

Abstract
Regulation of presynaptic voltage-gated calcium channels is critical for depolarization-evoked neurotransmitter release. Various studies attempted to determine the functional implication of Rim1, a component of the vesicle release machinery. Besides to couple voltage-gated Ca2+ channels to the presynaptic vesicle release machinery, it was evidenced that Rim1 also prevents voltage-dependent inactivation of the channels through a direct interaction with the ancillary β-subunits, thus facilitating neurotransmitter release. However, facilitation of synaptic activity may also be caused by a reduction of the inhibitory pathway carried by G-protein coupled receptors. Here, we explored the functional implication of Rim1 in G-protein regulation of Cav2.2 channels. Activation of μ-opioid receptors expressed in HEK-293 cells along with Cav2.2 channels produced a drastic current inhibition both in control and Rim1-expressing cells. In contrast, Rim1 considerably promoted the extent of current deinhibition following channel activation, favoring sustained Ca2+ influx under prolonged activity. Our data suggest that Rim1-induced facilitation of neurotransmitter release may come as a consequence of a decrease in the inhibitory pathway carried by G-proteins that contributes, together with the slowing of channel inactivation, to maintain Ca2+ influx under prolonged activity. The present study also furthers functional insights in the importance of proteins from the presynaptic vesicle complex in the regulation of voltage-gated Ca2+ channels by G-proteins.
Keywords: Calcium channel, Cav2.2 channel, N channel, beta subunit, Rim1, G-protein coupled receptor, mu opioid receptor, G-protein, beta/gamma subunit
Introduction
Presynaptic Cav2.2 voltage-gated calcium channels play an essential role in depolarization-evoked neurotransmitter release at nerve termini (29, 32). In turn, the released neurotransmitters produce channel inhibition through activation of G-protein coupled receptors (GPCRs) by a negative feedback loop (for review see (9, 30)). This rapid and spatially delimited inhibition, based on the direct binding of the Gβγ signaling complex onto the Cav2.2 pore-forming subunit (10, 37), is characterized at the whole-cell level by a number of distinct hallmarks. Namely, the binding of the Gβγ dimer onto Cav2.2 subunit produces the silencing of channel activity (“ON” effect), whereas Gβγ unbinding, which occurs following channel activation, induces an apparent set of biophysical modifications (“OFF” effects) that comprise (i) a slowing of the current activation and inactivation kinetics, (ii) a depolarizing shift of the voltage-dependence of channel activation, and (iii) a current recovery from G-protein inhibition (33). Hence, the kinetics of Gβγ dissociation from the channel determines to what extent a channel recovers from inhibition to contribute again to synaptic signaling under neuronal firing (3). Recently, we have evidenced that channel inactivation during membrane depolarization greatly influences Gβγ dissociation from the channel and hence the capability of the channel to be involved in synaptic activity in spite of maintained GPCR activation (35, 36).
Direct G-protein inhibition of Cav2.2 channels is regulated by many factors, including channel splicing (1, 25), pathological mutations (19, 35), channel phosphorylation (37), and ancillary channel β-subunit (5, 11, 17, 18). In some cases, these factors were found to influence G-protein regulation by modulation of channel inactivation properties (35, 36). Early reports also suggested that constituent proteins of the presynaptic vesicle release complex, known to be functionally coupled with neuronal voltage-gated Ca2+ channels (for review see (8)), influence G-protein inhibition. For instance, changes in G-protein regulation of Cav2.2 channels were reported in the presence of syntaxin 1A (12, 13, 16). Recently, it was evidenced that Rim1, besides coupling voltage-gated Ca2+ channels to the presynaptic vesicle release machinery and facilitating neurotransmitter release (15, 22, 27), also modify inactivation properties of Cav2.1 and Cav2.2 channels by interacting with the β subunit of these channels (14). This observation supports an important role of Rim1 in the control of neuronal voltage-gated Ca2+ channels activity and questions its implication in the modulation of G-protein regulation of neuronal voltage-gated channels.
Rim1 is part of the Rim superfamily of proteins whose members share a C2B domain at their C-termini. It forms a protein scaffold in presynaptic nerve terminals by interacting with numerous other protein components of the active zone, i.e. Munc13, ELKS (or CAST), Rim-binding protein, and liprins (4, 7, 24, 26, 31). At the physiological level, Rim1 was found to be essential to short- and long-term synaptic plasticity by affecting the readily releasable pool of vesicles (6, 26). Rim proteins were found to be required for normal Ca2+-triggering of exocytosis (27). In that respect, it was thus an interesting finding that Rim1 interacts with the β subunits of voltage-gated calcium channels (14). The main biophysical effect of this interaction is a significant slowing of channel inactivation thereby increasing Ca2+ influx during trains of action potentials. The molecular linkage between Rim1 and calcium channels contributes to anchor neurotransmitter-containing vesicles to voltage-dependent calcium channels. The increased vicinity and Ca2+ influx that result from the modification of channel inactivation kinetics presumably are involved in the facilitating effect of Rim1 on acetylcholine release in PC12 cells and glutamate release in cerebellar neurons (14). Besides, mutations in the gene coding for Rim1 is associated with autosomal dominant cone-rod dystrophy (CORD7), and is characterized by a progressive loss of photoreceptors along with retinal degeneration (2, 20). Interestingly, the R655H mutation of Rim1 was found to alter Rim1-mediated regulation of Cav2.1 channel (21).
The aim of the present study was to specifically test the influence of Rim1 in the direct G-protein inhibition of Cav2.2 channels. We investigated the effects of Rim1 on G-protein regulation of Cav2.2 channels expressed in HEK-293 cells, along with the ancillary β2a or β3 and α2δ subunits, and the μ-opioid receptor. As expected, Rim1 potently decreases the extent of Cav2.2 channel inactivation. Application of DAMGO ((D-Ala2,N-Me-Phe4,glycinol5)-Enkephalin) induced direct G-protein regulation whether Rim1 was expressed or not. Maximal G-protein inhibition produced by DAMGO application (“ON” effect) was similar for both Cav2.2/β3/α2δ and Cav2.2/β3/α2δ/Rim1 channels, suggesting that Rim1 does not alter the association of Gβγ onto the closed state of the channel. In contrast, the kinetic and extent of recovery from G-protein inhibition were found largely affected in Rim1-expressing cells. Interestingly, the effect of Rim1 on G-protein regulation of slow inactivating channels (i.e produced by co-expression of the β2a auxiliary subunit) was found decreased, indicating that Rim1 preferentially influence G-protein regulation of fast inactivating channels. Our findings thus provide novel evidence for an efficient Rim1-dependent modulation of direct G-protein regulation of Cav2.2 channels. More generally, the data stress out the importance of the constitutive proteins from the presynaptic vesicle machinery, not only for secretion, but also to fine-tune the regulation of presynaptic neuronal voltage-gated Ca2+ channels.
Materials and Methods
Plasmid cDNAs
The cDNAs used in this study were rabbit brain Cav2.2 (GenBank D14157), rat brain β2a (GenBank M80545) or β3 (GenBank M88751), rat brain α2δ1b subunit (GenBank M86621), mouse brain Rim1 (GenBank NM_053270) and the human μ-opioid receptor (hMOR, obtained from the UMR cDNA Resource Center www.cdna.org) (GenBank AY521028).
