

Abstract
Angiogenesis and the development of a vascular network is required for tumor progression, and involves release of angiogenic factors, including vascular endothelial growth factor (VEGF), from both malignant and stromal cell types1. Infiltration by cells of the myeloid lineage is a hallmark of many tumors, and in many cases the macrophages in these infiltrates express VEGF2. Here we show that deletion of inflammatory cell-derived VEGF attenuates the formation of a typical high-density vessel network, thus blocking the angiogenic switch in solid tumors. Vasculature in tumors lacking myeloid cell-derived VEGF was less tortuous, with increased pericyte coverage and decreased vessel length, indicating vascular normalization. In addition, loss of myeloid-derived VEGF decreases VEGFR2 phosphorylation in tumors, even though overall VEGF levels in the tumors are unaffected. However, myeloid deletion of VEGF resulted in an accelerated tumor progression in multiple subcutaneous isograft models and an autochthonous transgenic model of mammary tumorigenesis, with less overall tumor cell death and decreased tumor hypoxia. Furthermore, loss of myeloid cell VEGF increased tumor susceptibility to chemotherapeutic cytotoxicity. This demonstrates that myeloid-derived VEGF is essential for tumorigenic alteration of vasculature and signaling to VEGFR2, and that these changes act to retard, not promote, tumor progression.
To test the role of myeloid cell-derived VEGF in tumor progression, we created an in vivo, cell lineage-specific targeted deletion of VEGF via crosses of the loxP-flanked VEGF allele3 to the lysozyme M promoter-driven cre recombinase4; this expression is specific to cells of the myeloid lineage, including neutrophils and macrophages, but not dendritic cells (LysMCre/VEGF+f/+f)4. This results in VEGF gene excision in approximately 75% of isolated neutrophils, peritoneal macrophages5, and tumor associated macrophages (Supplementary Figure 1a).
To determine the function of myeloid cell-derived VEGF in an autochthonous mouse model of breast cancer, we then crossed these alleles to the MMTV-PyMT transgenic mouse strain6. To mitigate the influence of strain variation, individual transgenic alleles were backcrossed to >99% C57Bl/6J strain background (as assayed by SNP analysis).
As both genotypes carry the PyMT transgene, mice with a myeloid cell-specific deletion of VEGF-A (MMTV-PyMT/LysMCre+/VEGF+f/+f) will be termed mutant mice (Mut), whereas cre-negative mice (MMTV-PyMT/LysMCre-/VEGF+f/+f) will be termed wild-type (WT) (Supplementary Figure 1b).
To determine the time point of tumor onset, mammary glands of virgin WT and Mutant mice were palpated once a week. The first palpable tumor occurred at an average age of 11 weeks (Supplementary Figure 1d). Tumors were removed at two different time points, and total tumor burden from each mouse was evaluated. At 16 weeks, tumors from wild type and mutant animals showed no difference in total tumor mass (Supplemental Figure 1e). However, at 20 weeks, mutant mice had a significantly higher tumor burden than their wild type littermates (Figure 1a). At this point, tumors from wild type animals had increased their total mass by almost 400% relative to the 16 week time point; tumors from mutants showed an increase in tumor mass of almost 600% compared to the 16 week time point.
Figure 1.

Deletion of VEGF in myeloid cells results in reduced vascularization but accelerated progression of mammary tumors.
a, Total tumor mass of PyMT-WT mice (n=15) and PyMT-Mut mice (n=13) at the age of 20 weeks. b, Distribution of PyMT mammary tumors at prototypical premalignant (PM) lesions, malignant early carcinoma (EC) and late carcinoma (LC) stages in percent (±s.e.m.) between genotypes and at the age of 16 (n=4) and 20 weeks (PyMT-WT n=4, PyMT-Mut n=9). c, Quantitative analysis of the area covered by CD 31-positive cells within a tumor section for each stage (n=4). d, Development of vessel length in PyMT-tumors during malignant progression as determined by tracing CD 31-positive vessels that were exposed in a longitudinal cut (n=4). e, Analysis of vessel tortuosity based on CD 31-stained tumor sections (n=4). f, Quantitative analysis of the VEGF signal (PyMT-WT n=7, PyMT-Mut n=4). g, ratio of p-Tyr and VEGFR2 signal intensities as a measure of receptor activation (PyMT-WT n=7, PyMT-Mut n=4). Scale bars, 100 μm; **p<0.01, ***p<0.001; error bars, s.e.m.
