ligand E has empty π acceptor orbitals (akin to a CO or NO+ ligand, for example) then backbonding interactions will occur to stabilize the dxz/yz e set and lead to favorable metal-to-E multiple bonding. A degree of favorable net π-bonding can also be achieved even in the case of π-donating ligands such as oxos, imides, and nitrides if only partial population of the dxz/yz e set occurs. Perhaps counter-intuitively, the situation can therefore arise where a high-spin d-electron configuration can feature greater multiple bond character than an intermediate- or low-spin configuration for L’L3Fe(E) structure types (see Figure 2 and 7). This may be, for example, achieved using supporting ligands that confer a weak ligand field such that a high-spin state is conferred. This scenario directly contrasts that of L3M(E) species, where high spin configurations will always attenuate the degree of M(E) π bonding. Below we discuss select and pedagogically informative examples of each of these cases, beginning with systems that tend towards low-spin configurations.
Figure 7.
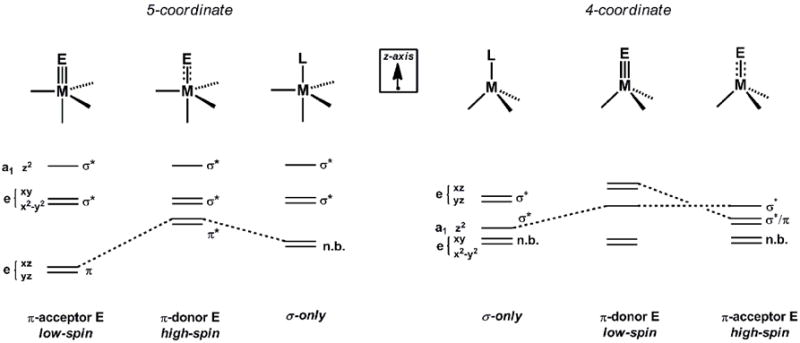
Qualitative d-orbital splitting diagrams for 4- and 5-coordinate C3v species, with the anticipated spin-state for each M(E) type noted.
3.2 Trigonal Bipyramidal L’L3Fe(NR) Species
The well-studied pseudotetrahedral L3FeIIINR complexes feature bona fide Fe≡NR triple bonds resulting from two highly destabilized, unoccupied σ*L3Feπ*FeN orbitals (Figures 2 and 7). Introducing a ligand trans to the imido group and shifting the Fe into the L3 plane of a TBP leads in principle to population of the π*FeN set, thereby obliterating a significant degree of the Fe-N multiple bonding character presumed responsible for the stability of pseudotetrahedral L3FeIIINR species. Accordingly, until very recently metal-ligand multiply bonded species in TBP configurations of the general type L’L3M(E), where E is a prototypical π-donor ligand, had been isolated only for d-electron counts of 0 or 1[26]. TBP systems with higher d-electron counts would be expected to dissociate the apical ligand and distort towards the more stable pseudotetrahedral geometry when accommodating an axial metal-ligand multiple bond, as evident from Meyer’s nitrides[(TIMENR)Fe(N)][BPh4][51] and imides [(TIMENR)Co(NAr)][BPh4][58].
To explore a system where such distortion of the axial ligand is prohibited, our group began to study the feasibility of L’L3Fe(E) species using an anionic tris(phosphino)silyl ligand, (2-R2PC6H4)3Si- ((SiPR3); R = Ph or iPr), as the L’L3tetradentate scaffold[135, 136]. These ligands feature a strongly boundsilyl donor in the axial position of a TBP, and three tightly chelated phosphine donors in the equatorial plane. Using this ligand scaffold, we have recently mapped the synthetic feasibility of iron imides of the type (SiPR3)Fe(NR) for direct comparison to [PhBP3]Fe(NR) and related 4-coordinate imides.
In contrast to the [PhBPR3]- ligand, which accommodates iron in both high- and low-spin configurations depending on the apically bound ligand (e.g., S = 3/2 [PhBPPh3]Fe(PPh3) and S = 1/2 [PhBPPh3]Fe(NtBu)) , the (SiPR3)- ligand appears to exclusively enforce low- and intermediate-spin configurations. Hence, 5-coordinate iron(I) d7(SiPR3)Fe-L complexes are S = ½, as for the representative complexes (SiPR3)Fe-N2 and (SiPR3)Fe(PMe3). Similarly, iron(II) d6 complexes of the type {(SiPR3)Fe-L}+ and (SiPR3)Fe-X(L = NH3, N2; X = Cl, Me) are invariably intermediate spin S = 1. Removal of an additional electron, as for the complex {(SiPR3)FeIII-Cl}{BArF24}, again results in a d5, S = 3/2 intermediate-spin configuration[135-137].
Using a synthetic protocol analogous to that described for the generation of [PhBPR3]MIII(NR) complexes (M = Fe, Co), we explored the generation of (SiPR3)Fe(NR) [89]. Our study showed that (SiPiPr3)Fe(NAr) species could be generated by addition of arylazides to (SiPiPr3)Fe-N2, but that they were unstable to subsequent decay pathways (Scheme 11). The azide adduct intermediates were more thermally stable and could be isolated for bulky alkyl azide derivatives, for example, (SiPiPr3)Fe(η1-N3Ad). The dominant decay pathway we have discerned for the (SiPiPr3)Fe(NAr) imide species concerned bimolecular ‘NAr’ coupling to generate azoarenes ArN=NAr. Such a pathway regenerated (SiPiPr3)FeI-N2, and hence arylazides could be catalytically degraded to the azoarene by products. Chemical trapping experiments were used to further corroborate the presence of the terminal imide moiety. For example, addition of tBuNC released the carbodiimide tBuN=C=N-p-tolyl and (SiPiPr3)Fe(CNtBu). This two electron nitrene transfer is diagnostic of isolable FeNR species (vide supra). Also, H-atom trapping by 9,10-dihydroanthracene afforded the d6 anilide (SiPiPr3)Fe(HN-p-tolyl). Related HAT chemistry has been observed in high-spin 4-coordinate FeNR species[65, 138] and invoked in other systems[45]. The ability of (SiPiPr3)FeNAr to fascilitate one (HAT), two (nitrene transfer), and four-electron (azoarene formation) transformations has hence been demonstrated.
Scheme 11.