Transient expression in HEK-293
Human embryonic kidney 293 (HEK-293) cells were grown in a Dulbecco’s modified Eagle’s culture medium containing 10% fetal bovine serum and 1% penicillin/streptomycin (all products were purchased from Invitrogen, Carlsbad, CA) and maintained under standard conditions at 37°C in a humidified atmosphere containing 5% CO2. Cells were transfected using Lipofectamine Plus transfection regent (Invitrogen) according to the manufacturer’s protocol with cDNAs encoding Cav2.2 channel along with β2a or β3, α2δ1b, hMOR, Rim1 and the Green-Lantern (CMV-GFP) expression vector (Life Technologies, Carlsbad, CA). Two days after transfection, cells were mechanically dissociated and patch-clamp recordings were performed 2 h later from fluorescent cells.
Patch-clamp recordings
Ba2+ currents were recorded in the whole-cell configuration of the patch-clamp technique at room temperature (22–24°C) in a bathing medium containing (in millimolar): BaCl2 10, TEA-Cl 125, D-glucose 10, HEPES 10 (pH 7.4 with TEA-OH). Patch pipettes were filled with a solution containing (in millimolar): CsCl 110, Mg-ATP 4, Mg-GTP 0.5, MgCl2 5, EGTA 10, HEPES 10 (pH 7.4 with CsOH), and had a resistance of 2–4 MΩ. Whole-cell patch-clamp recording were performed using an Axopatch 200B amplifier (Axon Instruments, Union City, CA). Acquisition and analyses were performed using pClamp software (Axon instruments). All traces were corrected on-line for leak and capacitance currents, digitized at 10 KHz and filtered at 2 KHz. DAMGO ((D-Ala2,N-Me-Phe4,glycinol5)-Enkephalin); purchased from Bachem, Budendorf, Germany) was applied at 10 μM and all recordings were performed within 1 min after DAMGO produced maximal current inhibition in order to minimize voltage-independent G-protein regulation and hMOR desensitization. The voltage-dependence of the peak Ba2+ current density was fitted with the following modified Boltzman equation:
with I(V) being the peak current density at the command potential V, Gmax the maximum conductance, Vrev the reversal potential, V1/2 the half-activation potential and k the steepness factor. The voltage-dependence of the whole-cell Ba2+ conductance was calculated using the following modified Boltzman equation:
with G(V) being the Ba2+ conductance at the command potential V.
Analyses of the parameters of G-protein regulation
Biophysical parameters of G-protein regulation (GIt0, the initial extent of G-protein inhibition before the start of the depolarization, τ, the time constant of G-protein unbinding from the channel, and RI, the maximal extent of recovery from inhibition) were measured and analyzed according to previously described procedures (34, 36). In brief, subtracting IDAMGO (the current recorded after DAMGO application) from IControl (the current recorded before DAMGO application) results in ILost, the evolution of the lost current during membrane depolarization under G-protein regulation. IControl and ILost are then extrapolated to t = 0 ms (the start of the depolarization) by fitting traces with an exponential function in order to determine GIt0, the maximal extent of G-protein inhibition. IDAMGO without unbinding (IDAMGO wo unbinding) represents an estimate of the amount of current that is present in IDAMGO and is obtained by the following equation:
The time dependence of G-protein dissociation (IG-protein unbinding) is then obtained by the following equation:
A fit of IG-protein unbinding by a mono-exponential function provides the time constant τ of G-protein dissociation from the channel and the maximal extent RImax of current recovery from G-protein inhibition.
Statistics
Least-squares fits were performed using an algorithm routine included in Clampfit 10. Data values are presented as mean ± S.E.M. for n recorded cells, were n is specified in Results. Statistical significance was determined using Student’s t test: *p < 0.05; **p < 0.01; ***p < 0.001; NS, statistically not different.
Results
Rim1 alters inactivation properties of Cav2.2/β3/α2δ1b channels expressed in HEK-293 cells
To characterize the functional impact of Rim1 on Cav2.2 calcium channel activity, Cav2.2 channels were transiently expressed in HEK-293 cells along with β3 and α2δ1b auxiliary subunits and Rim1, and whole-cell barium (Ba2+) currents were recorded two days after transfection. Representative Ba2+ current traces recorded in response to 500 ms depolarizing steps to values ranging between −40 mV and +60 mV, from a holding potential of −80 mV, are shown in Fig. 1A for Cav2.2/β3/α2δ1b (left panel) and Cav2.2/β3/α2δ1b/Rim1 channels (right panel). Figure 1B shows the mean normalized Ba2+ conductance versus membrane potential relationships for control (filled circles) and Rim1-expressing cells (open circles). The mean half-maximal activation potential (Fig. 1B, inset) remained unaltered (p = 0.89) in Rim1- expressing cells (−2.5 ± 2.2 mV, n = 9) compared to control cells (−2.8 ± 1.1 mV, n = 13). No significant difference in the maximal Ba2+ conductance was observed between control and Rim1-expressing cells (222 ± 35 pS/pF, n = 13, versus 219 ± 35 pS/pF, n = 9, p = 0.958). To further investigate the functional impact of Rim1 on the Cav2.2 channel, the kinetics of Ba2+ currents were analyzed in control and Rim1-expressing cells. Figure 1C shows normalized Ba2+ current traces obtained in response to 500 ms depolarizing steps to +20 mV from a holding potential of −80 mV, for Cav2.2/β3/α2δ1b and Cav2.2/β3/α2δ1b/Rim1 channels. The Ba2+ current activation kinetic remained unaltered (p > 0.05) in Rim1-expressing cells as evidenced by a similar time to peak of the current. Inactivation properties of the Ba2+ current was investigated by fitting the decay phase of the current by a single exponential function, providing the time constant (τ) of channel inactivation and the relative proportion of current contains I the inactivating and non-inactivating phases (Fig. 1D, top panel, red dashed lines). Mean corresponding values for the time constant (τ) are presented in Fig. 1D (bottom panel) for control (filled circles) and Rim1-expressing cells (open circles) and the relative proportion of current that inactivates (obtained from the exponential fits) is shown in Fig. 1E. Whereas the time constant of current inactivation remained unaffected in Rim1-expressing cells compared to control cells (p > 0.05), the extent of current inactivation was found strongly decreased in cells expressing Rim1. For instance, in response to a depolarizing step to +20 mV, the extent of current inactivation recorded from Rim1-expressing cells was, on average, 2.0-fold smaller than in control cells (45,3 ± 3.9%, n = 9, versus 92.0 ± 1.4%, n = 13, p < 0.001). This decrease in the extent of current inactivation induced by Rim1 remained significant at all depolarizing steps studied (from 0 mV to +50 mV). Taken together, these results indicate that Rim1 is a potent modulator of Cav2.2 channel inactivation. These findings are consistent with previous observations in experiments showing a Rim1-dependent modulation of Cav2.1 and Cav2.2 channel inactivation (14). Since we have previously shown that alterations in channel inactivation critically affect the direct G-protein regulation of voltage-gated calcium channels (34–36), this study was pursued to investigate whether and how Rim1 could affect G-protein inhibition of Cav2.2 channels.
Figure 1. Rim1 modulates inactivation properties of Cav2.2/β3 calcium channels.