A quantitative analysis of proliferating cells showed that the number of PCNA-positive cells increased with progression to malignancy (Supplementary Figure 1h) and that loss of myeloid cell-derived VEGF increased proliferation rates significantly in the early stages of malignant transformation (Supplementary Figure 1h). Detection of Polyoma middle T antigen and PCNA immunohistologically demonstrated that the proliferating cell types were primarily mammary epithelium (Supplementary Figure 1f).
In order to address whether the level of malignant progression mirrored tumor mass results, we histologically assessed tumor progression for each time point based on representative sections from each mammary gland using the criteria described in Lin, et al.7 in a blinded fashion. As depicted in Figure 1b, at the time point of 16 weeks, the majority of mammary gland tissues from both wild type and mutant animals are still at premalignant stages (96.2% and 89.5%, by area, respectively). After 20 weeks, the majority of tissues in mutant mice had progressed to malignant stages [EC (38.0%) and LC (52.5%)]. In wild type animals, mammary tissues were primarily at premalignant (30.9%) and early carcinoma (50.2%) stages; a minority of the tissue (18.9%) possessed late carcinomatous lesions (Figure 1b). These results indicated, surprisingly, that tumors develop at a more rapid pace in the absence of macrophage-derived VEGF.
Quantitative analysis of vessel density was then carried out (Figures 1c-1e, Supplementary Figure 1g). In wild-type mice, pre-malignant lesions have a low vascular density, which is reduced in myeloid VEGF null mutant mice (Figure 1c). Increased vascular density was found in tumors that had progressed to the malignant, early carcinoma (EC) stage, consistent with occurrence of an “angiogenic switch” during the transition to malignancy (Figure 1c). In late carcinoma stage tumors formed by multiple solid nodular areas, the overall vascular density was similar to that seen in the early carcinoma stage in wild type animals. However, in mutant mice, the malignancy-associated increased vascularization seen in wild type animals does not occur (Figure 1c). The length and the tortuosity of tumor blood vessels was also affected by the mutation: as depicted in Figure 1d, the average vessel length in tumors in wild type mice increased as tumors progressed to malignancy, indicating that vessel elongation plays a key role in the formation of a tumor vessel network in this model. Along with this increase, blood vessel tortuosity more than doubled in malignant lesions in wild type mice (Figure 1e). In tumors within mutant mice, there was no increase in vessel length prior to the late carcinoma stage (Figure 1d), and vessel tortuosity never changed in the mutants (Figure 1e).
Gene-expression analysis of lysates of mammary tumors from mice at the endpoint age of 20 weeks did not reveal any increase in expression of hypoxia markers, such as the PGK gene (Supplementary Figure 1i). However, this analysis of tumors did demonstrate that there was higher expression of VEGF mRNA in tumors from mutant animals (Supplementary Figure 1j). This was not mirrored by changes in VEGF protein, however, which did not vary in tumor lysates from wild type and mutant animals (Figure 1f).
We next wished to determine whether signaling to the tumor endothelium was affected by the loss of myeloid-derived VEGF. VEGFR2 is a primary endothelial cell-specific receptor tyrosine kinase that participates in VEGF signalling. By immunoprecipitating VEGFR2 from tumor lysates and probing with anti-phosphotyrosine followed by anti-VEGFR2 antibody via western blot, we were able to quantify activated and total VEGFR2 from wild type and mutant tumors. As shown in Figure 1g, loss of myeloid-derived VEGF causes an approximately 50% reduction in the ratio of phosphorylated VEGFR2 relative to total VEGFR2, when compared to wild type tumor lysates. Reduced VEGFR2 phosphorylation suggests that myeloid cell-derived VEGF plays a unique role in tumor vascularization that cannot be compensated for by VEGF from other sources within the tumor.
To analyze these effects in fully transformed cells, we next carried out experiments with subcutaneous isografts of Lewis Lung Carcinoma (LLC) cells. LLC tumors in myeloid VEGF null mutant mice had significantly greater tumor volumes at day 12 (Figure 2a), with no genotype-specific differences in tumor infiltration (Supplementary Figure 2). Flow cytometric analysis demonstrated that macrophages were the predominant myeloid infiltrate, and that levels of infiltrating macrophages and less abundant neutrophils were similar across genotypes (Supplementary figure 2).
Figure 2.
