

Abstract
Endometrial cancer is the most common gynecologic malignancy diagnosed among women in developed countries. One recent biomarker strongly associated with disease progression and survival is epithelial membrane protein-2 (EMP2), a tetraspan protein known to associate with and modify surface expression of certain integrin isoforms. In this study, we show using a xenograft model system that EMP2 expression is necessary for efficient endometrial tumor formation, and we have started to characterize the mechanism by which EMP2 contributes to this malignant phenotype. In endometrial cancer cells, the focal adhesion kinase (FAK)/Src pathway appears to regulate migration as measured through wound healing assays. Manipulation of EMP2 levels in endometrial cancer cells regulates the phosphorylation of FAK and Src, and promotes their distribution into lipid raft domains. Notably, cells with low levels of EMP2 fail to migrate and poorly form tumors in vivo. These findings reveal the pivotal role of EMP2 in endometrial cancer carcinogenesis, and suggest that the association of elevated EMP2 levels with endometrial cancer prognosis may be causally linked to its effect on integrin-mediated signaling.
Introduction
Endometrial cancer is a significant disease in women with nearly one out of every thirty-five women developing the disease over her lifetime [1], [2]. In the United States endometrial cancer is the most common malignancy of the female genital tract. It is generally accepted to be an endocrine-related neoplasm with a pronounced impact of sex hormone status, and an increased incidence with age [3].
One protein associated with the development of endometrial cancer is epithelial membrane protein-2 (EMP2). EMP2 is a member of the GAS-3/PMP22 subfamily, which together with tetraspanins and connexins, comprise the three subfamilies of the large 4-transmembrane family. In the endometrium, EMP2 functions as a prognostic marker which can help predict patients who will progress from endometrial hyperplasia to cancer [4]. In patients with endometrial cancer, EMP2 positive tumors have been shown to be more aggressive and invasive, and its expression within tumors correlates with poor prognosis and survival [4], [5].
Although the exact role of EMP2 in endometrial cancer remains poorly defined, EMP2 may function as a trafficking molecule for a variety of proteins and glycolipids to efficiently transfer from a post-Golgi endosomal compartment to the plasma membrane. Accordingly, modulation of EMP2 expression and localization causes pleiotrophic changes on the plasma membrane of several classes of molecules, including integrins, MHC class I, and GPI-linked proteins [6], [7], [8], and EMP2 appears to mediate trafficking of these molecules to detergent-insoluble glycosphingolipid-enriched (DIG) membrane domains. DIG domains are thought to be important for receptor complexing and resultant signal transduction (reviewed in [9], [10], [11]).
In this study, we examine the mechanism by which EMP2 contributes to the etiology of endometrial cancer. Previous studies have shown that EMP2 selectively interacts with integrins in endometrial cells [6]. As EMP2 expression also correlates with tumor invasiveness in patients with endometrial cancer, we examine if modulation of EMP2 alters integrin-associated signaling proteins. Specifically, we focus on focal adhesion kinase (FAK), one of the critical focal adhesion signaling proteins and a protein that EMP2 has been shown to associate with in other cell systems [12], [13]. In endometrial cancer cells, EMP2 promotes FAK and Src phosphorylation, and contributes to their localization within lipid raft domains. As cell adhesion and spreading is a crucial step in the formation and invasion cascade of cancer cells, our results suggest that EMP2 is a critical regulator of metastatic potential of endometrial cancer cells.
Materials and Methods
Cell lines
The human endometrial adenocarcinoma cell lines HEC-1A (HTB112, ATCC, Manassas, VA) and Ishikawa (generous gift from Dr. Carmen Williams, NIH) cells were cultured in McCoys or DMEM media supplemented with 10% fetal calf serum at 37°C in a humidified 5% CO2. Cell lines were used within 2 months after resuscitation of frozen aliquots and were authenticated based on viability, recovery, growth, morphology, and isoenzymology. Both cell lines were also tested for murine pathogens including mycoplasma by the Division of Laboratory Animal Medicine at the University of California, Los Angeles. Cells were passaged every 3–4 days. Stably transfected HEC-1A cells containing a human EMP2-GFP fusion protein (48 kD), control GFP, or EMP2 specific ribozyme have been previously described [6]. Endogenous EMP2 is expressed at 20 kD. Ishikawa cells were infected with a MSCV-EMP2 or control retrovirus, creating the cell lines Ishikawa/EMP2 and Ishikawa/V [14]. Infected cells were sorted by GFP using flow cytometry.
Preparation of Xenografts
Ethical Treatment of Animals Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The procedures used were approved by the Animal Research Committee at the University of California, Los Angeles under protocol #2004-182. All efforts were made to minimize animal suffering.
Four to six-week-old nude BALB/c female mice were obtained from Charles River Laboratories (Wilmington, MA) and maintained at the University of California, Los Angeles. Animals were inoculated subcutaneously (s.c.) with 1×106 HEC-1A/OE, HEC-1A/V, or HEC-1A/RIBO cells into the right and left shoulder flanks, respectively. Six mice were used per group. Tumors were measured twice a week, and tumor volumes were calculated by the formula π/6 x larger diameter x smaller diameter [15], and data expressed as mean ± standard error of the mean (SEM). Two-way ANOVA was used to evaluate overall differences in the means between different groups as a function of time, and significance was calculated using p<0.05. At day 30, tumors were excised, fixed in formalin, and then processed for hematoxylin and eosin staining by the Tissue Procurement Laboratory at UCLA.
Immunohistochemistry
Tumor samples were stained with human EMP2 antisera or a preimmune control. Briefly, for antigen retrieval, sections were incubated at 95°C for 20 minutes in 0.1 M citrate, pH 6.0. EMP2 expression was detected using rabbit human EMP2 antisera at a dilution of 1∶400 as previously described [5] followed by visualization using the Vector ABC kit (Vector Labs, Burlingame, CA) according to the manufacturer's instructions. DAB staining was quantified using Photoshop 7 [16].