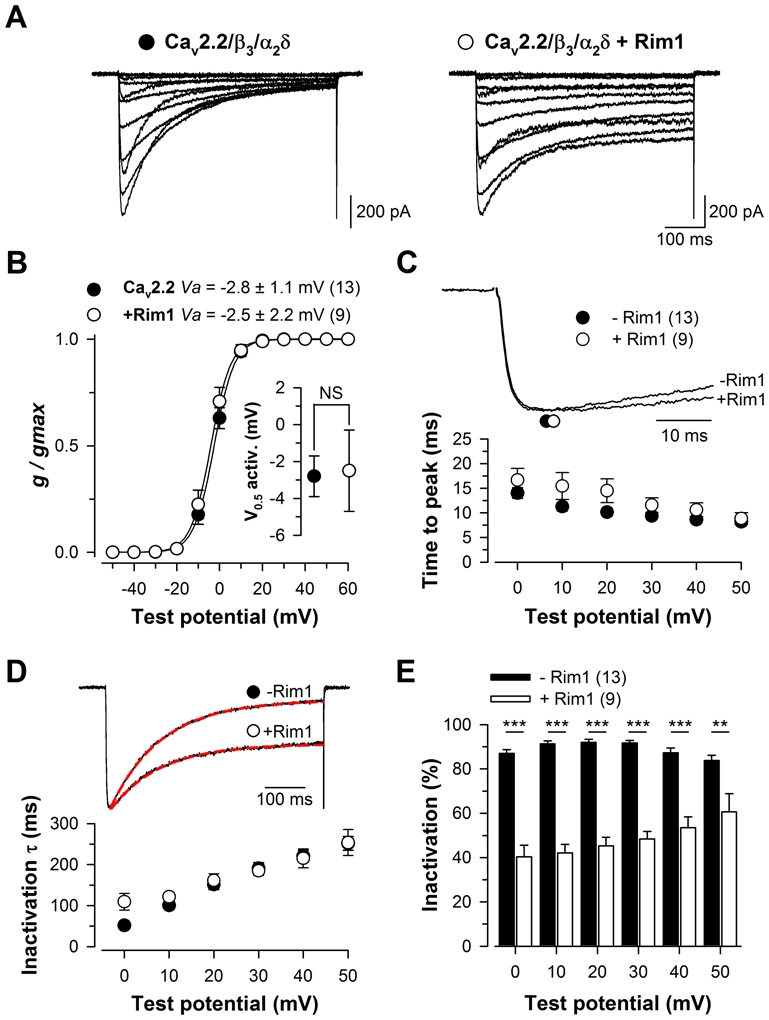
A, Representative set of whole cell Ba2+ current traces recorded from a Cav2.2/β3/α2δ1b (left panel) and Cav2.2/β3/α2δ1b/Rim1-expressing cell (right panel) in response to 500 ms depolarizing steps to values ranging between −40 mV and +60 mV from a holding potential of −80 mV. B, Corresponding mean normalized values of the voltage-dependence of Ba2+ conductance in the two populations. Inset presents the mean half-maximal activation potential for Cav2.2/β3/α2δ1b (filled circles) and Cav2.2/β3/α2δ1b/Rim1 channels (open circles). C, Representative normalized Ba2+ current traces recorded at +20 mV for Cav2.2/β3/α2δ1b and Cav2.2/β3/α2δ1b/Rim1 channels showing activation kinetics (top panel). Symbols illustrate the time to peak of the current. Mean values for the time to peak of the Ba2+ current as a function of membrane potential for Cav2.2/β3/α2δ1b (filled circles) and Cav2.2/β3/α2δ1b/Rim1 (open circles) channels (bottom panel). D, Representative normalized Ba2+ current traces recorded at +20 mV for Cav2.2/β3/α2δ1b and Cav2.2/β3/α2δ1b/Rim1 channels showing differences in inactivation kinetics (top panel). The superimposed red dashed lines correspond to the result from fitting a single exponential function to the inactivating phase of the current. Mean values for the time constant τ of Ba2+ current inactivation for both conditions as a function of membrane potential (bottom panel). E, Corresponding mean values for the extent of current inactivation for Cav2.2/β3/α2δ1b (filled bars) and Cav2.2/β3/α2δ1b/Rim1 channels (open bars). Values were significantly decreased in the presence of Rim1 at all potential values studied.
Rim1 modulates direct G-protein regulation of Cav2.2/β3 channels
To investigate the effect of Rim1 on direct G-protein regulation of Cav2.2 channels, Ba2+ currents were recorded under 10 μM DAMGO application, and common landmarks of the regulation (extent of current inhibition, depolarizing shift of the activation curve (commonly called reluctance), slowing of activation kinetics and prepulse facilitation) were analyzed. Representative Ba2+ current traces recorded in response to 500 ms depolarizing steps to values ranging between −40 mV and +60 mV, from a holding potential of −80 mV, are shown in Fig. 2A for Cav2.2/β3/α2δ1b (left panels) and Cav2.2/β3/α2δ1b/Rim1 channels (right panels), before (top panels) and after 10 μM DAMGO application (bottom panels). Figure 2B shows the corresponding mean peak Ba2+ current density as a function of membrane potential before (filled circles) and after DAMGO application (open circles), for control (left panel) and Rim1-expressing cells (right panel). The voltage-dependence of Ba2+ current activation was also determined in control and Rim1-expressing cells (Fig. 2B, insets). The mean half-activation potential is significantly shifted towards depolarized potentials (p < 0.001) in control cells after DAMGO application (from −2.8 ± 1.1 mV to 3.2 ± 1.1 mV, n = 13), whereas it remained more largely unaffected (p = 0.51) in cells expressing Rim1 (from −2.5 ± 2.2 mV to −0.2 ± 2.6 mV, n = 9). Hence, the DAMGO-induced depolarizing shift of the voltage-dependence of the activation curve was found significantly reduced (p = 0.027) in Rim1-epressing cells (2.3 ± 0.7 mV, n = 9) compared to control cells (6.0 ± 0.9 mV, n = 13). The extent of current inhibition produced by DAMGO application (the “ON” effect of the direct G-protein regulation) was measured at the peaks of the currents. Representative normalized current traces recorded in response to a depolarizing step to +10 mV for Cav2.2/β3/α2δ and Cav2.2/β3/α2δ/Rim1 channels before and after DAMGO application are shown in Fig. 2C and illustrate that the extent of the peak current inhibition in Rim1-expressing cells is strongly reduced compared to control cells. Figure 2D shows the percentage of current inhibition in control (filled bars) and Rim1-expressing cells (open bars) as a function of membrane potential. Globally, G-protein inhibition was found voltage-dependent and more pronounced at lowest voltages. For instance, the extent of current inhibition decreases from 63.2 ± 5.4% (0 mV) to 21.2 ± 2.3% (50 mV) in control cells, and from 33.1 ± 5.7% (0 mV) to 5.7 ± 3.2% (50 mV) in Rim1-expressing cells. This voltage-dependent decrease in G-protein inhibition reflects G-proteins unbinding from the channel at higher voltages. The extent of current inhibition was found drastically reduced in cells expressing Rim1 compared to control cells. For instance, in response to a depolarizing pulse to +20 mV, the mean peak current inhibition recorded from Rim1-expressing cells was, on average, 2.5-fold smaller than in control cells (13.5 ± 4.1%, n = 9, versus 33.3 ± 3.5%, n = 13, p = 0.002). Rim1-produced decrease in G-protein inhibition of Cav2.2 channels remained significant at all depolarizing steps studied (from 0 mV to +50 mV). The consequences of Rim1 expression on the “OFF” effects of G-protein regulation of Cav2.2 channels were also investigated. One characteristic “OFF” effect is the apparent slowing of current activation kinetics under DAMGO inhibition, which results from a channel opening- and time-dependent recovery from inhibition (33). The representative normalized current traces shown in Fig. 2E for Cav2.2/β3/α2δ and Cav2.2/β3/α2δ/Rim1 channels recorded in response to a depolarizing step to +10 mV before and after DAMGO application illustrate that G-protein inhibition produces a slight slowing of activation kinetics in control cells, whereas the extent of this effect appears greatly increased in cells expressing Rim1 as shown by larger time-to-peak values. Figure 2F shows the corresponding mean values for the shift of the current time-to peak produced by DAMGO application for control (filled bars) and Rim1-expressing cells (open bars). The shift of the current time-to-peak was found particularly pronounced at the lowest voltages tested, ranging from 12.7 ± 2.0 ms (0 mV) to 4.0 ± 0.5 ms (+50 mV) in control cells, and from 108.7 ± 42.0 ms (0 mV) to 8.6 ± 1.6 ms (+50 mV) in Rim1-expressing cells. Furthermore, for a given membrane potential, the slowing of the activation kinetics was found considerably increased in cells expressing Rim1 compared to control cells. For instance, in response to a depolarizing pulse of +10 mV, the mean shift of the current time-to-peak was, on average, 3.3-fold larger than in control