

Abstract
New nitric oxide synthase (NOS) inhibitors were designed de novo with knowledge gathered from the studies on the nNOS-selective dipeptide inhibitors. Each of the new inhibitors consists of three fragments: an aminopyridine ring, a pyrrolidine, and a tail of various length and polarity. The in vitro inhibitory assays indicate good potency and isoform selectivity for some of the compounds. Crystal structures of these inhibitors bound to either wild type or mutant nNOS and eNOS have confirmed design expectations. The aminopyridine ring mimics the guanidinium group of L-arginine and functions as an anchor to place the compound in the NOS active site where it hydrogen bonds to a conserved Glu. The rigidity of the pyrrolidine ring places the pyrrolidine ring nitrogen between the same conserved Glu and the selective residue nNOS Asp597/eNOS Asn368 which results in similar interactions observed with the α-amino group of dipeptide inhibitors bound to nNOS. These structures provide additional information to help in the design of inhibitors with greater potency, physico-chemical properties, and isoform selectivity.
Introduction
Three different mammalian isoforms of nitric oxide synthase (NOS) have been isolated and characterized: neuronal (nNOS), inducible (iNOS), and endothelial (eNOS). Although different isoforms have different cell and tissue distribution and are regulated through various mechanisms, they all catalyze the conversion of one guanidinium N atom of L-arginine (L-Arg) to nitric oxide. All three isoforms share a similar domain architecture with a N-terminal domain consisting of the catalytic heme active site and a cofactor, tetrahydrobiopterin, binding site, while the C-terminal domain containing FMN, FAD, and NADPH binding sites serves as an electron donating domain1,2. The linker between the two functional domains is a calmodulin binding motif. The binding of calmodulin enables electron flow from the flavins to the heme3.
Nitric oxide is an important signaling molecule involved in a wide range of physiological functions in the neuronal, immune, and cardiovascular system4,5. In order to exert appropriate functions, NO generation by the three different NOS isoforms is under tight regulation. The overproduction of NO by nNOS (or iNOS) and the underproduction by eNOS have been shown to lead to pathophysiological conditions such as neurodegenerative diseases6, stroke7,8, rheumatoid arthritis9, hypertension10, and atherosclerosis11. Inhibition of nNOS (or iNOS) can thus be of considerable therapeutic benefit. However, inhibition must be isoform selective so that only NO formation by the disease-associated NOS, (e.g. nNOS) will be inhibited by the treatment while the physiological function of the other isoform, often eNOS, is unaffected. Isoform-selective inhibition is a challenging problem given that the three isoforms have very few differences in their three-dimensional structures.
Previous structure-activity studies in our laboratories on a series of Nω-nitro-L-arginine containing dipeptide inhibitors12,13 (Fig. 1A) have uncovered some key structural features in the NOS active site that are responsible for the selective binding affinity of these inhibitors to nNOS over eNOS14–17. Most importantly, the single amino acid difference, Asp597 in nNOS and Asn368 in eNOS, had been identified as the major reason why inhibitors 1, 2 and 3 (Fig. 1A) bind more tightly to nNOS than eNOS. In nNOS these inhibitors adopt a curled binding mode which places the inhibitor α-amino group between Asp597 and Glu592 for optimal electrostatic stabilization. Since eNOS has Asn368 rather than Asp, there is no energetic incentive for the inhibitor to curl, and instead the inhibitors adopt an extended conformation. One obvious design principle to emerge from this work is to have a positively charged group corresponding to the α-amino group in 1, 2 or 3 on a rigid scaffold such that the inhibitor will not have to curl in order to optimize charge interactions with Asp597 and Glu592. One other design principle is to replace the nitro-guanidinium group with an aminopyridine primarily because the aminopyridine should mimic the H-bonding pattern of the nitro-guanidinium while the pKa of the aminopyridine group (≈ 6 – 7) should increase bioavailability18, 19. Based on these design principles, a new de novo design method was proposed, and a series of new inhibitors, 4, 5, 6, and 7 (Fig. 1B), have been synthesized, the in vitro inhibitory potency determined, and the inhibitors applied to an animal model18,19. Here we report the crystal structures of these inhibitors bound to both eNOS and nNOS. Unfortunately we were unable to obtain suitable crystals of eNOS in complex with 4 or 5 which often is the case for inhibitors that bind poorly to eNOS.
Fig. 1.


