

Abstract
AIM: To investigate the effects of schisandrin B (Sch B) on free fatty acid (FFA)-induced steatosis in L-02 cells.
METHODS: Cellular steatosis was induced by incubating L-02 cells with a FFA mixture (oleate and palmitate at the ratio of 2:1) for 24 h. Cytotoxicity and apoptosis were evaluated by 3-(4, 5-dmethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide assay and Annexin V/propidium iodide staining, respectively. Cellular total lipid was determined using a photocolorimetric method after Nile red staining, and triglyceride content was measured using an enzymatic kit. To study the effects of Sch B on steatosis, L-02 cells were treated with Sch B (1-100 μmol/L) in the absence or presence of 1 mmol/L FFA for 24 h, and cellular total lipid and triglyceride levels were measured. To explore the mechanisms of action of Sch B in the steatotic L-02 cells, mRNA levels of several regulators of hepatic lipid metabolism including adipose differentiation related protein (ADRP), sterol regulatory element binding protein 1 (SREBP-1), peroxisome proliferator-activated receptor (PPAR)-α and PPAR-γ were measured by quantitative real-time polymerase chain reaction (PCR), and protein levels of ADRP and SREBP-1 were measured by immunoblotting.
RESULTS: Treatment with 1 mmol/L FFA for 24 h induced intracellular lipid accumulation in L-02 cells comparable to that in human steatotic livers without causing apparent apoptosis and cytotoxicity. Sch B mitigated cellular total lipid and triglyceride accumulations in the steatotic L-02 cells in a dose-dependent manner. Quantitative real-time PCR and Western blot analyses revealed that treatment of L-02 cells with 100 μmol/L Sch B reverted the FFA-stimulated up-regulation of ADRP and SREBP-1.
CONCLUSION: Sch B inhibits FFA-induced steatosis in L-02 cells by, at least in part, reversing the up-regulation of ADRP and SREBP-1.
Keywords: Free fatty acid, Hepatic lipid metabolism, Hepatocellular steatosis, L-02 cells, Schisandrin B
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) has emerged as a serious and widespread metabolic disease, which entails a wide spectrum of liver disorders and damages ranging from simple steatosis through steatohepatitis and fibrosis to end stage liver diseases including cirrhosis and hepatocellular carcinoma[1]. The clinical significance of NAFLD is largely due to its high prevalence of around 20% in general populations and up to 80% in obese and diabetic individuals worldwide[2]. Although the pathogenesis of NAFLD has not been clearly defined, hepatic steatosis characterized by uncontrolled triglyceride accumulation in hepatocytes and oxidative stress are believed to play a crucial role[3]. Therefore, agents that are capable of lowering hepatic lipid levels and alleviating oxidative stress may be beneficial to the control of NAFLD.
Schisandrin B (Sch B) (Figure 1) is the most abundant and active dibenzocyclooctadiene derivative isolated from the fruits of Schisandra chinensis, a traditional Chinese medicinal herb commonly used in treatment of viral and chemical hepatitis. A growing body of evidence has shown that Sch B can protect liver from damage caused by oxidative stress. Sch B may inhibit oxygen free-radical lipoperoxidative damage to plasma membrane of rat hepatocytes in vitro[4]. Sch B pretreatment protects mouse livers against tumor necrosis factor α-induced apoptosis in a dose-dependent manner[5]. In addition, Sch B can protect mice against carbon tetrachloride-induced hepatic toxicity by inhibiting lipid peroxidation[6]. Recently, we have reported that Sch B has hepatic lipid lowering effects in mice fed a high-fat diet[7]. These lines of evidence underscore both hepatic lipid-lowering and antioxidant effects of Sch B, making it a promising candidate for the treatment of NAFLD. Although the antioxidant role of Sch B has been well investigated, the mechanism underlying its hepatic lipid-lowering action remains unknown. This study was designed to investigate the anti-hepatosteatotic effects and mechanisms of Sch B using cultured steatotic cells.
Figure 1.
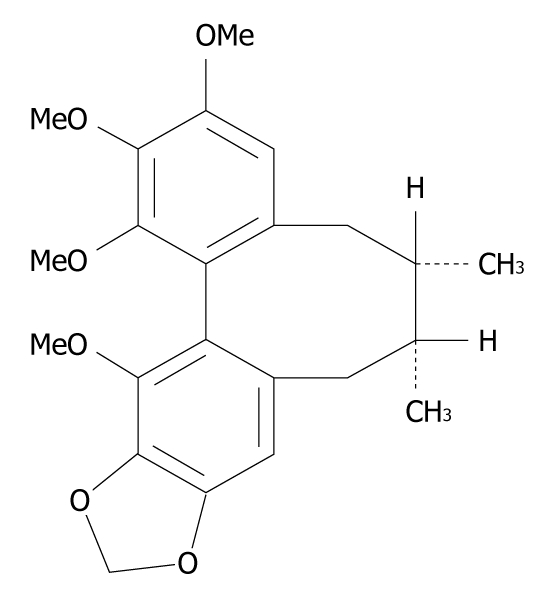
Chemical structure of schisandrin B.