Proliferation Assays
Cellular Proliferation was monitored using two independent assays. First, a standard static growth assay was performed as previously described [6], [17]. Briefly, 1×103 cells were plated in triplicates in a 96-well plates from 0–7 days. At each indicated day, cells were stained with toluidine blue, lysed with 2% SDS (Biowhittaker, Walkersville, MD), and absorbance measured at 595 nm. Each experiment was repeated at least three times, and groups were compared using an unpaired Student's t test.
Second, the BrdU Cell Proliferation Assay (EMD Chemicals, Gibbstown, NJ) was performed as per manufacturer's instructions. Briefly, triplicate cultures of 104 cells were cultured in a 96-well plate. Cells were then incubated in DMEM +0.5% FCS overnight to arrest the cells. Cells were then released in complete media containing BrdU for 2 or 24 hrs. Cells were then fixed, permeabilized and the DNA denatured. A detector anti-BrdU monoclonal antibody was then added and ultimately detected using a horseradish peroxidase-conjugated goat anti-mouse IgG1. To determine the amount of incorporated BrdU, a fluorogenic substrate was added and the absorbance quantitated at both 450 and 595 nm using a spectrophotometer.
Wound healing
105 HEC-1A and Ishikawa cells with modulated EMP2 expression were seeded in 35-mm tissue culture dishes. When cells were confluent, a “wound” was created using a 100-µl “yellow” pipette tip as described [13], [18]. Wound healing was monitored over 48 hrs with a 10x phase contrast objective, and images were collected using a Power Shot S80 camera (Canon, Lake Success, NY). Manual measurements were made to quantitate the wound healing process. Three independent experiments were performed, and the results averaged.
In some experiments, inhibitors were added to determine the contribution of specific pathways to wound healing. Wildtype HEC-1A or Ishikawa cells were treated with 10 µM of the FAK-Src small molecule inhibitor PP2 [19] or 5 µM of Akt inhibitor VIII ([20]; Calbiochem, San Diego, CA), the EGFR inhibitor Erlotinib ([21]; 10 µM, Genentech), or the Src family tyrosine kinase inhibitor Dasatinib ([22]; 10 nM, Bristol-Myers Squibb). Efficacy of the inhibitors was tested at the manufacturer's recommended dosage, and potential toxicity was measured using trypan-blue exclusion.
Immunoprecipitations
Cells were washed two times with PBS, placed in lysis solution (1% Nonidet P-40 containing 2 mM phenylmethylsulfonyl fluoride, 10 µg/ml aprotinin, 2 µg/ml, pepstatin, 10 mM iodoacetamide, 0.1 mM EDTA, 0.1 mM EGTA, 10 mM HEPES, and 10 mM KCl), and solubilized for 30 min at 4°C. Lysates were sonicated for 15 s, and the insoluble materials were pelleted at 10,000 rpm for 10 min. The cell lysates were precleared by incubation with protein A/G-agarose beads (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Precleared, equivalent lysates were incubated overnight with agarose beads bound to either anti-FAK rabbit polyclonal antisera (Santa Cruz Biotech), EMP2 antisera, or isotype rabbit sera. The beads were washed three times in lysis solution and finally in 50 mM Tris buffer (pH 8). Immune complexes were eluted from the beads using Laemmli sample buffer (62.5 mM Tris-Cl, pH 6.8, 10% glycerol, 2% SDS, 0.01% bromphenol blue, 2% β-mercaptoethanol). As EMP2 contains multiple glycosylation sites (13), N-linked glycans were cleaved using peptide N-glycanase (PNGase; New England Biolabs, Beverly, MA). Eluates were treated as per the manufacturer's instructions at 37°C for 2 h and analysed by Western blot analysis as described below. Blots were digitized using a flatbed scanner and the band density measured using NIH program Image J. EMP2:FAK stoichiometry was calculated by dividing the volume intensities of immunoprecipitation with anti-EMP2 sera over that with anti-FAK antisera. Experiments were repeated three times, averaged, and the SEM calculated.
Western blot analysis
Cells were lysed in Laemmli buffer. Proteins were separated by SDS-PAGE, transferred to a nitrocellulose membrane (Amersham Biosciences), and stained with Ponceau S (Sigma-Aldrich, St. Louis, MO) to determine transfer efficiency. Membranes were blocked with 10% low fat milk in PBS containing 0.1% Tween 20 and probed with EMP2 antisera (1∶1000), anti- 576/577p-FAK (Santa Cruz Biotechnology), anti- total FAK (BD Biosciences), anti-416 p-Src (Cell Signaling, Danvers, MA), anti-total Src (Cell Signaling) or β-actin (Sigma-Aldrich). In some experiments, blots were probed with anti-Flottillin-2 (BD Biosciences) or anti-EEA1 antibodies (BD Biosciences). Protein bands were visualized using a horseradish peroxidase-labeled secondary antibody (BD Biosciences; Southern Biotechnology Associates, Birmingham, AL) followed by chemiluminescence (ECL; Amersham Biosciences). Band intensities were quantified using the NIH program Image J as above. To account for loading variability, β-actin was used to normalize each sample. At least three independent experiments were performed and, where indicated, the results were evaluated for statistical significance using a Student's t-test (unpaired, one-tail). A level of p<0.05 was considered to be statistically significant.
Immunofluorescence
HEC-1A or Ishikawa cells were plated onto glass coverslips (Fisher Scientific, Pittsburgh, PA). Cells were fixed in cold methanol, blocked in 1% normal goat serum, and then incubated overnight at 4°C with combinations of EMP2, anti-397p-FAK (BD Biosciences, 1∶250) or a total FAK antibody (BD Biosciences, 1∶250) in a humidified chamber. Cells were rinsed with PBS +0.01% Triton X-100, then incubated (2–4 hours. at RT) with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG (1∶50) or rhodamine-conjugated donkey anti-mouse IgG (1∶500; Jackson ImmunoResearch Laboratories, West Grove, PA). Negative controls included incubation of cells with secondary antibody alone. Cells were washed in PBS +0.01% Triton, rinsed briefly with double deionized H20, and mounted in a 3.5% n-propyl gallate-glycerol solution.