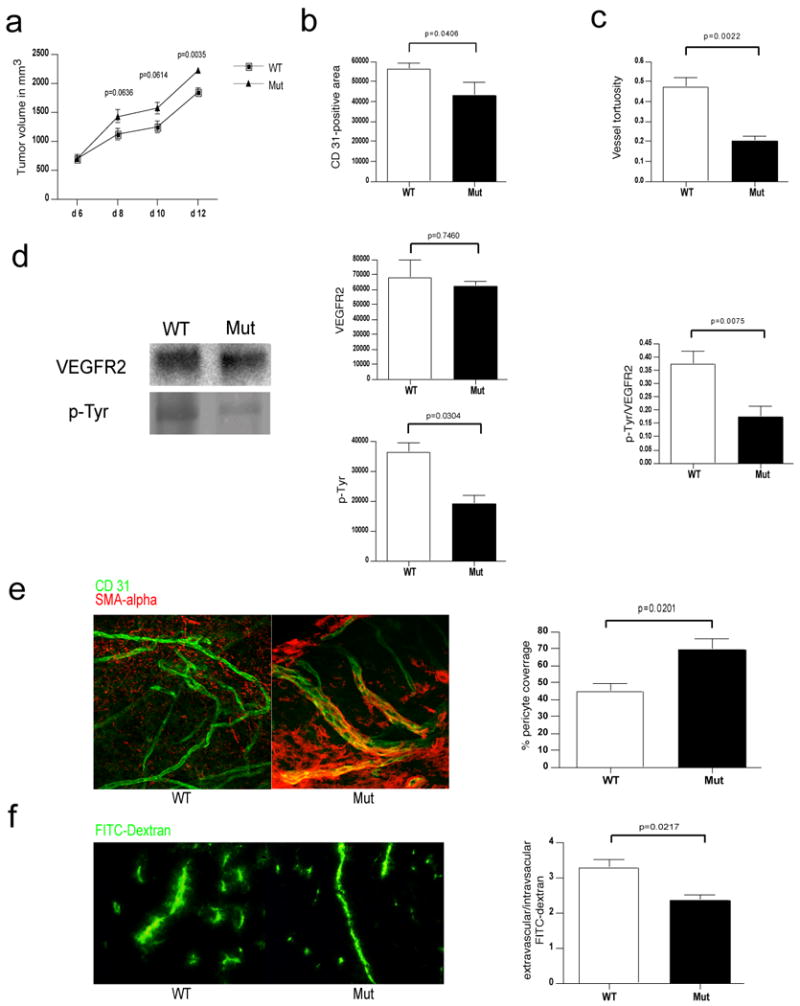
Deletion of VEGF in myeloid cells leads to a normalized vasculature and higher tumor volumes in Lewis Lung Carcinoma isografts.
a, Growth curve analysis of Lewis Lung Carcinoma (LLC) tumors injected subcutaneously in WT and Mut mice (n>7 for each group). b, Quantitative analysis of CD 31-positive endothelial cells (n=4). c, Determination of blood vessel tortuosity in LLC tumors. d, Left: Immunoblotting for VEGFR2 and phosphotyrosine (p-Tyr) after Immunoprecipitation of VEGFR2 from LLC tumor lysates. Center: Quantitative analysis of VEGFR2 (upper) and phosphotyrosine (lower) signals. Right: Ratio of p-Tyr and VEGFR2 signal intensities as a measure of receptor activation (WT n=8, Mut n=7). e, Left: Confocal microscopy images of simultaneous immunodetection of endothelial cells and pericytes in LLC tumors with the specific markers CD 31 and smooth muscle actin-alpha (SMA-alpha). Right: Pericyte coverage as assessed by SMA-alpha/CD 31 co-localization (n=4). f, Left: Fluorescent microscopy images of a FITC-dextran angiography on LLC isografts. Right: Ratio of extravascular over intravascular FITC-dextran as a measure of vascular permeability (WT n=6, Mut n=4). Scale bar, 100 μm; error bars, s.e.m.
A significant reduction in vascular density and vessel tortuosity was again observed in mutant mice (Figures 2b and 2c). An analysis of VEGF protein and mRNA (Supplementary Figures 3a and 3b) showed no significant differences between genotypes. However, when VEGFR2, phosphorylated VEGFR2, and the ratio of phosphorylated VEGFR2 to total VEGFR2 were quantified (Figure 2d), activation of VEGFR2 was once again reduced approximately 50% in mutant tumors relative to wild type tumors.