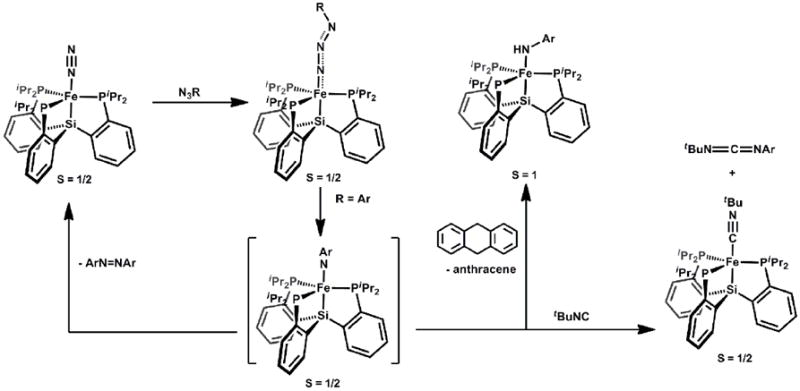
Direct detection of S = ½ [SiPiPr3]Fe(N-p-tolyl) was accomplished using low temperature EPR spectroscopy via in situ photolysis of the azide adduct precursor, (SiPiPr3)Fe(N3-p-tolyl), in a frozen glass[89]. These data, in addition to an accompanying DFT study, placeda majority of the unpaired spin density on iron for (SiPiPr3)Fe(N-p-tolyl). A distortion away from three-fold symmetry was predicted in the DFT minimized S = ½ structure. The low-spin configuration seems to be energetically preferred in silico, but was not appreciably lower in energy than the intermediate-spin S = 3/2 species, which also shows a distortion from three-fold symmetry. For comparison, a ruthenium congener, (SiPiPr3)Ru(N-p-trifluoromethylphenyl), could be prepared, isolated, and thoroughly characterized, including XRD analysis[139]. However, its EPR spectrum suggested an electronic structure distinct from that of (SiPiPr3)Fe(N-p-tolyl), and combined with accompanying X-ray data and DFT calculations, indicated that (SiPiPr3)Ru(N-p-trifluoromethylphenyl) is best described as a ruthenium(II) species with an imidyl radical. This may account for its enhanced stability relative to (SiPiPr3)Fe(NAr).
Very recently our group has explored a ligand closely related to (SiPiPr3)-, but where the Si-atom is replaced by a B-atom. The tris(phosphino)borane ligand (2-iPr2PC6H4)3B, was originally introduced by Bourissou and coworkers[140], and has been abbreviated as TPB (not to be confused with TBP!). We adopt the same convention here. The tris(phosphino)borane TPB ligand might better accommodate the terminal Fe(NR) linkage due to its ability to undergo an axial distortion that would weaken the Fe-B interaction, akin to that observed by Meyer in the tris(carbene)amine ligand scaffold. While studies of (TPB)Fe species are in their early days in our labs, we have determined that the complexes {(TPB)Fe(N2)}- and (TPB)Fe(N-p-MeO-phenyl) can be prepared (Scheme 12) and structurally characterized, and that the Fe-to-B bond distance adjusts dramatically to accommodate a π-acidic N2 ligand (2.29 Å in {(TPB)Fe(N2)}{Na(12-crown-4)2}) relative to a π-basic imide ligand (2.61 Å in (TPB)Fe(N-p-MeO-phenyl)). Indeed, the latter species is perhaps best formulated as trigonal pyramidal, as the iron sits 0.68 Å above the plane defined by the three phosphine donor ligands. However, a weak Fe-B interaction is indicated by DFT analysis and the triarylborane framework remains pyramidalized in the structure of (TPB) Fe(N-p-MeO-phenyl). The iron imide functionality displays similar metrical parameters in the solid-state (Fe-N: 1.668 Å, Fe-N-C: 170.2°) to L3Fe(NR) species in rigorously 4-coordinate geometries[141].
Scheme 12.
3.3 Trigonal Bipyramidal L’L3Fe(CR) and L’L3Fe(N2R) Species
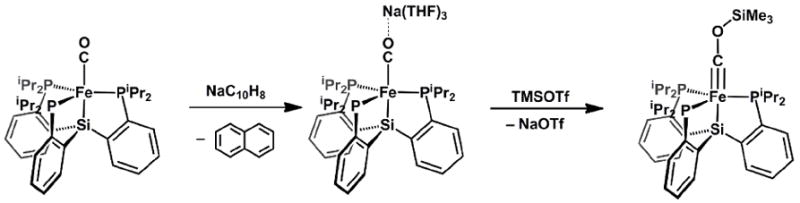
An alternative means of stabilizing metal-to-ligand multiple bonding in complexes of the L’L3Fe(E) structure type concerns coordination of a Lewis-acid (e.g., SiMe3+) to a π-acidic ligand (N2 or CO), which should in turn enhance its π-acceptor character. This is well-illustrated by our group’s recent characterization of the terminal carbyne complex (SiPiPr3)Fe(C-OSiMe3), generated via silylation of the coordinated CO ligand in(SiPiPr3)Fe(CO)- (Scheme 13)[142]. X-ray, Mössbauer, and DFT analysis of this carbyne have established that the iron center is best described as d8 with two electron pairs strongly backdonating into the carbyne C-atom to affordan Fe-C bond distance of 1.67 Å. The one caveat to this d8 assignment concerns the presence of a filled Fe-Si σ bonding orbital within the d-orbital energy manifold. This orbital is not shown in Figure 2, but if included, suggests that a d10 assignment is also plausible. Regardless, in contrast to the Fe-N interaction in the imide species (SiPiPr3)Fe(N-p-tolyl), the Fe-C interaction in (SiPiPr3)Fe(COSiMe3) definitely has triple bond character. Hence, one way to circumvent the apparent incompatibility of a high d-electron count within the TBP framework and an axial multiply bonded ligand is to reverse the polarity of the multiply bonded ligand so that it is a π-acceptor rather than a π-donor (Figure 7). Such a situation should thereby lead to stable complexes with high d-counts, and finds analogy in the stability of d8 FeL5 structure types such as Fe(CO)5[74].
Scheme 13.
Using a similar synthetic strategy (i.e. addition of an electrophilic silyl reagent to an Fe(0) precursor), the diazenido complex (SiPiPr3)Fe(N2SiMe3) could likewise be generated[137]. This species is isoelectronic with (SiPiPr3)Fe(COSiMe3) and also features a degree of Fe-to-N multiple bond character. The combined solid-state structure and diamagnetic ground-state, suggested that it too can be assigned a low-spin d8 configuration.
3.4 Trigonal Bipyramidal L’L3Fe(O) Species
Owing to their postulated mechanistic role in non-heme oxygen activating enzymes, there has been much recent interest in the development of synthetic iron(IV) oxo model species[5, 143]. The S = 2 high spin ground state of the biological non-hemeferryl intermediates is presumed to contribute to theirhighly reactive nature[144], and biomimetic analogues that are high-spin have likewise proven comparatively difficult to generate and characterize. The first generation of synthetic non-heme iron(IV) oxo complexes that were thoroughly characterized, including X-ray crystallography, were introduced by Que and Nam and featured local C4v symmetry and intermediate spin S = 1 ground states[106-109]. These have been thoroughly discussed elsewhere[8, 145]. A descent from four-fold symmetry to three-fold symmetry proved necessary to access well-defined and relatively stable high-spin iron oxo species that feature Fe=O multiple bond character[146].