Cell images were captured using a Zeiss LSM 510 confocal microscope at 600X magnification. The colocalization coefficient between EMP2 and 397p-FAK or total FAK was determined using Zeiss LSM 5 PASCAL software (Carl Zeiss MicroImaging GmbH, Germany), and values represent the relative number of colocalizing pixels compared to the total number of pixels above threshold. The mean colocalization coefficient was averaged from at least 3 independent images.
Lipid Raft Fractionation
Cells (5×107) were harvested and washed in PBS. Cells were resuspended in Tris-buffered saline (50 mM Tris, pH 7.5, 20 mM EDTA, complete protease inhibitors and PhosSTOP (both from Roche Diagnostics, Indianapolis, IN). Cells were lysed by sonication (Moran and Miceli, 1998; Lusa et al., 2001) and then dissolved in 1% Triton X-100 or 1% Brij-58 and incubated on ice for 60 min. The extract was mixed 1∶1 with 80% sucrose (40% final), followed by step overlays with 35 and 5% sucrose, and centrifuged at 46,000 rpm for 18 h with a Sorvall SW55 rotor (Global Medical Instrumentation, Albertville, MN). Fractions (500 µl) were collected from the top of the gradient, and analyzed by SDS-PAGE. In some cases, samples were treated with PNGase prior to protein resolution by SDS-PAGE.
Cholesterol depletion was performed as described previously (Claas et al., 2001). Briefly, cells were washed in PBS to remove serum. Cells were incubated in DMEM containing 20 mM methyl-β cyclodextrin (MβCD; Sigma-Aldrich) for 60 min at 37°C. Cells were analyzed by trypan blue exclusion to insure the lack of toxicity by MβCD before being fractionated as described above.
Statistical Analysis
All values in the text are mean + SEM. Differences between means were evaluated using a two-tailed Student's t test or ANOVA as indicted. Significant differences were taken at the p<0.05 level.
Results
EMP2 expression promotes endometrial tumor development
Endometrial tumors with high levels of EMP2 have been shown to be more aggressive and correlate with poor clinical outcome [5]. In order to determine if EMP2 expression directly contributed to tumor formation, we engineered endometrial carcinoma cells to express modulated levels of the protein (see Methods and Materials; [6]). 106 HEC-1A/EMP2, HEC-1A/V, or HEC-1A/RIBO were injected s.c. into nude mice, and tumor volume was then measured over 30 days (Figure 1A). EMP2 overexpression promoted a two fold increase in tumor growth over HEC-1A/V cells (Figure 1B) while tumors with reduced EMP2 expression exhibited a three fold reduction in growth compared to HEC-1A/EMP2 cells (Figure 1C). Two-way ANOVA indicated that a statistically significant difference existed in the effect of EMP2 on tumor growth (p = 0.019).
Figure 1. Tumor volume is increased in HEC-1A/EMP2 tumors.

(A) HEC-1A/EMP2, HEC-1A/V, and HEC-1A/RIBO cells were injected s.c. into nude mice. Tumor volume was determined using calipers. HEC-1A/EMP2 tumors were larger than tumors injected with HEC-1a/V and HEC-1a/RIBO cells. Values are averages (±SEM, n = 6). Comparison by ANOVA, p<0.05 (B) Representative nude Balb/c mouse displaying HEC-1A/EMP2 and HEC-1A/V subcutaneous tumors. (C) Representative Balb/c nude mouse displaying HEC-1A/V and HEC-1A/RIBO subcutaneous tumors. At day 30, HEC-1A/EMP2 (D), HEC-1A/V (E), and HEC-1A/RIBO (F) tumors were excised, fixed, and stained by hematoxylin and eosin or EMP2. H/E Magnification, 40X; EMP2 Staining Magnification, 400X. Scale bar = 10 µM.
After 30 days, tumors were excised, fixed, and stained by hemotoxylin/eosin or for EMP2 expression by immunohistochemistry (Figure 1D–F). Hemotoxylin and eosin staining confirmed the size of the tumors, where HEC-1A/RIBO tumors were significantly smaller than HEC-1A/EMP2 and HEC-1A/V tumors. To correlate the change in tumor size with EMP2 expression in vivo, immunohistochemistry was performed. As expected, two and four fold higher levels of EMP2 were observed in HEC-1A/EMP2 tumors compared to HEC-1A/V and HEC-1A/RIBO tumors, respectively (Figure 1D–F).
EMP2 expression does not alter cellular proliferation
One possible mechanism by which EMP2 could contribute to the malignant phenotype of endometrial cancer cells would be through alterations in cell proliferation. In order to test this, we first characterized the growth kinetics of HEC-1A/EMP2, HEC-1A/V, and HEC-1A/RIBO cells. Equivalent numbers of cells were plated on day 1 and then manually counted over the next 7 days. No significant differences in total cell numbers were observed between the three cell lines (Figure 2A). In order to validate this result, a BrdU cell proliferation assay was performed. Cells were incubated from 0–24 hours with BrdU to monitor its incorporation in proliferating cells (Figure 2B). No statistical differences were found in the kinetics of proliferation between the three cell lines.
Figure 2. EMP2 expression and FAK/Src signaling promote wound healing.

(A) HEC-1A/EMP2, HEC-1A/V, and HEC-1A/RIBO cells were analyzed quantitatively for total cell numbers from 0–7 days or (B) BrdU incorporation after 2 or 24 hours. No significant differences in cellular proliferation or total cell numbers were observed between the three cell lines. The experiment was repeated at least 3 times with similar results. (C) HEC-1A/EMP2, HEC-1A/V, or HEC-1A/RIBO cells were grown in a monolayer and then a “wound” created. After 24 h, wound closure was measured. Experiments were performed at least three times, and the results averaged. Comparison by Student's t test, * p = 0.03; ** p = 0.05. (D) The same experiment was also performed on Ishikawa/EMP2 and Ishikawa/V. Comparison by Student's t test, * p = 0.02. (E) HEC-1A cells were grown to reach a confluent monolayer. Cells were incubated with the PP2, Dasatinib, Erlotinib, AKTi VIII, or a vehicle control, and a wound created. After 36 h, plates were imaged, and the percentage of wound closure was calculated. Values are averages (±SEM, n = 3). Comparison by Student's t test, * p<0.05. (F) The same experiment was also performed on Ishikawa cells. Values are averages (±SEM, n = 3). Comparison by Student's t test, * p<0.05.