We next wished to determine the relationship between expression of myeloid cell VEGF and vessel normalization; to assay this, we visualized pericyte coverage of tumor vessels, and assayed levels of vessel pericyte coverage in the tumors (Figure 2e). As shown, the loss of VEGF expression in the myeloid cells resulted in a marked increase in the level of vessel pericyte association, indicating that VEGF expression from infiltrating myeloid cells is essential for intratumoral loss of vessel pericyte coverage. Vessel permeability also decreased in tumors from mutant animals (Figure 2f).
Hypoxia is associated with solid tumor growth and with tumorigenic vasculature. We found that the more slowly growing wild type tumors had higher levels of hypoxia (Figure 3a and Supplementary Figure 3c); and that this occurred even in areas that were extensively vascularized (Supplementary Figure 3c). Interestingly, staining of tumor sections with VEGF and F4/80, a macrophage marker, demonstrates that intense VEGF immunostaining in wild type tumors occurs in regions of high infiltration, whereas in mutant tumors VEGF staining is only found outside of infiltrated regions (Supplementary Figure 3d); this depicts how VEGF from myeloid cells could have an acute and localized effect on tumor vasculature.
Figure 3.
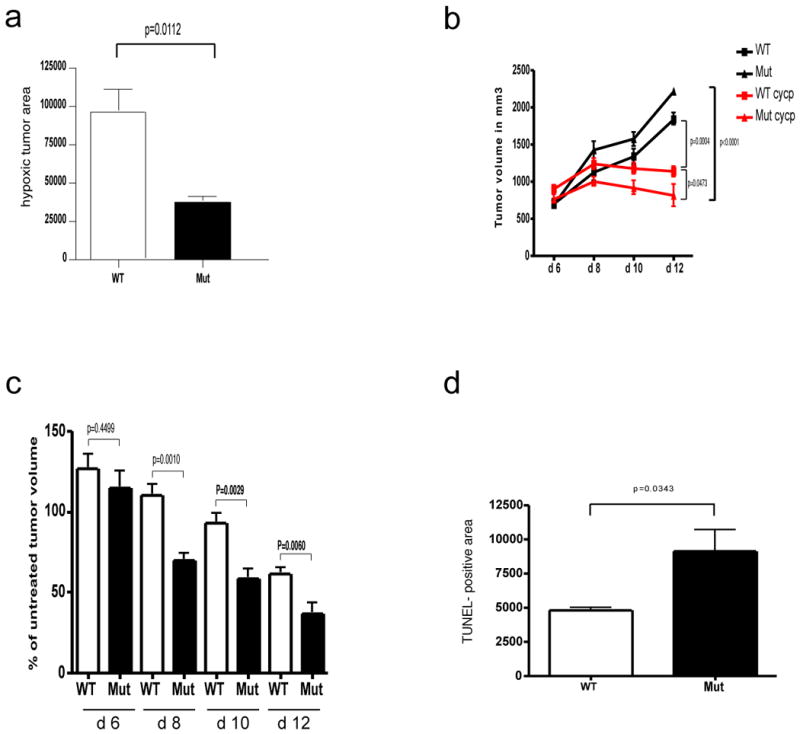
Deletion of VEGF myeloid cells results in reduced hypoxia and increased susceptibility of LLC tumors to cytotoxic agents.
a, Quantitative analysis of hypoxic tumor areas (WT n=6, Mut n=4). b, Growth curve analysis of LLC isografts from WT and Mut animals (n>4 for each group) treated with Cyclophosphamide (CYCP) (170mg/kg) at day 6, 8 and 10 after tumor implantation. c, Response of tumors from WT and Mut mice to CYCP treatment expressed as percentage of treated tumor volume to untreated tumor volume at specified time points. d, Quantitative analysis of TUNEL positive on CYCP-treated LLC isografts (n=4). Scale bar, 100 μm; error bars, s.e.m.
To determine whether the vascular changes we describe confer an increased susceptibility to cytotoxic agents, we administered the chemotherapy agent cyclophosphamide to wild type and mutant LLC tumor-bearing mice. Loss of myeloid-derived VEGF increased the chemotherapeutic efficacy of the treatment (Figures 3b,c). The cyclophosphamide treatment also resulted in a significantly increased level of tumor cell death in myeloid VEGF null tumor-bearing mice, as can be seen in Supplemental Figure 3e and as quantified in Figure 3d. Similar experiments carried out with cis-platinum gave comparable results (Supplementary figures 3f and 3g).
