

Abstract
Cytotoxicity-directed purification of a Symploca cf. hydnoides sample from Cetti Bay, Guam, afforded seven new cyclic depsipeptides, veraguamides A–G (1–7), together with the known compound dolastatin 16. The planar structures of 1–7 were elucidated using NMR and MS experiments, while enantioselective HPLC and Mosher's analysis of acid and base hydrolysates, respectively, were utilized to assign the absolute configurations of the stereocenters. Veraguamides A–G (1–7) are characterized by the presence of an invariant proline residue, multiple N-methylated amino acids, an α-hydroxy acid, and a C8-polyketide derived β-hydroxy acid moiety with a characteristic terminus as either an alkynyl bromide, alkyne, or vinyl group. These compounds and a semisynthetic analog (8) showed moderate to weak cytotoxic activity against HT29 colorectal adenocarcinoma and HeLa cervical carcinoma cell lines. Preliminary structure-activity relationship analysis identified several sensitive positions in the veraguamide scaffold that affect the cytotoxic activity of this compound class. Dolastatin 16 showed only weak cytotoxic activity on both cell lines tested. The complete stereostructure of dolastatin 16 was proposed for the first time through degradation followed by a combination of advanced Marfey's analysis and modified Mosher's analysis using phenylglycine methyl ester as a chiral anisotropic reagent.
Marine cyanobacteria have provided both structurally diverse and potent cytotoxic compounds with varying mechanisms of action as lead structures for drug discovery. Most of these natural products are peptide–polyketide hybrids and are derived from the genus Lyngbya, marked by N-methylation, modified amino acids, α- and β-hydroxy acids, and fatty acid-type moieties.1,2 We have had particular success with our drug discovery efforts based on the genus Symploca. From Micronesian collections of Symploca spp. we previously isolated the linear peptides dolastatin 10 and its analog symplostatin 1.3,4 These compounds are microtubule disrupting agents that interfere with mitosis and have pico- to nanomolar IC50 values against cancer cells.5 From a Floridian Symploca sp. collection we recently discovered the HDAC inhibitor, largazole, which showed strong cytotoxic activity against cancer cells with superior selectivity over nontransformed cells.6,7 In this light, we screened several Symploca sp. collections from Guam and Florida to find new cytotoxic agents. In this paper, we present the cytotoxicity-directed fractionation of a S. cf. hydnoides collection from Cetti Bay, Guam, which afforded the known compound dolastatin 16,8 together with seven new cyclic depsipeptides, given the trivial names veraguamides A–G (1–7).9
Results and Discussion
The freeze-dried cyanobacterium was extracted with EtOAc–MeOH (1:1) and subsequently solvent-partitioned into hexanes-, n-BuOH, and H2O-soluble fractions. The n-BuOH soluble fraction showed biological activity at 10 μg/mL in an initial cytotoxicity screen using the HT29 colorectal adenocarcinoma cell line. This fraction was further purified by silica column chromatography, with the cytotoxic activity concentrated in the fraction eluting with 20% i-PrOH in CH2Cl2. Reversed-phase HPLC purification of this silica fraction yielded dolastatin 16 and veraguamides A–G (1–7).
HRESIMS of the major compound in this series, veraguamide A (1), showed the distinctive 1:1 isotopic cluster for a Br-containing compound for the [M + H]+ peak at m/z 767.3675/769.3660, suggesting a molecular formula of C37H59BrN4O8. The 1H NMR spectrum of 1 displayed characteristic peptide resonances for a secondary amide proton (δH 6.25), two tertiary amide N-CH3s (δH 3.00, δH 2.94) and several α-protons (δH 3.85 – 4.95). 2D NMR analysis (Table 1) in CDCl3 using HSQC, COSY, TOCSY, and HMBC established the presence of four amino acids (Pro, Val, 2 × N-Me-Val) and an α-hydroxy acid [(2-hydroxy-3-methylpentanoic acid (Hmpa)]. The last spin system consisted of a CH3 doublet (δH 1.25) that showed a COSY correlation to a methine (δH 3.11) and HMBC correlations to a carbonyl (δC 170.8) and an oxymethine (δC 76.4). Further extension of this unit using HMBC and COSY established the presence of a 8-bromo-3-hydroxy-2-methyl-7-octynoic acid (Br-Hmoya) moiety in 1. This was supported by HMBC correlations of the methylene (δC-6 19.2/δH2–6 2.23) with two quaternary carbons at δC 38.4 and δC 79.3 and by the large difference in chemical shifts between these quaternary carbons characteristic for an alkynyl bromide.10 The linear sequence of N-Me-Val-1–Pro–Hmpa–N-Me-Val-2–Val–Br-Hmoya was established based on HMBC correlations between α-protons and carbonyl groups (Table 1) and was verified by MS/MS fragmentation (Figure 1). The deshielded C-3 methine of the Br-Hmoya unit suggested acylation with the carbonyl of N-Me-Val-1 to form a cyclic hexadepsipeptide, corroborated by HMBC and consistent with the molecular formula requirements based on HRESIMS.
Table 1.
NMR Data for Veraguamide A (1) in CDCl3
| unit | C/H no | δ Ca | δH (J in Hz)b | COSYb | HMBCb |
|---|---|---|---|---|---|
| Br-Hmoya | 1 | 170.8, C | |||
| 2 | 42.4, CH | 3.11, br q (7.4) | H-3, H3-9 | 1, 3, 4, 9 | |
| 3 | 76.4, CH | 4.85, dt (10.2, 2.5) | H-2, H-4a, H-4b | 1, 1 (N-Me-Val-1) | |
| 4a | 27.4, CH2 | 2.06, m | H-3, H-4b, H-5a, H-5b | 5, 6 | |
| 4b | 1.59, m | H-3, H-4a, H-5a, H-5b | |||
| 5a | 25.0, CH2 | 1.60, m | H-4a, H-4b, H-5b, H2-6 | 7, 8 | |
| 5b | 1.41, m | H-4a, H-4b, H-5a, H2-6 | 6, 8 | ||
| 6 | 19.2, CH2 | 2.23, m | H-5a, H-5b | 7, 8 | |
| 7 | 38.4, C | ||||
| 8 | 79.3, C | ||||
| 9 | 14.1, CH3 | 1.25, d (7.4) | H-2 | 1, 2, 3 | |
| N-Me-Val-1 | 1 | 170.6, C | |||
| 2 | 65.0, CH | 3.93, d (10.3) | H-3 | 1, 3, 4, N-Me, 1 (Pro) | |
| 3 | 28.3, CH | 2.30, m | H-2, H3-4, H3-5 | 2, 4, 5 | |
| 4 | 19.56, CH3 | 0.98, d (6.6) | H-3 | 2 | |
| 5 | 19.51, CH3 | 0.91, d (6.6) | H-3 | 2 | |
| N-Me | 28.6, CH3 | 3.00, s | 2, 1 (Pro) | ||
| Pro | 1 | 172.1, C | |||
| 2 | 57.3, CH | 4.94 dd (8.9, 4.5) | H-3a, H-3b | 1, 3, 4, 1 (Hmpa) | |
| 3a | 29.5, CH2 | 2.30, m | H-2, H-3b, H-4a | 1, 2, 5 | |
| 3b | 1.79, m | H-2, H-3a, H-4a | 1, 2, 5 | ||
| 4a | 24.9, CH2 | 2.03, m | H-3a, H-3b, H-4b, H-5a, H-5b | 2, 3, 5 | |
| 4b | 1.98, m | H-4a, H-5a, H-5b | 2, 3, 5 | ||
| 5a | 47.3, CH2 | 3.84, dt (−17.0, 7.1) | H-4a, H-4b, H-5b | 2, 3, 4 | |
| 5b | 3.61, dt (−17.0, 7.1) | H-4a, H-4b, H-5a | 3, 4 | ||
| Hmpa | 1 | 165.9, C | |||
| 2 | 76.1, CH | 4.90, d (9.1) | H-3 | 1, 3, 4, 5, 1 (N-Me-Val-2) | |
| 3 | 35.7, CH | 1.97, m | H-2, H3-6 | ||
| 4a | 24.81, CH2 | 1.54, m | H-4b, H3-5 | ||
| 4b | 1.13, m | H-4a, H3-5 | |||
| 5 | 10.5, CH3 | 0.87, t (7.3) | H-4a, H-4b | 3, 4 | |
| 6 | 13.8, CH3 | 1.01, d (6.8) | H-3 | 2, 3, 4 | |
| N-Me-Val-2 | 1 | 169.6, C | |||
| 2 | 66.0, CH | 4.15, d (9.5) | H-3 | 1, 3, 5, N-Me, 1(Val) | |
| 3 | 28.5, CH | 2.27, m | H-2, H3-4, H3-5 | 1 | |
| 4 | 20.4, CH3 | 1.00, d (7.0) | H-3 | 2 | |
| 5 | 20.2, CH3 | 1.11, d (7.0) | H-3 | 2, 3 | |
| N-Me | 30.0, CH3 | 2.94, s | 2, 1 (Val) | ||
| Val | 1 | 173.4, C | |||
| 2 | 52.8, CH | 4.70, dd (8.7, 6.5) | H-3, NH | 1, 3, 4, 5, 1 (Br-Hmoya) | |
| 3 | 32.1, CH | 1.96, m | H-2, H3-4, H3-5 | 5 | |
| 4 | 20.3, CH3 | 0.95, d (6.7) | H-3 | 2, 3, 5 | |
| 5 | 17.5, CH3 | 0.88, d (6.7) | H-3 | 2, 3 | |
| NH | 6.25, d (8.7) | H-2 | 1, 1 (Br-Hmoya) |
100 MHz
600 MHz
Figure 1.