EMP2 expression promotes cellular migration
In previous studies examining its expression in clinical endometrial cancer specimens, EMP2 correlated with invasive and more aggressive tumors [5]. To determine the contribution of EMP2 expression to endometrial cancer cell migration, HEC-1A/EMP2, HEC-1A/V, or HEC-1A/RIBO were grown to confluence. The cell layer was then wounded by using a sterile pipette tip, and images of the wound healing response were recorded over 48 hours. As shown in Figure 2C, HEC-1A/EMP2 cells exhibited a 33.5% increase in wound healing compared to HEC-1A/V (p = 0.03) and a 47.5% increase compared to HEC-1A/RIBO cells (p = 0.01). To confirm that this effect was not specific to HEC-1A cells, another endometrial cancer cell line was tested. Ishikawa cells were engineered to over express EMP2, and consistent with the previous results, Ishikawa/EMP2 exhibited a significant increase in wound healing over Ishikawa/V cells (Figure 2D; p = 0.02).
To determine the contribution of select signaling pathways on endometrial cancer cell migration, inhibitors were added to HEC-1A (Figure 2E) or Ishikawa cells (Figure 2F). Cells were then assayed for their response to migrate and close the wound. Both inhibition of Akt phosphorylation and EGFR using erlotinib had no significant effect on both HEC-1A and Ishikawa cell migration. In contrast, both the FAK-Src small molecule inhibitor PP2 and the Src family inhibitor dasatinib significantly inhibited migration. Importantly, minimal to no toxicity was observed for any of these inhibitors at the dosages indicated as determined by trypan blue exclusion (data not shown). These results suggested that activation of FAK and Src promoted migration in these cells.
EMP2 correlates with increased FAK and Src phosphorylation
Given the requirement for the FAK/Src pathway and EMP2 for migration of endometrial cancer cells, we investigated the relationship of these proteins in HEC-1A and Ishikawa cells. HEC-1A cells with modulated EMP2 levels were analyzed for EMP2, FAK, and Src phosphorylation. Total levels of FAK, Src, and β-Actin were used as controls. HEC-1A/EMP2 cells express a EMP2-GFP fusion protein at 48 kD and the endogenous protein at 20 kD. As expected, HEC-1A/RIBO cells reduced EMP2 expression by two-fold compared with the vector control. Increased expression of EMP2 resulted in an increase of activated FAK (phosphorylation at Y-576/577) as well as activated Src (phosphorylation at Y-416). Conversely, a reduction of EMP2 expression significantly reduced the expression of activated FAK and Src (Figure 3A). Similarly, Ishikawa cells with modulated EMP2 levels were analyzed for activated FAK and Src expression, and concordantly, upregulation of EMP2 promoted a two fold increase in the activation of FAK and Src (Figure 3B).
Figure 3. EMP2 promotes activated FAK and Src expression.

Expression of EMP2, 576/577 P-FAK, and 416 P-Src were assessed in (A) HEC-1A/EMP2, HEC-1A/V and HEC-1A/RIBO cells or (B) Ishikawa/EMP2 and Ishikawa/V cells. Semi-quantitative analysis of 576/577 P-FAK after correction for total FAK in both HEC-1A (C) and Ishikawa cells (D) from three independent experiments, respectively. β-actin expression was used as an additional loading control. Comparison by Student's t test, * p<0.05. EMP2 and β-actin panels of Figure 3A are excluded from this article's CC-BY license. See the accompanying retraction notice for more information.
EMP2 associates with FAK
In order to understand the relationship of FAK and EMP2 in endometrial cells, confocal microscopy was utilized to compare the localization of EMP2 and total FAK. In both HEC-1A and Ishikawa cells, EMP2 was expressed within the cytoplasm and at the plasma membrane (Figure 4A) [6], [23]. FAK displayed a similar localization pattern within cells, with intense cytoplasmic and membrane staining, and significant colocalization between EMP2 and FAK was observed. In HEC-1A cells, 34.5±1.3% of EMP2 colocalized with FAK (Figure 4A), and in Ishikawa cells, 42.6±8% colocalization was observed (data not shown).
Figure 4. Total FAK and EMP2 associate with each other.