A) Chemical structures and nomenclature for the inhibitors discussed in the paper. 1. L-Nω-nitroarginine-2,4-L-diaminobutyramide; 2. (4S)-N-(4-amino-5-[aminoethyl]aminopentyl)-N'-nitroguanidine; 3. L-Nω-nitroarginine-(4R)-amino-L-proline amide; 4. (±)-3-{cis-4'-[(6“-aminopyridin-2”-yl)methyl]pyrrolidin-3'-ylamino}propan-1-ol; 5. (±)-N1-{cis-4'-[(6“-aminopyridin-2”-yl)methyl]pyrrolidin-3'-yl}ethane-1,2-diamine; 6. (±)-N1-{cis-4'-[(6“-amino-4”-methylpyridin-2“-yl)methyl]pyrrolidin-3'-yl}-N2-(4'-chlorobenzyl)ethane-1,2-diamine; 7. (±)-N1-{trans-4'-[(6“-amino-4”-methylpyridin-2”-yl)methyl]pyrrolidin-3'-yl}-N2-(3'-chlorobenzyl)ethane-1,2-diamine. B) Structure of 1 complexed to nNOS and eNOS12. In nNOS the inhibitor adopts a curled conformation in order to enable the inhibitor α-amino group to optimally interact with both Glu592 and Asp597. In eNOS the residue corresponding to Asp597 is Asn368 and as a result, the inhibitor adopts an extended conformation.
Results
Binding of 4 and 5 to nNOS
Although all the new inhibitors 4 through 7 (Fig. 1A) used for crystallographic studies are the racemic mixture, the electron density of all four inhibitor complex structures show that only one enantiomer in each case is bound in the NOS active site. As expected, the two nitrogen atoms of the aminopyridine moiety of both 4 and 5 are involved in H-bonding with the Glu592 carboxylate (Fig. 2) similar to the bifurcated H-bonding found between two guanidino nitrogens of dipeptide inhibitor and the Glu592 side chain oxygens in the nNOS dipeptide complex structures12. The pyridine ring is roughly parallel to and stacking against the heme plane with the closest distance from the C3 atom of pyridine to the NB atom of heme in the range of 3.3 – 3.4 Å. The rigid pyrrolidine ring indeed brings its nitrogen within H-bonding distance to the Glu592 side chain resembling the curled binding conformation of dipeptide inhibitor where the α-amino group forms a H-bond to Glu592. This sp3-hybridized nitrogen cation also forms a H-bond with the conserved structural water, which then forms H-bonding interactions with the selective residue Asp597 of nNOS. There are two hydrogen atoms attached to this nitrogen atom. One forms a H-bond with Glu592. Another one forms a H-bond with a conserved structural water which then forms H-bond interactions with Asp597. The amino group next to the pyrrolidine ring (N8 in Fig 2), mimicking the peptide amide nitrogen in the dipeptide inhibitor, makes a direct H-bond with the heme propionate group of pyrrole ring A. The tails of compounds 4 and 5 adopt quite different conformations. The amino group of 5 curls back to make a H-bond to the second heme propionate while the hydroxyl group of 4, owing to its one carbon longer arm, can no longer make any direct contact with the heme (Fig. 2). This additional charge-charge interaction between the terminal amine of 5 and heme propionate is probably a major factor contributing to the tighter binding of inhibitor 5 (Ki = 0.388 μM) compared to 4 (Ki = 9.4 μM).
Fig. 2.

Active site structures of the wild type nNOS with inhibitor 4 (panel A) or 5 (panel B) bound viewed side by side in an identical orientation. Shown also the Fo – Fc omit map contoured at 3.0σ for each inhibitor. Hydrogen bonds are drawn with the dashed lines. The atomic color scheme for amino acids is: carbon, cyan or green; nitrogen, blue; oxygen, red; sulfur, yellow. The figures are made with PyMol (http:://pymol.sourceforge.net).
Binding of 6 and 7 to nNOS
Inhibitors 6 and 7 were derived from 5 with two modifications (Fig. 1A). First, a methyl group was introduced in the aminopyridine ring to provide additional contacts with a small hydrophobic pocket surrounded by Val567 and Phe584. Second, a chlorobenzyl group was attached to the terminal amino position in order to reach into a region where different NOS isoforms start to show sequence diversity. Inhibitor 6, similar to 4 and 5, has a (3'S, 4'S) cis-conformation at the two chiral carbons on the pyrrolidine ring. Its aminopyridine and pyrrolidine rings, therefore, bind to the active site the same as 4 and 5, as described previously19 (Fig. 3A). Inhibitor 7, on the other hand, possesses a (3'R, 4'S) trans-conformation. However, the H-bonding interactions from both its aminopyridine and pyrrolidine rings to the Glu592 side chain are still retained (Fig. 3B). The newly added methyl group in both 6 and 7 makes van der Waals contact with Phe384 with a closest distance of 3.6Å. Larger differences between 6 and 7 are seen in the third fragment beyond the pyrrolidine ring. The 3'S conformation in the pyrrolidine of 6 places the neighboring amino group (N8 in Fig. 3) downward towards the heme where it H-bonds with the heme propionate (Fig. 3A), whereas the 3'R conformation in 7 brings N8 away from the propionate (Fig. 3B). Lack of this H-bond in 7 might be one of the reasons 7 (Ki = 0.25 μM) binds more poorly to nNOS than does 6 (Ki = 0.085 μM). The remaining chain leads the chlorophenyl moiety to a hydrophobic pocket defined by Met336, Leu337, Tyr706, and Trp306 of the neighboring subunit. However, the exact orientation of the chlorophenyl ring is somewhat ambiguous owing to the poor density quality in the region, especially in the structure of the 7 complex.
