

Abstract
Cells within Bacillus subtilis biofilms are held in place by an extracellular matrix that contains cell-anchored amyloid fibers, composed of the amyloidogenic protein TasA. As biofilms age they disassemble because the cells release the amyloid fibers. This release appears to be the consequence of incorporation of D-tyrosine, D-leucine, D-tryptophan and D-methionine into the cell wall. Here, we characterize the in vivo roles of an accessory protein TapA (TasA anchoring/assembly protein; previously YqxM) that serves both to anchor the fibers to the cell wall and to assemble TasA into fibers. TapA is found in discrete foci in the cell envelope and these foci disappear when cells are treated with a mixture of D-amino acids. Purified cell wall sacculi retain a functional form of this anchoring protein such that purified fibers can be anchored to the sacculi in vitro. In addition, we show that TapA is essential for the proper assembly of the fibers. Its absence results in a dramatic reduction in TasA levels and what little TasA is left produces only thin fibers that are not anchored to the cell.
Keywords: B. subtilis, biofilm, amyloid, TasA, matrix
Introduction
When presented with a surface or an interface, bacteria will often grow as biofilms in which cells are held together by an extracellular matrix. For Bacillus subtilis this matrix is primarily composed of an exopolysaccharide and the amyloid protein TasA (Romero et al., 2010, Branda et al., 2006, Branda et al., 2001). Amyloid proteins are characterized by their ability to acquire a secondary structure enriched in β-sheets that facilitates the formation of extremely stable fibers (Kayed et al., 2003, Sunde et al., 1997). While originally studied in the context of human pathologies, amyloid fibers are now recognized as ubiquitous extracellular structures in the microbial world, where they have functional roles (Maury, 2009, Fowler et al., 2007). These roles include mediating interactions with biotic and abiotic surfaces, raising aerial structures and providing structural integrity to the biofilm matrix (Barnhart & Chapman, 2006, Badtke et al., 2009, Claessen et al., 2002, Collinson et al., 1993, Gebbink et al., 2005, Oh et al., 2007).
Because of their importance to bacterial physiology and their possible relevance as model systems for the study of human disease, the study of bacterial amyloid fibers has become an active area of research (Greenwald & Riek, 2010). While there is still much to understand about how amyloid fibers form and how they attach to the cell surface, some progress has been made by studying amyloid proteins produced by model bacteria. Polymerization of monomeric soluble subunits into complex and insoluble amyloid fibers can be aided by dedicated accessory proteins (Ferrone, 1999, Kayed et al., 2003, Tompa, 2009, Epstein & Chapman, 2008). This is the case for the curli fibers of Escherichia coli where the major structural subunit CsgA needs the accessory protein CsgB to polymerize in vivo (Chapman et al., 2002, Barnhart & Chapman, 2006, Hammar et al., 1996, Hammer et al., 2007). Streptomyces coelicolor also produces amyloid fibers. In this bacterium the fibers are composed of eight different chaplins, three of which can be covalently attached to the cell wall through the action of a sortase (Claessen et al., 2006, Claessen et al., 2003). Recently, it was found that some of the chaplins interact with cellulose in order to attach to the cell wall (de Jong et al., 2009). To date no nucleating factor for the formation of S. coelicolor fibers has been identified but proteins known as rodlins appear to facilitate the assembly process (de Jong et al., 2009).
The TasA amyloid fibers provide structural integrity to B. subtilis biofilms (Romero et al., 2010). The fibers are anchored to cells and form a robust protein scaffold that holds the cells together. However, as biofilms senesce they gradually fall apart because the fibers are released from the cell. Recent results revealed some aspects of the mechanism of this disassembly. At late stages in a biofilm life cycle, cells begin producing a mixture of D-amino acids that includes D-Tyr, D-Met, D-Leu and D-Trp. As these amino acids are incorporated into the peptidoglycan the TasA amyloid fibers are released from the cells, leading to biofilm disassembly (Kolodkin-Gal et al., 2010). Thus, it appears that the amyloid fibers are anchored to the cell wall and that this anchoring can be disrupted by incorporating unusual D-amino acids. Importantly, we obtained mutants that were resistant to the disassembling effect of D-Tyr (Kolodkin-Gal et al., 2010). These mutants carried altered forms of a protein, YqxM, that we had previously shown to be important for placing TasA in the extracellular matrix (Branda et al., 2006).
TasA is encoded by tasA, the third gene in a three-gene operon. The two other proteins encoded in this operon are also involved in the process of amyloid fiber formation. The second gene encodes SipW, a signal sequence peptidase with two known substrates. One substrate is TasA (Stover & Driks, 1999b, Tjalsma et al., 1998). The other substrate is the first gene product of the operon. Up to now we have referred to that first gene product as YqxM, based on the prior “unknown function” designation of its gene product. Indeed, this is the protein involved in placing TasA in the matrix and mutants of which yield D-Tyr resistant biofilms (Kolodkin-Gal et al., 2010, Branda et al., 2006). Here we show that this protein is required for the proper anchoring and polymerization of TasA at the cell surface. Furthermore, we show that much of this protein is found in discrete foci on the cell surface and that these foci disappear upon treatment with D-amino acids. In addition, we found that this protein is a minor component of the amyloid fibers and that in cells lacking it, TasA appears to be degraded. Thus, this protein has multiple functions. It serves to anchor the amyloid fibers to the cell as well as to nucleate fiber polymerization and is a component of the fibers. Therefore, we name it TapA for TasA anchoring/assembly protein and refer to it as such throughout this paper.
Results
TapA and TasA must be synthesized in the same cell to produce functional amyloid fibers
B. subtilis NCIB3610 forms floating biofilms, known as pellicles, at the liquid-air interface of standing liquid cultures (Branda et al., 2006). When fully formed, pellicles of wild-type cells are characterized by robust wrinkling (Fig. 1A, WT). This phenotype requires the expression of two operons: epsA-O (henceforth referred to simply as eps) and tapA-sipW-tasA (formerly referred to as the yqxM-sipW-tasA operon). The products of these operons form the two major components of the biofilm matrix: the exopolysaccharide and the TasA amyloid fibers (Branda et al., 2006, Branda et al., 2001). The fragile pellicles of eps mutants are clearly different from the flat pellicles of tasA and tapA mutants (Fig. 1A). Previously, we showed that extracellular complementation of biofilm formation occurs when eps and tasA mutants are mixed (Branda et al., 2006). We repeated these extracellular complementation experiments and added the observation that mixing of eps and tapA mutants yielded a wild-type pellicle as well (Fig. 1B, tasA + eps and tapA + eps). However, no complementation was obtained when tapA and tasA mutants were mixed (Fig. 1B, tapA + tasA). These data suggest that the TapA and TasA proteins must be produced in the same cell in order to assemble functional fibers. An alternative possibility is that the TasA fibers from tapA mutants are not attached to cells and are therefore diluted in the pellicle medium. We therefore also assessed extracellular complementation of the tapA and tasA mutants on agar surfaces and were unable to observe complementation as determined by colony wrinkling and Congo Red binding (Fig. S1). Thus, we favor the hypothesis that the two proteins must be produced in the same cell to form functional amyloids.
Figure. 1. tasA and tapA mutants do not complement each other extracellularly.

A) Pellicle formation of wild-type and mutant strains. B) Pellicles from two strains mixed at a 1:1 ratio, designated with a “+” between the two genotypes. Pellicles were grown in MSgg medium for 24 h prior to imaging.