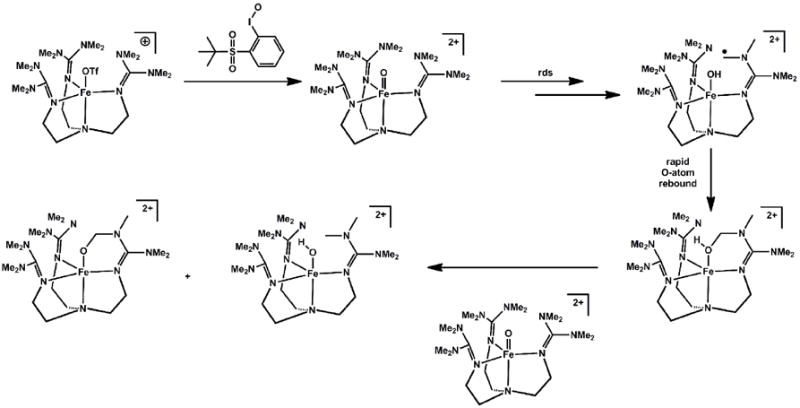
Que and coworkers described the preparation of the first synthetic example of a high-spin non-heme iron(IV) oxo species[147]. Using the neutral tetrapodal ligand TMG3tren (TMG3tren = 1,1,1-tris{2-[N2-(1,1,3,3-tetramethylguanidino)]ethyl}amine), the trigonal bipyramidal precursor species (TMG3tren)FeII(OTf)+ was exploited. Treatement of (TMG3tren)FeII(OTf)+ with 2-(tert-butylsulfonyl)-iodosylbenzene resulted in the formation of the unusual terminal oxo species (TMG3tren)FeIV(O)2+ (Scheme 14). Its high-spin state was determined by Mössbauer spectroscopy, and further corroborated by XAS and DFT calculations. The high spin nature of (TMG3tren)FeIV(O)2+ appeared to contribute to its high degree of thermal instability. A facile intra-molecular self-decay pathway afforded it a half-life of only ca. 30 s at 25 °C[148]. The self-decay pathway was thoroughly examined, and the proposed mechanism of decay is shown in Scheme 14. Kinetic studies on the decay of (TMG3tren)FeIV(O)2+ and (d36-TMG3tren)FeIV(O)2+ revealed a large and primary KIE of 24 at 25 °C. This isotope effect lent added stability to the deuterated analogue, and allowed for crystals suitable for X-ray data collection to be obtained at -80 °C. The Fe-O bond distance in (d36-TMG3tren)FeIV(O)2+ was found to be 1.661(2) Å, in agreement with that predicted by DFT and the distance obtained by EXAFS. This bond distance is very similar to that of S = 1 Fe=O species[56, 107, 149]. Taking into account the different d-orbital splitting diagrams under C4v and C3v symmetry this is to be expected, as in both cases the π* orbitals are populated by only two electrons.
Scheme 14.
The group of Borovik has also been able to prepare and isolate 5-coordinate high-spin iron(IV) and iron(III) oxo complexes that reside in local three-fold symmetric environments. With the idea that H-bonding to a terminal oxo ligand may stabilize it, the trianionic, tetrapodal ligand, tris[(N’-tert-butylureaylato)-N-ethyl)]aminato ([NNH33]3- (where H3 indicates the number of H-bond donors), was developed[150]. This ligand scaffold enforces a TBP geometry, and features three acidic C(O)NH(tBu) groups above the plane of the anionic nitrogen donors, which can act as H-bond donors to an oxo ligand.
Treatment of the ligand, [NNH33]3- with an equivalent of KH, followed by treatment with Fe(OAc)2 and half an equivalent of O2, afforded the terminal oxo [NNH33]FeIII(O)2-[151]. Its proposed mechanism of formation is shown in Scheme 15, which accounts for the additional equivalent of base. This complex is remarkable in that it is high-spin, and yet relatively stable. The solid-state structure of [NNH33]FeIII(O)2- revealed that the oxo ligand is only 0.04 Å displaced from the plane defined by the three urea nitrogen atoms and supported by H-bonding. The Fe-O bond distance of 1.813(3) Å in [NNH33]FeIII(O)2- is comparable to those found in FeIII2(μ-O) complexes[152], and hence is consistent with partial multiple bond character. This bond distance is however longer than that found in the d2 [FeO4]2-, which features Fe-O bond distances that range from 1.660(2) to 1.671(2) Å[153]. The stability of [NNH33]FeIII(O)2- is in part due to the formation of hydrogen bonds, which decreases the covalency in the Fe-O bond and results in elongation of the Fe-O bond[154].
Scheme 15.
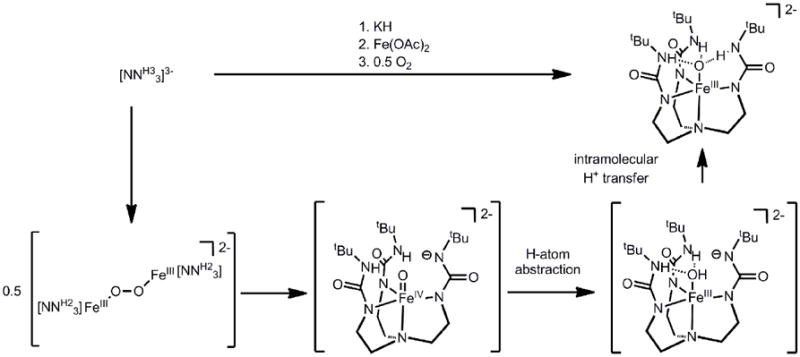
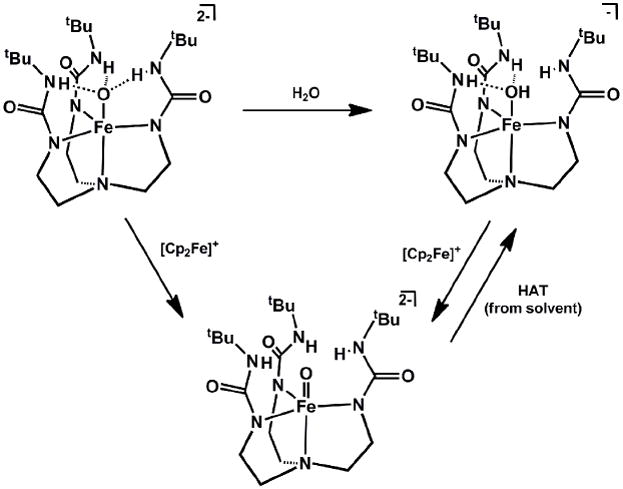
The iron(III) oxo species displayed modest HAT reactivity to give [NNH33]FeII(OH)2-, though the acidity of [NNH33]FeIII(OH)- allowed for facile protonation as an alternative reaction pathway[155]. Both the iron(III) oxo and hydroxo species underwent oxidation with [Cp2Fe]+ to generate the S = 2 [NNH33]FeIV(O)- (Scheme 16)[156]. This latter species was not thermally stable, and underwent H-atom abstraction from solvent to give [NNH33]FeIII(OH)- at room temperature, with a half-life of ca. 2.2 hours. The strong NH bond dissociation energy of the urea nitrogen atoms likely precluded intramolecular degradation pathways, which resulted in heightened stability compared to (TMG3tren)FeIV(O)2+. The solid-state structure of [NNH33]FeIV(O)- was obtained, and showed several discernable differences from its reduced congener. Now, the Fe-O bond distance has decreased by ca. 0.13 Å to 1.680(1) Å, which suggests a similar bonding description to that of (TMG3tren)FeIV(O)2+ (i.e. two electron population of π* orbitals). The oxo ligand is 0.262 Å displaced from the plane defined by the three urea H-bonding nitrogen atoms, and combined vibrational spectroscopy and DFT calculations corroborate the absence of H-bonds interactions with the oxo moiety [154, 156].