NAFLD patients exhibit an elevated lipolysis and high circulating free fatty acid (FFA) levels[8]. High circulating FFA levels can trigger a series of biological changes in hepatic lipid metabolism, thus ultimately leading to hepatic steatosis[9]. Therefore, cellular FFA loading is commonly utilized to develop in vitro models of steatosis. These models can reliably reproduce the key features of hepatic steatosis in human beings[10-12], rendering them useful for the identification of potential therapeutic targets and effective intervention approaches against NAFLD. Human hepatocytes in primary culture represent the model closest to human liver tissues. Nevertheless, their use is often greatly hampered due to scarcity of liver samples[13]. HepG2 and Huh-7, two human hepatoma cell lines, are frequently used in establishing in vitro steatosis models. However, the validity of cancer cell-based models is concerned because metabolic regulation is often altered in cancer cells. For example, it has been highlighted that cancer cells may carry out an increased fatty acid de novo synthesis irrespective of the extracellular lipid levels[13]. Therefore, in this study, we first established FFA-induced steatotic cells using an immortalized normal human hepatocytes-derived cell line L-02[14,15]. Then, we investigated the in vitro effects of Sch B on hepatosteatosis in the steatotic L-02 cells, and explored the underlying mechanisms.
MATERIALS AND METHODS
Cell culture and treatment
L-02 (Institute of Biochemistry and Cell Biology, Shanghai Institute for Biological Sciences, Shanghai, China) and HepG2 (ATCC) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, GIBCO) supplemented with 10% fetal bovine serum (FBS, GIBCO, USA) and 1% penicillin/streptomycin (P/S, GIBCO, USA) at 37°C in an atmosphere containing 5% CO2. When FFA mixture (sodium salts of oleate and palmitate, Sigma, Malaysia) was added, bovine serum albumin (BSA) was supplemented to a final concentration of 1% in the culture medium. Cell cultures were used in experiments when they reached 75% confluence.
Sch B was purchased from Ningli Technology Co. Ltd. (Kunming, China) with a purity of 98% as determined by HPLC. A stock solution of Sch B (100 mmol/L) was prepared in dimethylsulfoxide (DMSO). The concentration of vehicle DMSO was 0.1% in treated cell cultures.
Cell viability assay
Cytotoxicity of FFA to L-02 cells was assessed by 3-(4, 5-dmethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) assay. L-02 cells in 96-well plates were treated with FFA. After incubated for 24 h, 20 μL of MTT solution (5 mg/mL, USB, Austria) was added to each well and the plates were further incubated at 37°C for 4 h. After medium removal, 100 μL of DMSO was added to each well of the plates which were then gently shaken for 5 min. Optical absorbance was determined at 570 nm with a microplate spectrophotometer (BD Bioscience, USA). Each treatment was performed in triplicate.
Quantification of apoptosis
Early and late phase apoptotic cells were assessed using the Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit I (BD Bioscience, USA) following the manufacturer’s instructions. After treatment with FFA, L-02 cells were harvested and rinsed twice with cold PBS, resuspended in the binding buffer, and incubated with Annexin V-FITC and propidium iodide (PI) staining solution. Samples of 10 000 stained cells were analyzed using a flow cytometer (BD Bioscience, USA).
Nile red staining
L-02 cells in F96 microwell black plates (Nunc, Denmark) were treated with FFA in the presence or absence of Sch B for 24 h. Photocolorimetric measurement of intracellular lipid contents in Nile red stained cells was performed as previously described[16]. Each treatment was performed in triplicate.
Phase-contrast and fluorescence microscope imaging
L-02 cells in 6-well plates were treated with FFA for 24 h, washed with PBS, stained with 1 μmol/L Nile red in HBSS for 15 min and then examined under phase-contrast (Leica, Germany) and fluorescence (Nikon, Japan) microscopes.
Measurement of intracellular triglyceride content
Cellular triglyceride content was measured using an enzymatic kit (Zhongsheng Beikong Biotechnology and Science Inc, China) following the manufacturer’s instructions. Triglyceride content was expressed in microgram of triglycerides per microgram protein. Protein concentration was measured by Bio-Rad protein assay (Bio-Rad, USA). Each treatment was performed in triplicate.
Semi-quantitative and real-time quantitative polymerase chain reaction analyses
Total RNA was extracted with Trizol reagent (Invitrogen, USA), and 2 μg of RNA was reverse-transcribed with oligo-dT using the M-MLV reverse transcriptase (Promega, USA) according to the manufacturer’s protocol. For semi-quatitative polymerase chain reaction (PCR), the resultant cDNA was subjected to 25-30 cycles of PCR amplification (denaturing at 95°C for 30 s, annealing at 55-60°C for 30 s, extension at 72°C for 60 s). The PCR products were separated by electrophoresis on a 2% agarose gel and visualized with ethidium bromide staining. Quantitative real-time PCR was performed using SYBR green reaction mixture in the ABI 7500 fast real-time PCR system (Applied Biosystems). The PCR conditions were one cycle at 55°C for 2 min and at 95°C for 10 min, followed by 40 cycles of amplification at 95°C for 15 s and at 60°C for 1 min. The fluorescent signals were detected using the ABI Prism 7500HT sequence detection system (Applied Biosystems). The gene expression data were normalized to the endogenous control β-actin. The relative expression levels of genes were measured according to the formula 2-ΔCt, where ΔCt is the difference in threshold cycle values between the targets and β-actin. All samples were analyzed in triplicate. The specific primer pairs used for detecting messenger RNA are listed in Table 1.
Table 1.