Punctate localization of TapA on the cell wall
The finding that TapA and TasA must be produced in the same cell to yield wild-type biofilms prompted us to initiate TapA localization studies. In the study showing that different D-amino acids incorporated into the cell wall led to biofilm disassembly, we obtained mutant forms of TapA that resulted in biofilms that were resistant to D-tyrosine (Kolodkin-Gal et al., 2010). This provided initial indirect evidence that TapA was somehow associated with the cell wall. The fluorescence and electron microscopy results we present now further suggest that TapA is a cell-wall associated protein. Using anti-TapA antibodies and a FITC-conjugated secondary antibody it was possible to visualize TapA clusters in wild-type cells from pellicles after 24 hours of growth, when pellicles have reached maturity. Interestingly, most of the TapA is observed as puncta located on the cell surface (Fig. 2A). Treatment of wild-type pellicles with 3 μM D-Tyr led to the disappearance of the fluorescent puncta (Fig. 2B). In a prior work, we had shown that treatment with D-Tyr does not alter transcription of the tapA operon (Kolodkin-Gal et al., 2010). Rather, D-Tyr treatment leads to the release of the fibers from the cells (Kolodkin-Gal et al., 2010). Thus it appears that loss of puncta correlates with release of fibers. Importantly, puncta remained after treatment with 3 μM D-Tyr of a strain bearing a D-Tyr-resistant mutation in tapA (Fig. 2C) and no puncta were visible in a tapA mutant (Fig. 2D).
Figure. 2. Localization of TapA is punctate.

A-D) Immunocytochemistry with Anti-TapA antibody and FITC-conjugated secondary antibody performed on intact cells from 24 hr pellicles grown in MSgg broth. A) Wild-type cells show foci of TapA. B) Wild-type cells grown in the presence of 3 μM D-Tyr C) Strain IKG44, a spontaneous mutant resistant to D-amino acids, grown in the presence of 3 μM D-Tyr and D) tapA mutant cells, that do not have signal. E and F) Electron micrographs of thin sections of resin-embedded wild-type cells after 24 hr growth and immunogold labeling with anti-TapA antibody. E) Wild-type. Arrow points to the accumulation of gold particles in the electron-dense cell wall. F) tapA mutant does not show any signal. Scale bars, 2 μm in panels AD and 100 nm in panels E and F.
The results discussed above suggest an association of TapA with the peptidoglycan, but they are insufficient to show that the two macromolecules interact physically. However, two additional lines of evidence point to the existence of a close association between TapA and peptidoglycan. First, immunogold electron microscopic analyses revealed large concentrations of TapA at foci on the peptidoglycan (Fig. 2E). Second, cell fractionation studies localized a functional TapA-YFP fusion protein to the peptidoglycan fraction and not on the protoplast fraction (Figs. S2 and S3). Altogether, the results indicate that TapA is closely associated with the peptidoglycan. This led us to ask whether TapA co-purifies with peptidoglycan.
TapA is associated with purified sacculi and retains functionality
To obtain purified peptidoglycan sacculi, wild type, tasA and tapA mutants were grown for 24 h in pellicle biofilms and then subjected to several washes with a solution of SDS and β-mercaptoethanol and high temperatures to dissolve the cellular content (Fig. 3). To determine if the purified sacculi retained TapA, we analyzed the samples by TEM and immunogold labeling. Strikingly, despite the harsh treatment, TapA remained associated to the sacculi (Fig. 3). Importantly, only sacculi from wild type and the tasA mutant were decorated with anti-TapA antibodies. No signal was associated to sacculi purified from a tapA mutant (Fig. 3A). We previously demonstrated that purified TasA fibers added to a tasA mutant became cell associated and restored the formation of biofilms (Romero et al., 2010). We therefore wondered whether TapA in purified sacculi would retain the ability to associate with TasA fibers. To test this, a solution of purified TasA fibers was mixed with the different sacculi and incubated for 10 h at room temperature. Excess TasA was removed by centrifugation and the samples were suspended in PBS prior to TEM and immunogold labeling with anti-TasA antibody (Fig. 3B). Electron microscopy showed that the added TasA fibers were tightly associated with sacculi from wild type and tasA mutant cells but not with those of a tapA mutant (see Fig. 3B top panels with the bottom panels showing close-up views). Quantification of the gold-labeled particles showed that the tapA mutant sacculi bound about 10% of the fibers relative to the wild-type sacculi (Fig. S4A). As a complement to this experiment, we used western analysis with anti-TasA antibody to quantify the amount of TasA that remained associated with the sacculi in the above experiment. Using these analyses, we found the tapA mutant sacculi bound about 16 × fewer TasA fibers than the wild-type sacculi (Fig. S4B). Thus, we conclude that TapA is present in the peptidoglycan where it functions as an anchor point for TasA fibers. Moreover, TapA retains functionality as an anchor even after isolation of sacculi.
Figure. 3. TapA is associated with the cell wall.

Transmission electron micrographs and immunolabelling of B. subtilis sacculi. A) Anti-TapA antibody. Arrows point to gold particles present in wild-type and tasA mutant cells that are absent in the tapA mutant. B) Fibers of TasA (+ TasA) purified from B. subtilis were added to sacculi from the designated strains and immunogold labeling was performed with anti-TasA antibody. Bottom panel is a magnified view of the boxes marked in panel B. Scale bars, 200 nm.
TapA is a minor component of TasA fibers in the extracellular matrix
While the above experiments indicate that TapA is present in the peptidoglycan, they do not rule out the possibility that the protein might also be present in the extracellular matrix. To determine if TapA is present in the matrix, we separated the biofilm into medium (Med), cell and matrix (Mat) fractions and performed immunoblot analyses with anti-TapA antibody. Because biofilm formation is dynamic, we analyzed samples collected over time at a constant incubation temperature (30°C). Under these conditions pellicles reached maturity at 24 hr and by 48 hr began to disassemble. Results of these experiments are shown in Fig. 4. At 24 hr, two TapA-related bands were detected in both cells (Cell) and matrix (Mat); no TapA-related proteins were detected in the medium (Med). The larger band corresponded to 28 kDa, in close agreement with the predicted mass of secreted TapA, whose signal peptide has been cleaved off. The small band, corresponding to 24 kDa, probably represents a further processing of TapA. Importantly, neither of the two bands was detected in the tapA mutant (Fig. 4). After 48 hr of incubation, when disassembly was just beginning, only the smaller 24 kDa band remained in the extracellular matrix (Mat) and no signal could be detected in the cell fraction. Thus, TapA localization varies during biofilm development. In addition, it appears that TapA undergoes processing beyond the removal of the signal peptide and that this processed form of the protein remains matrix-associated at 48 hr of biofilm development. A similar processed product of TapA was observed previously in shaken cultures, before there was knowledge of its involvement in the production of an extracellular matrix (Stover & Driks, 1999a).
Figure. 4. TapA localizes to the cell and the matrix.

Biochemical fractionation of wild-type and tapA pellicles analyzed by immunoblot with anti-TapA antibody. Pellicles of cells grown in MSgg medium after A) 24 hr and B) 48 hr were mildly sonicated and separated into three fractions: medium (Med), cell and matrix (Mat). Each fraction was concentrated prior to analysis by SDS-PAGE.