Scheme 16.
4. Concluding remarks
As is hopefully now evident, the field of mid-to-late transition metals that feature metal-to-ligand multiple bonds has emerged as a rich and exciting area of coordination chemistry over the past decade. While the scope of this review has been limited to systems of iron and cobalt that reside in local three-fold symmetry, owing to our own specific expertise and research interests, mid-to-late metals in a host of local geometries and coordination numbers (e.g., linear 2-coordinate, C2v 3-coordinate, C4v 5- and 6-coordinate) have now been demonstrated to be compatible with metal-to-ligand multiple bonds to terminal imides, oxos, and nitrides. Figure 8 displays a few select examples from the recent literature.
Figure 8.
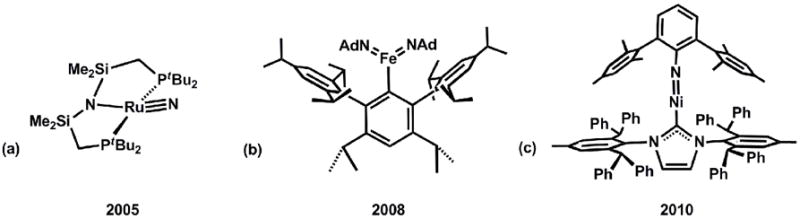
Select examples of M(E) species with low coordination numbers. (a) [(tBu2PCH2SiMe2)2N]RuIVN[157], (b)(Ar)FeV(NAd)2[46], (c) (IAr)NiII(NAr*)[158].
Pseudotetrahedral iron and cobalt systems of local three-fold symmetry have proven pedagogically informative in establishing how rich the redox chemistry of the L3M(E) structural unit can be. Species of the type L3CoII(X) and L3CoIII(E) established the feasibility of accessing d7 and d6 species in pseudotetrahedral low spin environments, and suggested the more general possibility of stabilizing the M≡E linkage for mid-to-late first row ionsin 4-coordinate pseudotetrahedral geometries. This possibility has been broadly realized for the case of iron. indeed, if one considers pseudotetrahedral complexes of the type L3Fe(NR) and L3Fe(N), one can now find representative complexes in the formal oxidation states Fe(2+), Fe(3+), Fe(4+), and Fe(5+) in which a bona fide Fe≡Nx triple bond is maintained (Figure 2). These complexes constitute four spectroscopically distinct d-electron configurations. If one disrupts the π manifold by replacing the Nx ligand by a chemically distinct L donor (e.g., N2, PMe3) then one can also access Fe(1+) (e.g., [PhBP3]Fe(PMe3)) and even Fe(0) (e.g., {[PhBP3]Fe(N2)}-) as spectroscopically distinct species with different d-orbital pictures (d7 and d8, respectively). The lesson is a simple one. A pseudotetrahedral L3Fe-X center can span six formal oxidation states, with six unique d-orbital electronic structures, simply by varying the identity of X! If one visitsiron centers of higher coordination number, one now finds examples of Fe(4+), Fe(5+), and even Fe(6+). Once again, the d-orbital structures of these species are rich in terms of the spin-states that are compatible with multiply bonded ligands. The overall conclusion is at this stage clear. Whereas a decade ago the chemistry of well-defined iron complexes featuring bona fide and terminally bonded imides, oxos, and nitrides was in its infancy, this area of chemistry, which now spans every oxidation state from Fe(2+) to Fe(6+) and also a host of electronic states ranging from low- to intermediate- to high-spin configurations, is as rich and perhaps even richer in terms of electronic diversity than that of any other transition element.
Scheme 10.