Primers used for polymerase chain reaction amplification of mRNA
| Gene | Forward primer | Reverse primer |
| SREBP-1 | ACGGCAGCCCCTGTAACGACCACTGTGA | TGCCAAGATGGTTCCGCCACTCACCAGG |
| ADRP | GGGATCCCTGTCTACCAAGC | AGATGTCGCCTGCCATCACC |
| PPAR-α | CCAGTATTTAGGAAGCTGTCCTG | CGTTGTGTGACATCCCGACAG |
| PPAR-γ | TGGTGACTTTATGGAGCCCAA | GGCAAACAGCTGTGAGGACTCAG |
| β-actin | GACTACCTCATGAAGATC | GATCCACATCTGCTGGAA |
SREBP-1: Sterol regulatory element binding protein 1; ADRP: Adipose differentiation related protein; PPAR: Peroxisome proliferator-activated receptor.
Western blot analysis
L-02 cells were harvested and lysed on ice with the RIPA buffer consisting of 50 mmol/L Tris-Cl, 1% NP-40, 0.35% sodium-deoxycholate, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, pH 7.4, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L NaF, 1 mmol/L Na3VO4 and 10 μg/mL each of aprotinin, leupetin and pepstatin A. Protein concentration in each sample was measured by the Bio-Rad protein assay. Protein samples (each 15 μg) were separated by SDS-PAGE and then electro-transferred onto nitrocellulose membranes (Amersham Biosciences, USA), which were blocked for 30 min with 5% skim milk in the TBST buffer containing 50 mmol/L Tris (pH 7.6), 150 mmol/L NaCl and 0.1% Tween-20 and incubated with specific antibodies against adipose differentiation related protein (ADRP) (Abcam), sterol regulatory element binding protein 1 (SREBP-1) (Santa Cruz) or β-actin (Santa Cruz) overnight at 4°C. The membranes were then incubated with HRP-conjugated secondary antibodies, and immunoreactive bands were visualized using the ECL detection kit (Amersham Biosciences, USA) following the manufacturer’s instructions.
Statistical analysis
All results were expressed as mean ± SE. The difference between two groups was analyzed using the Student’s t test.
RESULTS
Cytotoxic effect of FFA treatment on L-02 cells
L-02 cells were treated with 0.5-2 mmol/L a FFA mixture (oleate and palmitate at the ratio of 2:1) for 24 h and the cytotoxicity of FFA to L-02 cells was detected by MTT assay. No apparent cytotoxic effect of FFA was observed on L-02 cells after treatment with FFA at the concentration of 0.5 or 1 mmol/L, while the cell viability was decreased by 40% when L-02 cells were treated with FFA at the concentration of 2 mmol/L (Figure 2A). These results suggest that 0.5 or 1 but not 2 mmol/L FFA can be used to prepare steatotic L-02 cells.
Figure 2.

Cytotoxic and apoptotic effects of free fatty acid treatment on cultured cells. A: L-02 cells were treated with a free fatty acid (FFA) mixture (oleate and palmitate at the ratio of 2:1) at various concentrations for 24 h. Cell viability was determined by the 3-(4,5)-dimethylthiahiazo (-z-y1)-3,5-di- phenytetrazoliumromide (MTT) assay. bP < 0.01 vs control group; B: L-02 and HepG2 cells were treated with 1 mmol/L FFA mixture (oleate and palmitate at the ratio of 2:1) for 24 h and stained with Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide. Apoptotic and necrotic cells were monitored by flow cytometry. Normal, early and late apoptotic cells as well as necrotic cells were shown in Q3, Q4, Q2 and Q1 quadrants, respectively. The percentage of cells in each quadrant was displayed. Results were the representative of three independent experiments; C: Quantification of early phase apoptotic cells in response to FFA treatment. bP < 0.01 vs HepG2 control group.
Apoptotic effect of FFA treatment on L-02 cells
To evaluate the apoptotic effect of FFA treatment on L-02 cells, the L-02 cells were treated with 1 mmol/L FFA (oleate and palmitate at the ratio of 2:1) for 24 h, and then stained with Annexin V/PI. Apoptosis of L-02 cells was monitored by flow cytometry. For comparison, apoptosis of HepG2 cells induced by FFA treatment was also analyzed. FFA treatment did not trigger early- or late-stage apoptosis of L-02 cells but significantly induced early-stage apoptosis of HepG2 cells (Figure 2B and C). The percentage of apoptotic HepG2 cells was increased from 1.500% ± 0.473% in control cells to 9.267% ± 0.203% in FFA-treated cells (Figure 2C), which is consistent with previous reports showing that FFA causes apoptosis of HepG2 cells under the same conditions[11,12]. These results suggest that L-02 and HepG2 cell lines do have different responses to FFA treatment.
FFA treatment induced lipid accumulation in L-02 cells
We next examined the cellular lipid accumulation in L-02 cells treated with the FFA mixture for 24 h at the concentration of 0.5 mmol/L or 1 mmol/L using Nile red staining. The results showed that FFA induced lipid accumulation (Figure 3A) in L-02 cells in a dose-dependent manner, which was confirmed by fluorescent microscopy (Figure 3B). When the L-02 cells were treated with FFA mixture at the concentration of 1 mmol/L, the intracellular lipid content was increased by 5.34 ± 0.65-fold in L-02 cells compared to that in FFA-untreated controls (Figure 3A). The cellular lipid accumulation level in L-02 cells treated with FFA at the concentration of 1 mmol/L was comparable to that in human steatotic livers, which is 5.5-fold over non-steatotic livers[13].