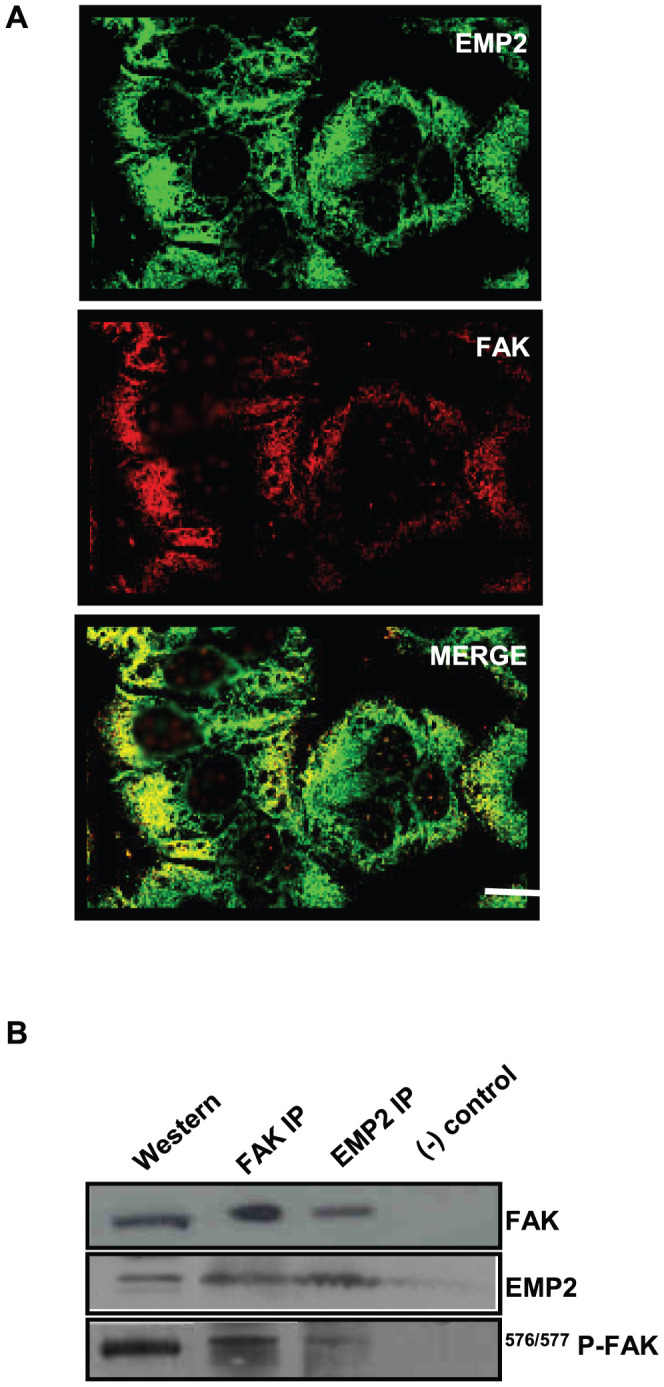
(A) Cellular images of HEC-1A cells. Cells were stained for EMP2 (FITC) and total FAK (Rhodamine) expression and imaged using confocal microscopy. EMP2 and FAK colocalize (yellow) in the cytoplasm and on the membrane of HEC-1A cells. Scale bar, 20 µM. (B) In order to assess if EMP2 and FAK immunoprecipitate together, HEC-1A cells were lysed in 1% NP-40 and an interaction assessed using immunoprecipitation/SDS-PAGE analysis. Both α-FAK antibodies and EMP2 antisera pulled down EMP2, FAK, and 576/577 p-FAK. Normal rabbit antisera served as the negative isotype control. Experiments were repeated three times with similar results; a representative image is displayed.
As microscopy experiments suggested that EMP2 and FAK resided within similar cellular compartments, we next determined if the two proteins associated with each other. Immunoprecipitations were preformed in wildtype HEC-1A cells using 1% Nonidet P-40 to prevent nonspecific hydrophobic interactions [24], [25]. Anti-EMP2 antisera pulled down both EMP2 and FAK (Figure 4B). Conversely, pull-down with anti-FAK antibodies resulted in detectable FAK and EMP2 (Figure 4B). We next determined the proportion of activated FAK that associated with EMP2. Approximately 35±3% of activated FAK immunoprecipitated with EMP2.
EMP2 colocalizes with activated FAK
Biochemical studies suggested that a subset of EMP2 immunoprecipitated with activated FAK. To confirm and extend this observation, the association of EMP2 and activated FAK was examined by confocal microscopy. Cells were stained for EMP2 (FITC) and activated FAK (Rhodamine). Approximately 30% of EMP2 in HEC-1A/EMP2 and 40% of EMP2 in Ishikawa/EMP2 colocalized with activated FAK.(Figure 5A, C). In HEC-1A/V and Ishikawa/V cells, a two fold reduction in colocalization was observed. As summarized in Figures 5B and D respectively, approximately 15 or 20% of EMP2 and activated FAK colocalized with HEC-1A/V and Ishikwawa/V cells, respectively. Finally, little colocalization (<5%) was observed between EMP2 and activated FAK in HEC-1A/RIBO cells.
Figure 5. EMP2 and activated FAK colocalize.

(A) Cellular images of HEC-1A/EMP2 cells stained for confocal microscopy using EMP2 antisera (FITC) and FAK (activated at tyrosine 397; Rhodamine). (B) Data from at least four separate samples were quantitated using Pascal software to calculate pixel intensity, and the resultant data was evaluated by Student's unpaired t-test. Overexpression of EMP2 demonstrated a two-fold increase in colocalization with p-FAK compared to vector control cells. Less than 5% colocalization was observed in HEC-1A/RIBO cells. Student's t test, * p = 0.01; ** p = 0.0001. Scale bar, 25 µM. (C) Ishikawa/EMP2 cells were stained using EMP2 antisera (FITC) and FAK (activated at tyrosine 397; Rhodamine) and analyzed by confocal microscopy. (D) Data from at least four separate samples were quantitated and analyzed as above. Student's t test, * p = 0.01. Scale bar, 25 µM.
As elevated EMP2 expression correlated with increased FAK activation, we next characterized the distribution of p-FAK within endometrial carcinoma cells. Increased EMP2 levels promoted activated FAK expression on the plasma membrane, and in contrast, the EMP2 specific ribozyme appeared to disrupt the organization of activated FAK (data not shown). HEC-1A/RIBO cells expressed little activated FAK on the plasma membrane, and instead, p-FAK was localized within intracellular compartments (data not shown).
EMP2 resides within lipid raft domains
Previous studies have shown that in NIH3T3 fibroblast cells, EMP2 localized within DIG membrane domains [7]. We thus hypothesized that EMP2 may create a functional signaling complex with activated FAK and Src within a DIG lipid raft membrane domain that regulated the migratory potential of endometrial cancer cells. To test this hypothesis, we initially confirmed the localization of EMP2 to lipid raft domains in wildtype HEC-1A cells. Using both 1% Brij-96 (Figure 6A) or 1% Triton X-100 (Figure 6B), EMP2 localized to the lipid fractions identified by a common raft protein ganglioside GM1 [26]. Cholesterol depletion of rafts using methyl-β cyclodextrin (MβCD) redistributed EMP2 into the non-raft soluble fractions.