Finding TapA present in the matrix led us to ask whether, similar to EPS and TasA, TapA might also be a structural component of the matrix. We hypothesized that TapA might be incorporated into the TasA fibers similar to CsgB in Curli fibers or other minor pilin proteins in pili of Gram-positive bacteria (Kline et al., 2010, Bian & Normark, 1997). To test this hypothesis, we carried out immunogold electron microscopy using gold particles of different diameters to simultaneously detect TapA and TasA in biofilm samples. Initial microscopy studies were done with strain DR4, which harbors a non-polar mutation in tapA and a functional TapA-YFP translational fusion (fusion functionality is demonstrated in supplemental figure Fig. S2). Biofilms of this strain were analyzed after 24 hr of growth by transmission electron microscopy (TEM) and coimmunogold labeling with anti-TasA and anti-YFP antibodies (Fig. 5). For these localization experiments, gold particles of 10 nm were used to detect TasA and gold particles of 15 nm were used to detect TapA-YFP. Indeed, fibers were decorated with both antibodies, indicating that TapA is incorporated into the TasA fibers (Fig. 5). Negative controls were performed with the anti-YFP antibody on wild type samples that did not harbor any YFP fusion and little to no signal was detected, indicating that the YFP antibody does not react non-specifically with the cells (data not shown).
Figure. 5. TapA and TasA localize to fibers.
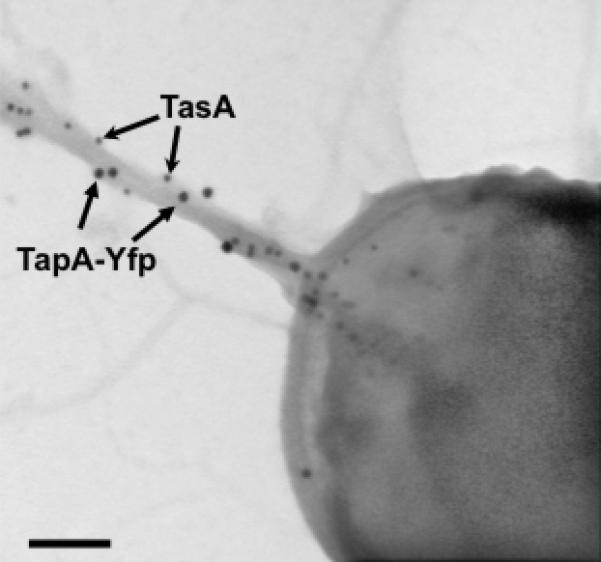
Electron micrograph of negatively stained, immunogold labeled samples from pellicles of strain DR4 after 24 hr of incubation in MSgg medium at 30°C. Two sizes of gold particles were used to detect TasA (10 nm) or YFP (15 nm). Arrows point to different sized gold particles. Scale bar, 100 nm.
The finding of TapA in the amyloid fibers was surprising because prior analyses had suggested that the fibers were composed largely of TasA. When fiber preparations were subjected to SDS-PAGE there was only one band visible - TasA - after Coomasie Blue staining (see Fig. S2 in (Romero et al., 2010)). We repeated those immunoblot experiments but now with both anti-TasA and anti-TapA antibodies. TapA was only detected in western blot analysis of purified TasA fibers after treatment with formic acid and a 200-fold concentration of the sample (see Fig. S5A). Interestingly, the size of the reacting band coincided with the molecular weight of TapA present in the matrix in fractionation experiments (see Fig. 4). We quantified the ratio of TasA to TapA in fibers purified from B. subtilis by performing quantitative immunoblot experiments using purified recombinant His6-TasA and His6-TapA as standards (Fig. S5B). We calculated that the ratio of TasA to TapA monomers was approximately 100:1.
A tapA mutant produces fewer and altered fibers
The findings presented thus far show that (i) TapA and TasA must be made in the same cell to be functional, (ii) TapA accumulates as a focus in the cell wall, and (iii) TapA is a minor component of the amyloid fibers. All of these data are consistent with the hypothesis that TapA might be a nucleator for TasA polymerization. To begin to explore this possibility, we examined the fate of TasA in a strain harboring a non-polar tapA mutation. We used TEM and immunogold labeling with anti-TasA antibodies to analyze wild-type and tapA mutant biofilms after 24 h of growth. In wild-type biofilms we observed TasA forming large fibers anchored to cells (Fig. 6A). In contrast, in the tapA mutant, most cells showed anti-TasA signal near the surface in the cytosol (Fig. 6B) and only rarely did we observe a few disorganized fibrils decorated with anti-TasA antibody and these were invariably dissociated from cells (Fig. 6C). In addition, we examined the amyloid properties of the residual TasA fibrils found in the tapA mutant. When grown on solid medium containing the amyloid binding dye Congo Red, the wild-type colonies stained red whereas the mutant colonies were brown, suggesting a lack of amyloids (Fig. 7A). Next, we subjected the tapA mutant to the same 1 M NaCl extraction procedure that we routinely use to prepare TasA fibers from wild-type cells (Romero et al., 2010). We examined the extract for its ability to bind the amyloid-binding dye Congo Red and measured the absorption at four wavelengths from 400 nm to 650 nm (Fig. 7B). The extract from wild-type cells shows a characteristic peak at 540 nm. This peak was much reduced in the tapA mutant, resembling more the sample that contained Congo Red alone. Together these data suggest that while a few TasA fibers polymerize in the absence of TapA, these are thin and short, and do not appear to retain the amyloid properties characteristic of the fibers formed by wild-type cells. Furthermore, as seen in the flat pellicle in Fig. 1, the fibers produced in the tapA mutant were incapable of forming wrinkled biofilms.
Figure. 6. TapA is required for TasA fiber formation.

Electron micrographs of negatively stained, anti-TasA immunogold labeled samples from pellicles grown for 24 hr in MSgg medium. A) Wild-type cells. B and C) tapA mutant cells did not harbor TasA-labeled fibers, but instead had cell-associated TasA signal (B). In rare occasions small and disorganized fibers of TasA were observed detached from cells (C). The bottom panel represents a magnified view of areas marked in figures A-C. Scale bars, 200 nm.
Figure. 7. tapA mutant cells and extracts do not bind Congo Red.

A) Colonies grown for 72 hr on Congo Red indicator plates. B) Absorbance of a 10 mM solution of Congo Red incubated alone or with extracts from wild-type or tapA mutant cells.
The amount of TasA is dramatically reduced in a tapA mutant
Mutants lacking tapA produce few fibers and these do not display obvious amyloid properties (See Figs. 6 & 7). It is possible that the decreased ability to bind Congo Red is simply due to a much lower total protein concentration. To determine if the decrease in amyloid properties was due to an overall decrease in TasA protein, similar quantities of wild-type and tapA mutant pellicles were subjected to SDS-PAGE and the gels were analyzed by immunoblot with anti-TasA antibody (Fig. 8A). We observed much less TasA protein in the tapA mutant compared to wild type. As expected, the tasA mutant cell extracts did not react with the antibody. To make sure that the decrease in TasA labeling was not due to secretion of TasA in the absence of TapA, we assayed the medium, as well as the matrix and cell fractions from a pellicle assay. We only observed TasA labeling in the cell fraction suggesting that there is not excess TasA secreted from that strain (Fig. S6). A reasonable explanation for the decreased amount of TasA in a tapA mutant could be reduced transcription from the promoter of the tapA-sipW-tasA operon. To test this hypothesis, we analyzed wild-type and tapA mutant cells harboring a PtapA-yfp reporter by flow cytometry (Fig. 8B). Both strains showed similar fluorescence profiles of expression at both 24 hr and 48 hr indicating that there is not decreased transcription of tasA in the tapA mutant. In addition, we constructed a strain in which the expression of tasA was uncoupled from the PtapA promoter and instead was placed under the control of the IPTG-inducible Phyperspank promoter. Induction of tasA transcription by the addition of 0.2 mM IPTG was enough to complement a tasA mutant biofilm morphology, however, this was not sufficient to complement a tapA mutant (Fig. S7). These findings taken together suggest that in the absence of TapA, the TasA protein does not form amyloid fibers and is thus less stable and may be a substrate for proteolytic degradation (Tjalsma et al., 2004).