Figure 3.

Free fatty acid induced lipid accumulation in L-02 cells. A: L-02 cells were incubated with a free fatty acid (FFA) mixture (oleate and palmitate at the ratio of 2:1) for 24 h. Intracellular lipid accumulation was evaluated after Nile red staining. Results were expressed as mean ± SE of three independent experiments. bP < 0.01 vs control group; B: Representative micrographs showing intracellular lipid accumulation in L-02 cells as observed by phase-contrast microscopy (panels a-c) and fluorescence microscopy (panels d-f). Panels a/d, b/e and c/f are control cells, cells treated with 0.5 and 1 mmol/L FFA, respectively; C: Triglyceride levels in L-02 cells treated with 1 mmol/L FFA. Results were expressed as mean ± SE of three independent experiments. bP < 0.01 vs control group.
We also measured the intracellular triglyceride levels in L-02 cells treated with FFA mixture at the concentration of 1 mmol/L. The triglyceride content was increased by about 2.5-fold from 0.108 ± 0.027 μg/μg protein in control cells to 0.241 ± 0.030 μg/μg protein in FFA-treated cells (Figure 3C), which is similar to the results obtained from human liver samples. The triglyceride content is about 2.7-fold higher in human steatotic livers than in non-steatosis livers[13].
The above data indicate that steatotic cells can be prepared by incubating L-02 cells with a FFA mixture (oleate and palmitate at the ratio of 2:1) at the concentration of 1 mmol/L for 24 h, in which lipid accumulation can reach a level similar to that in human steatotic livers in the absence of apoptosis.
Sch B treatment alleviated FFA-induced lipid accumulation in L-02 cells
To investigate the anti-steatotic effect of Sch B in L-02 cells, L-02 cells were exposed to various concentrations of Sch B (1-100 μmol/L) in the absence or presence of FFA mixture at the concentration of 1 mmol/L for 24 h. The intracellular total lipid levels in L-02 cells were measured after Nile red staining and the triglyceride contents were assayed using an enzymatic kit. Nile red staining assay showed that Sch B at all concentrations had no significant effect on the cellular lipid content in L-02 cells in the absence of FFA, but substantially ameliorated the lipid accumulation induced by FFA in a dose-dependent manner (Figure 4A). The lipid-lowering effect of Sch B at the concentration of 100 μmol/L was further confirmed by microscopic examination of the fluorescence of Nile red-stained L-02 cells (Figure 4B). The intracellular triglyceride measurements showed that Sch B inhibited the fat accumulation in a dose-dependent manner, and exerted a significant inhibitory effect in L-02 cells treated with FFA at the concentration of 1 mmol/L (Figure 4C). It was noteworthy that Sch B at each tested concentration did not elicit apparent cytotoxicity or apoptosis in L-02 cells in the presence or absence of 1 mmol/L FFA at 24 h (data not shown).
Figure 4.

Effect of schisandrin B on free fatty acid-induced fat accumulation in L-02 cells. A: L-02 cells were treated with schisandrin B (Sch B) (1 μmol/L, 10 μmol/L or 100 μmol/L) in the presence or absence of 1 mmol/L free fatty acid (FFA) mixture (oleate and palmitate at the ratio of 2:1) for 24 h. Intracellular total lipid levels were measured after Nile red staining; B: Representative micrographs showing intracellular lipid accumulation in Nile red stained L-02 cells after treatment with 100 μmol/L Sch B in the presence of 1 mmol/L FFA examined by fluorescent microscopy; C: L-02 cells were treated with Sch B at the indicated concentrations for 24 h and cellular triglyceride levels were measured using an enzymatic kit. bP < 0.01 vs FFA-untreated control groups; cP < 0.05, dP <0.01 vs FFA-treated groups. Data are from three independent experiments.
Sch B decreased mRNA and protein expression levels of ADRP and SREBP-1 in FFA-induced steatotic L-02 cells
To explore the mechanisms underlying Sch B-mediated lipid-lowering action in L-02 cells, the mRNA expression levels of ADRP, SREBP-1, peroxisome proliferator-activated receptor (PPAR)-α and PPAR-γ, four important regulators of hepatic lipid metabolism, were first measured by semi-quantitative PCR and quantitative real-time PCR, respectively. Sch B attenuated the FFA-induced fat accumulation most effectively at the concentration of 100 μmol/L, thus this dosage of Sch B was used in this experiment. The mRNA expression level of PPAR-α remained unchanged in FFA-treated L-02 cells, while the mRNA expression levels of the other three genes were up-regulated (Figure 5A and B). Concurrent treatment with Sch B at the concentration of 100 μmol/L for 24 h restored the FFA-upregulated ADRP and SREBP-1 expression to normal levels, but did not affect FFA-stimulated PPAR-γ mRNA expression. In addition, Sch B treatment did not obviously influence the expression of PPAR-α in L-02 cells in the presence of FFA.
Figure 5.