MS/MS fragmentation of veraguamide A (1), veraguamide D (4), and veraguamide E (5).
Veraguamide B (2) showed a 1:1 isotopic pattern for the pseudomolecular ion [M + H]+ at m/z 753.3517/755.3508, suggesting the presence of a Br as in 1 with a negative difference of 14 amu corresponding to one less CH2 unit and thus a molecular formula of C36H57BrN4O8. Comparison of the 1H NMR spectrum of 1 and 2 showed differences in the splitting pattern in the CH3 region atδH 0.93 ppm and the chemical shift of the α-proton (δH 4.85) of the α-hydroxy acid (Table 2). The vicinal methine (δH 2.17) of the α-hydroxy acid showed COSY correlations to two methyl groups (δH 0.93, δH 1.02) instead of COSY correlations to methylene and methyl protons in 1. Therefore, 2 possesses a 2-hydroxyisovaleric acid (Hiva) instead of the Hmpa unit as in 1.
Table 2.
NMR Data for Veraguamides B (2) and C (3) in CDCl3
| 2 | 3 | ||||
|---|---|---|---|---|---|
| unit | C/H no. | δ Ca | δH (J in Hz)b | δ Ca | δH (J in Hz)b |
| Br-Hmoyac/Hmoyad | 1 | 170.8, C | 170.8, C | ||
| 2 | 42.3, CH | 3.13, br q (7.4) | 42.4, CH | 3.10, br q (7.2) | |
| 3 | 76.4, CH | 4.85, d (8.7) | 76.4, CH | 4.86, dt (10.4, 2.5) | |
| 4a | 27.5, CH2 | 2.07, m | 27.4, CH2 | 2.07, m | |
| 4b | 1.60, m | 1.62, m | |||
| 5a | 24.93, CH2 | 1.61, m | 25.2, CH2 | 1.62, m | |
| 5b | 1.42, m | 1.44, m | |||
| 6 | 19.2, CH2 | 2.21, m | 18.0, CH2 | 2.19, m | |
| 7 | 38.4, C | 83.6, C | |||
| 8 | 79.4, C | 68.8, CH | 1.93 t (2.5) | ||
| 9 | 14.6, CH3 | 1.25, d (7.4) | 14.5, CH3 | 1.25, d (7.2) | |
| N-Me-Val-1 | 1 | 170.7, C | 170.7, C | ||
| 2 | 65.0, CH | 3.94, d (10.4) | 65.0, CH | 3.93, d (11.0) | |
| 3 | 28.26, CH | 2.29, m | 28.3, CH | 2.28, m | |
| 4 | 19.57, CH3 | 0.98, d (6.5) | 19.58, CH3 | 0.98, d (6.8) | |
| 5 | 19.55, CH3 | 0.92, d (6.5) | 19.56, CH3 | 0.91, d (6.8) | |
| N-Me | 28.7, CH3 | 3.00, s | 28.7, CH3 | 3.00, s | |
| Pro | 1 | 172.1, C | 172.2, C | ||
| 2 | 57.2, CH | 4.95 dd (8.6, 4.8) | 57.3, CH | 4.94 dd (8.4, 5.0) | |
| 3a | 29.4, CH2 | 2.28, m | 29.5, CH2 | 2.28, m | |
| 3b | 1.79, m | 1.79, m | |||
| 4a | 24.99, CH2 | 2.04, m | 24.89, CH2 | 2.03, m | |
| 4b | 1.98, m | 1.99, m | |||
| 5a | 47.3, CH2 | 3.80, dt (−16.4, 6.9) | 47.3, CH2 | 3.84, dt (−16.8, 7.1) | |
| 5b | 3.60, dt (−16.4, 6.9) | 3.60, dt (−16.8, 7.1) | |||
| Hivac/Hmpad | 1 | 165.8, C | 165.9, C | ||
| 2 | 77.2, CH | 4.85, d (8.7) | 76.0, CH | 4.89, d (9.4) | |
| 3 | 29.6, CH | 2.17, m | 35.7, CH | 1.98, m | |
| 4 | 18.1, CH3 | 1.02, t (6.6) | 24.81, CH2 | 1.54, m | |
| 1.13, m | |||||
| 5 | 18.5, CH3 | 0.93, d (6.6) | 10.5, CH3 | 0.86, t (7.3) | |
| 6 | 13.8, CH3 | 1.01, d (6.7) | |||
| N-Me-Val-2 | 1 | 169.6, C | 169.6, C | ||
| 2 | 66.1, CH | 4.15, d (10.2) | 66.0, CH | 4.13, d (10.0) | |
| 3 | 28.34, CH | 2.28, m | 28.5, CH | 2.28, m | |
| 4 | 20.3, CH3 | 0.99, d (6.8) | 20.4, CH3 | 0.99, d (6.4) | |
| 5 | 20.1, CH3 | 1.11, d (6.8) | 20.2, CH3 | 1.10, d (6.4) | |
| N-Me | 30.0, CH3 | 2.94, s | 30.0, CH3 | 2.93, s | |
| Val | 1 | 173.5, C | 173.4, C | ||
| 2 | 52.8, CH | 4.71, dd (8.6, 6.4) | 52.8, CH | 4.70, dd (8.6, 6.2) | |
| 3 | 32.2, CH | 1.98, m | 32.1, CH | 1.96, m | |
| 4 | 20.3, CH3 | 0.94, d (6.4) | 20.3, CH3 | 0.94, d (6.7) | |
| 5 | 17.5, CH3 | 0.88, d (6.4) | 17.6, CH3 | 0.87, d (6.7) | |
| NH | 6.26, d (8.6) | 6.26, d (8.6) | |||
100 MHz
600 MHz
Refers to Compound 2
Refers to Compound 3
The HRESIMS spectrum of veraguamide C (3) showed a negative deviation of 79 amu compared with 1 which indicated the lack of Br and a molecular formula of C37H60N4O8. This was supported by the absence of the 1:1 isotopic pattern for the [M + H]+ peak when compared to 1. The 1H NMR spectrum of 3 showed an additional triplet at δH 1.93 with JH,H = 2.5 Hz (Table 2); otherwise it was virtually identical to that of 1. This proton correlated to a methine at δC 68.8 and a quaternary C (δC 83.6) in the HSQC and HMBC spectra, respectively. These signals are indicative of a terminal alkyne; hence 3 had to bear a 3-hydroxy-2-methyl-7-octynoic acid (Hmoya) moiety in lieu of the Br-Hmoya present in 1 and 2.
Veraguamide D (4) appeared closely related to 3 as its 1H NMR spectrum showed the acetylenic proton at δH 1.93 (Table 3). In comparison to 3, the HRESIMS spectrum of 4 showed a positive difference of 14 amu, corresponding to an additional CH2 unit and in agreement with a molecular formula of C38H62N4O8. The 1H and 13C NMR spectra of 4 showed a high-field CH3 at δC 11.4/δH 0.93, characteristic of an Ile or Ile-derived moiety. COSY (δH/δH 0.93/1.46, 1.46/2.02, 2.02/4.01) and (δC/δH 15.8/4.01, 28.7/4.01) correlations established that the N-Me-Val-1 residue is replaced by an N-Me-Ile in veraguamide D (4).
Table 3.