Figure. 8. TasA protein levels are low in a tapA mutant.

A) Immunoblot with anti-TasA antibodies of whole cells obtained from 24 hr or 48 hr MSgg pellicles from wild type, tapA and tasA mutant strains. B) Flow cytometry analysis showed no variations in the dynamics of PtapA-yfp expression in wild type (black line) and tapA mutant cells (red line). Grey peak corresponds to background of cells with no fluorescent reporter. Y-axis is number of cells and X-axis is fluorescence intensity in arbitrary units.
Purified amyloid fibers cannot complement a tapA mutant extracellularly
Given that the tapA mutant is defective in TasA fiber production, we needed to re-think the experiment shown in Figure 1 where the tapA and tasA mutants were unable to complement each other extracellularly. This inability could simply be due to the fact that both mutants are unable to produce functional TasA amyloids. Thus, we asked whether purified TasA amyloid fibers could restore biofilm formation to a tapA mutant. Using the same experimental approach we reported previously, we reproduced the ability of purified TasA fibers to restore pellicle formation to a tasA mutant (Romero et al., 2010) (Fig. 9A). However, we were unable to observe complementation when TasA fibers were added to the tapA mutant (Fig. 9B). Next, the entire complemented pellicles shown in Fig. 9A and B were harvested and cells were pelleted to remove excess fibers prior to TEM immunogold analysis with anti-TasA antibodies. We observed TasA-labeled fibers emanating from cells of the tasA mutant (Fig. 9C). However, in the tapA mutant, the purified TasA appeared as small and disorganized fibrils (Fig. 9D). Thus, the tapA mutant cells cannot bind pre-formed amyloid filaments. This observation is in agreement with the results shown in Fig. 3 in which sacculi from tapA mutant cells could not bind purified amyloid fibers.
Figure. 9. Purified TasA fibers cannot restore pellicle formation to tapA mutant cells.

A and B) Pellicles grown in MOLP broth for 48 hr. Purified TasA fibers were included in the + TasA panels at the beginning of the experiment. A) tasA mutant. B) tapA mutant. C and D) Anti-TasA immunogold-labeled electron micrographs of cells from the + TasA panels. C) TasA fibers are present in the “tasA mutant + TasA” pellicle and D) absence of signal in the tapA mutant after addition of purified TasA. Scale bars 500 nm.
Discussion
Amyloid fibers are produced by bacteria to serve as a major structural component of the extracellular matrix that holds biofilm-associated cells together (Collinson et al., 1993, Chapman et al., 2002, Claessen et al., 2003, Dueholm et al., 2010, Romero et al., 2010). In B. subtilis biofilms the amyloid fibers are primarily composed the TasA protein, which is encoded in the tapA-sipW-tasA operon. While we had studied TasA in some detail, the precise function of TapA was not clear. We had previously obtained evidence suggesting that TapA was involved in getting TasA to the matrix (Branda et al., 2006). Recently, we found that at late stages in the life cycle of a biofilm, specific D-amino acids, including D-tyrosine, are produced and incorporated in the cell wall (Kolodkin-Gal et al., 2010). This results in biofilm disassembly due to a release of the cells from the TasA fibers. Interestingly, mutants in the C-terminal domain of TapA confer resistance to the biofilm-dissociating activity of D-tyrosine. This gave rise to the hypothesis that TapA might function to anchor TasA fibers to the cell wall. In this paper we tested this hypothesis and showed that, indeed, TapA serves as an anchor and assembly factor for TasA fibers. We found that TapA localizes to the cell wall where it is necessary to support TasA fiber formation and attachment. Furthermore, in the absence of TapA, TasA appears to be degraded. Finally, in addition to its cell-wall localization, TapA is also present as a minor component of the TasA fibers.
Genetic, fractionation and microscopy experiments all indicate that TapA is associated with the cell wall. Interestingly, TapA remained associated with the peptidoglycan in sacculi preparations. This procedure involves numerous washes in 1% SDS, 0.5% beta mercaptoethanol, 2 M NaCl and treatment at 80°C. Because of its ability to stay associated with peptidoglycan through this harsh treatment, we suspect TapA is covalently linked to the cell wall. One way that proteins become covalently linked to the cell wall in Gram-positive bacteria is via sortase activity. Sortases are enzymes that recognize C-terminal sorting sequences, frequently an LPXTG motif, and catalyze attachment of these secreted proteins to the peptidoglycan (Tjalsma et al., 2004, Marraffini et al., 2006). However, TapA does not have a sequence that is similar to a known sortase domain. Nonetheless, we tested whether TapA functionality depended on the two predicted sortases of B. subtilis, the products of the yhcS and ywpE genes (Tjalsma et al., 2000a), by mutating both of these genes. The double mutant was not defective in biofilm formation, suggesting they are not involved in cell wall anchoring of TapA (data not shown). How TapA is linked to the cell wall, and whether it is through a covalent bond are questions that are currently under investigation.
In addition to their role in linking proteins to the cell wall, some sortases have also been shown to function in assembling of pili in gram-positive bacteria (Marraffini et al., 2006). In B. cereus, pili are composed of a major subunit, BcpA, and a minor subunit BcpB. BcpA harbors an LPXTG motif that is recognized by Sortase A and allows covalent linkage to the cell wall (Budzik et al., 2008). In addition, BcpA has a YPKN motif that is recognized by a different sortase, Sortase D, which allows for BcpA to be linked to BcpB (Budzik et al., 2009). Of note, TasA harbors a YPKN motif in its sequence that tempted us to speculate a similar mechanism might be occurring between TasA and TapA during fiber assembly in B. subtils. However, as stated above, even a double mutant in the two predicted sortase genes of B. subtilis did not alter biofilm morphology. Once again, we are left with an elusive mechanism of assembly.
TasA is an example of a growing number of bacterial proteins that form surface structures with amyloid properties. These amyloid-like fibers have now been described in diverse species such as E. coli, S. enterica, S. coelicolor, Pseudomonas fluorescens, and B. subtilis (Collinson et al., 1993, Chapman et al., 2002, Claessen et al., 2003, Dueholm et al., 2010, Romero et al., 2010). There is vast diversity in primary sequence of these amyloidogenic proteins and they are found in Gram-positive and Gram-negative organisms, which have dramatic differences in their cell wall structures. Thus, it is likely that there are also major differences in the mechanisms of fiber assembly and attachment to cells in the different organisms. Fibers of TasA appear to polymerize in vitro following a kinetics that matches a nucleation-polymerization model, described previously for curli in E. coli (Romero et al., Naiki & Gejyo, 1999). Similar to the protein subunits of curli, CsgA and CsgB, we hypothesized that polymerization of TasA on cell surfaces could be promoted by TapA. In the absence of TapA, we observed some auto-assembly of TasA, but only in small fibers that were detached from cell. This is different from curli assembly in E. coli, where in the absence of CsgB there is no fiber formation (Barnhart & Chapman, 2006). Our data also indicate that TasA is less stable in the absence of TapA, suggesting that TapA might play a role in stabilizing TasA. This is similar to what is observed in E. coli curli, where an accessory factor, CsgG, has been shown to stabilize CsgA and CsgB, the subunits composing the amyloid fibers (Hammar et al., 1996).