Effect of schisandrin B on mRNA and protein expression levels of several lipid metabolism-related molecules in free fatty acid-treated L-02 cells. A, B: Semi-quantitative polymerase chain reaction (PCR) and real-time quantitative PCR showing mRNA levels of adipose differentiation related protein (ADRP), sterol regulatory element binding protein 1 (SREBP-1), peroxisome proliferator-activated receptor (PPAR)-α and PPAR-γ. Results shown are the representative of three independent experiments. aP < 0.05, bP < 0.01 vs control group; cP < 0.05 vs free fatty acid-treated groups; C: Immunoblotting showing expression levels of ADRP and SREBP-1 proteins. The representative blots out of three experiments are shown. FFA: Circulating free fatty acid; Sch B: Schisandrin B.
Immunoblot analysis showed that the protein levels of ADRP and SREBP-1 were dramatically elevated in L-02 cells in response to FFA treatment, but returned to normal levels after concurrent treatment with Sch B (Figure 5C).
DISCUSSION
FFA-induced hepatocellular steatosis models have been widely applied in studies on NAFLD pathogenesis and anti-NAFLD drugs[13,17]. Although human hepatocytes in primary culture represent the most stringent model of the human liver, they are tedious to prepare and the reproducibility of experimental results is often a big problem[18,19]. On the other hand, the use of liver cancer cell lines including HepG2 and Huh-7 is often questioned about their acquired genetic and epigenetic alterations which may endow them with numerous properties including metabolism regulation distinct from normal hepatocytes[20]. In the present study, we successfully prepared FFA-induced steatotic cells using a normal human hepatocytes-derived cell line L-02. The steatotic L-02 cells behave similarly to human steatotic livers in two aspects. First, 24 h treatment with a FFA mixture (oleate and palmitate at the ratio of 2:1) at the concentration of 1 mmol/L caused no apparent toxicity to L-02 cells. Second, the induced fat accumulation in L-02 cells was comparable to that in human steatotic livers[13]. These aforementioned attributes make FFA-induced steatotic L-02 cells suitable for the investigations of NAFLD pathogenesis and anti-NAFLD agents.
High circulating FFA concentration may aggravate hepatic fat accumulation by disrupting lipid metabolism in NAFLD patients, and thus studying how FFA overload influences metabolic regulators will further improve our understanding about the pathogenesis of NAFLD. Given that palmitic and oleic acids are the most abundant fatty acids in liver triglycerides in both normal subjects and NAFLD patients[21], clarification of their effect on hepatocytes is of great importance. In the present study, treatment of L-02 cells with a FFA mixture (oleate and palmitate at the ratio of 2:1) did not significantly affect PPAR-α mRNA expression level but increased the mRNA expression levels of ADRP, SREBP-1 and PPAR-γ. The unchanged PPAR-α mRNA expression in L-02 cells treated with FFA suggests that PPAR-α-mediated mitochondria fatty acid β-oxidation may not been affected by FFA in our experimental conditions. The increased SREBP-1 and PPAR-γ mRNA expression levels in response to FFA treatment are in good accord with a recent report assuming that the up-regulations of SREBP-1 and PPAR-γ are linked to the steatogenic property of oleic acid[17]. Moreover, the ADRP mRNA expression was elevated in L-02 cells challenged with FFA, which is in agreement with the reported observations in other cell lines treated with FFA[22,23]. ADRP is a lipid storage droplet-associated protein found in most cells and tissues, and has been suggested to be a marker of lipid accumulation, because the cellular level of ADRP is proportional to the total mass of neutral lipids within the cells[24]. Fatty acids have been implicated as ligands for PPAR family members including PPAR-α and PPAR-γ, it is thus believed that the stimulation of ADRP gene by FFA is at least in part due to PPAR activation. It has been shown that the activated PPAR can complex with retinoid X receptor and bind to the PPAR response element in the promoter of ADRP gene[25,26]. As both PPAR-α and PPAR-γ were detectable in L-02 cells, further studies are needed to ascertain whether one or both of them are required for the up-regulation of ADRP gene expression induced by FFA treatment. These findings suggest that exposure to exogenous FFA may interfere with lipid metabolism through the modulation of metabolic regulators in L-02 cell line derived from normal human hepatocytes.
We demonstrated that Sch B exerted a drastic inhibitory effect on FFA-induced steatosis in L-02 cells. This finding and the hepatic lipid-lowering action of Sch B observed in high-fat diet-fed mice[7] strongly highlight the anti-steatosis potential of Sch B. Since FFA overloading contributed to hepatic fat accumulation through modulation of lipid metabolism- related genes in our established steatotic L-02 cells, it is conceivable that Sch B may attenuate fat accumulation by counteracting or reversing the unfavorable changes in expression of genes evoked by FFA. In this study, Sch B treatment restored the FFA-induced up-regulation of both mRNA and protein levels of ADRP and SREBP-1 to normal levels, indicating that ADRP and SREBP-1 are the potential targets of Sch B in relation to its lipid-lowering property.