NMR Data for Veraguamides D (4) and E (5) in CDCl3
| 4 | 5 | ||||
|---|---|---|---|---|---|
| unit | C/H no. | δ Ca | δH (J in Hz)b | δ a | δ (J in Hz)b |
| Hmoya | 1 | 170.8, C | 170.71, C | ||
| 2 | 42.4, CH | 3.13, br q (7.2) | 42.4, CH | 3.08, br q (7.4) | |
| 3 | 76.4, CH | 4.86, dt (10.8, 2.6) | 76.5, CH | 4.85, d (9.0) | |
| 4a | 27.5, CH2 | 2.07, m | 27.5, CH2 | 2.06, m | |
| 4b | 1.63, m | 1.62, m | |||
| 5a | 25.2, CH2 | 1.63, m | 25.2, CH2 | 1.61, m | |
| 5b | 1.47, m | 1.43, m | |||
| 6 | 17.5, CH2 | 2.18, m | 18.0, CH2 | 2.18, m | |
| 7 | 83.6, C | 83.6, C | |||
| 8 | 68.8, CH | 1.93, t (2.5) | 68.8, CH | 1.93, t (2.3) | |
| 9 | 14.4, CH3 | 1.24, d (7.2) | 14.5, CH3 | 1.23, d (7.4) | |
| N-Me-Ilec/N-Me-Vald | 1 | 170.7, C | 170.69, C | ||
| 2 | 64.0, CH | 4.01, d (10.6) | 64.9, CH | 3.93, d (10.0) | |
| 3 | 34.6, CH | 2.02, m | 28.3, CH | 2.29, m | |
| 4 | 25.7, CH2 | 1.46, m | 19.59, CH3 | 0.91, d (6.4) | |
| 5 | 11.4, CH3 | 0.93, t (6.5) | 19.56, CH3 | 0.98, d (6.4) | |
| 6 | 15.8, CH3 | 0.94, d (6.8) | |||
| N-Me | 28.7, CH3 | 2.99, s | 28.6, CH3 | 3.00, s | |
| Pro | 1 | 172.7, C | 172.2, C | ||
| 2 | 57.2, CH | 4.94 dd (8.9, 4.8) | 57.3, CH | 4.94 dd (9.0, 5.3) | |
| 3a | 28.8, CH2 | 2.26, m | 29.5, CH2 | 2.29, m | |
| 3b | 1.78, m | 1.78, m | |||
| 4a | 24.9, CH2 | 2.03, m | 24.89, CH2 | 2.01, m | |
| 4b | 1.97, m | 1.99, m | |||
| 5a | 47.2, CH2 | 3.82, dt (−17.0, 7.3) | 47.3, CH2 | 3.86, dt (−17.0, 7.0) | |
| 5b | 3.60, dt (−17.0, 7.3) | 3.60, dt (−17.0, 7.0) | |||
| Hmpa | 1 | 165.9, C | 166.0, C | ||
| 2 | 76.0, CH | 4.90, d (9.2) | 76.1, CH | 4.85, d (9.0) | |
| 3 | 35.7, CH | 1.98, m | 35.1, CH | 1.98, m | |
| 4 | 24.8, CH2 | 1.53, m | 24.86, CH2 | 1.54, m | |
| 1.12, m | 1.13, m | ||||
| 5 | 10.5, CH3 | 0.86, t (7.6) | 10.5, CH3 | 0.86, t (7.0) | |
| 6 | 13.9, CH3 | 0.99, d (6.9) | 13.8, CH3 | 1.01, d (6.8) | |
| N-Me-Valc/N-Me-Iled | 1 | 169.6, C | 169.7, C | ||
| 2 | 66.0, CH | 4.15, d (9.4) | 65.2, CH | 4.22, d (9.6) | |
| 3 | 28.4, CH | 2.28, m | 35.7, CH | 1.98, m | |
| 4 | 20.3, CH3 | 1.10, d (6.8) | 26.6, CH2 | 1.54, m | |
| 1.06, m | |||||
| 5 | 20.2, CH3 | 0.99, d (6.8) | 11.7, CH3 | 0.96, t (7.2) | |
| 6 | 16.5, CH3 | 1.04, d (6.9) | |||
| N-Me | 30.0, CH3 | 2.92, s | 30.1, CH3 | 2.93, s | |
| Valc/Iled | 1 | 173.4, C | 173.5, C | ||
| 2 | 52.8, CH | 4.70, dd (8.6, 6.6) | 52.4, CH | 4.70, dd (8.4, 6.7) | |
| 3 | 32.1, CH | 1.98, m | 38.6, CH | 1.69, m | |
| 4 | 19.0, CH3 | 0.94, d (6.6) | 23.9, CH2 | 1.47, m | |
| 5 | 17.6, CH3 | 0.87, d (6.6) | 1.05, m | ||
| 11.5, CH3 | 0.85, d (6.6) | ||||
| 6 | 16.3, CH3 | 0.91, d (6.6) | |||
| NH | 6.24, d (8.6) | 6.26, d (8.4) | |||
125 MHz
600 MHz
Refers to Compound 4
Refers to Compound 5
Compound 5 (C39H64N4O8) exhibited a close relationship to both 3 and 4, showing a positive deviation of 28 amu and 14 amu, respectively, and also having the Hmoya moiety. The NMR data of 5 (Table 3) indicated the presence of two high-field methyl (δC 11.7/δH 0.96, δC 11.5/δH 0.85) and additional methylene (δC 26.6/δH 1.54, 1.06; δC 23.9/δH 1.47, 1.05) groups in comparison with 3, which suggested that two isopropyl groups in the latter are replaced with sec-butyl groups in the former. This is further corroborated by HMBC and COSY correlations which established the replacement of Val and N-Me-Val-2 moieties with Ile and N-Me-Ile, respectively, in veraguamide E (5). The N-Me-Val residue that was replaced by N-Me-Ile was located at different positions in 4 and 5; with N-Me-Val-1 replaced in the former and N-Me-Val-2 in the latter. This NMR result was verified by MS/MS fragmentation of both 4 and 5 (Figure 1).
The 1H NMR spectrum of veraguamide F (6) (Table 4) showed additional resonances for aromatic protons at δH 7.2 – 7.4 ppm, upfield-shifted N-Me protons to δH 2.60 ppm presumably due to shielding by the aromatic ring, and a low-field α-proton of the hydroxy acid (δH 5.47), with the acetylenic proton still present (δH 1.93). COSY correlations of δH 5.47 to diastereotopic CH2 protons at δH 3.17/δH 2.91, together with HMBC correlations of the latter to aromatic carbons at δC 136.2, δC 129.3 (Table 4) established the presence of phenyllactic acid (Pla) as the α-hydroxy acid in 6. These NMR-derived conclusions fulfilled the molecular formula requirements for C40H58N4O8 based on HRESIMS of 6.
Table 4.
NMR Data for Veraguamide F (6) in CDCl3
| unit | C/H no. | δCa | δ (J in Hz)b |
|---|---|---|---|
| Hmoya | 1 | 170.9, C | |
| 2 | 42.2, CH | 3.24 br q (7.3) | |
| 3 | 76.6, CH | 4.89, dt (10.6, 2.4) | |
| 4a | 27.5, CH2 | 2.08, m | |
| 4b | 1.64, m | ||
| 5a | 25.2, CH2 | 1.65, m | |
| 5b | 1.45, m | ||
| 6 | 18.0, CH2 | 2.20, m | |
| 7 | 83.5, C | ||
| 8 | 68.9, CH | 1.93, t (2.5) | |
| 9 | 14.6, CH3 | 1.29, d (7.3) | |
| N-Me-Val-1 | 1 | 170.7, C | |
| 2 | 65.3, CH | 3.95, d (10.4) | |
| 3 | 28.3, CH | 2.32, m | |
| 4 | 19.6, CH3 | 1.00, d (6.5) | |
| 5 | 19.7, CH3 | 0.93, d (6.5) | |
| N-Me | 28.7, CH3 | 3.06, s | |
| Pro | 1 | 172.3, C | |
| 2 | 57.1, CH | 4.97, dd (9.0, 4.5) | |
| 3a | 29.1, CH2 | 2.27, m | |
| 3b | 1.83, m | ||
| 4a | 25.0, CH2 | 2.07, m | |
| 4b | 1.97, m | ||
| 5a | 47.0, CH2 | 3.62, m | |
| 5b | 3.56, m | ||
| Pla | 1 | 165.4, C | |
| 2 | 72.8, CH | 5.47, dd (9.8, 4.0) | |
| 3 | 36.7, CH2 | 3.17, m | |
| 2.91, m | |||
| 4 | 136.2, C | ||
| 5//9 | 129.3, CH | 7.18, d (8.0) | |
| 6/8 | 128.5, CH | 7.28, m | |
| 7 | 126.7, CH | 7.20, m | |
| N-Me-Val-2 | 1 | 168.9, C | |
| 2 | 65.8, CH | 4.05, d (10.5) | |
| 3 | 27.4, CH | 2.08, m | |
| 4 | 19.8, CH3 | 0.89, d (6.8) | |
| 5 | 20.0, CH3 | 0.91, d (6.8) | |
| N-Me | 29.0, CH3 | 2.60, s | |
| Val | 1 | 173.4, C | |
| 2 | 52.7, CH | 4.76, dd (8.5, 6.1) | |
| 3 | 32.3, CH | 1.98, m | |
| 4 | 20.3, CH3 | 0.91, d (6.6) | |
| 5 | 17.7, CH3 | 0.88, d (6.6) | |
| NH | 6.28, d (8.5) |
100 MHz
600 MHz
Veraguamide G (7) lacked the acetylenic signal (δC 68.8/δH 1.93) observed for 3–6 and instead showed downfield resonances of a terminal methylene (δC 114.9/δH 4.97) and a methine (δC 138.2/δH 5.74). These signals indicated that the terminal alkyne group of the C8-polyketide derived moiety is replaced by a terminal vinyl group. This conclusion was further supported by the positive deviation of 2 amu compared to 3 and a molecular formula of C37H62N4O8. Hence,the Hmoya unit present in 3–6 was replaced by 3-hydroxy-2-methyl-7-octenoic acid (Hmoea) in 7.
Enantioselective HPLC analysis coupled with mass spectrometry or UV detection of the acid hydrolysates of 1–7 allowed us to assign the absolute configuration of all the amino acids and α-hydroxy acid components as L and S, respectively. To determine the absolute configuration at C-2 and C-3 of the Br-Hmoya unit, veraguamide A (1) was subjected to methanolysis to yield the linear fragment 9 (Figure 2). The observed coupling constant of 3.2 Hz was characteristic for a syn configuration, whereas a coupling constant near 6.3 Hz would have been expected for the anti configuration.11 The absolute configuration at C-3 and consequently for C-2 of the Br-Hmoya unit of 9 was determined using Mosher's analysis. The derived Δδ values (Figure 2) predicted an R configuration at C-3 and hence from the relative configuration, C-2 should have an S configuration. Of note, comparison of the 3JH,H values of H-2 and H-3 with a model system to assign the relative configuration could only be applied when C-3 bears a free hydroxy group. This moiety is involved in intramolecular H-bonding with the adjacent carbonyl group, thus hindering free bond rotation across C-2 and C-3.12,13 Accordingly, the corresponding MTPA-esters (10, 11) did not show the same 3JH,H values for H-2 and H-3 as that of 9. The same absolute configuration at C-2 and C-3 for Hmoya, Hmoea and 3-hydroxy-2-methyl-octanoic acid (Hmoaa) is expected based on virtually identical 13C NMR shifts and specific rotations observed for 1–8.