Fig. 3.

Active site structures of the wild type nNOS with inhibitor 6 (panel A) or 7 (panel B) bound. Density around each inhibitor is the Fo – Fc omit map contoured at 3.0σ. Residue Trp306 belongs to the neighboring subunit.
Binding of 6 and 7 to eNOS
As expected, inhibitors 6 and 7 bind very much the same to eNOS. Part of the inhibitor design effort was to rigidify the inhibitor such that the key amino group interacting with Asp597 in nNOS would bind the same in eNOS18,19. This is exactly what happens. The pyrrolidine N atom is positioned the same in both eNOS and nNOS even to the extent that in both NOS isoforms a water molecule bridges between the pyrrolidine N atom and Asp597 in nNOS (Asn368 in eNOS). However, the pyrrolidine N atom should experience greater electrostatic stabilization in nNOS since nNOS has two negative charges nearby, Asp597 and Glu592, while eNOS has only Glu363. A second difference involves the chlorophenyl tail. In nNOS the aromatic ring can potentially form closer contacts with Met336 compared to the smaller corresponding residue, Val 106, in eNOS. However, the electron density for the chlorophenyl group is more well-ordered in eNOS than in nNOS and thus, it is doubtful that differences in interaction between the inhibitor aromatic ring and protein is a major contributor to isoform selectivity. The primary source of the 1,000-fold selectivity is more likely due to the greater electrostatic stabilization to the pyrrolidine N atom of 6 in nNOS.
Inhibitor binding to the nNOS Asp597Asn/Met336Val mutant
In previous work we converted Asp597 and Met336 in nNOS to the corresponding residues in eNOS, Asn and Val, in order to test our hypothesis that both of these residues form more favorable contacts with the dipeptide inhibitors in nNOS than in eNOS13. Consistent with the finding that the binding mode of the new inhibitors reported here is unchanged between nNOS and eNOS, they also maintain the identical binding mode when wild type and mutant nNOS structures are compared. Inhibitor 5 found in the nNOS Asp597Asn/Met336Val double mutant structure has its aminopyridine and pyrrolidine rings H-bonded to Glu592 in a manner similar to that observed in wild type nNOS (Fig. 5). The only difference is that the terminal amino group in the mutant structure has weak density and seems not to form a H-bond with the heme propionate from pyrrole ring D in contrast to the case in the wild type nNOS-5 complex structure. As expected, the Ki values for the nNOS double mutant increase for all inhibitors but the mutant still binds these inhibitors better than eNOS. Although we were not able to obtain suitable crystals of the eNOS-5 complex, it is probably safe to assume that 5 forms a complex with eNOS very similar to the one observed in nNOS and the nNOS double mutant. What then needs to be explained is why wild type nNOS binds 5 about 1,070-fold better than eNOS but only 90-fold better than the nNOS double mutant, a difference of 11-fold in Ki. In terms of free energy a factor of 11 accounts to ≈ 1.4 kcal/mol. It is possible that the tail primary amino group may be the source of this difference. In the wild type nNOS structure the primary amino group of 5 has strong and continuous electron density with a heme propionate, but this is not the case in the nNOS double mutant. An even weaker interaction between this amino group and the heme propionate in eNOS could easily account for 1.4 kcal/mol.
Fig. 5.