What is the biological function of TapA? TapA accumulates in the cell wall in young biofilms, when active polymerization of TasA fibers is needed. At this point, TapA appears to be involved in the formation and anchoring of TasA fibers. Perhaps TasA does not polymerize well in the absence of TapA and is thus rendered unstable. As the biofilms age it may become advantageous for cells to dissociate from the network of amyloid fibers. Because of the intrinsic stability of amyloid fibers, it is possible that depolymerizing the fibers is an inefficient process at best. Therefore, the “Achilles Heel” of the matrix may be the point where the fibers are anchored to the cell envelope. Maintaining an amyloid anchoring protein such as TapA provides an ideal mechanism for eventual disassembly of the biofilm. There is no need to break down the amyloid fibers. Instead, they are simply released when they are not required anymore. To do this, D-amino acids are produced and incorporated into the cell wall. This disrupts the anchoring of TapA, and cells are freed from the network of amyloid fibers. We propose that at the time of biofilm disassembly, the role of TapA is to provide a weak link that makes disassembly far simpler than destruction of the amyloid fibers. Thus, TapA is a bifunctional protein that allows for both assembly and detachment of the TasA amyloid fibers during biofilm formation. It will be interesting if other amyloid fiber anchoring proteins have properties similar to TapA.
Experimental procedures
Growth media and culture conditions
Luria–Bertani (LB) broth: 1% tryptone (Difco), 0.5% yeast extract (Difco), 0.5% NaCl. MSgg broth: 100 mM morpholinepropane sulphonic acid (MOPS) (pH 7), 0.5% glycerol, 0.5% glutamate, 5 mM potassium phosphate (pH 7), 50 μg/ml tryptophan, 50 μg/ml phenylalanine, 2 mM MgCl2, 700 μM CaCl2, 50 μM FeCl3, 50 μM MnCl2, 2 μM thiamine, 1 μM ZnCl2 (Branda et al., 2001). MOLP broth as described previously (Romero et al., 2010). Media were solidified with the addition of 1.5% agar. For colony architecture, 3 μL of starting culture were spotted onto MSgg agar plates and incubated at 30 °C for the indicated times. For pellicle formation, 12 μL of starting culture were added to 4 mL or 1 mL of MSgg or MOLP broth in a 6- or 24-well microtiter dish, respectively, and incubated without agitation at 30 °C for the indicated time. For Congo red binding assay, MSgg plates containing 20 μg/ml Congo Red and 10 μg/ml Coomassie Brilliant Blue G were spotted with cells as described above and assayed after 72 h at 30 °C (Romero et al., 2010). For extracellular complementation, starting cultures were mixed with 40 μg of purified protein and the mixtures were used to form pellicles, as described above.
Antibiotic concentrations (final): MLS (1 μg/ml erythromycin, 25 μg/ml lincomycin); spectinomycin (100 μg/ml); tetracycline (10 μg/ml); chloramphenicol (5 μg/ml); kanamycin (10 μg/ml).
Strain construction
Strains used and generated in this study are listed in Table 1. Plasmids were constructed and amplified in Escherichia coli XL-1 Blue (Strategene) following manufacturer protocols. To construct the plasmid pDFR2 (lacA::PtapA-tapA-yfp, spc) a 1,250 base-pair (bp) DNA fragment containing the promoter sequence of tapA and the open reading frame of the tapA gene, without stop codon, was amplified using the primers PtapA-F (5’-tggcGAATTCtcagagttaaatggtattgcttcact-3’) and tapA-R (5’-ctgatcagcttcattgcttttttcatc-3’). A fragment of 600 bp containing the whole sequence of yfp was amplified from plasmid pKM003 using the primers yfp-F (5’-gatgaaaaaagcaatgaagctgatcagatgagtaaaggagaagaactt-3’) a n d y f p-R (5’-gcgctcaGGATCCttatttgta-3’). Long flanking homology PCR (LFH-PCR) was used to join the PCR fragments. The resulting PCR product was digested with EcoRI and BamHI enzymes and cloned into the plasmid pDR183 (Doan et al., 2005), cut with the same enzymes.
Table 1.
Strains used in this study
| Strain | Genotype | Reference |
|---|---|---|
| B. subtilis 168 | Prototroph | (Branda et al., 2001) |
| SSB149 | ı68 (tapA-sipW-tasA)::spc | (Branda et al., 2004) |
| NCIB 3610 | Wild type. Undomesticated strain | (Branda et al., 2001) |
| SSB488 | eps::tet | (Branda et al., 2006) |
| CA017 | tasA::km | (Vlamakis et al., 2008) |
| FC268 |
(tapA-sipW-tasA)::spc
amyE::(tapA(13-234)-sipW-tasA) (cm) |
(Chu et al., 2006) |
| ZK3755 | lacA::PtapA-yfp | (Lopez et al., 2009) |
| IKG44 | NCIB 3610 TapA6 | (Kolodkin-Gal et al., 2010) |
| DR4 |
(tapA-sipW-tasA)::spc
amyE::(tapA(13-234)-sipW-tasA) (cm), lacA::PtapA-tapA-yfp |
This study |
| DR5 |
(tapA-sipW-tasA)::spc
amyE::(tapA(13-234)-sipW-tasA) (cm), lacA::Phyperspank-tasA |
This study |
| DR6 | tasA::km, lacA::Phyperspank-tasA | This study |
| DR7 |
(tapA-sipW-tasA)::spc
amyE::(tapA(13-234)-sipW-tasA) (cm), lacA::PtapA-yfp |
This study |
The plasmid pDFR4 (lacA::Phyperspank-tasA) was constructed in two steps. First, the open reading frame of tasA was amplified using the primers TasA-F (5’-aataaaaGTCGACataaaaggggagcttaccatgggtatgaa-3’) and Tas A-R (5’-tttGCATGCttattaatttttatcctcgctatgcgc-3’). The PCR product was digested with SalI and SphI enzymes and cloned into the plasmid pDR111 cut with the same enzymes to generate the plasmid pDFR3 (amyE::Phyperspank-tasA). Then pDFR3 was digested with XhoI and BamHI and the fragment containing Phyperspank-tasA was sub-cloned into the plasmid pDR183 digested with the same enzymes, resulting in pDFR4 (lacA::Phyperspank-tasA).
To construct the plasmids pDFR5 (pET22b-TapA) and pDFR6 (pET22b-TasA), a fragment of DNA containing the open reading frame without signal peptide or stop codon of the tapA or tasA genes was amplified by using the primers TapAH-F (5’-aaaaaaaaa-CATATGatatgcttacaatttttc-3’) and TapAH-R (5’-aaaaaaaaa-CTCGAGctgatcagcttcattgct-3’) or TasAH-F (5’-aaaaaaaaa-CATATGgcatttaacgacattaaa-3’) and TasAH-R (5’-aaaaaaa-CTCGAGatttttatcctcgctatgcgc-3’), respectively. Both fragments were digested with NdeI and XhoI enzymes and cloned into the plasmid pET22-b, cut with the same enzymes.