Figure 2.
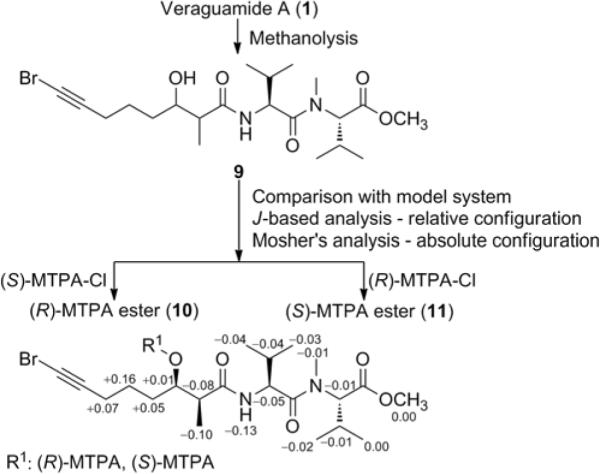
Assignment of absolute configuration of veraguamide A (1) using methanolysis and subsequent Mosher's analysis. Δδ = δ(S-MTPA ester) − δ(R-MTPA ester).
To gain insight into structure–activity relationships, veraguamide A (1) was partially (Lindlar catalyst, H2) and fully (Pd/C, H2) hydrogenated to yield the semisynthetic veraguamide G (7) and tetrahydroveraguamide A (8), respectively. The cytotoxic activities of 1–8, semisynthetic veraguamide G (7), and dolastatin 16 were evaluated for effects on viability of HT29 colorectal and HeLa cervical adenocarcinoma cells (Table 6). The IC50 values of the natural and semisynthetic veraguamide G (7) were comparable suggesting the activities of these compounds were not likely due to traces of highly biologically active impurities. The most active in this series of compounds are veraguamides D (4) and E (5), with IC50 values more than 5-fold lower than those for their related congener veraguamide C (3). This suggested that increased hydrophobicity of specific units (II, IV, V, VI) increased the cytotoxicity of this compound class, with the position having minimal effect on the bioactivity as 4 and 5 showed comparable IC50s. However, modification with bulkier groups is detrimental to the activity, as exemplified by a close to 10-fold decrease in the cytotoxic activity of 6 (Table 6) compared to 3, where a phenyllactic acid (Pla) moiety is introduced at position IV of the former instead of the Hmpa unit. The C8-polyketide derived moiety also plays a role in the cytotoxicity of these compounds. Comparing the biological activities of related compounds 1, 3, 7, and 8, weaker cytotoxicity was observed for compounds with a Br-Hmoya or Hmoaa unit, while compounds with Hmoya or Hmoea were about equally potent. This then suggests the importance of a π-system combined with the presence of acetylenic or vinylic protons in this moiety for cytotoxic activity.
Table 6.
Cytotoxic Activity (IC50, μM) of Natural and Semisynthetic Veraguamides
| Compound | HT29 | HeLa |
|---|---|---|
| Veraguamide A (1) | 26 | 21 |
| Veraguamide B (2) | 30 | 17 |
| Veraguamide C (3) | 5.8 | 6.1 |
| Veraguamide D (4) | 0.84 | 0.54 |
| Veraguamide E (5) | 1.5 | 0.83 |
| Veraguamide F (6) | 49 | 49 |
| Veraguamide G (7) | 2.7 | 2.3 |
| Tetrahydroveraguamide A (8) | 33 | 48 |
| Dolastatin 16 | 78 | 58 |
The veraguamides are reminiscent of other cyanobacterial compounds such as hantupeptins,13,14 antanapeptins,15 and trungapeptins.16 These compounds are also cyclic hexadepsipeptides with the characteristic C8-polyketide derived units as Hmoya, Hmoea, or Hmoaa. Veraguamide F (6) is a constitutional isomer of antanapeptin D,15 where an N-Me-Phe and Hiva are present in the latter instead of Pla and N-Me-Val as in 6. It is interesting that subtle changes in structure of these compounds have a profound effect on the cytotoxicity. Antanapeptins A–D (brine shrimp) as well as trungapeptin A (KB and LoVo cells) did not display cytotoxicity at the reported concentrations (10 μg/mL),15,16 while hantupeptins A–C were cytotoxic against MOLT-4 leukemia and MCF7 breast cancer cells, with hantupeptin A being the most active in this series with IC50s of 32 nM and 4.0 μM, respectively.13 Trungapeptins B and C were not tested for cytotoxicity.16
Although dolastatin 16 has been isolated previously from sea hares8 and cyanobacteria,15,17 its stereostructure has been only partially assigned. Therefore we analyzed the remaining dolaphenvaline (Dpv) and dolamethylleucine (Dml) units. The absolute configuration of the Dpv unit in dolastatin 16 was determined as 2S,3R using enantioselective HPLC-MS analysis of the FDLA-derivatized acid hydrolysate in comparison with that for pitiprolamide.18 The configuration of the Dpv unit in dolastatin 16 is the same as in pitiprolamide and kulokekahilide-1 determined from X-ray crystallography and chemical synthesis, respectively.18,19 To determine the configuration of the stereocenters in the Dml unit, we carried out acid and base hydrolysis of dolastatin 16 to yield the Dml unit and the linear fragment 12, respectively. The Dml unit is related to the 3-amino-2-methylpentanoic acid (Map) unit, present in dolastatin 1220,21 and majusculamide C,22 as well as the 3-amino-2-methylhexanoic acid (Amha) unit in kulokekahilide-1,19 lyngbyastatin 1,21 and the ulongamides.23 For all these α-substituted β-amino acids, the corresponding Marfey's adducts of the two 3R isomers (2S,3R and 2R,3R) consistently eluted later by reversed-phase C18 HPLC than those of the two 3S isomers (2S,3S and 2R,3S), while the C-2 stereoisomers are poorly resolved and variable.19–23 Extrapolation to the elution order we obtained for the Dml suggested the presence of 3R in the Dml unit of dolastatin 16.24 To assign the configuration at C-2 of Dml, modified Mosher's analysis using phenylglycine methyl ester (PGME)25 derivatization of 12 (Figure 3) suggested a 2R configuration. The applicability of this chiral derivatization technique has been demonstrated for α- and β- substituted carboxylic acids,25 but may not have been widely used for α,β-disubstituted carboxylic acids. From the Δδ values that we obtained (Figure 3), it is predicted that there is some deviation from the presumed conformation of the PGME amide, but nonetheless a systematic arrangement of positive and negative Δδ values was obtained. Therefore, we propose a 2R,3R configuration for the Dml unit of dolastatin 16. The 13C NMR chemical shifts for the Dpv and Dml unit in both dolastatin 16 and homodolastatin 16 were similar, suggesting the latter would also have a (2S,3R)-Dpv and (2R,3R)-Dml units.8,26 Dolastatin 16 showed weak cytotoxic activity with IC50 of 78 μM and 58 μM for HT29 and HeLa cell lines, respectively. These values are significantly higher than those reported by Pettit et al. against a different panel of cell lines.8
Figure 3.

Assignment of absolute configuration dolastatin 16 by base hydrolysis and modified Mosher's analysis using PGME. Δδ = δ(S-PGME amide) − δ(R-PGME amide).
Experimental Section
General Experimental Procedures
Optical rotations were measured on a Perkin-Elmer 341 polarimeter. UV spectra were recorded on SpectraMax M5 (Molecular Devices). 1H and 2D NMR spectra were recorded in CDCl3 on a Bruker Avance II 600 MHz spectrometer equipped with a 5-mm TXI cryogenic probe using residual solvent signals (δH 7.26; δC 77.0) as internal standards. HSQC and HMBC experiments were optimized for 1JCH = 145 and nJCH = 7 Hz, respectively. TOCSY experiments were done using a mixing time of 100 ms. 13C NMR spectra were recorded on Varian 400 MHz or Bruker 500 MHz NMR spectrometers. HRESIMS data was obtained using an Agilent LC-TOF mass spectrometer equipped with an APCI/ESI multimode ion source detector. LRESIMS measurements and MS/MS fragmentation were done on an ABI 3200Q TRAP.
Biological Material
The Symploca cf. hydnoides cyanobacterium was collected by hand while snorkeling in the shallow waters of the southern fore-reef (1–3 m) of Cetti Bay, Guam on April 17, 2009. A voucher specimen, which is preserved in 100% EtOH, is deposited in the University of Guam Herbarium (accession no. GUAM-GH11446). A voucher specimen is also retained at the Smithsonian Marine Station, Fort Pierce, FL.
Extraction and Isolation
The freeze-dried cyanobacterium (142.0 g) was extracted with EtOAc–MeOH (1:1) to yield the nonpolar extract (11.6 g). This was partitioned between hexanes and 20% aqueous MeOH, the latter concentrated under reduced pressure and further partitioned between n-BuOH and H2O. The n-BuOH fraction was concentrated to dryness (2.84 g) and chromatographed on Si gel eluting first with CH2Cl2, followed by increasing concentrations of i-PrOH, while after 100% i-PrOH, increasing gradients of MeOH were used. The 20% i-PrOH fraction was subjected to a C18 SPE eluting with 25%, 50%, 75%, and 100% MeOH in H2O. The 100% MeOH fraction was purified by semipreparative reversed-phase HPLC (Phenomenex Synergi-Hydro RP, 250 × 10 mm, 4 μm; flow rate, 2.0 mL/min) using a linear gradient of MeOH–H2O (70%–100% MeOH in 60 min and then 100% MeOH for 10 min) to yield dolastatin 16 (tR 32.6 min, 21.6 mg), semipure veraguamide C (tR 36.4 min, 15.1 mg), semipure veraguamide F (tR 37.4 min, 10.0 mg), a mixture of veraguamides B and D (tR 40.0 min, 25.0 mg), veraguamide A (1) (tR 42.6 min, 25.9 mg), and a mixture of veraguamides E and G (tR 43.7 min, 9.0 mg).