Figure 6. EMP2 forms a complex with FAK and Src in DIG lipid raft membrane domains.

(A) EMP2 expression in HEC-1A lipid raft membrane domains was verified by Brij-58 insolubility. Cells were lysed in 1% Brij-58 and centrifuged in a sucrose density gradient. Nine fractions (500 µl each) were collected from the top of the gradient and tested for GM1 by a cholera toxin-HRP dot blot and for EMP2 (∼Mr 20 kDa) by using SDS-PAGE and Western blot analysis. (B) To verify the cholesterol dependence of EMP2 expression within the lipid raft. HEC-1A cells were preincubated in the presence (+) or absence (–) of MβCB. Cells were lysed in 1% Triton X-100, gradient fractionated, and EMP2 expression detected by Western blot analysis. (C) HEC-1A/EMP2, HEC-1A/V, and HEC-1A/RIBO cells were lysed in 1% Triton X-100 and centrifuged as above. Samples were probed by Western blot analysis for EMP2, activated FAK, activated Src, total FAK, total Src, Flotillin-2, and EEA1. Experiments were performed independently three times with similar results.
We next determined if modulation of EMP2 expression altered the distribution of Src, FAK, or their activated forms into lipid raft domains (Figure 6C). In both HEC-1A/EMP2 and HEC-1A/V, the distribution of total FAK and Src was similar, with a significant localization within lipid raft fractions (fractions 2–5) as well as in the 1% Triton X-100 detergent soluble fractions (fractions 6–9). In contrast, decreased levels of total FAK and Src were present in lipid raft fractions in HEC-1A/RIBO cells. Similar results were observed with the activated forms of these proteins. Upregulation of EMP2 expression resulted in increased localization of activated FAK and Src in lipid rafts. In contrast, reduced EMP2 expression decreased reduced the levels of activated FAK and eliminated all activated Src within lipid domains. The redistribution of the FAK-Src signaling complex appeared to be specific as EMP2 did not alter the distribution of a known raft protein Flotillin-2 or the detergent soluble endosome protein EEA1 [27], [28].
Discussion
The tetraspan protein EMP2 has been implicated as a prognostic and survival marker for endometrial cancer [4], [5]. In this study, we examine the tumor growth of endometrial carcinoma cells with increased or reduced EMP2 expression to determine the contribution of EMP2 to endometrial cancer tumorigenicity. Dramatically, HEC-1A tumors with increased EMP2 expression (HEC-1A/EMP2) exhibit a two fold increase in tumor volume compared to HEC-1A/V and a three fold increase compared to cells with reduced EMP2 expression (HEC-1A/RIBO). Mechanistically, we have started to characterize how EMP2 expression affects endometrial tumor progression. We demonstrate that EMP2 is sufficient to activate FAK and its signal transduction cascade. This effect may be direct as EMP2 and FAK associate with each other, and a significant proportion of activated FAK and Src reside within an EMP2-lipid raft complex.
Intact lipid structures have been shown to play critical roles for proper signal transduction and migration [29], [30]. DIG membrane microdomains are enriched in cholesterol and sphingolipids and make up two types of membrane compartments. The first are flask-shaped invaginations called caveolae which contain caveolin-1 [31], and the second type are flat domains which lack caveolins called lipid rafts [32]. Endometrial epithelial cells do not have caveolae as endometrial epithelial cells lack expression of both caveolin-1 and caveolin-2 (Wadehra and Braun, unpublished results). Our data supports the conclusion that EMP2 complexes with FAK and Src leading to enhanced protein phosphorylation, and that this association occurs within lipid raft microdomains. We predict that EMP2 expression is necessary to traffic and/or stabilize FAK and Src phosphorylation within a lipid raft domain with the functional consequence of this interaction being improved cell migration.
FAK acts as a receptor-proximal protein that bridges the growth-factor-receptor and integrin signaling pathways [33]. A previous study has shown that in endometrial epithelial cells, EMP2 regulates αvβ3 integrin expression [6], and it is known that in epithelial cells β3 integrin activation directly leads to Src and FAK phosphorylation resulting in stable focal adhesions [34], [35]. Analysis of EMP2-β3 integrin-FAK association data suggest that in the endometrium EMP2 may act as a molecular adaptor for efficient integrin-mediated FAK activation. In this way, we predict that EMP2 works to link select integrin isoforms and their associated signaling modules within DIG lipid raft membrane domains [25], [36].
This is similar to the role proposed for other members of the tetraspan superfamily as several groups have hypothesized that tetraspanins create a scaffold to regulate signaling, trafficking, and structural characteristics of their membrane protein constituents [37], [38]. Tetraspanins which have been shown to participate in the formation of a variety of signaling complexes important for integrin signaling, B-cell receptor signaling, and fertilization [24], [25]. With regard to integrin signaling, for example, it has been shown that the interaction of CD151 with integrin α3β1 helps modulate cell adhesion and motility on the extracellular matrix laminin [39], [40], suggesting that the GAS-3 and tetraspanins families of proteins may have more in common than was previously appreciated. Interestingly, tetraspanins are not found in a lipid raft domains [41]. Thus, it may be that GAS-3 proteins such as EMP2 help organize lipid raft domain signaling while the tetraspanins organize non-raft domain signaling.
EMP2 is an early prognostic biomarker in endometrial hyperplasia, and high expression of EMP2 in endometrial cancer is associated with a poor prognosis. Similarly, we would predict that in endometrial cancer, β3 integrin expression will be upregulated with a concomitant increase in FAK and Src activation. Clinical data supports this hypothesis. First, β3 integrin expression has been shown to be upregulated in a number of cancers, including endometrial [42]. Second, activation of FAK and Src kinase has been documented in the progression of benign to malignant endometrium [42], [43]. We predict that a better understanding of the mechanism of EMP2 may help predict endometrial cancer incidence, prognosis and treatment.