cells (49.4 ± 14.0 ms, n = 9, versus 15.1 ± 1.6 ms, n = 13, p = 0.005). The difference in the slowing of the current activation kinetics between Cav2.2/β3/α2δ and Cav2.2/β3/α2δ/Rim1 channels remained significant at all membrane potential studied (from 0 mV to +50 mV). Finally, current prepulse facilitation under G-protein regulation was investigated with the common double pulse protocol, by comparing the amplitude of the Ba2+ current before (P1) and following (P2) application of a strong depolarizing prepulse (PP) to +100 mV. Representative current traces are shown in Fig. 2G, illustrating an increase of the extent of prepulse facilitation in Rim1-expressing cells. Fig. 2H shows the corresponding mean normalized values of prepulse facilitation in control (black symbol) and Rim1-expressing cells (open symbol) as a function of prepulse duration. For instance, in response to a 45 ms long prepulse duration, current facilitation was found 1.6-fold (p = 0.04) increased in Rim1-expressing cells. Taken together, these results strongly suggest that Rim1 modulates G-protein regulation of Cav2.2 channels, either by modulating the capability of G-proteins to inhibit the channel (“ON” effect), or by modulating the kinetics and extent of recovery from G-protein inhibition (“OFF” effect).
Figure 2. Rim1 modulates direct G-protein regulation of Cav2.2/β3 calcium channels.
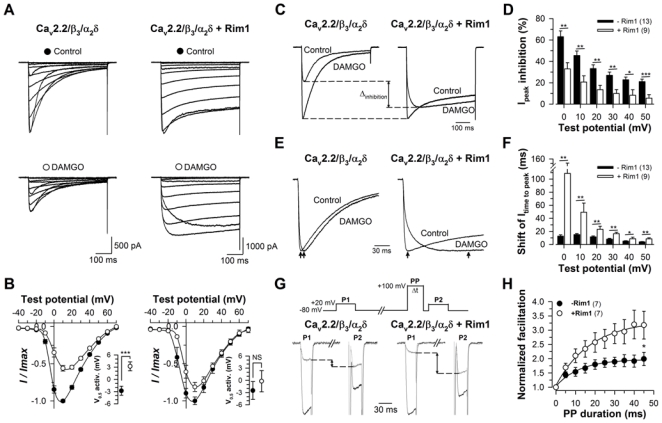
A, Representative set of whole cell Ba2+ current traces recorded from a Cav2.2/β3/α2δ1b (left panel) and Cav2.2/β3/α2δ1b/Rim1-expressing cell (right panel) in response to 500 ms depolarizing steps to values ranging between −40 mV and +60 mV from a holding potential of −80 mV, before (top panels) and after (bottom panels) 10 μM DAMGO application. B, Corresponding normalized mean voltage-dependence of the peak Ba2+ current density for Cav2.2/β3/α2δ1b (left panel) and Cav2.2/β3/α2δ1b/Rim1 channels (right panel). Insets represent the mean half-maximal activation potential before (filled circles) and after 10 μM DAMGO application (open circles). C, Representative normalized Ba2+ current traces recorded at +10 mV for Cav2.2/β3/α2δ1b (left panel) and Cav2.2/β3/α2δ1b/Rim1 channels (right panel) before and after DAMGO application showing the inhibition of the peak Ba2+ current under direct G protein regulation. Arrows indicate the decrease of the current inhibition in the presence of Rim1. D, Mean values for the peak Ba2+ current inhibition produced by DAMGO application in the absence (filled bars) and presence of Rim1 (open bars) as a function of membrane potential. Values were significantly decreased in the presence of Rim1 for all potential values studied. E, Representative normalized Ba2+ current traces recorded at +10 mV for Cav2.2/β3/α2δ1b (left panel) and Cav2.2/β3/α2δ1b/Rim1 channels (right panel) before and after DAMGO application showing the slowing of Ba2+ current activation kinetics under direct G-protein regulation. Arrows indicate the time to peak of the current. F, Corresponding mean values of the shift of the current time to peak in the absence (filled bars) and presence of Rim1 (open bars). Values were significantly increased in the presence of Rim1 for all potential values studied. G, Representative Ba2+ current traces for control (black traces) and DAMGO (grey traces) conditions elicited at +20 mV before (P1) and following (P2) a 20 ms long depolarizing prepulse (PP) to +100 mV recorded from a Cav2.2/β3/α2δ1b (left panel) and Cav2.2/β3/α2δ1b/Rim1-expressing cell (right panel). D, Corresponding normalized prepulse facilitation values ((P2/P1DAMGO)/(P2/P1Control)) as a function of the prepulse duration. Values were significantly increased by the presence of Rim1.
Rim1 does not affect the maximal G-protein inhibition of Cav2.2/β3 channel
In order to differentiate the contribution of Rim1 to the “ON” effect of G-protein regulation from the “OFF” effect, the maximal G-protein inhibition was measured at the start of the depolarization to avoid the important and confounding fraction of recovery from inhibition that has already occurred during depolarization when current amplitudes are measured at their peak. Representative current traces elicited at +10 mV and +30 mV from a holding potential of −80 mV are shown for Cav2.2/β3/α2δ (Fig. 3A, top panel) and Cav2.2/β3/α2δ/Rim1 channels (Fig. 3B, top panel), before (IControl) and after DAMGO application (IDAMGO). According to our developed method of analysis of G-protein regulation (34, 36), the Lost current traces were extracted (ILost) by subtracting IDAMGO from IControl (Fig. 3A and 3B, bottom panels). ILost provides the time course of the Lost current following G-protein activation. Hence, the current inhibition measured from the levels of ILost and IControl when extrapolated at t = 0 ms (the start of the depolarization) provides the net maximal G-protein inhibition (GIt0) before the process of current recovery has taken place (Fig. 3A and 3B, bottom panels). Figure 3C shows the average GIt0 values for control (filled bars) and Rim1-expressing cells (open bars) as a function of membrane potential. As expected for an inhibition at t = 0 ms, almost no voltage-dependence of the maximal G-protein inhibition was observed. This inhibition varies between 70.5 ± 5.0% (0 mV) and 57.5 ± 4.3% (+30 mV) for control cells, and between 68.3 ± 2.8% (0 mV) and 54.4 ± 4.1% (+30 mV) for Rim1-expressing cells. This non significant voltage-dependence is certainly linked to the difficulty in precisely estimating the maximal extent of G-protein inhibition at higher voltages when the kinetics of current recovery become very fast (see Fig. 4). The data are thus more reliable at lower voltages. More importantly, contrary to what was observed when measuring inhibition at the peaks of the currents, no difference was observed between Cav2.2/β3/α2δ and Cav2.2/β3/α2δ/Rim1 channels. For instance, in response to a depolarizing step to +20 mV, GIt0 remained unaffected (p = 0.58) in Rim1-expressing cells (57.2 ± 3.0%, n = 9) compared to control cells (60.5 ± 4.0%, n = 13). This suggests that Rim1 does not affect binding of Gβγ onto the closed state of Cav2.2 channels (“ON” effect). Because important differences where however observed in the landmarks of Cav2.2 channels under G-protein regulation in the presence of Rim1 (Fig. 2), we next investigated the possibility that Rim1 could alter “OFF” effects of the regulation.