Acknowledgments
The authors are indebted to our many coworkers and colleagues in the field that have contributed to the work that has been described in this review. JCP is particularly grateful to those students and postdocs that shaped the intellectual principles that now form the basis of our ideas concerning L3M(E) and L’L3M(E) systems of iron and cobalt. JCP is also grateful to his friend and colleague Professor Harry Gray, who continues to be an inspirational mentorand has made discussions of the electronic structures of metal oxos, imides, and nitrides at once so enjoyable and insightful over the course of the past decade. Happy Birthday Chief! We also wish to acknowledge the various sources of funding that have, over the years, made the relevant work in our labs possible. This includes support from the NIH (GM-070757), the NSF (CHE-0132216 and 0750234), the Dreyfus Foundation, and the Alfred P. Sloan Foundation. CTS is grateful for an NSF Graduate Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nugent WA, Mayer JM. Metal Ligand Multiple Bonds. Wiley & Sons; New York, NY: 1988. [Google Scholar]
- 2.Holm RH, Berg JM. Acc Chem Res. 1986;19:363–370. [Google Scholar]
- 3.Doyle MP. Chem Rev. 1986;86:919–939. [Google Scholar]
- 4.Groves JT, Han Y-Z. In: Cytochrome P450: Structure, Mechanism, and Biochemistry. Ortiz de Montellano PR, editor. Plenum Press; New York, NY: 1995. pp. 3–48. [Google Scholar]
- 5.Costas M, Mehn MP, Jensen MP, Que L. Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 6.Mehn MP, Peters JC. J Inorg Biochem. 2006;100:634–643. doi: 10.1016/j.jinorgbio.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 7.Green MT. Curr Opin Chem Biol. 2009;13:84–88. doi: 10.1016/j.cbpa.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 8.Que L. Acc Chem Res. 2007;40:493–500. doi: 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]
- 9.Collman JP, Zhang XM, Lee VJ, Uffelman ES, Brauman JI. Science. 1993;261:1404–1411. doi: 10.1126/science.8367724. [DOI] [PubMed] [Google Scholar]
- 10.Gao Y, Hanson RM, Klunder JM, Ko SY, Masamune H, Sharpless KB. J Am Chem Soc. 1987;109:5765–5780. [Google Scholar]
- 11.Joergensen KA. Chem Rev. 1989;89:431–458. [Google Scholar]
- 12.Groves JT, Takahashi T. J Am Chem Soc. 1983;105:2073–2074. [Google Scholar]
- 13.Li Z, Quan RW, Jacobsen EN. J Am Chem Soc. 1995;117:5889–5890. [Google Scholar]
- 14.Evans DA, Woerpel KA, Hinman MM, Faul MM. J Am Chem Soc. 1991;113:726–728. [Google Scholar]
- 15.Müller P, Fruit C. Chem Rev. 2003;103:2905–2919. doi: 10.1021/cr020043t. [DOI] [PubMed] [Google Scholar]
- 16.Davies HML, Hansen T, Churchill MR. J Am Chem Soc. 2000;122:3063–3070. [Google Scholar]
- 17.Davies HML, Manning JR. Nature. 2008;451:417–424. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunay A, Theopold KH. Chem Rev. 2010;110:1060–1081. doi: 10.1021/cr900269x. [DOI] [PubMed] [Google Scholar]
- 19.Rittle J, Green MT. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 20.Schrock RR. Acc Chem Res. 2005;38:955–962. doi: 10.1021/ar0501121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crossland JL, Tyler DR. Coord Chem Rev. 2010;254:1883–1894. [Google Scholar]
- 22.Shaik S, Hirao H, Kumar D. Acc Chem Res. 2007;40:532–542. doi: 10.1021/ar600042c. [DOI] [PubMed] [Google Scholar]
- 23.Shaik S, Lai W, Chen H, Wang Y. Acc Chem Res. 2010;43:1154–1165. doi: 10.1021/ar100038u. [DOI] [PubMed] [Google Scholar]
- 24.Cundari TR, Dinescu A, Kazi AB. Inorg Chem. 2008;47:10067–10072. doi: 10.1021/ic801337f. [DOI] [PubMed] [Google Scholar]
- 25.Cundari TR. J Am Chem Soc. 1992;114:7879–7888. [Google Scholar]
- 26.Betley TA, Wu Q, Van Voorhis T, Nocera DG. Inorg Chem. 2008;47:1849–1861. doi: 10.1021/ic701972n. [DOI] [PubMed] [Google Scholar]
- 27.Berry JF. Comments Inorg Chem. 2009;30:28–66. [Google Scholar]
- 28.Ballhausen CJ, Gray HB. Inorg Chem. 1962;1:111–122. [Google Scholar]
- 29.Mayer JM. Inorg Chem. 1988;27:3899–3903. [Google Scholar]
- 30.Cummins CC, Schrock RR, Davis WM. Organometallics. 1992;11:1452–1454. [Google Scholar]
- 31.Odom AL, Cummins CC, Protasiewicz JD. J Am Chem Soc. 1995;117:6613–6614. [Google Scholar]
- 32.Anderson TM, Neiwert WA, Kirk ML, Piccoli PMB, Schultz AJ, Koetzle TF, Musaev DG, Morokuma K, Cao R, Hill CL. Science. 2004;306:2074–2077. doi: 10.1126/science.1104696. [DOI] [PubMed] [Google Scholar]
- 33.Mayer JM, Tulip TH. J Am Chem Soc. 1984;106:3878–3879. [Google Scholar]
- 34.Mayer JM. Comments Inorg Chem. 1988;8:125–135. [Google Scholar]
- 35.Spaltenstein E, Conry RR, Critchlow SC, Mayer JM. J Am Chem Soc. 1989;111:8741–8742. [Google Scholar]
- 36.Rouschias G. Chem Rev. 1974;74:531–566. [Google Scholar]
- 37.Mayer JM, Thorn DL, Tulip TH. J Am Chem Soc. 1985;107:7454–7462. [Google Scholar]
- 38.Moyer BA, Meyer TJ. Inorg Chem. 1981;20:436–444. [Google Scholar]
- 39.Moyer BA, Meyer TJ. J Am Chem Soc. 1978;100:3601–3603. [Google Scholar]
- 40.Thompson MS, Meyer TJ. J Am Chem Soc. 1982;104:4106–4115. [Google Scholar]
- 41.Glueck DS, Wu JX, Hollander FJ, Bergman RG. J Am Chem Soc. 1991;113:2041–2054. [Google Scholar]
- 42.Haymotherwell RS, Wilkinson G, Hussainbates B, Hursthouse MB. Polyhedron. 1993;12:2009–2012. [Google Scholar]