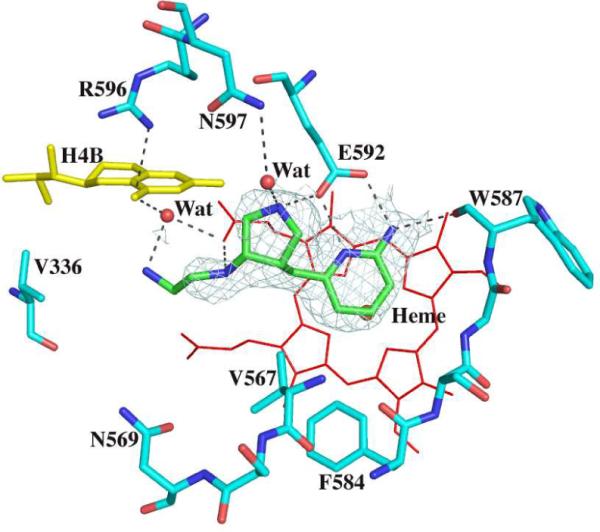
Active site structure of the nNOS Asp597Asn/Met336Val mutant with inhibitor 5 bound. The Fo–Fc omit map is shown around the inhibitor contoured at 3.0σ.
MM-PBSA to estimate ΔG of binding
It would be advantageous to employ a rapid computational method to estimate which inhibitors not only bind best but exhibit greater selectivity for nNOS over eNOS. Toward this end we have been using the MM-PBSA methodology as implemented in Amber 9.0 to compute ΔGbind. The procedure described in Materials and Methods was used to compute ΔGbind for four different NOS inhibitors in five different crystal structures. As shown in Fig. 6A these are all aminopyridine inhibitors similar to inhibitors used for the crystal structures described in this study. However, because the Ki values from which ΔGexp are derived for inhibitors 4, 5, 6, and 7 used in the present study are mixtures of optical isomers, these were not included in the training set for generating Fig. 6B. Fig. 6B is a plot of computed free energy obtained from single energy minimized structures20,21 vs. experimental free energies derived from Ki measurements. The computed free energies are much larger since we have not included inhibitor entropy corrections. Even so, the fit is quite good. Although the inhibitors used in Fig. 6 are very similar, they span a Ki range of over 103, yet the relative ΔGcalc agrees well with ΔGexp.
Fig. 6.

A) Structures of the various inhibitors complexed with either eNOS, nNOS, or both used to construct the plot in panel B. B) Plot of experimental ΔGexp vs computed ΔGcalc using 5 different crystal structures.
Inhibitors 4 and 5 were used with the plot in Fig. 6B as test cases. Since we do not have crystal structures of these inhibitors bound to eNOS, 4 and 5 were modeled into the eNOS active site pocket assuming that they adopt the same binding mode as in nNOS. ΔGcalc was calculated using the protocol outlined in the Methods section and converted to the same scale as experimental, ΔGexp, using the straight line equation derived from Fig. 6B.
ΔΔG is defined as the difference in binding free energy between nNOS and eNOS, ΔΔG = ΔGbind(nNOS) − ΔGbind(eNOS). For inhibitor 4 ΔΔGexp= 2.2 kcal/mol and ΔΔGcalc= 1.3 kcal/mol while for 5 ΔΔGexp = 4.2 kcal/mol and ΔΔGcalc= 4.1 kcal/mol (Table 1). It thus appears that the simplified MM-PBSA method used here is capable of correctly estimating NOS isoform selectivity and should prove useful in pre-screening novel NOS inhibitors of similar structure.
Table 1.
In vitro NOS inhibition (Ki in μM)
| Inhibitor | nNOS | eNOS | nNOS DM | eNOS/nNOS | ΔΔGexp (kcal/mol) | ΔΔGcalc (kcal/mol) |
|---|---|---|---|---|---|---|
| 1 | 0.3 | 107.0 | 357 | |||
|
| ||||||
| 2 | 0.15 | 80.0 | 533 | |||
|
| ||||||
| 3 | 0.10 | 110.0 | 1100 | |||
|
| ||||||
| 4(±) | 9.4 | 366.6 | 49.2 | 39 | 2.2 | 1.3 |
| 5(±) | 0.388 | 416.7 | 36.7 | 1068 | 4.2 | 4.1 |
| 6(±) | 0.085 | 85.2 | 1.2 | 1002 | - | - |
| 7(±) | 0.25 | 95.2 | 6.1 | 381 | - | - |
Discussion
The present work illustrates that fairly simple design principles derived from our previous work12, 22 can be used to prepare novel NOS inhibitors that retain isoform selectivity but might exhibit improved pharmacological properties. As in the earlier dipeptide inhibitor work, the key to isoform selectivity is the Asp597 (nNOS) vs. Asn368 (eNOS) difference. The pyrrolidine ring N atom and its close proximity to two negative charges in the nNOS active site compared to only one negative charge in the eNOS active site appears to be the key factor controlling selectivity. The advantage of these newer pyrrolidine inhibitors is that they are fairly rigid compared to the dipeptide inhibitors used in our previous work, and all bind the same to eNOS and nNOS; therefore, understanding the structural basis for selectivity is not complicated by differences in inhibitor conformation as in the case of the dipeptide inhibitors. The simplified MM-PBSA procedure, where only a single energy minimized structure is used and entropic factors ignored, does a reasonably good job in estimating isoform selectivity and is very rapid.