Figure 3. Rim1 does not affect the maximal G-protein inhibition of Cav2.2 channel.
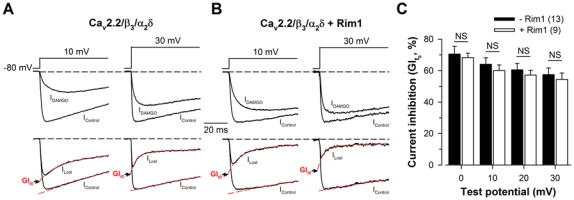
A, Representative normalized Ba2+ current traces elicited at +10 mV and +30 mV before (IControl) and after 10 μM DAMGO application (IDAMGO) for Cav2.2/β3/α2δ1b channels (top panel). Corresponding traces allowing the measurement of the maximal DAMGO inhibition at the start of the depolarization (GIt0) (bottom panel). IControl and ILost (obtained by subtracting IDAMGO from IControl) were fitted (red dashed lines) by a mono- and a double-exponential function, respectively, in order to better estimate GIt0. B, Legend as in (a) but for cells expressing Cav2.2/β3/α2δ1b/Rim1 channels. C, Corresponding mean values of GIt0 for Cav2.2/β3/α2δ1b (filled bars) and Cav2.2/β3/α2δ1b/Rim1 channels (open bars) as a function of membrane potential. Values were not altered by the presence of Rim1.
Figure 4. Rim1 promotes Cav2.2 channel recovery from G-protein inhibition.
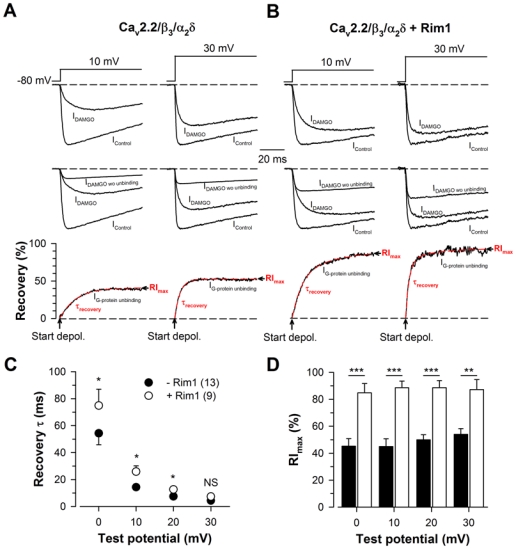
A, Representative normalized Ba2+ current traces elicited at +10 mV and +30 mV before (IControl) and after 10 μM DAMGO application (IDAMGO) for Cav2.2/β3/α2δ1b channels (top panel). Corresponding traces showing the amount of current that is present in IDAMGO (IDAMGO wo unbinding) are also shown (middle panel) and used to calculate IG-protein unbinding, the time dependence of G-protein dissociation from the channel (bottom panel). IG-protein unbinding was fitted with a mono-exponential function (red dashed lines) in order to determine the time constant of recovery from G-protein inhibition (τrecovery) and the maximal extent of recovery (RImax). B, Legend as in (A) but for cells expressing Cav2.2/β3/α2δ1b/Rim1 channels. Corresponding mean values of τrecovery (C) and RImax (D) for Cav2.2/β3/α2δ1b (filled symbols) and Cav2.2/β3/α2δ1b/Rim1 channels (open symbols) as a function of membrane potential. RImax values were significantly increased in the presence of Rim1 for all potential values studied.
Rim1 promotes Cav2.2/β3 channel recovery from G-protein inhibition
The two parameters that characterize the “OFF” components of G-protein regulation of Cav2.2 channels, i.e. the time constant of current recovery from inhibition (τrecovery) following channel activation, and the maximal extent of current recovery (RImax), were extracted. Representative currents traces before and after DAMGO application are shown for Cav2.2/β3/α2δ (Fig. 4A, top panel) and Cav2.2/β3/α2δ/Rim1 channels (Fig. 4B, top panel) in response to depolarizing steps to +10 mV and +30 mV from a holding potential of −80 mV. Corresponding current traces that describe the evolution of the current under G-protein inhibition if no current recovery took place (IDAMGO wo unbinding), and obtained by applying GIt0 at the control current (IControl) recorded before activation of G-proteins, are shown in Fig. 4A and 4B (middle panels) and used to extract the evolution of the current recovery from G-protein inhibition (IG-protein unbinding) (obtained by dividing the difference between IDAMGO and IDAMGO wo unbinding by the difference between IControl and IDAMGO wo unbinding) (Fig. 4A and 4B, bottom panels). Corresponding IG-protein unbinding traces were best fitted with a mono-exponential function, providing both the time constant of current recovery from G-protein inhibition (τrecovery) and the maximal extent of recovery (RImax). Corresponding average values for τrecovery and RImax are shown in Fig. 4C and 4D, respectively, as a function of membrane potential. We observed a slight but significant slowing of τrecovery in Rim1-expressing cells compared to control cells. For instance, in response to a depolarizing step to +10 mV, the time constant of current recovery from G-protein inhibition in Rim1-expressing cells is 1.8-fold slower than in control cells (25.9 ± 4.3 ms, n = 9, versus 14.3 ± 1.9 ms, n = 13, p = 0.014) (Fig. 4C). Also, significantly larger RImax values were observed in cells expressing Rim1. For instance, at a membrane potential of +10 mV, RImax values are on average 2.0-fold larger in Rim1-expressing cells than in control cells (88.6 ± 4.9%, n = 9, versus 44.9 ± 5.8%, n = 13, p < 0.001) (Fig. 4D).