The final purification of the semipure veraguamide C (3) was achieved using semipreparative HPLC (Phenomenex Phenyl-hexyl, 250 × 10 mm, 4 μm; flow rate, 2.0 mL/min) using a linear gradient of MeOH–H2O (85%–100% MeOH in 40 min and then 100% MeOH for 5 min) to yield veraguamide C (3) (tR 19.9 min, 10.7 mg). Using the same chromatographic conditions, purification of the semipure veraguamide F yielded veraguamide F (6) (tR 21.7 min, 6.8 mg). The mixture of veraguamides B and D was resolved using the same chromatographic condition with a different linear gradient (70%–100% MeOH in 45 min and then 100% MeOH for 10 min) to yield veraguamide D (4) (tR 36.6 min, 4.0 mg) and veraguamide B (2) (tR 37.3 min, 11.5 mg). The mixture of veraguamides E and G was further purified using the same chromatographic conditions to yield veraguamide G (7) (tR 39.9 min, 4.4 mg) and veraguamide E (5) (tR 40.5 min, 3.6 mg).
Hydrogenation of 1
A catalytic amount of 10% Pd/C was added to a methanolic solution of 1 (1.8 mg/mL). The reaction was left to stir for 6 h under a hydrogen balloon. The catalyst was filtered through a Celite pad, and the filtrate, upon concentration, was purified by semipreparative HPLC (Phenomenex Phenyl-hexyl, 250 × 10 mm, 4 μm; flow rate, 2.0 mL/min) using a linear gradient of MeOH–H2O (70%–100% MeOH in 45 min and then 100% MeOH for 10 min) to yield 8 (tR 40.9 min, 1.2 mg).
Partial hydrogenation of 1 was carried out with Lindlar catalyst, using the same reaction and chromatographic conditions stated above. This afforded the semisynthetic veraguamide G (tR 39.9 min, 1.7 mg). The LRESIMS and 1H NMR spectra of the semisynthetic veraguamide G were in good agreement with the spectra for the natural product (7).
Veraguamide A (1)
colorless, amorphous solid; [α]20D −44 (c 0.44, MeOH); UV (MeOH); λmax (log ε) 202 (6.29); IR (film) νmax 3428, 2966, 2877, 2361, 2334, 1736, 1647, 1190 cm−1; 1H NMR ,13C NMR, COSY, and HMBC data, see Table 1; HRESIMS m/z 767.3675 [M + H]+ (calcd for C37H6079BrN4O8, 767.3594), m/z [M + H]+ 769.3660 (calcd for C37H6081BrN4O8, 769.3574) (100:100 [M + H]+ ion cluster).
Veraguamide B (2)
colorless, amorphous solid; [α]20D −40c 0.16, MeOH); UV (MeOH); λmax (log ε) 202 (4.30); IR (film) νmax 3378, 2966, 2886, 1738, 1657, 1190 cm−1; 1H NMR and 13C NMR data, see Table 2; HRESIMS m/z 753.3517 [M + H]+ (calcd for C36H5879BrN4O8, 753.3438), m/z [M + H]+ 755.3508 (calcd for C36H5881BrN4O8, 755.3418) (100:100 [M + H]+ ion cluster).
Veraguamide C (3)
colorless, amorphous solid; [α]20D −44 (c 0.31, MeOH); UV (MeOH); λmax (log ε) 202 (4.17); IR (film) ν 3429, 2965, 2871, 1732, 1650, 1190 cm−1;1H NMR and 13C NMR data, see Table 2; HRESIMS m/z 689.4486 [M + H]+ (calcd for C37H61N4O8, 689.4490).
Veraguamide D (4)
colorless, amorphous solid; [α]20D −57 (c 0.11, MeOH); UV (MeOH); λmax (log ε) 202 (4.30); IR (film) νmax 3378, 2958, 2878, 1732, 1647 cm−1; 1H NMR and 13C NMR data, see Table 3; HRESIMS m/z 703.4639 [M + H]+ (calcd for C38H63N4O8, 703.4646).
Veraguamide E (5)
colorless, amorphous solid; [α]20D −56 (c 0.22, MeOH); UV (MeOH); λmax (log ε) 202 (4.30); IR (film) νmax 3320, 2958, 2877, 1733, 1647 cm−1; 1H NMR and 13C NMR data, see Table 3; HRESIMS m/z 717.4799 [M + H]+ (calcd for C39H65N4O8, 717.4802).
Veraguamide F (6)
colorless, amorphous solid; [α]20D −41 (c 0.13, MeOH); UV (MeOH); λmax (log ε) 206 (4.33); IR (film) νmax 3450, 3306, 2965, 2871, 1737, 1661, 1647, 1490 cm−1; 1H NMR and 13C NMR data, see Table 4; HRESIMS m/z 723.4411 [M + H]+ (calcd for C40H59N4O8, 723.4333).
Veraguamide G (7)
colorless, amorphous solid; [α]20D −41 (c 0.17, MeOH); UV (MeOH); λmax (log ε) 202 (4.26); IR (film) νmax 3436, 2965, 2879, 1740, 1661, 1646 cm−1; 1H NMR and 13C NMR data, see Table 5; HRESIMS m/z 691.4649 [M + H]+ (calcd for C40H59N4O8, 691.4646).
Table 5.
NMR Data for Veraguamide G (7) and Tetrahydroveraguamide A (8) in CDCl3
| 7 | 8 | ||||
|---|---|---|---|---|---|
| unit | C/H no. | δ Ca | δH (J in Hz)b | δCb,c | δH (J in Hz)b |
| Hmoead/Hmoaae | 1 | 170.9, C | 170.7, C | ||
| 2 | 42.4, CH | 3.10, br q (7.4) | 42.1, CH | 3.08, br q (7.4) | |
| 3 | 76.8, CH | 4.85, dt (10.6, 2.4) | 76.8, CH | 4.86, dt (10.1, 2.1) | |
| 4a | 27.9, CH2 | 1.98, m | 31.0, CH2 | 1.21, m | |
| 4b | 1.45, m | 1.26, m | |||
| 5a | 25.5, CH2 | 1.48, m | 28.2, CH2 | 1.39, m | |
| 5b | 1.30, m | ||||
| 6a | 33.2, CH2 | 2.05, m | 25.8, CH2 | 1.39, m | |
| 6b | 1.20, m | ||||
| 7 | 138.2, CH | 5.74, m | 22.2, CH2 | 1.26, m | |
| 8 | 114.9, CH2 | 4.97, m | 13.6, CH3 | 0.85, t (6.9) | |
| 9 | 14.4, CH3 | 1.22, d (7.4) | 14.0, CH3 | 1.23, d (7.4) | |
| N-Me-Val-1 | 1 | 170.7, C | 170.7, C | ||
| 2 | 65.0, CH | 3.93, d (9.8) | 64.9, CH | 3.93, d (10.7) | |
| 3 | 28.3, CH | 2.28, m | 28.3, CH | 2.28, m | |
| 4 | 19.59, CH3 | 0.98, d (6.4) | 19.2, CH3 | 0.98, d (6.5) | |
| 5 | 19.54, CH3 | 0.92, d (6.4) | 19.3, CH3 | 0.91, d (6.5) | |
| N-Me | 28.6, CH3 | 3.01, s | 28.4, CH3 | 3.00, s | |
| Pro | 1 | 172.2, C | 172.0, C | ||
| 2 | 57.3, CH | 4.95, dd (8.7, 5.0) | 57.1, CH | 4.94, dd (9.0, 5.0) | |
| 3a | 29.4, CH2 | 2.29, m | 29.1, CH2 | 2.28, m | |
| 3b | 1.79, m | 1.79, m | |||
| 4a | 24.9, CH2 | 2.03, m | 24.6, CH2 | 2.03, m | |
| 4b | 1.98, m | 1.99, m | |||
| 5a | 47.3, CH2 | 3.84, dt (−16.7, 7.1) | 47.0, CH2 | 3.84, dd (−17.0, 7.3) | |
| 5b | 3.61, dt (−16.7, 7.1) | 3.60, dd (−17.0, 7.3) | |||
| Hmpa | 1 | 165.9, C | 165.7, C | ||
| 2 | 76.0, CH | 4.90, d (8.7) | 76.6, CH | 4.90, d (8.8) | |
| 3 | 35.7, CH | 1.98, m | 35.4, CH | 1.98, m | |
| 4 | 24.8, CH2 | 1.54, m | 24.5, CH2 | 1.54, m | |
| 1.13, m | 1.13, m | ||||
| 5 | 10.5, CH3 | 0.86, t (7.3) | 10.2, CH3 | 0.86, t (7.1) | |
| 6 | 13.8, CH3 | 1.00, d (6.0) | 13.5, CH3 | 1.00, d (6.4) | |
| N-Me-Val-2 | 1 | 169.6, C | 169.5, C | ||
| 2 | 66.0, CH | 4.15, d (10.2) | 65.8 CH | 4.14, d (9.6) | |
| 3 | 28.6, CH | 2.28, m | 28.1, CH | 2.28, m | |
| 4 | 20.4, CH3 | 1.00, d (6.1) | 20.0, CH3 | 0.99, d (6.6) | |
| 5 | 20.2, CH3 | 1.10, d (6.1) | 19.9, CH3 | 1.10, d (6.6) | |
| N-Me | 30.0, CH3 | 2.93, s | 29.7, CH3 | 2.93, s | |
| Val | 1 | 173.4, C | 173.3, C | ||
| 2 | 52.7, CH | 4.70, dd (8.6, 6.7) | 52.5, CH | 4.70, dd (8.6, 6.2) | |
| 3 | 32.1, CH | 1.98, m | 31.7, CH | 1.96, m | |
| 4 | 20.3, CH3 | 0.93, d (6.8) | 19.3, CH3 | 0.93, d (6.3) | |
| 5 | 17.6, CH3 | 0.86, d (6.8) | 17.2, CH3 | 0.87, d (6.3) | |
| NH | 6.23, d (8.6) | 6.26, d (8.6) | |||
125 MHz
600 MHz
Based on HSQC and HMBC
Refers to Compound 7
Refers to Compound 8
Tetrahydroveraguamide A (8)
colorless, amorphous solid; [α]20D −43 (c 0.05, MeOH); UV (MeOH); λmax (log ε) 202 (4.33); 1H NMR and 13C NMR data, see Table 5; HRESIMS m/z 693.4791 [M + H]+ (calcd for C37H65N4O8, 693.4802).