ADRP expression is closely associated with intracellular lipid droplets and up-regulated in hepatic steatosis in human and mouse models[27,28]. It has been reported that ADRP overexpression may promote lipid accumulation in fibroblasts and macrophages without changing the expression of adipogenic genes and genes involved in lipid efflux[29,30]. Intriguingly, ADRP overexpression may facilitate the uptake of long chain FFA in COS-7 cells[31]. Moreover, Edvardsson et al[32] have recently proposed that ADRP may enhance cellular triglyceride accumulation in hepatocytes by increasing fatty acid uptake, driving fatty acids to triglyceride formation as well as preventing the use of triglyceride in VLDL assembly. In this connection, the down-regulation of ADRP may contribute to the anti-hepatosteatotic effect of Sch B by inhibiting the uptake of exogenous long chain FFA, decreasing the incorporation of FFA into triglyceride and increasing the availability of triglyceride for VLDL assembly. It has been demonstrated that ADRP-deficient mice produced by either knock-out or anti-sense oligonucleptide technology do not acquire diet-induced hepatic steatosis[26,33,34], raising the possibility that ADRP may become a putative molecular target for the prevention of NAFLD,thus screening for compounds that can repress hepatic ADRP expression may provide a new direction for the identification of potential therapeutic agents against NAFLD. It has been recently demonstrated that pycnogenol, a French maritime pine bark extract, can reduce oleic acid-induced lipid droplet formation in mouse liver epithelial cells MMuLi by inhibiting ADRP expression, and interestingly the suppression of ADRP expression is mediated in part by facilitating mRNA degradation[23]. How Sch B impairs ADRP expression in steatotic L-02 cells remains to be evaluated.
SREBP-1 is the most important transcription factor regulating de novo lipogenesis in the liver. There is compelling evidence that supports the involvement of SREBP-1 in NAFLD development. It has been reported that SREBP-1 expression is significantly elevated in livers from NAFLD and obesity patients, and from insulin-resistant and hyperinsulinemic ob/ob mice[27,35,36]. Overexpression of SREBP-1 in cultured hepatocytes or mouse livers can increase hepatic triglyceride deposition and mRNA expression of genes involved in lipogenesis[37-39]. Moreover, in Lepob/ob mice deficient in SREBP-1, hepatic steatosis is markedly attenuated, which is accompanied by decreased mRNA levels of lipogenic enzymes[40]. These lines of evidence strongly suggest that SREBP-1 plays a pivotal role in the regulation of hepatic lipid metabolism, thus pharmacological manipulation of SREBP-1 may be beneficial to the management of NAFLD. In this study, Sch B could reverse FFA-induced up-regulation of SREBP-1. Therefore it is plausible to infer that the down-regulation of SREBP-1 may partly contribute to the lipid-lowering activity of Sch B by inhibiting de novo lipogenesis. Since SREBP-1 may transcriptionally activate a variety of genes required for lipogenesis in the liver[41], it is of interest to investigate which SREBP-1 target genes are regulated by Sch B. Another question is how Sch B regulates the expression of SREBP-1. A most recently study showed that resveratrol inhibits palmitate-induced lipid accumulation in HepG2 cells by reducing the up-regulation of SREBP-1 via the Sirt1-FOXO1 pathway[42]. Whether the Sirt1-FOXO1 pathway is involved in Sch B-mediated down-regulation of SREBP-1 remains to be clarified.
In summary, Sch B has an inhibitory effect on FFA-induced steatosis in L-02 cells, and the decreased expression of ADRP and SREBP-1 may account for the inhibitory effect of Sch B by reducing FFA uptake, incorporation of FFA into triglycerides and de novo fatty acid synthesis, as well as by increasing VLDL assembly. Changes in ADRP and SREBP-1 expression may also provide mechanistic explanations for the hepatic lipid-lowering effect of Sch B in mice fed a high-fat diet as reported previously by us. The results of this study provide the molecular evidence for developing Sch B as a therapeutic agent against NAFLD.
COMMENTS
Background
Non-alcoholic fatty liver disease (NAFLD), characterized by fatty infiltration of the liver (hepatic steatosis), is posing a definite threat to global human health. So far, no satisfactory therapeutic agent is available against NAFLD. Schisandrin B (Sch B), a bioactive constituent isolated from Fructus Schisandrae, has been recently reported to exhibit hepatic lipid-lowering effect in mice fed with a high-fat diet. However, the mechanisms of action remain to be elucidated.
Research frontiers
The circulating free fatty acid (FFA) levels are often elevated in NAFLD patients, which may promote the disease progression by interfering with hepatic lipid metabolism through modulating the expression of important metabolic regulators. Cellular FFA loading is commonly utilized to develop in vitro models of steatosis valuable for the study of anti-steatosis agents.
Innovations and breakthroughs
The use of human hepatocarcinoma cell lines in developing cellular steatosis models has been questioned due to the altered metabolic regulation in cancerous cells. In this study, FFA-induced steatotic cells were successfully prepared using a normal human hepatocytes-derived cell line L-02. Using this model, the authors demonstrated that Sch B effectively attenuated FFA-induced fat accumulation by abrogating up-regulations of two key metabolic regulators, namely adipose differentiation related protein (ADRP) and sterol regulatory element binding protein 1 (SREBP-1).
Applications
FFA-induced steatotic L-02 cells can be used in investigations of anti-steatosis agents and NAFLD pathogenesis. By understanding how Sch B alleviates cellular steatosis, this study provides the molecular evidence for developing Sch B as a therapeutic agent against NAFLD.
Terminology
ADRP and SREBP-1 are important proteins involved in lipid metabolism in normal livers, and their aberrant expression is believed to contribute to the development of NAFLD.