Rim1 also modulates G-protein regulation of Cav2.2 channels expressed along with the β2a auxiliary subunit
In order to better understand the importance of channel inactivation in Rim1-mediated modulation of G-protein regulation of Cav2.2 channels, G-protein regulation was investigated on a slow inactivating channel as resulting from the co-expression with the β2a auxiliary subunit. Representatives Ba2+ current traces of Cav2.2/β2a/α2δ1b (left panel) and Cav2.2/β2a/α2δ1b/Rim1 channels (right panel) are shown in Fig. 5A. Consistent with what was observed with the β3 auxiliary subunit, the mean half activation potential remained unaltered (p = 0.30) in Rim1-expressing cell (0.5 ± 1.2 mV, n = 10) compared to control cells (1.5 ± 1.4 mV, n = 10) (Fig. 5B). Because of the difficulty to precisely fit the extremely slow inactivating phase of the current in Rim1-expressing cells (particularly at lower potentials), effect of Rim1 on Cav2.2/β2a channel inactivation was investigated by comparing the amplitude of the current measured at the peak to the amplitude measured at the end of the 500 ms depolarizing steps. Mean values of the extent of current inactivation of Cav2.2/β2a/α2δ and Cav2.2/β2a/α2δ/Rim1 channels as a function of the membrane potential are shown in Fig. 5C. As expected, co-expression of the β2a auxiliary subunit decreases Cav2.2 channel inactivation compared to the inactivation produced in the presence of the β3 subunit. For instance, in response to a depolarizing step to +20 mV, the extent of current inactivation in the presence of the β2a subunit was, on average, 1.5-fold smaller than in the presence of the β3 subunit. Moreover, co-expression of Rim1 with Cav2.2/β2a channels produced an additional effect on channel inactivation. For instance, the extent of current inactivation recorded from Rim1-expressing cells at +20 mV was, on average, 3.9-fold smaller than in control cells (15.0 ± 3.7%, n = 10, versus 58.4 ± 5.0%, n = 10, p < 0.001) and remained significant at all depolarizing step studied (from 0 mV to +30 mV). Similar to what was observed in the presence of the β3 subunit, the maximal extent of G-protein inhibition of Cav2.2/β2a channels was found unaffected by the presence of Rim1 (Fig. 6C), whereas the maximal extent of current recovery from G-protein inhibition (RImax) was found increased in Rim1-expressing cells (Fig. 6E). For instance, at a membrane potential of +10 mV, RImax values are on average 1.3-fold larger in Rim1-expressing cells than in control cells (98.4 ± 1.6%, n = 10, versus 67.8 ± 6.4%, n = 10, p = 0.008). However, in contrast to what was observed with the β3 subunit, this effect is less pronounced in the presence of the β2a subunit. For instance, at a membrane potential of +10 mV, the difference in RImax values between control and Rim1-expressing cells is on average 1.4-fold smaller in the presence of the β2a subunit than in the presence of the β3 subunit. Moreover, no statistical difference in the time constant of recovery was observed (Fig. 6D).
Figure 5. Rim1 also decreases inactivation of Cav2.2 channel in the presence of the β2a auxiliary subunit.
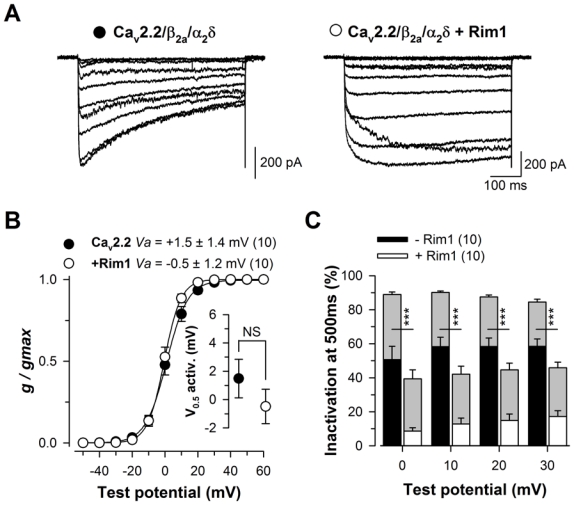
A, Representative set of whole cell Ba2+ current traces recorded from a Cav2.2/β2a/α2δ1b (left panel) and Cav2.2/β2a/α2δ1b/Rim1-expressing cell (right panel) in response to 500 ms depolarizing steps to values ranging between −40 mV and +60 mV from a holding potential of −80 mV. B, Corresponding mean normalized values of the voltage-dependence of Ba2+ conductance in the two populations. Inset presents the mean half-maximal activation potential for Cav2.2/β2a/α2δ1b (filled circles) and Cav2.2/β2a/α2δ1b/Rim1 channels (open circles). C, Mean values for the extent of current inactivation for Cav2.2/β2a/α2δ1b (filled bars) and Cav2.2/β2a/α2δ1b/Rim1 channels (open bars) measured at the end of the 500 ms long depolarizing steps. Values were significantly decreased in the presence of Rim1 for all potential values studied. For comparison, the extent of current inactivation measured in the presence of the β3 auxiliary subunit is shown in grey.
Figure 6. Rim1 also modulates direct G-protein regulation of Cav2.2 calcium channel in the presence of the β2a auxiliary subunit.
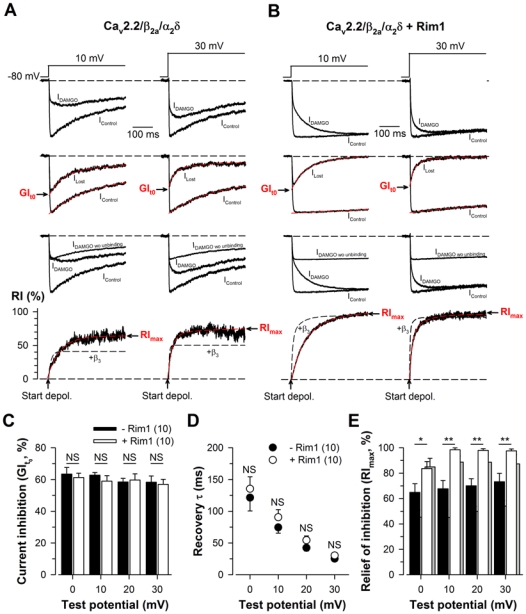
A, Representative normalized Ba2+ current traces elicited at +10 mV and +30 mV before (IControl) and after 10 μM DAMGO application for Cav2.2/β2a/α2δ1b channels (first panel). Corresponding traces allowing the measurement of the maximal DAMGO inhibition at the start of the depolarization (GIt0) (second panel). Corresponding traces showing the amount of current that is present in IDAMGO (IDAMGO wo unbinding) are also shown (third panel) and used to calculate IG-protein unbinding, the time dependence of G-protein dissociation from the channel (fourth panel). IG-protein unbinding was fitted with a mono-exponential function (red dashed lines) in order to determine the time constant of recovery from G-protein inhibition (τrecovery) and the maximal extent of recovery (RImax). For comparison, the time course and extent of current recovery from inhibition measured in the presence of the β3 auxiliary subunit is shown in black dashed lines. B, Legend as in (A) but for cells expressing Cav2.2/β2a/α2δ1b/Rim1 channels. C, Corresponding mean values of GIt0 for Cav2.2/β2a/α2δ1b (filled bars) and Cav2.2/β2a/α2δ1b/Rim1 channels (open bars) as a function of membrane potential. Corresponding mean values of τrecovery (D) and RImax (E) for Cav2.2/β2a/α2δ1b (filled symbols) and Cav2.2/β2a/α2δ1b/Rim1 channels (open symbols) as a function of membrane potential. For comparison, RImax values obtained in the presence of the β3 subunit are indicated in grey.
Discussion
Earlier findings from our group have shown that channel inactivation greatly influences G-protein regulation. In particular, it was found, using a series of β-subunit constructs (36), or familial hemiplegic migraine mutations of Cav2.1 channels (35), that decrease of channel inactivation significantly enhances membrane depolarization-induced recovery from G-protein inhibition. Other findings have reported that syntaxin 1A also modifies G-protein regulation of presynaptic calcium channels (12, 13, 16), further suggesting that studying the effect of Rim1 on G-protein regulation is of prime importance.