Dolastatin 16
colorless, amorphous solid; [α]20D +22 (c 0.18, MeOH) {lit.8 [α]20D +15.5 (c 0.20, MeOH)}; UV (MeOH); λmax (log ε) 202 (4.51); IR (film) νmax 3407, 2965, 2871, 1733, 1647 cm−1; 1H NMR and 13C NMR data are identical with literature values8 (Supporting Information); LRESIMS m/z 879.6 [M + H]+, 901.3 [M + Na]+.
Acid Hydrolysis of Veraguamides and Enantioselective Analysis
Portions of 1–7 (100 μg) were acid-hydrolyzed (200 μL 6 N HCl, 110 °C, 20 h), the product mixtures dried, reconstituted in 100 μL H2O and analyzed by enantioselective HPLC-UV and enantioselective HPLC-MS. The absolute configurations of the amino acids N-Me-Ile, Ile, N-Me-Val, Val, and Pro were determined by enantioselective HPLC-MS [column, Chirobiotic TAG (250 × 4.6 mm), Supelco; solvent, MeOH–10 mM NH4OAc (40:60, pH 5.30); flow rate, 0.5 mL/min; detection by ESIMS in positive ion mode (MRM scan)]. The acid hydrolysates of 1–4, 6, and 7 showed retention times at 7.8, 11.6, and 13.6 min corresponding to L-Val, N-Me-L-Val, and L-Pro, respectively. The acid hydrolysate of 4 in addition showed a retention time at 12.4 min corresponding to N-Me-L-Ile. The acid hydrolysate of 5 showed retention times at 8.4, 11.6, 12.4, and 13.6 min, corresponding to L-Ile, N-Me-L-Val, N-Me-L-Ile, and L-Pro, respectively. The retention times (tR, min; MRM ion pair) of the authentic amino acids were as follows: NMe-L-Val (11.6; 132→86), N-Me-D-Val (34.3), L-Val (7.8; 118→72), D-Val (13.7), N-Me-L-Ile (12.4; 146→100), N-Me-L-allo-Ile (15.0), N-Me-D-Ile (49.0), N-Me-D-allo-Ile (51.0), L-Ile (8.4; 132→86), L-allo-Ile (8.6), D-allo-Ile (17.6), D-Ile (20.2), L-Pro (13.6; 116→70), D-Pro (36.0). Compound-dependent parameters used were as follows: N-Me-Val: DP 29.4, EP 4.2, CE 17.4, CXP 2.7, CEP 10.6; Val: DP 5.7, EP 9.0, CE 40.0, CXP 8.0, CEP 10.0; N-Me-Ile: DP 35.0, EP 7.0, CE 17.0, CXP 2.0, CEP 10.0; Ile: DP 40.0, EP 9.0, CE 15.0, CXP 3.0, CEP 8.0; Pro: DP 35.0, EP 7.7, CE 22.7, CXP 5.0, CEP 10.3. Source gas parameters used were as follows: CUR 40, CAD Medium, IS 4500, TEM 750, GS1 65, GS2 65.
The absolute configurations of the α-hydroxy acids [2-hydroxyisovaleric acid (Hiva), 2-hydroxy-3-methylpentanoic acid (Hmpa), and phenyllactic acid (Pla)] were determined using enantioselective HPLC [column, CHIRALPAK MA (+) (50 × 4.6 mm); solvent, CH3CN–2 mM CuSO4 (10:90); flow rate, 1.0 mL/min; detection by UV (254 nm)]. The acid hydrolysates of 1, 3–5, and 7 each showed peaks at 33.0 min corresponding to (2S,3S)-Hmpa. The acid hydrolysate of 2 contained a component that had a retention time at 10.0 min corresponding to (2S)-Hiva, while 6 gave a peak at 51.0 min corresponding to (2S)-Pla. The retention times of the authentic standards were as follows: (2R)-Hiva (6.0), (2S)-Hiva (10.0), (2R,3S)-Hmpa (16.0), (2R,3R)-Hmpa (19.0), (2S,3R)-Hmpa (26.0), (2S,3S)-Hmpa (33.0), (2R)-Pla (33.5), (2S)-Pla (51.0). All other amino acid units eluted within less than 5.0 min using this chromatographic condition.
Methanolysis of 1
Compound 1 (5.0 mg) was dissolved in 2.0 mL of 5% (w/w) methanolic KOH solution and stirred for 24 h at room temperature. The solvent was evaporated and the residue was partitioned between CH2Cl2 and H2O. The organic layer was collected, dried over anhydrous MgSO4 and concentrated to dryness under nitrogen. The crude methanolysis product was further purified by semipreparative reversed-phase HPLC (Phenomenex Synergi-Hydro RP, 250 × 10 mm, 4 μm; flow rate, 2.0 mL/min) using a linear gradient of MeOH–H2O (70%–100% MeOH in 60 min and then 100% MeOH for 10 min) to yield 9 (tR 19.3 min, 1.4 mg).
9
colorless, amorphous solid; 1H NMR (CDCl3) δ 6.30 (d, J = 8.1 Hz, 1H), 4.92 (d, J = 10.5 Hz, 1H), 4.76 (dd, J = 9.1, 6.6 Hz, 1H), 3.78 (ddd, J = 8.6, 4.4, 3.2 Hz, 1H), 3.49 (s, 3H), 3.08 (s, 3H), 2.42 (qd, J = 6.8, 3.2 Hz , 1H), 2.23 (m, 2H), 2.03 (m, 1H), 1.74 (m, 2H), 1.56 (m, 1H), 1.46 (m, 2H), 1.01 (d, J = 6.8 Hz, 3H), 0.98 (d, J = 6.8 Hz, 3H), 0.93 (d, J = 6.8 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H); HRESIMS m/z 497.1640 [M + Na]+ (calcd for C21H3579BrN2O5Na, 497.1627), m/z [M + Na]+ 499.1616 (calcd for C21H3581Br N2O5Na, 499.1607) (100:100 [M + Na]+ ion cluster).
Preparation of MTPA Esters
The methanolysis product 9 was dissolved in 50 μL CDCl3, and was divided into two equal portions and to each was added 0.75 mL triethylamine. To one portion was added 10 μL of (R)-MTPA-Cl and to another was added 10 μL of (S)-MTPA-Cl to give the (S)-MTPA ester (10) and (R)-MTPA ester (11), respectively. Each reaction was allowed to stir for 24 h and 10 μL of N,N-dimethylaminopropylamine was added to quench the reactions. The reaction products were dried under N2 and applied onto silica SPE eluting with EtOAc – hexanes (1:1). The semipure product was further purified by semipreparative HPLC (Phenomenex Phenyl-hexyl, 250 × 10 mm, 4 μm; flow rate, 2.0 mL/min) using a linear gradient of MeOH–H2O (70%–100% MeOH in 45 min and then 100% MeOH for 10 min) to yield 10 (tR 38.0 min, 0.1 mg) or 11 (tR 37.8 min, 0.1 mg).
10
colorless, amorphous solid; 1H NMR (CDCl3) δ 7.57 (dd, J = 6.4, 2.7 Hz, 2H), 7.41 (m, 3H), 6.14 (d, J = 9.2 Hz, 1H), 5.28 (q, J = 6.9 Hz, 1H), 4.93 (d, J = 10.6 Hz, 1H), 4.68 (dd, J = 9.1, 7.6 Hz, 1H), 3.69 (s, 3H), 3.58 (s, 3H), 3.06 (s, 3H), 2.46 (quintet, J = 7.1 Hz , 1H), 2.21 (m, 1H), 2.15 (t, J = 6.8 Hz, 2H), 1.95 (m, 1H), 1.68 (m, 1H), 1.47 (m, 2H), 1.09 (d, J = 7.0 Hz, 3H), 1.01 (d, J = 6.6 Hz, 3H), 0.92 (d, J = 6.9 Hz, 3H), 0.86 (d, J = 6.9 Hz, 3H), 0.81 (d, J = 6.9 Hz, 3H); HRESIMS m/z 729.1746 [M + K]+ (calcd for C31H4279BrF3N2O7K, 729.1759), m/z [M + K]+ 731.1747 (calcd for C31H4281BrF3N2O7K, 731.1755) (100:100 [M + K]+ ion cluster); LRESIMS m/z 691/693 (100:100 [M + H]+ ion cluster), 713/715 (100:100 [M + Na]+ ion cluster).