Funding Statement
The authors acknowledge the following agencies who have helped support this work: National Institutes of Health (NIH) grants HD48540 (J. Braun), R21 CA131756 (M. Wadehra), CA016042 (University of California at Los Angeles Jonsson Comprehensive Cancer Center flow cytometry core); Early Detection Research Network NCI CA86366 (L. Goodglick); American Cancer Society # RSG-03-160-01-LIB (L. Goodglick). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Creasman WT, Odicino F, Maisonneuve P, Beller U, Benedet JL, et al. Carcinoma of the corpus uteri. J Epidemiol Biostat. 2001;6:47–86. [PubMed] [Google Scholar]
- 2.Denschlag D, Tan L, Patel S, Kerim-Dikeni A, Souhami L, et al. Stage III endometrial cancer: preoperative predictability, prognostic factors, and treatment outcome. Am J Obstet Gynecol. 2007;196:546 e541–547. doi: 10.1016/j.ajog.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 3.Gossett DR, Alo P, Bristow RE, Galati M, Kyshtoobayeva A, et al. Inability of immunohistochemistry to predict clinical outcomes of endometrial cancer patients. IntJ GynecolCancer. 2004;14:145–151. doi: 10.1111/j.1048-891x.2004.014028.x. [DOI] [PubMed] [Google Scholar]
- 4.Habeeb O, Goodglick L, Soslow RA, Rao RG, Gordon LK, et al. Epithelial membrane protein-2 expression is an early predictor of endometrial cancer development. Cancer. 2010;116:4718–4726. doi: 10.1002/cncr.25259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wadehra M, Natarajan S, Seligson DB, Williams CJ, Hummer AJ, et al. Expression of epithelial membrane protein-2 is associated with endometrial adenocarcinoma of unfavorable outcome. Cancer. 2006;107:90–98. doi: 10.1002/cncr.21957. [DOI] [PubMed] [Google Scholar]
- 6.Wadehra M, Forbes A, Pushkarna N, Goodglick L, Gordon LK, et al. Epithelial membrane protein-2 regulates surface expression of alphavbeta3 integrin in the endometrium. DevBiol. 2005;287:336–345. doi: 10.1016/j.ydbio.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 7.Wadehra M, Goodglick L, Braun J. The tetraspan protein EMP2 modulates the surface expression of caveolins and glycosylphosphatidyl inositol-linked proteins. Mol Biol Cell. 2004;15:2073–2083. doi: 10.1091/mbc.E03-07-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wadehra M, Su H, Gordon LK, Goodglick L, Braun J. The tetraspan protein EMP2 increases surface expression of class I major histocompatibility complex proteins and susceptibility to CTL-mediated cell death Clinical Immunology. 2003;107:129–136. doi: 10.1016/s1521-6616(03)00048-2. [DOI] [PubMed] [Google Scholar]
- 9.Lajoie P, Goetz JG, Dennis JW, Nabi IR. Lattices, rafts, and scaffolds: domain regulation of receptor signaling at the plasma membrane. J Cell Biol. 2009;185:381–385. doi: 10.1083/jcb.200811059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patra SK. Dissecting lipid raft facilitated cell signaling pathways in cancer. Biochim Biophys Acta. 2008;1785:182–206. doi: 10.1016/j.bbcan.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 11.Poveda JA, Fernandez AM, Encinar JA, Gonzalez-Ros JM. Protein-promoted membrane domains. Biochim Biophys Acta. 2008;1778:1583–1590. doi: 10.1016/j.bbamem.2008.01.021. [DOI] [PubMed] [Google Scholar]
- 12.Morales SA, Mareninov S, Coulam P, Wadehra M, Goodglick L, et al. Functional consequences of interactions between FAK and epithelial membrane protein 2 (EMP2). Invest Ophthalmol Vis Sci. 2009;50:4949–4956. doi: 10.1167/iovs.08-3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morales SA, Mareninov S, Wadehra M, Zhang L, Goodglick L, et al. FAK activation and the role of epithelial membrane protein 2 (EMP2) in collagen gel contraction. Invest Ophthalmol Vis Sci. 2009;50:462–469. doi: 10.1167/iovs.07-1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimazaki K, Wadehra M, Forbes A, Chan AM, Goodglick L, et al. Epithelial membrane protein 2 modulates infectivity of Chlamydia muridarum (MoPn). Microbes Infect. 2007;9:1003–1010. doi: 10.1016/j.micinf.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 15.Agus DB, Scher HI, Higgins B, Fox WD, Heller G, et al. Response of Prostate Cancer to Anti-Her-2/neu Antibody in Androgen-dependent and -independent Human Xenograft Models. Cancer Res. 1999;59:4761–4764. [PubMed] [Google Scholar]
- 16.Brey EM, Lalani Z, Johnston C, Wong M, McIntire LV, et al. Automated selection of DAB-labeled tissue for immunohistochemical quantification. J Histochem Cytochem. 2003;51:575–584. doi: 10.1177/002215540305100503. [DOI] [PubMed] [Google Scholar]
- 17.Wadehra M, Iyer R, Goodglick L, Braun J. The tetraspan protein epithelial membrane protein-2 interacts with beta1 integrins and regulates adhesion. J Biol Chem. 2002;277:41094–41100. doi: 10.1074/jbc.M206868200. [DOI] [PubMed] [Google Scholar]
- 18.Acconcia F, Barnes CJ, Kumar R. Estrogen and tamoxifen induce cytoskeletal remodeling and migration in endometrial cancer cells. Endocrinology. 2006;147:1203–1212. doi: 10.1210/en.2005-1293. [DOI] [PubMed] [Google Scholar]
- 19.Morales S, Mareninov S, Prasad P, Wadehra M, Braun J, et al. Collagen Gel Contraction by ARPE-19 is mediated by a FAK-Src dependent pathway. Experimental Eye Research. 2007;85:790–798. doi: 10.1016/j.exer.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 20.Barnett SF, Defeo-Jones D, Fu S, Hancock PJ, Haskell KM, et al. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem J. 2005;385:399–408. doi: 10.1042/BJ20041140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang MY, Lu KV, Zhu S, Dia EQ, Vivanco I, et al. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer Res. 2006;66:7864–7869. doi: 10.1158/0008-5472.CAN-04-4392. [DOI] [PubMed] [Google Scholar]