In the present study, we demonstrate that Rim1, besides to modulate biophysical properties of Cav2.2 channels, also greatly influences direct G-protein inhibition of Cav2.2 channels. These results provide strong support for the ability of the proteins of the presynaptic vesicle complex to modulate G-protein regulation of Cav2.2 channels.
Rim1 differentially affects “ON” and “OFF” G-protein regulation of Cav2.2 channels
One of the major inhibitory pathways controling voltage-gated Ca2+ channels at the presynaptic level is mediated by G-protein coupled receptor activation. This inhibition is recognized by a strong current inhibition (“ON” effect), whereas the process of channel activation induced by membrane depolarization produces deinhibition, even under the maintained presence of the GPCR agonist. This deinhibition is characterized by an apparent depolarized shift of the voltage-dependence of activation curve, a slowed current kinetics, and a more or less pronounced extent of current recovery (“OFF” effects). Hence, if current inhibition finally only represents an index of the total amount of channels subjected to direct G-protein inhibition, current deinhibition really reflects the importance of this regulation under neuronal activity. In this study, “ON” and “OFF” G-protein regulation parameters were analyzed using our recently developed method (34–36). Hence, the extent of G-protein inhibition, measured at the start of the depolarization, before the initiation of the recovery process, was found unaffected by Rim1, suggesting that Rim1 does not affect the binding of Gβγ dimer onto the closed-state of the channel. This result is consistent with the fact that Rim1 modulates channel activity by interacting with the β-subunit and not directly with the Cav2 subunit. Thus, it is not expected to affect one of the structural channel determinants known to be involved in Gβγ binding (i.e. the I–II loop, the amino-, and carboxy-terminal regions of the Cav2 subunit). In contrast, we observed that Rim1 critically affects the “OFF” effects of Cav2.2 channel regulation by G-proteins. Whereas the time constant of current recovery from inhibition was slowed in Rim1-expressing cells, the maximal extent of current recovery was found drastically increased. These results are consistent with earlier reports suggesting that the molecular process of channel inactivation accelerates the recovery from inhibition, but reduces the temporal window in which the process can take place (35, 36). Hence, by preventing Cav2.2 channel inactivation, Rim1 slows down the recovery from G-protein inhibition but drastically improves the recovery process by increasing the time window during which it takes place. Interestingly, our observation that the effect of Rim1 on G-protein regulation was found less pronounced in the presence of the β2a subunit than in the presence of the β3 subunit suggest that Rim1 modulation could particularly affect fast inactivating channels. It is well know that Cav2.2 channels may associate with one of four ancillary β-subunits (β1-4), and that the specific Cav2.2/β combination assembled determines channel properties, while variation in the proportion of each combination contributes to the functional diversity of neurons (28). Hence, our results suggest that Rim1 could play an important role in synapses expressing fast inactivating Cav2.2 channels.
Potential implication in Rim1-induced facilitation of neurotransmitter release
It is well know that neuronal voltage-gated Ca2+ channels are in close association with several members of SNARE proteins (i.e. syntaxin 1A/1B, SNAP-25, synaptotagmin 1 and synaptobrevin 2), linking Ca2+ influx to the presynaptic vesicle release machinery, critical for a fast and spatially delimited neurotransmitter release (23). Previous works have shown that Rim1 plays an important role in this coupling, facilitating neurotransmitter release (15, 22, 27). Additionally, by preventing channel inactivation, Rim1 contributes to maintain a sustained Ca2+ influx during neuronal firing, which also contributes to promote neurotransmitter release (14). In addition, a facilitation of neurotransmitter release could also be triggered by a diminution of the inhibitory pathway carried by G-proteins. Thus, Rim1, by promoting channel deinhibition, contributes to maintain Ca2+ influx through Cav2.2 channels, especially during prolonged synaptic activity even under continuous activation of the inhibitory GPCR. Hence, combined, the increase in functional coupling between voltage-gated Ca2+ channels and vesicle machinery, the slowing of channel inactivation kinetics, and the decrease in the inhibitory pathway may altogether contribute to Rim1-induced facilitation of evoked neurotransmitter release.
References
- 1.Andrade A, Denome S, Jiang YQ, Marangoudakis S, Lipscombe D. Opioid inhibition of N-type Ca2+ channels and spinal analgesia couple to alternative splicing. Nat Neurosci. 2010;13:1249–1256. doi: 10.1038/nn.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barragan I, Marcos I, Borrego S, Antinolo G. Molecular analysis of RIM1 in autosomal recessive Retinitis pigmentosa. Ophthalmic Res. 2005;37:89–93. doi: 10.1159/000084250. [DOI] [PubMed] [Google Scholar]
- 3.Bertram R, Swanson J, Yousef M, Feng ZP, Zamponi GW. A minimal model for G protein-mediated synaptic facilitation and depression. J Neurophysiol. 2003;90:1643–1653. doi: 10.1152/jn.00190.2003. [DOI] [PubMed] [Google Scholar]
- 4.Betz A, Thakur P, Junge HJ, Ashery U, Rhee JS, Scheuss V, Rosenmund C, Rettig J, Brose N. Functional interaction of the active zone proteins Munc13-1 and RIM1 in synaptic vesicle priming. Neuron. 2001;30:183–196. doi: 10.1016/s0896-6273(01)00272-0. [DOI] [PubMed] [Google Scholar]
- 5.Canti C, Bogdanov Y, Dolphin AC. Interaction between G proteins and accessory subunits in the regulation of α1B calcium channels in Xenopus oocytes. J Physiol. 2000;527:419–432. doi: 10.1111/j.1469-7793.2000.t01-1-00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castillo PE, Schoch S, Schmitz F, Sudhof TC, Malenka RC. RIM1alpha is required for presynaptic long-term potentiation. Nature. 2002;415:327–330. doi: 10.1038/415327a. [DOI] [PubMed] [Google Scholar]
- 7.Coppola T, Magnin-Luthi S, Perret-Menoud V, Gattesco S, Schiavo G, Regazzi R. Direct interaction of the Rab3 effector RIM with Ca2+ channels, SNAP-25, and synaptotagmin. J Biol Chem. 2001;276:32756–32762. doi: 10.1074/jbc.M100929200. [DOI] [PubMed] [Google Scholar]
- 8.Davies JN, Zamponi GW. Old proteins, developing roles: The regulation of calcium channels by synaptic proteins. Channels (Austin) 2008;2:130–138. doi: 10.4161/chan.2.2.6214. [DOI] [PubMed] [Google Scholar]
- 9.De Waard M, Hering J, Weiss N, Feltz A. How do G proteins directly control neuronal Ca2+ channel function? Trends Pharmacol Sci. 2005;26:427–436. doi: 10.1016/j.tips.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 10.De Waard M, Liu H, Walker D, Scott VE, Gurnett CA, Campbell KP. Direct binding of G-protein betagamma complex to voltage-dependent calcium channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- 11.Feng ZP, Arnot MI, Doering CJ, Zamponi GW. Calcium channel beta subunits differentially regulate the inhibition of N-type channels by individual Gbeta isoforms. J Biol Chem. 2001;276:45051–45058. doi: 10.1074/jbc.M107784200. [DOI] [PubMed] [Google Scholar]