- 43.Jensen MP, Mehn MP, Que L. Angew Chem Int Ed. 2003;42:4357–4360. doi: 10.1002/anie.200351605. [DOI] [PubMed] [Google Scholar]
- 44.King ER, Betley TA. Inorg Chem. 2009;48:2361–2363. doi: 10.1021/ic900219b. [DOI] [PubMed] [Google Scholar]
- 45.Lucas RL, Powell DR, Borovik AS. J Am Chem Soc. 2005;127:11596–11597. doi: 10.1021/ja052952g. [DOI] [PubMed] [Google Scholar]
- 46.Ni C, Fettinger JC, Long GJ, Brynda M, Power PP. Chem Commun. 2008:6045–6047. doi: 10.1039/b810941a. [DOI] [PubMed] [Google Scholar]
- 47.Bart SC, Lobkovsky E, Bill E, Chirik PJ. J Am Chem Soc. 2006;128:5302–5303. doi: 10.1021/ja057165y. [DOI] [PubMed] [Google Scholar]
- 48.Scepaniak JJ, Young JA, Bontchev RP, Smith JM. Angew Chem Int Ed. 2009;48:3158–3160. doi: 10.1002/anie.200900381. [DOI] [PubMed] [Google Scholar]
- 49.Scepaniak JJ, Fulton MD, Bontchev RP, Duesler EN, Kirk ML, Smith JM. J Am Chem Soc. 2008;130:10515–10517. doi: 10.1021/ja8027372. [DOI] [PubMed] [Google Scholar]
- 50.Nieto I, Ding F, Bontchev RP, Wang HB, Smith JM. J Am Chem Soc. 2008;130:2716–2717. doi: 10.1021/ja0776834. [DOI] [PubMed] [Google Scholar]
- 51.Vogel C, Heinemann FW, Sutter J, Anthon C, Meyer K. Angew Chem Int Ed. 2008;47:2681–2684. doi: 10.1002/anie.200800600. [DOI] [PubMed] [Google Scholar]
- 52.Betley TA, Peters JC. J Am Chem Soc. 2004;126:6252–6254. doi: 10.1021/ja048713v. [DOI] [PubMed] [Google Scholar]
- 53.Brown SD, Betley TA, Peters JC. J Am Chem Soc. 2003;125:322–323. doi: 10.1021/ja028448i. [DOI] [PubMed] [Google Scholar]
- 54.Brown SD, Peters JC. J Am Chem Soc. 2005;127:1913–1923. doi: 10.1021/ja0453073. [DOI] [PubMed] [Google Scholar]
- 55.Thomas CM, Mankad NP, Peters JC. J Am Chem Soc. 2006;128:4956–4957. doi: 10.1021/ja0604358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rohde J-U, In JH, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L. Science. 2003;299:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 57.Jenkins DM, Betley TA, Peters JC. J Am Chem Soc. 2002;124:11238–11239. doi: 10.1021/ja026852b. [DOI] [PubMed] [Google Scholar]
- 58.Hu X, Meyer K. J Am Chem Soc. 2004;126:16322–16323. doi: 10.1021/ja044271b. [DOI] [PubMed] [Google Scholar]
- 59.Cowley RE, Bontchev RP, Sorrell J, Sarracino O, Feng YH, Wang HB, Smith JM. J Am Chem Soc. 2007;129:2424–2425. doi: 10.1021/ja066899n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dai XL, Kapoor P, Warren TH. J Am Chem Soc. 2004;126:4798–4799. doi: 10.1021/ja036308i. [DOI] [PubMed] [Google Scholar]
- 61.Kogut E, Wiencko HL, Zhang LB, Cordeau DE, Warren TH. J Am Chem Soc. 2005;127:11248–11249. doi: 10.1021/ja0533186. [DOI] [PubMed] [Google Scholar]
- 62.Mindiola DJ, Hillhouse GL. J Am Chem Soc. 2001;123:4623–4624. doi: 10.1021/ja010358a. [DOI] [PubMed] [Google Scholar]
- 63.Melenkivitz R, Mindiola DJ, Hillhouse GL. J Am Chem Soc. 2002;124:3846–3847. doi: 10.1021/ja017787t. [DOI] [PubMed] [Google Scholar]
- 64.Mindiola DJ, Hillhouse GL. J Am Chem Soc. 2002;124:9976–9977. doi: 10.1021/ja0269183. [DOI] [PubMed] [Google Scholar]
- 65.Eckert NA, Vaddadi S, Stoian S, Lachicotte RJ, Cundari TR, Holland PL. Angew Chem Int Ed. 2006;45:6868–6871. doi: 10.1002/anie.200601927. [DOI] [PubMed] [Google Scholar]
- 66.Cowley RE, DeYonker NJ, Eckert NA, Cundari TR, DeBeer S, Bill E, Ottenwaelder X, Flaschenriem C, Holland PL. Inorg Chem. 2010;49:6172–6187. doi: 10.1021/ic100846b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cowley RE, Eckert NA, Elhaïk J, Holland PL. Chem Commun. 2009:1760–1762. doi: 10.1039/b820620a. [DOI] [PubMed] [Google Scholar]
- 68.Holland PL. Acc Chem Res. 2008;41:905–914. doi: 10.1021/ar700267b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jenkins DM, Di Bilio AJ, Allen MJ, Betley TA, Peters JC. J Am Chem Soc. 2002;124:15336–15350. doi: 10.1021/ja026433e. [DOI] [PubMed] [Google Scholar]
- 70.Jenkins DM, Peters JC. J Am Chem Soc. 2003;125:11162–11163. doi: 10.1021/ja036198f. [DOI] [PubMed] [Google Scholar]
- 71.Jenkins DM, Peters JC. J Am Chem Soc. 2005;127:7148–7165. doi: 10.1021/ja045310m. [DOI] [PubMed] [Google Scholar]
- 72.Shapiro IR, Jenkins DM, Thomas JC, Day MW, Peters JC. Chem Commun. 2001:2152–2153. doi: 10.1039/b104447h. [DOI] [PubMed] [Google Scholar]
- 73.Betley TA, Peters JC. J Am Chem Soc. 2003;125:10782–10783. doi: 10.1021/ja036687f. [DOI] [PubMed] [Google Scholar]
- 74.Cotton FA, Wilkinson G. Advanced Inorganic Chemistry. 5. Wiley; New York: 1988. [Google Scholar]
- 75.Garrett BB, Goedken VL, Quaglian JV. J Am Chem Soc. 1970;92:489. [Google Scholar]
- 76.Gerloch M, Hanton LR. Inorg Chem. 1980;19:1692–1698. [Google Scholar]
- 77.Gerloch M, Manning MR. Inorg Chem. 1981;20:1051–1056. [Google Scholar]
- 78.Banci L, Benelli C, Gatteschi D, Mani F. Inorg Chem. 1982;21:1133–1136. [Google Scholar]
- 79.Gray HB. Chemical Bonds: An Introduction to Atomic and Molecular Structure. University Science Books; Mill Valley, CA: 1994. [Google Scholar]
- 80.Strauss SH, Silver ME, Long KM, Thompson RG, Hudgens RA, Spartalian K, Ibers JA. J Am Chem Soc. 1985;107:4207–4215. [Google Scholar]
- 81.Goedken VL, Pluth JJ, Peng S-M, Bursten B. J Am Chem Soc. 1976;98:8014–8021. [Google Scholar]