To determine the differential contributions of tumor cell and myeloid cell-derived VEGF, we next created fibrosarcoma cell lines derived from C57Bl/6J embryos homozygous for the conditional allele of the VEGF gene. We then used cre recombinase expression from adenovirus to create matching wild type (infected with control adenovirus) and VEGF null (infected with cre recombinase-expressing adenovirus) tumor cell lines, and injected them subcutaneously into mice wild type or null for myeloid VEGF expression. As can be seen in Figure 4a, fibrosarcomas that were wild type for VEGF expression grew faster and significantly larger in mice lacking VEGF in myeloid cells, which is similar to what we observed in the other models described above. However, consistent with previous findings from our laboratory8, tumors lacking VEGF expression grew more slowly than those expressing VEGF when implanted into wild type mice (Figure 4a).
Figure 4.
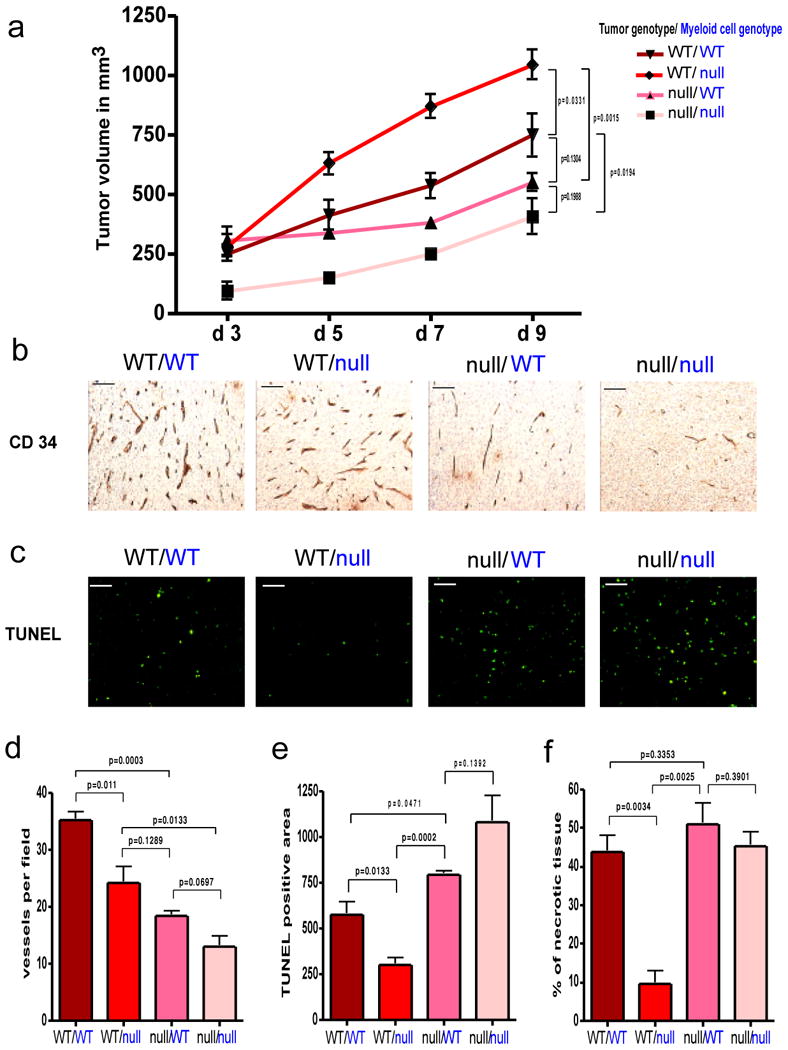
Effect of tumor cell-derived versus myeloid cell-derived VEGF on tumor angiogenesis and growth.
a, Growth curve analysis of wildtype (WT) and VEGF nullizygous (null) fibrosarcoma isografts (genotype labeled in black) implanted into WT-mice or Mut-mice (null) with a myeloid cell-specific deletion of VEGF (genotype labeled in blue) (n=4 for each group). b, CD 34 immunostaining on fibrosarcoma isografts. c, Detection of apoptotic cells in fibrosarcomas by TUNEL-staining. d, Quantitative analysis of CD 34-positive blood vessels (n=4). e, Quantification of TUNEL-positive cells (n=4). f, Assessment of tumor necrosis on fibrosarcoma midline sections. Shown is the perimeter of necrotic areas expressed as percentage of total tumor perimeter (n=4). Scale bar, 100 μm; error bars, s.e.m.