11
colorless, amorphous solid; 1H NMR (CDCl3) δ 7.56 (dd, J = 4.3, 3.6 Hz, 2H), 7.41 (m, 3H), 6.27 (d, J = 8.3 Hz, 1H), 5.27 (q, J = 6.0 Hz, 1H), 4.94 (d, J = 10.4 Hz, 1H), 4.73 (dd, J = 9.1, 7.5 Hz, 1H), 3.69 (s, 3H), 3.57 (s, 3H), 3.07 (s, 3H), 2.54 (quintet, J = 6.7 Hz, 1H), 2.22 (m, 1H), 2.08 (td, J = 7.2, 2.9 Hz, 2H), 2.00 (m, 1H), 1.63 (m, 1H), 1.31 (m, 2H), 1.19 (d, J = 6.7 Hz, 3H), 1.01 (d, J = 6.3 Hz, 3H), 0.95 (d, J = 6.8 Hz, 3H), 0.90 (d, J = 7.0 Hz, 3H), 0.83 (d, J = 6.5 Hz, 3H); HRESI/APCIMS m/z 691.2202 [M + H]+ (calcd for C31H4379BrF3N2O7, 691.2206), m/z [M + H]+ 693.2186 (calcd for C31H4381BrF3N2O7, 693.2186) (100:100 [M + H]+ ion cluster).
Advanced Marfey's Analysis of Dpv Unit in Dolastatin 16
To liberate the authentic (2S,3R)-Dpv standard, pitiprolamide (500 μg) was hydrolyzed (400 μL of 6 N HCl, 110 °C, 20 h). The mixture was dried and reconstituted in 400 μL H2O. Half of this sample was used to epimerize the α-amino acids in this mixture (40 μL Et3N and 40 μL Ac2O, heating at 60 °C for 1 h)27 to yield a mixture of (2S,3R)-Dpv and (2R,3R)-Dpv standards. These standards were derivatized with 1% (w/v) Marfey's reagent (L-FDLA and DL-FDLA).28 Dolastatin 16 was hydrolyzed and derivatized with 1% (w/v) L-FDLA according to the procedure stated above. The absolute configuration of the Dpv unit was determined by reversed-phase HPLC-MS [column, Phenomenex Synergi Hydro-RP (150 × 4.6 mm) using a linear gradient of 0.1% HCOOH in MeOH– 0.1% aqueous HCOOH (40%–100% for 45 min and then 100% MeOH for 10 min); flow rate, 0.5 mL/min; detection by ESIMS in negative ion mode (MRM scan)] of the FDLA adducts. The acid hydrolysate of dolastatin 16 showed a peak at 36.1 min corresponding to (2S,3R)-Dpv. Coinjection of the acid hydrolysate with the DL-FDLA derivatized epimerization product showed enrichment of the peak corresponding to (2S,3R)-Dpv. The retention times (tR, min; MRM ion pair) of the authentic amino acid L-FDLA adducts were as follows: (2S,3R)-Dpv (36.1; 486→191), (2S,3S)-Dpv [(36.6); (= D-FDLA derivatized (2R,3R)-Dpv)], (2R,3S)-Dpv [(43.5); (= D-FDLA derivatized (2S,3R)-Dpv)], (2R,3R)-Dpv (44.3). Compound-dependent parameters used were as follows: DP −95.0, EP −6.0, CE −31.0, CXP −2.0, CEP −26.0. Source gas parameters used were as follows: CUR 40, CAD High, IS 4500, TEM 450, GS1 40, GS2 40.
Advanced Marfey's Analysis of β-Amino Acid Unit in Dolastatin 12 and Dolastatin 16
Samples of dolastatin 12 and dolastatin 16 (0.2 mg each) were hydrolyzed (200 μL of 6 N HCl, 110 °C, 20 h) to liberate the β-amino acids Map and Dml, respectively. Another portion of dolastatin 12 and dolastatin 16 (0.2 mg each) was first heated with 3 N NaOH (80 °C, 3 h), the resulting product mixture acidified with 5% (v/v) HCl, dried under N2, and hydrolyzed (200 μL of 6 N HCl, 110 °C, 20 h) to liberate the partially C-2 epimerized β-amino acid mixture. The reaction products were derivatized using L-FDLA and DL-FDLA.28 The order of elution of the β-amino acids Dml and Map liberated from dolastatin 16 and dolastatin 12, respectively, was determined by reversed-phase HPLC-MS [column, Phenomenex Synergi Hydro-RP (150 × 4.6 mm) using a linear gradient of 0.1% HCOOH in MeOH− 0.1% aqueous HCOOH (25%–80% for 50 min and then 100% MeOH for 5 min); flow rate, 0.5 mL/min; detection by ESIMS in negative ion mode (MRM scan)] of the FDLA adducts. The enantiomers bearing the 3R configuration consistently eluted later than those with the 3S configuration. The retention times (tR, min; MRM ion pair) of the Dml-L-FDLA adducts were as follows: (2S,3S)-Dml [(29.3; 438.2→310.3); (= D-FDLA derivatized (2R,3R)-Dml)], (2R,3S)-Dml [(30.8); (= D-FDLA derivatized (2S,3R)-Dml)], (2R,3R)-Dml (33.7), (2S,3R)-Dml (35.6). The retention times (tR, min; MRM ion pair) of the Map-L-FDLA adducts were as follows: (2R,3S)-Map [(27.5; 424.5→310.4); (= D-FDLA derivatized (2S,3R)-Map)], (2S,3S)-Map [(29.6); (= D-FDLA derivatized (2R,3R)-Dml)], (2R,3R)-Dml (31.0), (2S,3R)-Dml (32.5). Compound-dependent parameters used were as follows: DP −95.0, EP −6.0, CE −31.0, CXP −2.0, CEP −26.0. Source gas parameters used were as follows: CUR 40, CAD Medium, IS 4500, TEM 450, GS1 40, GS2 40.
Base Hydrolysis of Dolastatin 16
Dolastatin 16 (4.0 mg) was hydrolyzed using 2 N KOH–MeOH (1:1) for 3 h at 80 °C. The base hydrolysate was neutralized with 2 N HCl, partitioned between EtOAc:H2O, the organic layer collected and dried under N2. The crude base hydrolysate product was purified by semipreparative HPLC (Phenomenex Synergi Hydro-RP, 250 × 10 mm, 4 μm; flow rate, 2.0 mL/min) using a linear gradient of ACN–H2O (40%–100% MeOH in 45 min) to yield 12 (tR 22.7 min, 1.0 mg).
12
colorless, amorphous solid; 1H NMR (CDCl3) δ 7.13 – 7.26 (m), 6.81 (d, J = 9.0 Hz, 1H), 6.65 (d, J = 10.4 Hz, 1H), 4.83 (d, J = 10.6 Hz, 1H), 4.73 (dd, J = 8.7, 3.8 Hz, 1H), 4.59 (m, 2H), 4.48 (m, 1H), 4.28 (m, 1H), 3.79 (m, 1H), 3.56 – 3.69 (m, 2H), 3.50 (m, 1H), 3.36 (m, 1H), 3.16 (s, 3H), 2.70 (m, 2H), 2.24 – 2.48 (m, 6H), 1.94 – 2.19 (m, 2H), 1.77 – 1.94 (m), 1.14 (d, J = 6.8 Hz, 3H), 1.03 (d, J = 7.3 Hz, 3H), 0.96 (m, 6H), 0.90 (m, 6H), 0.80 (m, 6H); HRESI/APCIMS m/z 750.4407 [M + Na]+ (calcd for C39H61N5O8Na, 750.4418).
Modified Mosher's Analysis Using PGME
The base hydrolysate product was dissolved in 600 μL DMF-d7 and divided into two portions. To one portion was added 0.7 mg (S)-PGME, and the solution cooled and stirred at 0 °C before the successive addition of PyBop (1.8 mg), HOBt (0.5 mg), and N-Me morpholine (5.0 μL). The reaction was left to stir for another 3 h at room temperature and quenched with the addition of EtOAc. The resulting solution was successively washed with 5% HCl, saturated NaHCO3, and brine. The organic layer was collected and dried over anhydrous MgSO4 before drying under N2 to yield 13 (0.8 mg). The same procedure was used to prepare the (R)-PGME derivatized product 14 (0.6 mg). 1H NMR chemical shifts of the Dml spin system were assigned by COSY and TOCSY analyses.