To generate the strains DR4, ((tapA-sipW-tasA)::spc, amyE::(tapA(13-234)-sipW-tasA) (cm), lacA::PtapA-tapA-yfp, mls) and DR5, ((tapA-sipW-tasA)::spc, amyE::(tapA(13-234)-sipW-tasA) (cm), lacA::Phyperspank-tasA, mls), the mutant strain lacking the tapA-sipW-tasA operon (SSB149) was transformed by natural competence with pDFR2 and pDFR4 respectively, and positive clones were used as donor strains for transferring the constructs into B. subtilis strain mutant (FC268) by means of SPP1-mediated generalized transduction (Yasbin & Young, 1974). To generate DR6 (tasA::km, lacA::Phyperspank-tasA, mls), B. subtilis strain 168 was transformed with plasmid pDFR4 and subsequently transduced into CA017. For construction of the strain DR7, the strain ZK3755 (B. subtilis 3610 containing the translational fusion lacA::PtapA-yfp (Lopez et al., 2009)) was used as donor and transduced into strain FC268 (Chu et al., 2006).
Protein purification
TasA produced by B. subtilis was purified as previously described (Romero et al., 2010). Briefly, cells were grown in MSgg broth at 30°C for 20 hr. After centrifugation, the pellet containing cells was extracted twice with saline extraction buffer supplemented with a protease inhibitor mixture (Complete; Roche). Supernatant containing the protein was separated from cells by centrifugation and filtered through a 0.4 μm polyethersulfone bottle-top filter. This procedure purified fibers containing TasA and the product was stored at -20ºC prior to use.
The plasmids pDFR5 and pDFR6 were used for the production of His6-TapA and His6-TasA fusion proteins respectively. These plasmids were transformed in BL21 competent E. coli cells. Cultures in 100 ml of LB supplemented with Amp were grown shaking at 37°C to an O.D. 600 of 0.5. IPTG was added to a final concentration of 1 mM and the cultures were incubated for additional 3 hr. Cells were harvested by centrifugation, resuspended in 15 ml of lysis buffer, 1 × CelLytic B cell lysis reagent (Sigma) diluted in 20mM Tris, 500mM NaCl, 20mM Imidazol and 1mM PMSF, and supplemented with 100 μg ml-1 of freshly prepared lysozyme solution, and incubated for 15 minutes. Further disruption and reduction of viscosity was done by sonication.
1.5 ml of nickel chelating resin (G-Bioscience) were washed and conditioned as indicated by the manufacturer prior to adding to the samples and incubating with gentle agitation for 1 h at room temperature. The mixture was centrifuged and after decanting the supernatant, the lysate/resin mixture was washed with 5 volumes of binding buffer (20mM Tris, 500mM NaCl, 20mM Imidazol, 1mM PMSF), 3 volumes of washing buffer-I (20mM Tris, 500mM NaCl, 40mM Imidazol, 1mM PMSF), and 3 volumes of washing buffer-II (20mM Tris, 500mM NaCl, 100mM Imidazol, 1mM PMSF). The proteins were eluted with elution buffer (20mM Tris, 500mM NaCl, 500mM Imidazol, 1mM PMSF). Purified proteins were stored at -20° until used.
Cell fractionation assay
Cell fractionation assays were performed as described previously (Tjalsma et al., 2000b). Cells grown in biofilm conditions were separated from the extracellular matrix by sonication as described previously (Branda et al., 2006). Protoplasts were obtained from by treatment of cells with 0.5 mg ml-1 lysozyme in protoplast buffer (Tjalsma et al., 2000b) for 30 minutes. Protoplast supernatant containing cell-wall-associated proteins was separated from protoplasts by mild centrifugation, and the protoplasts were resuspended in fresh protoplast buffer. A fraction of the protoplast preparation was additionally treated with 1% Triton X 100 to release cytoplasmic proteins and spores.
Immunoblot and Quantitative immunoblot
Samples were analyzed by SDS-PAGE with either 12% or 10% polyacrylamide and blotted onto PVDF membrane using standard protocols. For detection of TasA, blots were probed with anti-TasA antibodies raised in rabbits used at dilution of 1:20,000. In the fractionation assays, blots were probed with anti-YFP polyclonal antibody (Clontech) at dilution 1:5,000 or anti-sigmaA antibody (a gift from D. Rudner, Harvard Medical School, Boston, MA) as a control for detecting cytoplasmic content. A secondary anti-rabbit IgG antibody conjugated to horseradish peroxidase (BioRad) was used at a dilution of 1:20,000. Blots were developed using the Pierce super signal detection system (Pierce, Thermo Scientific). TapA antibody was kindly provided by A. Driks, Loyola University Medical Center, Maywood, IL.
Quantification of TasA and TapA proteins in purified fibers was done by quantitative immunoblot as previously described (Chai et al., 2008). The ratio TasA:TapA was estimated to be 100:1 by comparing the pixel density of supplementary figure 3B fibers from B. subtilis to known amounts of purified proteins from E. coli. In order to visualize the TapA, fibers were concentrated 20-fold for the anti-TapA western relative to the anti-TasA blot.
Preparation of sacculi
Sacculi from the B. subtilis strains NCIB 3610 (WT), tasA and tapA were obtained as previously described (Reusch, 1982). Briefly, cells were grown in MSgg broth standing liquid cultures for 24 hr. Bacteria where harvested by centrifugation and the pellets were treated 5 times for 15 minutes per treatment with extraction buffer (1% SDS and 0.5% beta mercaptoethanol) at 80°C. The remaining pellet was washed in distilled water and 2 M NaCl, for a total of 12 rinses. The obtained sacculi were stored at -20°C prior to use. For mixing experiments, a solution of TasA fibers (in buffer Tris 20mM NaCl 50mM, ph 7) purified from B. subtilis were mixed with different sacculi and incubated for 10 h at room temperature. Then the sacculi were recovered by soft centrifugation, resuspended in a minimal volume of PBS and analyzed by TEM and immunogold labeling with anti-TasA antibody.
Transmission electron microscopy and immunolabeling
Observation of intact cells was done from 24 hr pellicles. Samples were adsorbed onto carbon or formvar/carbon coated grids which were previously treated to make the grids hydrophilic. The excess sample was blotted off using filter paper and the grids were floated on 5 mL of 1 - 2% uranyl acetate for a few minutes. The samples were dried prior to examination.
For immunolocalization studies, samples were floated on blocking buffer (1% nonfat dry milk in PBS with 0.1% Tween 20) for 30 min and on anti-TasA or anti-YFP primary antibodies diluted 1:150 in blocking buffer for 2 hr, rinsed in PBST and exposed to goat-anti-rabbit 20-nm gold secondary antibody diluted 1:50 (TedPella, Inc.) for 1hr, and rinsed. For double labeling, samples were fixed in 1% glutaraldehyde for followed by quenching in 4 drops of 0.15 M Glycine / PBS prior to proceeding with primary and secondary antibodies conjugated to different sized gold particles as indicated in the text. All grids were stained with uranyl acetate and lead citrate for visualization.