Interestingly, when VEGF was absent from both the tumor and the myeloid compartment, tumor growth essentially collapsed, with greatly increased apoptosis and necrosis (Figure 4a-4f). Further, although there was a reduction in tumor vessel density (Figures 4b and 4d) ranging from the highest vessel count in tumors wild type in all tissue compartments, to the lowest in those lacking VEGF in tumor cells and myeloid cells, these values did not directly correspond to changes in tumor growth. Rather, they demonstrate the unique role for myeloid-derived VEGF in facilitating changes in tumor vessel function and normalization.
Tumor-associated macrophages have been shown to be crucial for malignant progression in a mouse model of breast cancer7,9; however, these studies removed macrophages completely from tumors, and thus were removing all of the many aspects of macrophage involvement in tumor progression. In isolating one of those aspects experimentally, we have shown that myeloid-derived VEGF is required for the formation of a high-density vessel network, that loss of VEGF from this source greatly reduces activation of VEGFR2 in spite of little or no loss in total tumor VEGF, and that the absence of this increased vascular density does not hamper, but rather accelerates tumor progression.
When administered as single agents, anti-angiogenic drugs have produced modest objective responses in clinical trials, but overall, these single agent approaches have not yielded significant long-term survival benefits10. In contrast, when given in combination with chemotherapy, antibodies targeted against VEGF result in an increase in survival in colorectal cancer patients11. Our observations argue that a critical mediator of this sensitization to chemotherapy lies in the infiltrating innate immune cells, rather than in the malignant cells of the tumor. These findings open an important new avenue for the investigation of anti-angiogenic therapies targeted to tumor-associated cells of the immune system.
Methods Summary
Transgenic mice (C57Bl/6J) expressing the polyoma middle T (PyMT) oncoprotein under the promoter of the mouse mammary tumor virus (MMTV)-long terminal repeat were bred to mice (C57Bl/6J) with both alleles of exon 3 of VEGF-A flanked by loxP sites (VEGF+f/+f)12. Myeloid cell-specific knock out of VEGF was achieved by breeding male mice homozygous for the floxed VEGF allele and hemizygous for the PyMT oncogene (MMTV-PyMT/VEGF+f/+f) with female mice (C57Bl/6J) homozygous for the floxed VEGF allele expressing Cre recombinase driven by the lysozyme M promoter (LysMCre+/VEGF+f/+f)4. For our studies, we used female mice hemizygous for the PyMT oncogene carrying two floxed VEGF alleles and positive for cre-expression (MMTV-PyMT/LysMCre+/VEGF+f/+f) designated as mutants (Mut) whereas female littermates negative for cre expression (MMTV-PyMT/LysMCre-/VEGF+f/+f) served as wildtype controls (WT). Loss of VEGF expression in myeloid cells had no effect on normal mammary development (Supplemental Figure 1c).
To generate isografts 1×107 Lewis lung carcinoma cells on a BL6 background (ATCC) were injected subcutaneously into LysMCre-/VEGF+f/+f (WT) and LysMCre+/VEGF+f/+f mice (Mut). Mouse embryonic fibroblasts (MEFs) were isolated from WT mice. The transgenic MEFs were immortalized by stable transfection with SV40 large T antigen and then transformed with a vector expressing oncogenic mutant H-Ras (Val-12). Subsequently, the VEGF+f/+f MEFs were infected with an adenovirus expressing Cre recombinase to delete exon 3 of the VEGF gene. A total of 5×106 of VEGF null or wildtype MEF's were injected subcutaneously into WT and Mut mice. Data are expressed as mean ± SEM. Statistical significance was determined by two-tailed unpaired t test.
Methods
Chemotherapy
The treatment with chemotherapy was started six days post-injection of 1×107Lewis lung carcinoma cells. Cisplatin (5mg/kg) and Cyclophosphamide (170mg/kg) were purchased from Sigma and given i.p. every second day until day 10.
Histology, immunohistochemistry and immunofluorescence
For whole-mount preparation of mammary glands, the inguinal gland was removed and fixed in 10% phosphate-buffered formalin (Fisher Scientific, Fair Lawn, NJ) and dehydrated through a series of graded acetone and ethanol washes. Glands were stained for 3 h with Harris hematoxylin (VWR Int., West Chester, PA), and excess stain was removed by washing in water. Glands were destained in acidic 50% ethanol and dehydrated and stored in methyl salicylate (Fisher Scientific).