- 12.Jarvis SE, Magga JM, Beedle AM, Braun JE, Zamponi GW. G protein modulation of N-type calcium channels is facilitated by physical interactions between syntaxin 1A and Gbetagamma. J Biol Chem. 2000;275:6388–6394. doi: 10.1074/jbc.275.9.6388. [DOI] [PubMed] [Google Scholar]
- 13.Jarvis SE, Zamponi GW. Distinct molecular determinants govern syntaxin 1A-mediated inactivation and G-protein inhibition of N-type calcium channels. J Neurosci. 2001;21:2939–2948. doi: 10.1523/JNEUROSCI.21-09-02939.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiyonaka S, Wakamori M, Miki T, Uriu Y, Nonaka M, Bito H, Beedle AM, Mori E, Hara Y, De Waard M, Kanagawa M, Itakura M, Takahashi M, Campbell KP, Mori Y. RIM1 confers sustained activity and neurotransmitter vesicle anchoring to presynaptic Ca2+ channels. Nat Neurosci. 2007;10:691–701. doi: 10.1038/nn1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lonart G, Tang X, Simsek-Duran F, Machida M, Sanford LD. The role of active zone protein Rab3 interacting molecule 1 alpha in the regulation of norepinephrine release, response to novelty, and sleep. Neuroscience. 2008;154:821–831. doi: 10.1016/j.neuroscience.2008.03.047. [DOI] [PubMed] [Google Scholar]
- 16.Lu Q, AtKisson MS, Jarvis SE, Feng ZP, Zamponi GW, Dunlap K. Syntaxin 1A supports voltage-dependent inhibition of alpha1B Ca2+ channels by Gbetagamma in chick sensory neurons. J Neurosci. 2001;21:2949–2957. doi: 10.1523/JNEUROSCI.21-09-02949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meir A, Bell DC, Stephens GJ, Page KM, Dolphin AC. Calcium channel beta subunit promotes voltage-dependent modulation of alpha1B by G beta gamma. Biophys J. 2000;79:731–746. doi: 10.1016/S0006-3495(00)76331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meir A, Dolphin AC. Kinetics and Gbetagamma modulation of Ca(v)2.2 channels with different auxiliary beta subunits. Pflugers Arch. 2002;444:263–275. doi: 10.1007/s00424-002-0803-3. [DOI] [PubMed] [Google Scholar]
- 19.Melliti K, Grabner M, Seabrook GR. The familial hemiplegic migraine mutation R192Q reduces G-protein-mediated inhibition of P/Q-type (Ca(V)2.1) calcium channels expressed in human embryonic kidney cells. J Physiol. 2003;546:337–347. doi: 10.1113/jphysiol.2002.026716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Michaelides M, Holder GE, Hunt DM, Fitzke FW, Bird AC, Moore AT. A detailed study of the phenotype of an autosomal dominant cone-rod dystrophy (CORD7) associated with mutation in the gene for RIM1. Br J Ophthalmol. 2005;89:198–206. doi: 10.1136/bjo.2004.050773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miki T, Kiyonaka S, Uriu Y, De Waard M, Wakamori M, Beedle AM, Campbell KP, Mori Y. Mutation associated with an autosomal dominant cone-rod dystrophy CORD7 modifies RIM1-mediated modulation of voltage-dependent Ca2+ channels. Channels (Austin) 2007;1:144–147. doi: 10.4161/chan.4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mittelstaedt T, Alvarez-Baron E, Schoch S. RIM proteins and their role in synapse function. Biol Chem. 2010;391:599–606. doi: 10.1515/BC.2010.064. [DOI] [PubMed] [Google Scholar]
- 23.Mochida S, Sheng ZH, Baker C, Kobayashi H, Catterall WA. Inhibition of neurotransmission by peptides containing the synaptic protein interaction site of N-type Ca2+ channels. Neuron. 1996;17:781–788. doi: 10.1016/s0896-6273(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 24.Ohtsuka T, Takao-Rikitsu E, Inoue E, Inoue M, Takeuchi M, Matsubara K, Deguchi- Tawarada M, Satoh K, Morimoto K, Nakanishi H, Takai Y. Cast: a novel protein of the cytomatrix at the active zone of synapses that forms a ternary complex with RIM1 and munc13-1. J Cell Biol. 2002;158:577–590. doi: 10.1083/jcb.200202083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raingo J, Castiglioni AJ, Lipscombe D. Alternative splicing controls G protein-dependent inhibition of N-type calcium channels in nociceptors. Nat Neurosci. 2007;10:285–292. doi: 10.1038/nn1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC, Sudhof TC. RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature. 2002;415:321–326. doi: 10.1038/415321a. [DOI] [PubMed] [Google Scholar]
- 27.Schoch S, Mittelstaedt T, Kaeser PS, Padgett D, Feldmann N, Chevaleyre V, Castillo PE, Hammer RE, Han W, Schmitz F, Lin W, Sudhof TC. Redundant functions of RIM1alpha and RIM2alpha in Ca(2+)-triggered neurotransmitter release. EMBO J. 2006;25:5852–5863. doi: 10.1038/sj.emboj.7601425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scott VE, De Waard M, Liu H, Gurnett CA, Venzke DP, Lennon VA, Campbell KP. Beta subunit heterogeneity in N-type Ca2+ channels. J Biol Chem. 1996;271:3207–3212. doi: 10.1074/jbc.271.6.3207. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi T, Momiyama A. Different types of calcium channels mediate central synaptic transmission. Nature. 1993;366:156–158. doi: 10.1038/366156a0. [DOI] [PubMed] [Google Scholar]
- 30.Tedford HW, Zamponi GW. Direct G protein modulation of Cav2 calcium channels. Pharmacol Rev. 2006;58:837–862. doi: 10.1124/pr.58.4.11. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Sugita S, Sudhof TC. The RIM/NIM family of neuronal C2 domain proteins. Interactions with Rab3 and a new class of Src homology 3 domain proteins. J Biol Chem. 2000;275:20033–20044. doi: 10.1074/jbc.M909008199. [DOI] [PubMed] [Google Scholar]
- 32.Weber AM, Wong FK, Tufford AR, Schlichter LC, Matveev V, Stanley EF. N-type Ca2+ channels carry the largest current: implications for nanodomains and transmitter release. Nat Neurosci. 2010;13:1348–1350. doi: 10.1038/nn.2657. [DOI] [PubMed] [Google Scholar]
- 33.Weiss N, Arnoult C, Feltz A, De Waard M. Contribution of the kinetics of G protein dissociation to the characteristic modifications of N-type calcium channel activity. Neurosci Res. 2006;56:332–343. doi: 10.1016/j.neures.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 34.Weiss N, De Waard M. Introducing an alternative biophysical method to analyze direct G protein regulation of voltage-dependent calcium channels. J Neurosci Methods. 2007;160:26–36. doi: 10.1016/j.jneumeth.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 35.Weiss N, Sandoval A, Felix R, Van den Maagdenberg A, De Waard M. The S218L familial hemiplegic migraine mutation promotes deinhibition of Ca(v)2.1 calcium channels during direct G-protein regulation. Pflugers Arch. 2008;457:315–326. doi: 10.1007/s00424-008-0541-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weiss N, Tadmouri A, Mikati M, Ronjat M, De Waard M. Importance of voltage-dependent inactivation in N-type calcium channel regulation by G-proteins. Pflugers Arch. 2007;454:115–129. doi: 10.1007/s00424-006-0184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]