The ultimate practical outcome of these efforts is to develop therapeutic agents that can treat neuro-pathological conditions associated with the overproduction of NO by nNOS. Very recently, our groups have shown that compounds closely related to 6 are capable of significant protection against neural damage in a rabbit cerebral palsy model19. Moreover, protection is associated with a decrease in brain NO production and brain NOS activity without affecting eNOS regulated blood pressure. Our efforts on NOS structure-based drug design thus add to a short but growing documented list where the close interaction between structural biology, medicinal chemistry, and computer modeling can result in the development of novel compounds with demonstrated in vivo therapeutic effects.
Experimental Section
Protein and crystal preparations
The full-length, wild type or mutant, nNOS and eNOS proteins were expressed in E. coli strain BL21 (DE3). The proteins were purified with Ni Sepharose and/or 2', 5'- ADP Sepharose columns as described previously12, 13, 23. The heme domain nNOS or eNOS was generated from the partially purified full-length enzymes by limited trypsin digest. The heme domain protein was further separated from the fragments of the reductase domain by gel filtration through a Superdex 200 column23.
The purified nNOS or eNOS heme domain protein was concentrated to 7–9 (nNOS) or 20 (eNOS) mg/ml. About 8–10 mM of inhibitor was added to the protein before the sitting drop vapor diffusion crystallization setups using the reservoir solutions reported earlier12, 13, 23. Crystallization plates were left in a 5 °C incubator for more than 2 days to allow crystals to reach full size. Fresh crystals of less than 10 days old were flash-cooled with liquid nitrogen after passing through a series of cyroprotectant solutions as described previously12, 13, 23.
Inhibitory assays
Nitric oxide formation was monitored by the hemoglobin capture assay24 with buffer components described previously25. The apparent Ki values were obtained by measuring percent inhibition in the presence of 10 μM L-arginine with various amount of inhibitor. The parameters of the following inhibition equation were fitted to the initial 1 min velocity data: % inhibition = 100[I]/{[I] + Ki (1 + [S]/Km)}. Km value for nNOS double mutant was determined to be 1.9 μM.
X-ray diffraction data collection and crystal structure determination
The X-ray diffraction data were collected under a liquid nitrogen stream (100 K) with CCD detectors either at Stanford Synchrotron Radiation Lightsource (SSRL, Menlo Park, CA) or at Advanced Light Source (ALS, Berkeley, CA). Raw data were processed with HKL200026. The binding of inhibitor was detected by difference Fourier synthesis. The inhibitor then was modeled into the density using O27 and refined with CNS28. Water molecules were added automatically with CNS and inspected visually in O. The refined structures were validated before deposition to the RSCB data bank (http://deposit.pdb.org/validate/). The data collection and refinement statistics are summarized in Table 2.
Table 2.
Data collection and refinement statistics
| Data set1 PDB code | nNOS 4 3B3M | nNOS 5 3B3N | nNOS 6 3B3O | nNOS 7 3B3P | nNOS DM 5 3DQR | eNOS 6 3DQS | eNOS 7 3DQT |
|---|---|---|---|---|---|---|---|
| Cell dimensions(Å) (SG: P212121 | a = 51.50 | a = 52.05 | a = 52.21 | a = 52.58 | a = 51.87 | a = 57.96 | a = 58.25 |
| b = 109.53 | b = 110.69 | b = 111.53 | b = 110.25 | b = 110.70 | b = 106.84 | b = 106.49 | |
| c = 163.25 | c = 164.84 | c = 164.84 | c = 164.39 | c = 164.36 | c = 156.77 | c = 156.42 | |
| Data resolution (Å) | 1.95 | 1.98 | 2.05 | 2.45 | 2.40 | 2.03 | 2.54 |
| Total observations | 279484 | 245363 | 217270 | 131153 | 155390 | 261113 | 140776 |
| Unique reflections | 65107 | 66436 | 60556 | 35031 | 37670 | 64663 | 32771 |
| Rsym2 | 0.040 (0.251)3 | 0.064 (0.532) | 0.052 (0.428) | 0.101 (0.592) | 0.094 (0.732) | 0.054 (0.504) | 0.089 (0.740) |
| < I/σ > | 15.9 (5.1)3 | 11.7 (1.9) | 10.7 (1.9) | 6.3 (1.6) | 7.2 (1.9) | 9.6 (2.1) | 9.2 (2.1) |
| Completeness (%) | 94.9 (65.1)3 | 98.7 (90.1) | 97.8 (96.1) | 98.7 (99.4) | 99.3 (99.0) | 99.9 (100.0) | 99.6 (99.9) |