After removal tumors were fixed in 10% Formalin, Zinc fixative (BD Pharmingen) or 4% paraformaldehyde, respectively and then embedded in paraffin. For hematoxylin-eosin (H&E) and immunostaining, 5μm sections were deparaffinized with xylene and rehydrated with graded ethanol. The sections were stained according to routine immunohistochemistry procedures and visualized by Vectastain ABC or ABC-AP kit (Vector Laboratories, Burlingame, CA).
Primary antibodies used for immmunohistochemistry and immunofluorescence: biotinylated rat anti-CD 31 at 1:100 dilution (BD Pharmingen), rat anti-CD 34 (Novus), biotinylated mouse anti-PCNA at 1:800 dilution (BD Pharmingen), biotinylated rat anti-F4/80 at 1:200 dilution (Serotec), rabbit anti-VEGF at 1:50 dilution (sc-152, Santa Cruz), mouse anti-SMA-alpha at 1:500 (Chemicon). The fluorochrome-conjugated Alexa 488 and Alexa 555 were used as secondary antibodies (1:200).
Pimonidazole Hydrochloride (Hypoxyprobe™-1) was injected intraperitoneally (60 mg/kg bodyweight) 1 hour prior to tumor removal and was detected with a Mouse IgG1 monoclonal antibody (Mab1, Natural Pharmacia International).
TUNEL-Assay (Promega) was performed according to the manufacturers instructions.
Quantitative analysis of histologic markers
For quantitative analysis of the distribution of immunohistochemical markers within the tumor, the midline sections of tumors were photographed into TIFF images using a Leica DMR microscope and SPOT™ RT color camera system (Diagnostic Instruments Inc.) and the area (number of pixels) with positive staining equal to or greater than a set threshhold was measured using the Image J program and such marked areas were normalized by the number of images for each tumor. For comparison at different stages of progression PyMT tumors were classified as previously described7.
To determine vessel density, the vasculature marked by CD 31 and CD 34 was skeletonized using the Image J program and the area covered by blood vessels was measured and the number of vessels per field was determined, respectively.
To estimate change in vessel length, the skeletonized blood vessels that showed up in a longitudinal cut within an image were traced along their midline and the number of pixels was converted into distance in millimeters using Image J.
The tortuosity (T) of blood vessel was defined by the following equation:
T=L/S − 1
where L is the length of the vessel of interest and S the straight line distance between its endpoints.
Confocal microscopy
Co-localization of pericytes (SMA-α) endothelial cells (CD 31) was imaged on 20-μm sections of excised LLC tumors. Images were acquired using laser scanning confocal microscopy under 60/1.4 NA oil objective (Nikon C1si, Nikon Instruments Inc).
Flow cytometry
Tumors were excised and cut into small pieces before incubation in a shaker at 37 degrees at 250 RPM for 2 hours in 0.1% Collagenase A (Roche) in RPMI 1640 (Invitrogen). The resulting single cell suspension was pelleted and rinsed in PBS before treatment with hypotonic lysis buffer and passing through a 40uM cell strainer (BD Biosciences). One million cells per condition were then incubated with Fc Block (BD Biosciences) to block nonspecific FcR binding before labeling with fluorescently conjugated antibodies (eBiosciences) using standard protocols. Data was acquired on a FACSCalibur flow cytometer (BD Biosciences) with propidium iodide to exclude non-viable cells and analyzed with FlowJo Software (Tree Star).
Determination of vascular permeability
200 μl of Fluorescein isothiocyanate-dextran (2,000,000 MW, Sigma) were injected intravenously at a concentration of 50mg/ml. After 10 minutes tumors were removed and immediately frozen in OCT compound. 10 μm cryosections were cut and vascular permeability was determined based on 10 randomly taken pictures from each tumor. By using Image J the edges of blood vessel were indentified and the extravascular as well as the intravascular area (number of pixels) covered by FITC-dextran were measured. In order to determine vascular permeability in a quantitative manner the amount of extravascular staining over the amount of intravascular FITC-dextran was calculated.
Immunoprecipitation and Immunoblotting
Tumors were lysed in RIPA buffer and 500 μg of lysate were used for immunoprecipitation of VEGFR2. The following antibodies were used in this study: rabbit anti-VEGFR2 (55B11, Cell signaling), HRP-conjugated anti-phosphotyrosine (4G10®, Millipore), rabbit anti-VEGF (sc-152, Santa Cruz) and mouse anti-β-actin (Sigma). For a quantitative analysis the membranes were scanned with a fluorescense scanner and the signal stength was determined by using ImageQuant® software.