- 82.Betley TA, Peters JC. Inorg Chem. 2003;42:5074–5084. doi: 10.1021/ic0343096. [DOI] [PubMed] [Google Scholar]
- 83.Albright TA, Burdett JK, Whangbo MH. Orbital Interactions in Chemistry. John Wiley & Sons; New York: 1985. [Google Scholar]
- 84.Verma AK, Nazif TN, Achim C, Lee SC. J Am Chem Soc. 2000;122:11013–11014. [Google Scholar]
- 85.Mehn MP, Brown SD, Jenkins DM, Peters JC, Que L. Inorg Chem. 2006;45:7417–7427. doi: 10.1021/ic060670r. [DOI] [PubMed] [Google Scholar]
- 86.Yap GPA. CCDC deposition number 253991. 2004 [Google Scholar]
- 87.Shay DT, Yap GPA, Zakharov LN, Rheingold AL, Theopold KH. Angew Chem Int Ed. 2005;44:1508–1510. doi: 10.1002/anie.200462529. [DOI] [PubMed] [Google Scholar]
- 88.Wasbotten IH, Ghosh A. Inorg Chem. 2007;46:7890–7898. doi: 10.1021/ic700543f. [DOI] [PubMed] [Google Scholar]
- 89.Mankad NP, Müller P, Peters JC. J Am Chem Soc. 2010;132:4083–4084. doi: 10.1021/ja910224c. [DOI] [PubMed] [Google Scholar]
- 90.Mindiola DJ, Hillhouse GL. Chem Commun. 2002:1840–1841. doi: 10.1039/b204846a. [DOI] [PubMed] [Google Scholar]
- 91.Shay DT, Yap GPA, Zakharov LN, Rheingold AL, Theopold KH. Angew Chem Int Ed. 2006;45:7870–7870. [Google Scholar]
- 92.Thyagarajan S, Shay DT, Incarvito CD, Rheingold AL, Theopold KH. J Am Chem Soc. 2003;125:4440–4441. doi: 10.1021/ja028267g. [DOI] [PubMed] [Google Scholar]
- 93.Nieto I, Cervantes-Lee F, Smith JM. Chem Commun. 2005:3811–3813. doi: 10.1039/b505985b. [DOI] [PubMed] [Google Scholar]
- 94.Burred AK, Steedman AJ. J Chem Soc Chem Commun. 1995:2109–2110. [Google Scholar]
- 95.Danopoulos AA, Wilkinson G, Hussain-Bates B, Hursthouse MB. Polyhedron. 1992;11:2961–2964. [Google Scholar]
- 96.Bell B, Chatt J, Dilworth JR, Leigh GJ. Inorg Chim Acta. 1972;6:635–637. [Google Scholar]
- 97.Michelman RI, Andersen RA, Bergman RG. J Am Chem Soc. 1991;113:5100–5102. [Google Scholar]
- 98.Lu CC, Saouma CT, Day MW, Peters JC. J Am Chem Soc. 2007;129:4–5. doi: 10.1021/ja065524z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kisko JL, Hascall T, Parkin G. J Am Chem Soc. 1998;120:10561–10562. [Google Scholar]
- 100.Lappert MF, MacQuitty JJ, Pye PL. J Chem Soc Dalton Trans. 1981:1583–1592. [Google Scholar]
- 101.Sohn YS, Hendrickson DN, Gray HB. J Am Chem Soc. 1970;92:3233–3234. [Google Scholar]
- 102.Sohn YS, Hendrickson DN, Gray HB. J Am Chem Soc. 1971;93:3603–3612. [Google Scholar]
- 103.Brown SD, Peters JC. J Am Chem Soc. 2004;126:4538–4539. doi: 10.1021/ja0399122. [DOI] [PubMed] [Google Scholar]
- 104.Daida EJ, Peters JC. Inorg Chem. 2004;43:7474–7485. doi: 10.1021/ic0488583. [DOI] [PubMed] [Google Scholar]
- 105.Sohn YS, Hendrickson DN, Hart Smith J, Gray HB. Chem Phys Lett. 1970;6:499–501. [Google Scholar]
- 106.Paine TK, Costas M, Kaizer J, Que L. J Biol Inorg Chem. 2006;11:272–276. doi: 10.1007/s00775-006-0089-6. [DOI] [PubMed] [Google Scholar]
- 107.Klinker EJ, Kaizer J, Brennessel WW, Woodrum NL, Cramer CJ, Que L. Angew Chem Int Ed. 2005;44:3690–3694. doi: 10.1002/anie.200500485. [DOI] [PubMed] [Google Scholar]
- 108.Rohde J-U, Torelli S, Shan XP, Lim MH, Klinker EJ, Kaizer J, Chen K, Nam WW, Que L. J Am Chem Soc. 2004;126:16750–16761. doi: 10.1021/ja047667w. [DOI] [PubMed] [Google Scholar]
- 109.Lim MH, Rohde J-U, Stubna A, Bukowski MR, Costas M, Ho RYN, Münck E, Nam W, Que L. Proc Natl Acad Sci U S A. 2003;100:3665–3670. doi: 10.1073/pnas.0636830100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Thomas JC, Peters JC. Inorg Chem. 2003;42:5055–5073. doi: 10.1021/ic034150x. [DOI] [PubMed] [Google Scholar]
- 111.Fjare DE, Gladfelter WL. Inorg Chem. 1981;20:3533–3539. [Google Scholar]
- 112.Bennett MV, Stoian S, Bominaar EL, Münck E, Holm RH. J Am Chem Soc. 2005;127:12378–12386. doi: 10.1021/ja052150l. [DOI] [PubMed] [Google Scholar]
- 113.Summerville DA, Cohen IA. J Am Chem Soc. 1976;98:1747–1752. doi: 10.1021/ja00423a019. [DOI] [PubMed] [Google Scholar]
- 114.Li M, Shang M, Ehlinger N, Schulz CE, Scheidt WR. Inorg Chem. 2000;39:580–583. doi: 10.1021/ic990900k. [DOI] [PubMed] [Google Scholar]
- 115.Wagner WD, Nakamoto K. J Am Chem Soc. 1988;110:4044–4045. [Google Scholar]
- 116.Wagner WD, Nakamoto K. J Am Chem Soc. 1989;111:1590–1598. [Google Scholar]
- 117.Meyer K, Bill E, Mienert B, Weyhermüller T, Wieghardt K. J Am Chem Soc. 1999;121:4859–4876. [Google Scholar]
- 118.Aliaga-Alcalde M, George SD, Mienert B, Bill E, Wieghardt K, Neese F. Angew Chem Int Ed. 2005;44:2908–2912. doi: 10.1002/anie.200462368. [DOI] [PubMed] [Google Scholar]
- 119.Berry JF, Bill E, Bothe E, George SD, Mienert B, Neese F, Wieghardt K. Science. 2006;312:1937–1941. doi: 10.1126/science.1128506. [DOI] [PubMed] [Google Scholar]
- 120.Berry JF, DeBeer George S, Neese F. Phys Chem Chem Phys. 2008;10:4361–4374. doi: 10.1039/b801803k. [DOI] [PubMed] [Google Scholar]
- 121.Du Bois J, Tomooka CS, Hong J, Carreira EM. Acc Chem Res. 1997;30:364–372. [Google Scholar]
- 122.Chang CJ, Low DW, Gray HB. Inorg Chem. 1997;36:270–271. doi: 10.1021/ic970824q. [DOI] [PubMed] [Google Scholar]
- 123.Petrenko T, George SD, Aliaga-Alcalde N, Bill E, Mienert B, Xiao Y, Guo Y, Sturhahn W, Cramer SP, Wieghardt K, Neese F. J Am Chem Soc. 2007;129:11053–11060. doi: 10.1021/ja070792y. [DOI] [PubMed] [Google Scholar]