| Reflection used in refinement | 64931 | 66359 | 60120 | 34892 | 37621 | 63716 | 32770 |
| R factor4 | 0.204 | 0.230 | 0.213 | 0.209 | 0.205 | 0.197 | 0.197 |
| R-free5 | 0.233 | 0.270 | 0.253 | 0.265 | 0.261 | 0.227 | 0.258 |
| No. protein atoms | 6663 | 6659 | 6677 | 6677 | 6819 | 6418 | 6439 |
| No. heterogen atoms | 165 | 163 | 181 | 181 | 163 | 209 | 199 |
| No. water molecules RMS deviation | 546 | 392 | 418 | 247 | 268 | 488 | 184 |
| bond length((Å) | 0.007 | 0.009 | 0.010 | 0.009 | 0.010 | 0.009 | 0.010 |
| bond angle (˚) | 1.4 | 1.5 | 1.4 | 1.5 | 1.5 | 1.5 | 1.6 |
nNOS DM refers to nNOS D597N/M336V double mutant. See Fig. 1A for chemical structures and nomenclature of inhibitors.
Rsym = Σ |I - <I> | /Σ I, where I is the observed intensity of a reflection and <I> the averaged intensity of multiple observations of the reflection and its symmetry mates.
The values in parentheses were obtained in the outermost resolution shell.
R factor = Σ ∥Fo|-|Fc∥ / Σ |Fo|, Fo and Fc are the observed and calculated structure factors, respectively.
R-free was calculated with the 5% of reflections set aside randomly throughout the refinement.
Computational approaches
The free energy of binding of various NOS inhibitors was estimated using the MM-PBSA method29 as implemented in Amber 9.0. In this method the total free energy of the NOS-inhibitor complex is taken as the sum of the following energy terms
where EMM = the total molecular mechanics energy computed with the Sander module in Amber 9.0, Gsolv is the solvation free energy estimated from the Poisson-Boltzman equation, Gnp = the nonpolar solvation energy estimated from the solvent accessible surface area, and TSsolute = the solute entropy. From a single energy minimized structure the free energy was computed for the NOS-inhibitor complex, NOS alone with the inhibitor removed, and the inhibitor alone. The overall free energy of binding was computed from the following equation
As others have done the solute entropy was ignored30. Given that the inhibitors used for these calculations are structurally very similar with a similar number of rotatable bonds, ignoring inhibitor entropy introduces little error in comparing relative calculated and experimental free energies but does, of course, preclude the calculation of absolute free energies.
Inhibitor parameters and charges were assigned using the GAFF force field31 and AM1-BCC charge scheme32,33 as implemented in the Antechamber module in Amber 9.0. Heme parameters developed for cytochrome P450 were provided by Dr. Dan Harris34. It was necessary to carefully check the Antechamber output to make sure the correct atom types were assigned. For some inhibitors it was necessary to increase the force constant on improper torsion angles from 1.1 to 10.1 kcal/Å in order to maintain planarity of the nitroguanidinium and aminopyridine groups. To prepare the models for energetic calculations all crystallographic waters were removed and TIP3 waters added back within 30Å of the inhibitor. The resulting solvated structure was first energy minimized using the steepest descent method for 1,000 cycles with the inhibitor and heme heavy atom restrained to the starting crystallographic positions. The restraints were relaxed to 10.0 kcal/Å2 for the inhibitor and heme followed by another 1,000 cycles of refinement. In the last step the restraints for the heme and inhibitor were relaxed to 1.0 kcal/Å2 followed by 1,000 cycles of minimization. In the case of some eNOS-inhibitor complexes where structures were not determined but the nNOS-inhibitor complex structure was available, the inhibitor was positioned into the eNOS active site assuming it adopts the same conformation and position as that found in nNOS.
Fig. 4.

Active site structures of the wild type eNOS with inhibitor 6 (panel A) or 7 (panel B) bound. Also shown around the inhibitor is the Fo – Fc omit map contoured at 3.0σ. Residue Trp76 belongs to the neighboring subunit. Alternate conformations of heme propionate off the pyrrole ring D are also depicted.
Acknowledgements
This research was supported by NIH grants GM57353 (T.L.P) and GM49725 (R.B.S.). We thank the beam line staff at SSRL and ALS for their assistance during data collections.