- 22.Lu KV, Zhu S, Cvrljevic A, Huang TT, Sarkaria S, et al. Fyn and SRC are effectors of oncogenic epidermal growth factor receptor signaling in glioblastoma patients. Cancer Res. 2009;69:6889–6898. doi: 10.1158/0008-5472.CAN-09-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wadehra M, Mainigi M, Morales SA, Rao RG, Gordon LK, et al. Steroid hormone regulation of EMP2 expression and localization in the endometrium. Reprod Biol Endocrinol. 2008;6:15. doi: 10.1186/1477-7827-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levy S, Shoham T. The tetraspanin web modulates immune-signalling complexes. NatRevImmunol. 2005;5:136–148. doi: 10.1038/nri1548. [DOI] [PubMed] [Google Scholar]
- 25.Maecker HT, Todd SC, Levy S. The tetraspanin superfamily: molecular facilitators. FASEB J. 1997;11:428–442. [PubMed] [Google Scholar]
- 26.Naroeni A, Porte F. Role of cholesterol and the ganglioside GM(1) in entry and short-term survival of Brucella suis in murine macrophages. Infect Immun. 2002;70:1640–1644. doi: 10.1128/IAI.70.3.1640-1644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salzer U, Prohaska R. Stomatin, flotillin-1, and flotillin-2 are major integral proteins of erythrocyte lipid rafts. Blood. 2001;97:1141–1143. doi: 10.1182/blood.v97.4.1141. [DOI] [PubMed] [Google Scholar]
- 28.Cortese K, Sahores M, Madsen CD, Tacchetti C, Blasi F. Clathrin and LRP-1-independent constitutive endocytosis and recycling of uPAR. PLoS One. 2008;3:e3730. doi: 10.1371/journal.pone.0003730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang C, Yoo Y, Fan H, Kim E, Guan KL, et al. Regulation of Integrin beta 1 recycling to lipid rafts by Rab1a to promote cell migration. J Biol Chem. 2010;285:29398–29405. doi: 10.1074/jbc.M110.141440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baillat G, Siret C, Delamarre E, Luis J. Early adhesion induces interaction of FAK and Fyn in lipid domains and activates raft-dependent Akt signaling in SW480 colon cancer cells. Biochim Biophys Acta. 2008;1783:2323–2331. doi: 10.1016/j.bbamcr.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 31.Hakomori SI. Glycosynaptic microdomains controlling tumor cell phenotype through alteration of cell growth, adhesion, and motility. FEBS Lett. 2010;584:1901–1906. doi: 10.1016/j.febslet.2009.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park EK, Park MJ, Lee SH, Li YC, Kim J, et al. Cholesterol depletion induces anoikis-like apoptosis via FAK down-regulation and caveolae internalization. J Pathol. 2009;218:337–349. doi: 10.1002/path.2531. [DOI] [PubMed] [Google Scholar]
- 33.Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, et al. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 34.Berditchevski F, Odintsova E. Characterization of integrin-tetraspanin adhesion complexes: role of tetraspanins in integrin signaling. JCell Biol. 1999;146:477–492. doi: 10.1083/jcb.146.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawaguchi T, Yamashita Y, Kanamori M, Endersby R, Bankiewicz KS, et al. The PTEN/Akt pathway dictates the direct alphaVbeta3-dependent growth-inhibitory action of an active fragment of tumstatin in glioma cells in vitro and in vivo. Cancer Res. 2006;66:11331–11340. doi: 10.1158/0008-5472.CAN-06-1540. [DOI] [PubMed] [Google Scholar]
- 36.Yunta M, Lazo PA. Tetraspanin proteins as organisers of membrane microdomains and signalling complexes. Cell Signal. 2003;15:559–564. doi: 10.1016/s0898-6568(02)00147-x. [DOI] [PubMed] [Google Scholar]
- 37.Kobayashi T, Vischer UM, Rosnoblet C, Lebrand C, Lindsay M, et al. The tetraspanin CD63/lamp3 cycles between endocytic and secretory compartments in human endothelial cells. MolBiolCell. 2000;11:1829–1843. doi: 10.1091/mbc.11.5.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berditchevski F. Complexes of tetraspanins with integrins: more than meets the eye. J Cell Sci. 2001;114:4143–4151. doi: 10.1242/jcs.114.23.4143. [DOI] [PubMed] [Google Scholar]
- 39.Stipp CS. Laminin-binding integrins and their tetraspanin partners as potential antimetastatic targets. Expert Rev Mol Med. 2010;12:e3. doi: 10.1017/S1462399409001355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zevian S, Winterwood NE, Stipp CS. Structure-Function Analysis of Tetraspanin CD151 Reveals Distinct Requirements for Tumor Cell Behaviors Mediated by α3{beta}1 versus α6{beta}4 Integrin. J Biol Chem. 2011;286:7496–7506. doi: 10.1074/jbc.M110.173583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hemler ME. Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol. 2005;6:801–811. doi: 10.1038/nrm1736. [DOI] [PubMed] [Google Scholar]
- 42.Park DW, Ryu HS, Choi DS, Park YH, Chang KH, et al. Localization of matrix metalloproteinases on endometrial cancer cell invasion in vitro. Gynecol Oncol. 2001;82:442–449. doi: 10.1006/gyno.2001.6294. [DOI] [PubMed] [Google Scholar]
- 43.Desouki MM, Rowan BG. SRC kinase and mitogen-activated protein kinases in the progression from normal to malignant endometrium. Clin Cancer Res. 2004;10:546–555. doi: 10.1158/1078-0432.ccr-0661-03. [DOI] [PubMed] [Google Scholar]