For subcellular localization studies of TapA, cells grown for 24 hr in pellicle assays were separated by mild sonication as previously described. Cell suspensions were fixed with an equal volume of 4% paraformaldehyde in 0.1 M sodium phosphate buffer, pH 7.4. Small pieces of cell pellet were infiltrated in 2.3 M sucrose in PBS containing 0.15 M glycine. The samples were high pressure frozen (HPF) in a Leica EM Pact2 high-pressure freezer and freeze-substituted in 0.2% glutaraldehyde + 0.1% uranyl acetate in Acetone at -90°C for 72 hr. Samples were slowly warmed up to room temperature and embedded in LR White resin. Ultrathin sections were cut at -120°C with a cryo-diamond knife. Sections were transferred to a formvar/carbon coated copper grid and stored at 4°C until immunogold labeled. For contrasting, grids were floated in mixture of 2% methyl cellulose with 3% uranyl acetate for 10 min. Excess liquid was removed and grids were allowed to dry before examining in a Tecnai G2 Spirit BioTWIN microscope at an accelerating voltage of 80 KV. Images were taken with an AMT 2k CCD camera.
Immunocytochemistry, image capture and analysis
Biofilms were harvested at 24 or 48 hr, resuspended in 1 ml PBS buffer and dispersed with three pulses of mild sonication. Cells were separated by centrifugation and fixed with a solution of 4% paraformaldehyde for 7 minutes, washed in PBS and resuspended in GTE buffer (50 mM Glucose, 10 mM EDTA pH 8, 20 mM Tris-HCl pH 8) (Vlamakis et al., 2008). The fixed cells were blocked with 2% BSA/PBS for 30 min at room temperature. Primary antibody, anti-TapA or anti-YFP was used at a dilution of 1:50 in 2% BSA/PBS for 1 hr at room temperature. Samples were washed 10X with PBS and exposed to the secondary goat anti-rabbit conjugated to FITC antibody (Invitrogen, Molecular Probes) at a dilution of 1:100 in 2% BSA/PBS for 1 hr at room temperature in the dark. Finally samples were washed 8x with PBS and resuspended in fresh GTE buffer before visualization.
Fluorescence microscopy images were taken on a Nikon Eclipse TE2000-U microscope equipped with an X-cite 120 illumination system, using a Hamamatsu digital camera model ORCA-ER. Fluorescence signal was detected with a Ex436/500 filter. Image processing was performed using MetaMorph® Software and Photoshop®.
Flow cytometry
Biofilms of cells harboring PtapA-yfp fusions (ZK3755 and DR7) were harvested at 24 or 48 hr were dispersed as previously described. Cells were fixed in 4% paraformaldehyde solution for 7 min, washed with PBS and re-suspended in GTE buffer. After fixation, single cells were obtained by mild sonication of the samples (Branda et al., 2006). For flow cytometric analysis, dilution of 1:100 of cell suspensions were measured on a BD LSR II flow cytometer (BD Biosciences) using a solid state laser. For YFP fluorescence, a laser excitation of 488 nm coupled with 530/30 and 505LP sequential filters were used. The photomultiplier voltage was set at 400-500 V. Every sample was analyzed by counting 50,000 events using FACS Diva (BD Biosciences) software to capture the data (Lopez et al., 2009). Further analysis was performed using FlowJo 8.7.2 software (http://www.flowjo.com).
Supplementary Material
Acknowledgements
We would like to thank members of the Kolter and Losick laboratories for helpful discussions. We thank Adam Driks (Loyola University Medical Center, Maywood, IL) for kindly providing antibodies against TasA and TapA (YqxM). We thank M. Ericsson, L. Trakimas, and E. Benecchi for help and guidance in the electron microscope. This work was funded by National Institutes of Health grants to R.K. (GM58213) and R.L. (GM18546) as well as grants to R.K and R.L. by the BASF Advanced Research Initiative at Harvard. D.R. is the recipient of a MEC/Fulbright post-doctoral fellowship from Secretaría General de Estado de Universidades e Investigación del Ministerio de Educación y Ciencia (Spain).
References
- Badtke MP, Hammer ND, Chapman MR. Functional amyloids signal their arrival. Sci Signal. 2009;2:pe43. doi: 10.1126/scisignal.280pe43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnhart MM, Chapman MR. Curli biogenesis and function. Annu Rev Microbiol. 2006;60:131–147. doi: 10.1146/annurev.micro.60.080805.142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian Z, Normark S. Nucleator function of CsgB for the assembly of adhesive surface organelles in Escherichia coli. EMBO J. 1997;16:5827–5836. doi: 10.1093/emboj/16.19.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branda SS, Chu F, Kearns DB, Losick R, Kolter R. A major protein component of the Bacillus subtilis biofilm matrix. Mol Microbiol. 2006;59:1229–1238. doi: 10.1111/j.1365-2958.2005.05020.x. [DOI] [PubMed] [Google Scholar]
- Branda SS, Gonzalez-Pastor JE, Ben-Yehuda S, Losick R, Kolter R. Fruiting body formation by Bacillus subtilis. Proc Natl Acad Sci U S A. 2001;98:11621–11626. doi: 10.1073/pnas.191384198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branda SS, Gonzalez-Pastor JE, Dervyn E, Ehrlich SD, Losick R, Kolter R. Genes involved in formation of structured multicellular communities by Bacillus subtilis. J Bacteriol. 2004;186:3970–3979. doi: 10.1128/JB.186.12.3970-3979.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzik JM, Marraffini LA, Souda P, Whitelegge JP, Faull KF, Schneewind O. Amide bonds assemble pili on the surface of bacilli. Proc Natl Acad Sci U S A. 2008;105:10215–10220. doi: 10.1073/pnas.0803565105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzik JM, Oh SY, Schneewind O. Sortase D forms the covalent bond that links BcpB to the tip of Bacillus cereus pili. J Biol Chem. 2009;284:12989–12997. doi: 10.1074/jbc.M900927200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Chu F, Kolter R, Losick R. Bistability and biofilm formation in Bacillus subtilis. Mol Microbiol. 2008;67:254–263. doi: 10.1111/j.1365-2958.2007.06040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S, Hultgren SJ. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science. 2002;295:851–855. doi: 10.1126/science.1067484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu F, Kearns DB, Branda SS, Kolter R, Losick R. Targets of the master regulator of biofilm formation in Bacillus subtilis. Mol Microbiol. 2006;59:1216–1228. doi: 10.1111/j.1365-2958.2005.05019.x. [DOI] [PubMed] [Google Scholar]
- Claessen D, de Jong W, Dijkhuizen L, Wosten HA. Regulation of Streptomyces development: reach for the sky! Trends Microbiol. 2006;14:313–319. doi: 10.1016/j.tim.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Claessen D, Rink R, de Jong W, Siebring J, de Vreugd P, Boersma FG, Dijkhuizen L, Wosten HA. A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes Dev. 2003;17:1714–1726. doi: 10.1101/gad.264303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claessen D, Wosten HA, van Keulen G, Faber OG, Alves AM, Meijer WG, Dijkhuizen L. Two novel homologous proteins of Streptomyces coelicolor and Streptomyces lividans are involved in the formation of the rodlet layer and mediate attachment to a hydrophobic surface. Mol Microbiol. 2002;44:1483–1492. doi: 10.1046/j.1365-2958.2002.02980.x. [DOI] [PubMed] [Google Scholar]