Abbreviations used
- NOS
nitric oxide synthase
- nNOS
neuronal NOS
- iNOS
inducible NOS
- eNOS
endothelial NOS
- 2',5'-ADP
adenosine 2',5'-diphosphate
- CCD
charge coupled device
- MM-PBSA
Molecular Mechanics with Poisson-Boltzman Surface Area methodology
- GAFF
general Amber force field
- AM1-BCC
Austin Model 1-Bond Charge Correction. Crystallographic coordinates have been deposited with the Protein Data Base with accession codes 3B3M, 3B3N, 3B3O, 3B3P, 3DQR, 3DQS, and 3DQT
References
- 1.Griffith OW, Stuehr DJ. Nitric oxide synthases: properties and catalytic mechanism. Annu. Rev. Physiol. 1995;57:707–736. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]
- 2.Raman CS, Martasek P, Masters BSS. Structural themes determining function in nitric oxide synthases. In: Kadish KM, Smith KM, Guilard R, editors. The porphyrin handbook. Vol. 4. Acadamic Press; San Diego: 2000. pp. 293–339. [Google Scholar]
- 3.Abu-Soud HM, Stuehr DJ. Nitric oxide synthases reveal a role for calmodulin in controlling electron transfer. Proc. Natl. Acad. Sci. U. S. A. 1993;90:10769–10772. doi: 10.1073/pnas.90.22.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 5.Kerwin JF, Jr., Lancaster JR, Jr., Feldman PL. Nitric oxide: a new paradigm for second messengers. J. Med. Chem. 1995;38:4343–4362. doi: 10.1021/jm00022a001. [DOI] [PubMed] [Google Scholar]
- 6.Dawson VL, Dawson TM. Nitric oxide in neurodegeneration. Prog. Brain Res. 1998;118:215–229. doi: 10.1016/s0079-6123(08)63210-0. [DOI] [PubMed] [Google Scholar]
- 7.Lipton P. Ischemic cell death in brain neurons. Physiol. Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 8.Sims NR, Anderson MF. Mitochondrial contributions to tissue damage in stroke. Neurochem. Int. 2002;40:511–526. doi: 10.1016/s0197-0186(01)00122-x. [DOI] [PubMed] [Google Scholar]
- 9.Bingham CO., 3rd The pathogenesis of rheumatoid arthritis: pivotal cytokines involved in bone degradation and inflammation. J. Rheunatol. Suppl. 2002;65:3–9. [PubMed] [Google Scholar]
- 10.Taddei S, Virdis A, Ghiadoni L, Sudano I, Salvetti A. Endothelial dysfunction in hypertension. J. Cardiovascular Pharmacol. 2001;38(Suppl. 2):S11–S14. doi: 10.1097/00005344-200111002-00004. [DOI] [PubMed] [Google Scholar]
- 11.Napoli C, de Nigris F, Williams-Ignarro S, Pignalosa O, Sica V, Ignarro LJ. Nitric oxide and atherosclerosis: An update. Nitric Oxide. 2006;15:265–279. doi: 10.1016/j.niox.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 12.Flinspach ML, Li H, Jamal J, Yang W, Huang H, Hah JM, Gomez-Vidal JA, Litzinger EA, Silverman RB, Poulos TL. Structural basis for dipeptide amide isoform-selective inhibition of neuronal nitric oxide synthase. Nat. Struct. Mol. Biol. 2004;11:54–59. doi: 10.1038/nsmb704. [DOI] [PubMed] [Google Scholar]
- 13.Li H, Flinspach ML, Igarashi J, Jamal J, Yang W, Gomez-Vidal JA, Litzinger EA, Huang H, Erdal EP, Silverman RB, Poulos TL. Exploring the binding conformations of bulkier dipeptide amide inhibitors in constitutive nitric oxide synthases. Biochemistry. 2005;44:15222–15229. doi: 10.1021/bi0513610. [DOI] [PubMed] [Google Scholar]
- 14.Huang H, Martasek P, Roman LJ, Masters BS, Silverman RB. N(omega)-Nitroarginine-containing dipeptide amides. Potent and highly selective inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 1999;42:3147–3153. doi: 10.1021/jm990111c. [DOI] [PubMed] [Google Scholar]
- 15.Huang H, Martasek P, Roman LJ, Silverman RB. Synthesis and evaluation of peptidomimetics as selective inhibitors and active site probes of nitric oxide synthases. J. Med. Chem. 2000;43:2938–2945. doi: 10.1021/jm000127z. [DOI] [PubMed] [Google Scholar]
- 16.Hah JM, Roman LJ, Martasek P, Silverman RB. Reduced amide bond peptidomimetics. (4S)-N-(4-amino-5-[aminoakyl]aminopentyl)-N'-nitroguanidines, potent and highly selective inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 2001;44:2667–2670. doi: 10.1021/jm0101491. [DOI] [PubMed] [Google Scholar]