13
colorless, amorphous solid; 1H NMR (CDCl3) δ 7.27 – 7.38 (m, 12H, incl. NH Dml at 7.27), 5.57 (dd, J = 14.0, 7.4 Hz, 1H), 5.02 (d, J = 10.2 Hz, 1H), 4.42 (m, 2H), 4.24 (d, J = 2.6 Hz, 1H), 3.74 (s, 3H), 3.56 – 3.65 (m, 3H, incl. H-3 Dml at 3.62), 3.45 (m, 2H), 3.29 (m, 1H), 2.99 (s, 3H), 2.75 (m, 2H, incl. H-2 Dml), 2.30 – 2.40 (m, 5H), 2.02 – 2.09 (m, 4H), 1.97 – 2.00 (m, 4H), 1.45 (m, 1H, H-5 Dml), 1.19 (d, J = 7.2 Hz, 3H, H3-7 Dml), 1.04 (d, J = 6.6 Hz, 3H), 0.95 (d, J = 6.8 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H, H3-6 Dml), 0.85 (d, J = 6.8 Hz, 3H), 0.82 (d, J = 6.7 Hz, 3H, H3-5 Dml), 0.80 (d, J = 6.6 Hz, 6H); HRESI/APCIMS m/z 897.5099 [M + Na]+ (calcd for C48H70F3N6O9Na, 897.5102).
14
colorless, amorphous solid; 1H NMR (CDCl3) δ 7.30 – 7.40 (m, 10H), 7.14 – 7.24 (m, 4H, incl. NH Dml at 7.21), 5.46 (d, J = 7.3 Hz, 1H), 4.99 (d, J = 10.2 Hz, 1H), 4.52 (dd, J = 6.4, 2.3 Hz, 1H), 4.42 (t, J = 6.4 Hz, 1H), 4.27 (d, J = 2.0 Hz, 1H), 4.01, (m, 1H), 3.72 (s, 3H), 3.56 – 3.67 (m, 3H, incl. H-3 Dml at 3.64), 3.02 (s, 3H), 2.65 – 2.68 (m, 2H, incl. H-2 Dml at 2.68), 2.30 – 2.40 (m, 5H), 1.89 – 2.02 (m, 8H), 1.62 (m, 1H, H-5 Dml), 1.13 (d, J = 6.8 Hz, 3H, H3-7 Dml), 1.03 (d, J = 6.7 Hz, 3H), 0.98 (d, J = 6.8 Hz, 3H, H3-6 Dml), 0.96 (d, J = 6.7 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H, H3-5 Dml), 0.86 (d, J = 6.8 Hz, 3H), 0.81 (d, J = 6.7 Hz, 6H); HRESI/APCIMS m/z 897.5101 [M + Na]+ (calcd for C48H70F3N6O9Na, 897.5102).
Cell Viability Assay
HT29 colorectal adenocarcinoma and HeLa cervical carcinoma cells were cultured in Dulbecco's modified Eagle medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Hyclone) under a humidified environment with 5% CO2 at 37 °C. HeLa (3,000) and HT29 (12,500) cells were seeded in 96-well plates and treated with varying concentrations of 1–8, and dolastatin 16, positive control (paclitaxel), and solvent control (DMSO) after 24 h of seeding. The cells were incubated for an additional 48 h before the addition of the MTT reagent. Cell viability was measured according to the manufacturer's instructions (Promega). IC50 values for positive control were 3.0 and 2.6 nM for HT29 and HeLa cells, respectively. IC50 calculations were done by GraphPad Prism 5.03 based on duplicate experiments.
Supplementary Material

Acknowledgment
This research was supported by the National Institutes of Health, NIGMS grant P41GM086210. We are grateful to J. Rocca for assistance in obtaining the NMR spectra, R. Montaser for providing an authentic standard of pitiprolamide, and J. Quiñata of the Cetti Bay Ecostation (Agat, Guam) for providing the UOGML with access for collections. This is a contribution of the University of Guam Marine Laboratory and contribution #843 from the Smithsonian Marine Station at Fort Pierce.
Footnotes
Supporting Information Available: NMR spectra for compounds 1–14 and dolastatin 16. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- (1).Tan LT. J. Appl. Phycol. 2010;22:659–676. [Google Scholar]
- (2).Nunnery JK, Mevers E, Gerwick WH. Curr. Opinion Biotechnol. 2010;21:787–793. doi: 10.1016/j.copbio.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Harrigan GG, Luesch H, Yoshida WY, Moore RE, Nagle DG, Paul VJ, Mooberry SL, Corbett TH, Valeriote FA. J. Nat. Prod. 1998;61:1075–1077. doi: 10.1021/np980321c. [DOI] [PubMed] [Google Scholar]
- (4).Luesch H, Moore RE, Paul VJ, Mooberry SL, Corbett TH. J. Nat. Prod. 2001;64:907–910. doi: 10.1021/np010049y. [DOI] [PubMed] [Google Scholar]
- (5).Mooberry SL, Leal RM, Tinley TL, Luesch H, Moore RE, Corbett TH. Int. J. Cancer. 2003;104:512–521. doi: 10.1002/ijc.10982. [DOI] [PubMed] [Google Scholar]
- (6).Taori K, Paul VJ, Luesch H. J. Am. Chem. Soc. 2008;130:1806–1807. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- (7).Liu Y, Salvador LA, Byeon S, Ying Y, Kwan JC, Law BK, Hong J, Luesch H. J. Pharmacol. Exp. Ther. 2010;335:351–361. doi: 10.1124/jpet.110.172387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Pettit GR, Xu J, Hogan F, Williams MD, Doubek DL, Schmidt JM, Cerny RL, Boyd MR. J. Nat. Prod. 1997;60:752–754. doi: 10.1021/np9700230. [DOI] [PubMed] [Google Scholar]
- (9).The trivial names were assigned to conform with the naming by W. Gerwick and co-workers, who concurrently isolated members of this compound class: Mevers E, Liu WT, Engene N, Mohimani H, Byrum T, Pevzner PA, Dorrestein PC, Gerwick WH. J. Nat. Prod. doi: 10.1021/np200077f. concurrently submitted for publication..
- (10).Edwards DJ, Marquez BL, Nogle LM, McPhail K, Goeger DE, Roberts MA, Gerwick WH. Chem. Biol. 2004;11:817–833. doi: 10.1016/j.chembiol.2004.03.030. [DOI] [PubMed] [Google Scholar]
- (11).Sone H, Kondo T, Kiryu M, Ishiwata H, Ojika M, Yamada K. J. Org. Chem. 1995;60:4774–4781. [Google Scholar]
- (12).Meyers AI, Yamamoto Y. Tetrahedron. 1984;40:2309–2315. [Google Scholar]
- (13).Tripathi A, Puddick J, Prinsep MR, Lee PP, Tan LT. Phytochemistry. 2010;71:307–311. doi: 10.1016/j.phytochem.2009.10.006. [DOI] [PubMed] [Google Scholar]
- (14).Tripathi A, Puddick J, Prinsep MR, Lee PP, Tan LT. J. Nat. Prod. 2009;72:29–32. doi: 10.1021/np800448t. [DOI] [PubMed] [Google Scholar]
- (15).Nogle LM, Gerwick WH. J. Nat. Prod. 2002;65:21–24. doi: 10.1021/np010348n. [DOI] [PubMed] [Google Scholar]
- (16).Bunyajetpong S, Yoshida WY, Sitachitta N, Kaya K. J. Nat. Prod. 2006;69:1539–1542. doi: 10.1021/np050485a. [DOI] [PubMed] [Google Scholar]
- (17).Tan LT, Goh BP, Tripathi A, Lim MG, Dickinson GH, Lee SS, Teo SL. Biofouling. 2010;26:685–695. doi: 10.1080/08927014.2010.508343. [DOI] [PubMed] [Google Scholar]
- (18).Montaser R, Abboud KA, Paul VJ, Luesch H. J. Nat. Prod. 2011;74:109–112. doi: 10.1021/np1006839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kimura J, Takada Y, Inayoshi T, Nakao Y, Goetz G, Yoshida WY, Scheuer PJ. J. Org. Chem. 2002;67:1760–1767. doi: 10.1021/jo010176z. [DOI] [PubMed] [Google Scholar]
- (20).Harrigan GG, Yoshida WY, Moore RE, Nagle DG, Park PU, Biggs J, Paul VJ, Moobery SL, Corbett TH, Valeriote FA. J. Nat. Prod. 1998;61:1221–1225. doi: 10.1021/np9801211. [DOI] [PubMed] [Google Scholar]
- (21).Williams PG, Moore RE, Paul VJ. J. Nat. Prod. 2003;66:1356–1363. doi: 10.1021/np0302145. [DOI] [PubMed] [Google Scholar]
- (22).Williams DE, Burgoyne DL, Rettig SJ, Andersen RJ. J. Nat. Prod. 1993;56:545–551. [Google Scholar]
- (23).Luesch H, Williams PG, Yoshida WY, Moore RE, Paul VJ. J. Nat. Prod. 2002;65:996–1000. doi: 10.1021/np0200461. [DOI] [PubMed] [Google Scholar]
- (24).We recorded the retention of the L-FDLA and D-FDLA derivatized Dml and its C-2 epimer (obtained by treatment with base) to obtain four different retention times.
- (25).Yabuuchi T, Kusumi T. J. Org. Chem. 2000;65:397–404. doi: 10.1021/jo991218a. [DOI] [PubMed] [Google Scholar]
- (26).Davies-Coleman MT, Dzeha TM, Gray CA, Hess S, Pannell LK, Hendricks DT, Arendse CE. J. Nat. Prod. 2003;66:712–715. doi: 10.1021/np030014t. [DOI] [PubMed] [Google Scholar]
- (27).Harada K-I, Fujii K, Mayumi T, Hibino Y, Suzuki M, Ikai Y, Oka H. Tetrahedron Lett. 1995;36:1515–1518. [Google Scholar]
- (28).Fujii K, Ika Y, Oka H, Suzuki M, Harada K. Anal. Chem. 1997;69:5146–5151. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.