Peer review
The authors examined the anti-steatotic effects of Sch B on human hepatocytes line treated in vitro with FFAs to induce steatosis. Sch B reduced cellular total lipid and triglyceride levels in a dose dependent manner, which is consistent with the earlier findings. The novel aspect of this study is that the expression of ADRP and SREBP-1, two lipid metabolism regulators, is reduced by Sch B, which provides some insights into the mechanism of action of Sch B.
Footnotes
Supported by The Hong Kong Baptist University, No. FRG/08-09/II-30
Peer reviewer: Debbie Trinder, PhD, School of Medicine and Pharmacology, University of Western Australia, Fremantle Hospital, PO Box 480, Fremantle 6959, Western Australia, Australia
S- Editor Wang JL L- Editor Wang XL E- Editor Zheng XM
References
- 1.Schreuder TC, Verwer BJ, van Nieuwkerk CM, Mulder CJ. Nonalcoholic fatty liver disease: an overview of current insights in pathogenesis, diagnosis and treatment. World J Gastroenterol. 2008;14:2474–2486. doi: 10.3748/wjg.14.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gentile CL, Pagliassotti MJ. The role of fatty acids in the development and progression of nonalcoholic fatty liver disease. J Nutr Biochem. 2008;19:567–576. doi: 10.1016/j.jnutbio.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang TM, Wang BE, Liu GT. Effect of schisandrin B on lipoperoxidative damage to plasma membrane of rat liver in vitro. Proc Natl Acad Sci USA. 1992;13:255–258. [PubMed] [Google Scholar]
- 5.Ip SP, Che CT, Kong YC, Ko KM. Effects of schisandrin B pretreatment on tumor necrosis factor-alpha induced apoptosis and Hsp70 expression in mouse liver. Cell Stress Chaperones. 2001;6:44–48. doi: 10.1043/1355-8145(2001)006<0044:EOSBPO>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiu PY, Leung HY, Siu AH, Poon MK, Ko KM. Schisandrin B decreases the sensitivity of mitochondria to calcium ion-induced permeability transition and protects against carbon tetrachloride toxicity in mouse livers. Biol Pharm Bull. 2007;30:1108–1112. doi: 10.1248/bpb.30.1108. [DOI] [PubMed] [Google Scholar]
- 7.Pan SY, Dong H, Zhao XY, Xiang CJ, Fang HY, Fong WF, Yu ZL, Ko KM. Schisandrin B from Schisandra chinensis reduces hepatic lipid contents in hypercholesterolaemic mice. J Pharm Pharmacol. 2008;60:399–403. doi: 10.1211/jpp.60.3.0017. [DOI] [PubMed] [Google Scholar]
- 8.Marra F, Gastaldelli A, Svegliati Baroni G, Tell G, Tiribelli C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol Med. 2008;14:72–81. doi: 10.1016/j.molmed.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Parekh S, Anania FA. Abnormal lipid and glucose metabolism in obesity: implications for nonalcoholic fatty liver disease. Gastroenterology. 2007;132:2191–2207. doi: 10.1053/j.gastro.2007.03.055. [DOI] [PubMed] [Google Scholar]
- 10.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 11.Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, Burgart LJ, Gores GJ. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. 2004;40:185–194. doi: 10.1002/hep.20283. [DOI] [PubMed] [Google Scholar]
- 12.Wu X, Zhang L, Gurley E, Studer E, Shang J, Wang T, Wang C, Yan M, Jiang Z, Hylemon PB, et al. Prevention of free fatty acid-induced hepatic lipotoxicity by 18beta-glycyrrhetinic acid through lysosomal and mitochondrial pathways. Hepatology. 2008;47:1905–1915. doi: 10.1002/hep.22239. [DOI] [PubMed] [Google Scholar]
- 13.Gómez-Lechón MJ, Donato MT, Martínez-Romero A, Jiménez N, Castell JV, O'Connor JE. A human hepatocellular in vitro model to investigate steatosis. Chem Biol Interact. 2007;165:106–116. doi: 10.1016/j.cbi.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Ye XZ, Zhu DH, Shen DW. Ultrastructure of continuously cultured adult human liver. Acta Biology Experimentalis Sinica. 1980;13:361–364. [Google Scholar]
- 15.Xu ZG, Du JJ, Zhang X, Cheng ZH, Ma ZZ, Xiao HS, Yu L, Wang ZQ, Li YY, Huo KK, et al. A novel liver-specific zona pellucida domain containing protein that is expressed rarely in hepatocellular carcinoma. Hepatology. 2003;38:735–744. doi: 10.1053/jhep.2003.50340. [DOI] [PubMed] [Google Scholar]
- 16.McMillian MK, Grant ER, Zhong Z, Parker JB, Li L, Zivin RA, Burczynski ME, Johnson MD. Nile Red binding to HepG2 cells: an improved assay for in vitro studies of hepatosteatosis. In Vitr Mol Toxicol. 2001;14:177–190. doi: 10.1089/109793301753407948. [DOI] [PubMed] [Google Scholar]
- 17.Ricchi M, Odoardi MR, Carulli L, Anzivino C, Ballestri S, Pinetti A, Fantoni LI, Marra F, Bertolotti M, Banni S, et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J Gastroenterol Hepatol. 2009;24:830–840. doi: 10.1111/j.1440-1746.2008.05733.x. [DOI] [PubMed] [Google Scholar]