Determination of deletion frequency and gene expression by quantitative PCR
Thioglycollate-elicited macrophages as well as FACS sorted F4/80-positive tumor-associated macrophages isolated from PyMT-Mut tumors were used for isolation of genomic DNA as previuously described5. Whole tumors were ground to a fine powder in liquid nitrogen and subsequently homogenized in TRIzol® reagent (Invitrogen). Total RNA was isolated following the manufacturer's instructions. For real-time PCR analysis, the isolated RNA was reverse transcribed. For PCR reactions, TaqMan-Universal Mastermix (Applied Biosytems) was used. 50 ng cDNA was used as template to determine the relative amount of mRNA by real-time PCR (ABI Prism 7700 sequence detection system), using specific primers and probes with the following sequences:
VEGF reverse 5′-ATCCGCATGATCTGCATGG-3′,
VEGF forward 5′-AGTCCCATGAAGTGATCAAGTTCA-3′,
VEGF probe (6FAM)TGCCCACGTCAGAGAGCAACATCAC(BHQ-1)
PGK reverse 5′-TTCTTGCTGCTCTCAGTACCACA-3′
PGK forward 5′-CAAATTTGATGAGAATGCCAAGACT-3′
PGK probe (6FAM)TATACCTGCTGGCTGGATGGGCTTGGACT(BHQ-1)
Cell Culture
Cells were cultured in Dulbecco's modified Eagle's medium (DMEM High Glucose), supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, 50 U/ml penicillin and 100μg/ml streptomycin.
Supplementary Material
Acknowledgments
Acknowledgements and Competing Interests statement: We acknowledge helpful conversations with J. DuRose, N. Ferrara, L. Iruela-Arispe, L. Coussens, M. Karin, and H.-P. Gerber, and the support of the Deutsche Forschungsgemeinschaft to C.S. (STO 787/1-1), a Susan G. Komen Dissertation Research Award (DISS0402406) to A.D., and NIH CA82515 to R.S.J.
Footnotes
Author contributions: C.S. generated isografts, conducted drug treatment studies, performed biochemical experiments, immunohistochemical procedures, histological analysis and prepared the manuscript. A.D. generated MMTV-PyMT/LysMCre/VEGF+f/+f mice, conducted related tumor palpation/mass studies, initial histology, flow cytometry, and assisted with the manuscript. A.W. generated VEGF null fibroblasts. N.Z. performed preliminary cDNA studies on MMTV-PyMT/LysMCre/VEGF+f/+f -tumors. N.T. assisted with the FITC-dextran angiography. J.I.G. and D.A.C. provided confocal microscopy images and technical advise. R.S.J. supervised and directed the project.
References
- 1.Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 2.Lewis JS, Landers RJ, Underwood JC, Harris AL, Lewis CE. Expression of vascular endothelial growth factor by macrophages is up-regulated in poorly vascularized areas of breast carcinomas. J Pathol. 2000;192(2):150–158. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH687>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 3.Gerber H, et al. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nature Medicine. 1999;5:623–628. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- 4.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8(4):265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 5.Cramer T, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112(5):645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12(3):954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin EY, et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163(5):2113–2126. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grunstein J, Roberts WG, Mathieu-Costello O, Hanahan D, Johnson RS. Tumor-derived expression of vascular endothelial growth factor is a critical factor in tumor expansion and vascular function. Cancer Research. 1999;59(7):1592–1598. [PubMed] [Google Scholar]
- 9.Lin EY, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66(23):11238–11246. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- 10.Mayer EL, Lin NU, Burstein HJ. Novel approaches to advanced breast cancer: bevacizumab and lapatinib. J Natl Compr Canc Netw. 2007;5(3):314–323. doi: 10.6004/jnccn.2007.0026. [DOI] [PubMed] [Google Scholar]
- 11.Hurwitz HI, Honeycutt W, Haley S, Favaro J. Long-term treatment with bevacizumab for patients with metastatic colorectal cancer: case report. Clin Colorectal Cancer. 2006;6(1):66–69. doi: 10.3816/CCC.2006.n.023. [DOI] [PubMed] [Google Scholar]
- 12.Gerber HP, et al. VEGF is required for growth and survival in neonatal mice. Development. 1999;126(6):1149–1159. doi: 10.1242/dev.126.6.1149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.