- 124.Mindiola DJ, Cummins CC. Angew Chem Int Ed. 1998;37:945–947. doi: 10.1002/(SICI)1521-3773(19980420)37:7<945::AID-ANIE945>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 125.Lu CC, Peters JC. Inorg Chem. 2006;45:8597–8607. doi: 10.1021/ic060735q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.MacBeth CE, Thomas JC, Betley TA, Peters JC. Inorg Chem. 2004;43:4645–4662. doi: 10.1021/ic049936p. [DOI] [PubMed] [Google Scholar]
- 127.Peters JC, Johnson AR, Odom AL, Wanandi PW, Davis WM, Cummins CC. J Am Chem Soc. 1996;118:10175–10188. [Google Scholar]
- 128.Laplaza CE, Cummins CC. Science. 1995;268:861–863. doi: 10.1126/science.268.5212.861. [DOI] [PubMed] [Google Scholar]
- 129.Yandulov DV, Schrock RR. J Am Chem Soc. 2002;124:6252–6253. doi: 10.1021/ja020186x. [DOI] [PubMed] [Google Scholar]
- 130.Nakamoto K. Infrared and Raman Spectra of Inorganic and Coordination Compounds Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry. 5. John Wiley & Sons, Inc; New York: 1997. [Google Scholar]
- 131.Hendrich MP, Gunderson W, Behan RK, Green MT, Mehn MP, Betley TA, Lu CC, Peters JC. Proc Natl Acad Sci U S A. 2006;103:17107–17112. doi: 10.1073/pnas.0604402103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rohde J-U, Betley TA, Jackson TA, Saouma CT, Peters JC, Que L., Jr Inorg Chem. 2007;46:5720–5726. doi: 10.1021/ic700818q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Scepaniak JJ, Vogel CA, Khusniyarov MM, Heinemann FW, Meyer K, Smith JM. 2010 manuscript submitted for publication. [Google Scholar]
- 134.Miskowski VM, Gray HB, Hopkins MD. Advances in Transition Metal Coordination Chemistry. 1996;1:159–186. [Google Scholar]
- 135.Whited MT, Mankad NP, Lee YH, Oblad PF, Peters JC. Inorg Chem. 2009;48:2507–2517. doi: 10.1021/ic801855y. [DOI] [PubMed] [Google Scholar]
- 136.Mankad NP, Whited MT, Peters JC. Angew Chem Int Ed. 2007;46:5768–5771. doi: 10.1002/anie.200701188. [DOI] [PubMed] [Google Scholar]
- 137.Lee YH, Mankad NP, Peters JC. Nature Chemistry. 2010;2:558–565. doi: 10.1038/nchem.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Eckert NA, Vaddadi S, Stoian S, Lachicotte RJ, Cundari TR, Holland PL. Angewandte Chemie-International Edition. 2006;45:6868–6871. doi: 10.1002/anie.200601927. [DOI] [PubMed] [Google Scholar]
- 139.Takaoka A, Gerber LCH, Peters JC. Angew Chem Int Ed. 2010;49:4088–4091. doi: 10.1002/anie.201001199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bontemps S, Bouhadir G, Dyer PW, Miqueu K, Bourissou D. Inorg Chem. 2007;46:5149–5151. doi: 10.1021/ic7006556. [DOI] [PubMed] [Google Scholar]
- 141.Moret M-E, Peters JC. 2010 submitted for publication. [Google Scholar]
- 142.Lee Y, Peters JC. 2010 submitted for publication. [Google Scholar]
- 143.Shan X, Que L., Jr J Inorg Biochem. 2006;100:421–433. doi: 10.1016/j.jinorgbio.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 144.Decker A, Rohde J-U, Klinker EJ, Wong SD, Que L, Solomon EI. J Am Chem Soc. 2007;129:15983–15996. doi: 10.1021/ja074900s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nam W. Acc Chem Res. 2007;40:522–531. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]
- 146.Pestovsky O, Stoian S, Bominaar EL, Shan X, Münck E, Que L, Bakac A. Angew Chem Int Ed. 2005;44:6871–6874. doi: 10.1002/anie.200502686. [DOI] [PubMed] [Google Scholar]
- 147.England J, Martinho M, Farquhar ER, Frisch JR, Bominaar EL, Münck E, Que L. Angew Chem Int Ed. 2009;48:3622–3626. doi: 10.1002/anie.200900863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.England J, Guo Y, Farquhar ER, Young VG, Jr, Münck E, Que L., Jr J Am Chem Soc. 2010;132:8635–8644. doi: 10.1021/ja100366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Thibon A, England J, Martinho M, Young VG, Frisch JR, Guillot R, Girerd J-J, Münck E, Que L, Banse F. Angew Chem Int Ed. 2008;47:7064–7067. doi: 10.1002/anie.200801832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Borovik AS. Acc Chem Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]
- 151.MacBeth CE, Golombek AP, Young VG, Yang C, Kuczera K, Hendrich MP, Borovik AS. Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]
- 152.Kurtz DM. Chem Rev. 1990;90:585–606. [Google Scholar]
- 153.Hoppe ML, Schlemper EO, Murmann RK. Acta Crystallogr Sect B. 1982;38:2237–2239. [Google Scholar]
- 154.Dey A, Hocking RK, Larsen P, Borovik AS, Hodgson KO, Hedman B, Solomon EI. J Am Chem Soc. 2006;128:9825–9833. doi: 10.1021/ja061618x. [DOI] [PubMed] [Google Scholar]
- 155.Gupta R, Borovik AS. J Am Chem Soc. 2003;125:13234–13242. doi: 10.1021/ja030149l. [DOI] [PubMed] [Google Scholar]
- 156.Lacy DC, Gupta R, Stone KL, Greaves J, Ziller JW, Hendrich MP, Borovik AS. J Am Chem Soc. 2010;132:12188–12190. doi: 10.1021/ja1047818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Walstrom A, Pink M, Yang XF, Tomaszewski J, Baik MH, Caulton KG. J Am Chem Soc. 2005;127:5330–5331. doi: 10.1021/ja050361k. [DOI] [PubMed] [Google Scholar]
- 158.Laskowski CA, Miller AJM, Hillhouse GL, Cundari TR. 2010 submitted for publication. [Google Scholar]