- 17.Gomez-Vidal JA, Martasek P, Roman LJ, Silverman RB. Potent and selective conformationally restricted neuronal nitric oxide synthase inhibitors. J. Med. Chem. 2004;47:703–710. doi: 10.1021/jm030297m. [DOI] [PubMed] [Google Scholar]
- 18.Ji H, Stanton BZ, Li H, Martásek P, Roman L, Poulos TL, Silverman RB. Minimal pharmacophoric elements and a pharmacophore-driven strategy for fragment-based de novo design, an approach directed at molecular diversity and isozyme selectivity. design of selective neuronal nitric oxide synthase inhibitors. J. Am. Chem. Soc. 2008;130:3900–3914. doi: 10.1021/ja0772041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji H, Tan S, Igarashi J, Li H, Derrick M, Martasek P, Roman LJ, Vasquez-Vivar J, Poulos TL, Silverman RB. Selective neuronal nitric oxide synthase inhibitors and the prevention of cerebral palsy. Ann. Neurol. 2008 doi: 10.1002/ana.21555. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuhn B, Gerber P, Schulz-Gasch T, Stahl M. Validation and use of the MMPBSA approach for drug discovery. J. Med. Chem. 2005;48:4040–4048. doi: 10.1021/jm049081q. [DOI] [PubMed] [Google Scholar]
- 21.Weis A, Katebzadeh K, Soderhjelm O, Nilsson I, Ryde U. Ligand affinities predicted with the MM/PBSA method: Dependence on the simulation method and the force field. J. Med. Chem. 2006;49:6596–6606. doi: 10.1021/jm0608210. [DOI] [PubMed] [Google Scholar]
- 22.Flinspach M, Li H, Jamal J, Yang W, Huang H, Silverman RB, Poulos TL. Structures of the neuronal and endothelial nitric oxide synthase heme domain with D-nitroarginine-containing dipeptide inhibitors bound. Biochemistry. 2004;43:5181–5187. doi: 10.1021/bi0361867. [DOI] [PubMed] [Google Scholar]
- 23.Li H, Shimizu H, Flinspach M, Jamal J, Yang W, Xian M, Cai T, Wen EZ, Jia Q, Wang PG, Poulos TL. The novel binding mode of N-alkyl-N'-hydroxyguanidine to neuronal nitric oxide synthase provides mechanistic insights into NO biosynthesis. Biochemistry. 2002;41:13868–13875. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]
- 24.Hevel JM, Marletta MA. Nitric-oxide synthase assays. Methods Enzymol. 1994;233:250–258. doi: 10.1016/s0076-6879(94)33028-x. [DOI] [PubMed] [Google Scholar]
- 25.Hah JM, Martasek P, Roman LJ, Silverman RB. Aromatic reduced amide bond peptidomimetics as selective inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 2003;46:1661–1669. doi: 10.1021/jm0202932. [DOI] [PubMed] [Google Scholar]
- 26.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 27.Jones TA, Zou J-Y, Cowan SW, Kjeldgaarrd M. Improved methods for building models in electron density and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 28.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR System: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 29.Massova I, Kollman PA. Computational alanine scanning to probe protein-protein interactions: A novel approach to evaluate binding free energies. J. Am. Chem. Soc. 1999;121:8133–8143. [Google Scholar]
- 30.Brown SP, Muchmore SW. High-throughput calculation of protein-ligand binding affinities: Modification and adaption of the MM-PBSA protocol to enterprise grid computing. J. Chem. Inf. Model. 2006;46:999–1005. doi: 10.1021/ci050488t. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case D. Development and testing of a general Amber force field. J. Am. Chem. Soc. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 32.Jakalian A, Bush BL, Jack DB, Bayly CI. Fast, efficient generation of high-quality atom charges. AM1-BCC model: I. Method. J. Comp. Chem. 2000;21:132–146. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 33.Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atom charges. AM1-BCC model: II. Parameterization and validation. J. Comp. Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 34.Harris DL, Park JY, Gruenke L, Waskell L. Theoretical study of the ligand-CYP2B4 complexes: Effect of structure on binding free energies and heme spin state. Proteins. 2004;15:895–914. doi: 10.1002/prot.20062. [DOI] [PubMed] [Google Scholar]