- Collinson SK, Doig PC, Doran JL, Clouthier S, Trust TJ, Kay WW. Thin, aggregative fimbriae mediate binding of Salmonella enteritidis to fibronectin. J Bacteriol. 1993;175:12–18. doi: 10.1128/jb.175.1.12-18.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong W, Wosten HA, Dijkhuizen L, Claessen D. Attachment of Streptomyces coelicolor is mediated by amyloidal fimbriae that are anchored to the cell surface via cellulose. Mol Microbiol. 2009 doi: 10.1111/j.1365-2958.2009.06838.x. [DOI] [PubMed] [Google Scholar]
- Doan T, Marquis KA, Rudner DZ. Subcellular localization of a sporulation membrane protein is achieved through a network of interactions along and across the septum. Mol Microbiol. 2005;55:1767–1781. doi: 10.1111/j.1365-2958.2005.04501.x. [DOI] [PubMed] [Google Scholar]
- Dueholm MS, Petersen SV, Sonderkaer M, Larsen P, Christiansen G, Hein KL, Enghild JJ, Nielsen JL, Nielsen KL, Nielsen PH, Otzen DE. Functional amyloid in Pseudomonas. Mol Microbiol. 2010;77:1009–1020. doi: 10.1111/j.1365-2958.2010.07269.x. [DOI] [PubMed] [Google Scholar]
- Epstein EA, Chapman MR. Polymerizing the fibre between bacteria and host cells: the biogenesis of functional amyloid fibres. Cell Microbiol. 2008;10:1413–1420. doi: 10.1111/j.1462-5822.2008.01148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrone F. Analysis of protein aggregation kinetics. Methods Enzymol. 1999;309:256–274. doi: 10.1016/s0076-6879(99)09019-9. [DOI] [PubMed] [Google Scholar]
- Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid--from bacteria to humans. Trends Biochem Sci. 2007;32:217–224. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Gebbink MF, Claessen D, Bouma B, Dijkhuizen L, Wosten HA. Amyloidsa functional coat for microorganisms. Nat Rev Microbiol. 2005;3:333–341. doi: 10.1038/nrmicro1127. [DOI] [PubMed] [Google Scholar]
- Greenwald J, Riek R. Biology of amyloid: structure, function, and regulation. Structure. 2010;18:1244–1260. doi: 10.1016/j.str.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Hammar M, Bian Z, Normark S. Nucleator-dependent intercellular assembly of adhesive curli organelles in Escherichia coli. Proc Natl Acad Sci U S A. 1996;93:6562–6566. doi: 10.1073/pnas.93.13.6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer ND, Schmidt JC, Chapman MR. The curli nucleator protein, CsgB, contains an amyloidogenic domain that directs CsgA polymerization. Proc Natl Acad Sci U S A. 2007;104:12494–12499. doi: 10.1073/pnas.0703310104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kline KA, Dodson KW, Caparon MG, Hultgren SJ. A tale of two pili: assembly and function of pili in bacteria. Trends Microbiol. 2010;18:224–232. doi: 10.1016/j.tim.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodkin-Gal I, Romero D, Cao S, Clardy J, Kolter R, Losick R. D-amino acids trigger biofilm disassembly. Science. 2010;328:627–629. doi: 10.1126/science.1188628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez D, Vlamakis H, Losick R, Kolter R. Cannibalism enhances biofilm development in Bacillus subtilis. Mol Microbiol. 2009;74:609–618. doi: 10.1111/j.1365-2958.2009.06882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, Dedent AC, Schneewind O. Sortases and the art of anchoring proteins to the envelopes of gram-positive bacteria. Microbiol Mol Biol Rev. 2006;70:192–221. doi: 10.1128/MMBR.70.1.192-221.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maury CP. The emerging concept of functional amyloid. J Intern Med. 2009;265:329–334. doi: 10.1111/j.1365-2796.2008.02068.x. [DOI] [PubMed] [Google Scholar]
- Naiki H, Gejyo F. Kinetic analysis of amyloid fibril formation. Methods Enzymol. 1999;309:305–318. doi: 10.1016/s0076-6879(99)09022-9. [DOI] [PubMed] [Google Scholar]
- Oh J, Kim JG, Jeon E, Yoo CH, Moon JS, Rhee S, Hwang I. Amyloidogenesis of type III-dependent harpins from plant pathogenic bacteria. J Biol Chem. 2007;282:13601–13609. doi: 10.1074/jbc.M602576200. [DOI] [PubMed] [Google Scholar]
- Reusch VM., Jr. Isolation and analysis of sacculi from Streptococcus sanguis. J Bacteriol. 1982;151:1543–1552. doi: 10.1128/jb.151.3.1543-1552.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero D, Aguilar C, Losick R, Kolter R. Amyloid fibers provide structural integrity to Bacillus subtilis biofilms. Proc Natl Acad Sci U S A. 2010;107:2230–2234. doi: 10.1073/pnas.0910560107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover AG, Driks A. Control of synthesis and secretion of the Bacillus subtilis protein YqxM. J Bacteriol. 1999a;181:7065–7069. doi: 10.1128/jb.181.22.7065-7069.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover AG, Driks A. Secretion, localization, and antibacterial activity of TasA, a Bacillus subtilis spore-associated protein. J Bacteriol. 1999b;181:1664–1672. doi: 10.1128/jb.181.5.1664-1672.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CC. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J Mol Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- Tjalsma H, Antelmann H, Jongbloed JD, Braun PG, Darmon E, Dorenbos R, Dubois JY, Westers H, Zanen G, Quax WJ, Kuipers OP, Bron S, Hecker M, van Dijl JM. Proteomics of protein secretion by Bacillus subtilis: separating the “secrets” of the secretome. Microbiol Mol Biol Rev. 2004;68:207–233. doi: 10.1128/MMBR.68.2.207-233.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjalsma H, Bolhuis A, Jongbloed JD, Bron S, van Dijl JM. Signal peptide-dependent protein transport in Bacillus subtilis: a genome-based survey of the secretome. Microbiol Mol Biol Rev. 2000a;64:515–547. doi: 10.1128/mmbr.64.3.515-547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjalsma H, Bolhuis A, van Roosmalen ML, Wiegert T, Schumann W, Broekhuizen CP, Quax WJ, Venema G, Bron S, van Dijl JM. Functional analysis of the secretory precursor processing machinery of Bacillus subtilis: identification of a eubacterial homolog of archaeal and eukaryotic signal peptidases. Genes Dev. 1998;12:2318–2331. doi: 10.1101/gad.12.15.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjalsma H, Stover AG, Driks A, Venema G, Bron S, van Dijl JM. Conserved serine and histidine residues are critical for activity of the ER-type signal peptidase SipW of Bacillus subtilis. J Biol Chem. 2000b;275:25102–25108. doi: 10.1074/jbc.M002676200. [DOI] [PubMed] [Google Scholar]
- Tompa P. Structural disorder in amyloid fibrils: its implication in dynamic interactions of proteins. FEBS J. 2009;276:5406–5415. doi: 10.1111/j.1742-4658.2009.07250.x. [DOI] [PubMed] [Google Scholar]
- Vlamakis H, Aguilar C, Losick R, Kolter R. Control of cell fate by the formation of an architecturally complex bacterial community. Genes Dev. 2008;22:945–953. doi: 10.1101/gad.1645008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasbin RE, Young FE. Transduction in Bacillus subtilis by bacteriophage SPP1. J Virol. 1974;14:1343–1348. doi: 10.1128/jvi.14.6.1343-1348.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.