- 18.Gómez-Lechón MJ, Donato MT, Castell JV, Jover R. Human hepatocytes as a tool for studying toxicity and drug metabolism. Curr Drug Metab. 2003;4:292–312. doi: 10.2174/1389200033489424. [DOI] [PubMed] [Google Scholar]
- 19.Gómez-Lechón MJ, Donato MT, Castell JV, Jover R. Human hepatocytes in primary culture: the choice to investigate drug metabolism in man. Curr Drug Metab. 2004;5:443–462. doi: 10.2174/1389200043335414. [DOI] [PubMed] [Google Scholar]
- 20.De Gottardi A, Vinciguerra M, Sgroi A, Moukil M, Ravier-Dall’Antonia F, Pazienza V, Pugnale P, Foti M, Hadengue A. Microarray analyses and molecular profiling of steatosis induction in immortalized human hepatocytes. Lab Invest. 2007;87:792–806. doi: 10.1038/labinvest.3700590. [DOI] [PubMed] [Google Scholar]
- 21.Araya J, Rodrigo R, Videla LA, Thielemann L, Orellana M, Pettinelli P, Poniachik J. Increase in long-chain polyunsaturated fatty acid n - 6/n - 3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci (Lond) 2004;106:635–643. doi: 10.1042/CS20030326. [DOI] [PubMed] [Google Scholar]
- 22.Wei P, Taniguchi S, Sakai Y, Imamura M, Inoguchi T, Nawata H, Oda S, Nakabeppu Y, Nishimura J, Ikuyama S. Expression of adipose differentiation-related protein (ADRP) is conjointly regulated by PU.1 and AP-1 in macrophages. J Biochem. 2005;138:399–412. doi: 10.1093/jb/mvi136. [DOI] [PubMed] [Google Scholar]
- 23.Fan B, Ikuyama S, Gu JQ, Wei P, Oyama J, Inoguchi T, Nishimura J. Oleic acid-induced ADRP expression requires both AP-1 and PPAR response elements, and is reduced by Pycnogenol through mRNA degradation in NMuLi liver cells. Am J Physiol Endocrinol Metab. 2009;297:E112–E123. doi: 10.1152/ajpendo.00119.2009. [DOI] [PubMed] [Google Scholar]
- 24.Brasaemle DL, Barber T, Wolins NE, Serrero G, Blanchette-Mackie EJ, Londos C. Adipose differentiation-related protein is an ubiquitously expressed lipid storage droplet-associated protein. J Lipid Res. 1997;38:2249–2263. [PubMed] [Google Scholar]
- 25.Kliewer SA, Sundseth SS, Jones SA, Brown PJ, Wisely GB, Koble CS, Devchand P, Wahli W, Willson TM, Lenhard JM, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc Natl Acad Sci U S A. 1997;94:4318–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang BH, Li L, Paul A, Taniguchi S, Nannegari V, Heird WC, Chan L. Protection against fatty liver but normal adipogenesis in mice lacking adipose differentiation-related protein. Mol Cell Biol. 2006;26:1063–1076. doi: 10.1128/MCB.26.3.1063-1076.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohjima M, Enjoji M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, Fujino T, Yada M, Yada R, Harada N, et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med. 2007;20:351–358. [PubMed] [Google Scholar]
- 28.Motomura W, Inoue M, Ohtake T, Takahashi N, Nagamine M, Tanno S, Kohgo Y, Okumura T. Up-regulation of ADRP in fatty liver in human and liver steatosis in mice fed with high fat diet. Biochem Biophys Res Commun. 2006;340:1111–1118. doi: 10.1016/j.bbrc.2005.12.121. [DOI] [PubMed] [Google Scholar]
- 29.Imamura M, Inoguchi T, Ikuyama S, Taniguchi S, Kobayashi K, Nakashima N, Nawata H. ADRP stimulates lipid accumulation and lipid droplet formation in murine fibroblasts. Am J Physiol Endocrinol Metab. 2002;283:E775–E783. doi: 10.1152/ajpendo.00040.2002. [DOI] [PubMed] [Google Scholar]
- 30.Larigauderie G, Furman C, Jaye M, Lasselin C, Copin C, Fruchart JC, Castro G, Rouis M. Adipophilin enhances lipid accumulation and prevents lipid efflux from THP-1 macrophages: potential role in atherogenesis. Arterioscler Thromb Vasc Biol. 2004;24:504–510. doi: 10.1161/01.ATV.0000115638.27381.97. [DOI] [PubMed] [Google Scholar]
- 31.Gao J, Serrero G. Adipose differentiation related protein (ADRP) expressed in transfected COS-7 cells selectively stimulates long chain fatty acid uptake. J Biol Chem. 1999;274:16825–16830. doi: 10.1074/jbc.274.24.16825. [DOI] [PubMed] [Google Scholar]
- 32.Edvardsson U, Ljungberg A, Lindén D, William-Olsson L, Peilot-Sjögren H, Ahnmark A, Oscarsson J. PPARalpha activation increases triglyceride mass and adipose differentiation-related protein in hepatocytes. J Lipid Res. 2006;47:329–340. doi: 10.1194/jlr.M500203-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Imai Y, Varela GM, Jackson MB, Graham MJ, Crooke RM, Ahima RS. Reduction of hepatosteatosis and lipid levels by an adipose differentiation-related protein antisense oligonucleotide. Gastroenterology. 2007;132:1947–1954. doi: 10.1053/j.gastro.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 34.Varela GM, Antwi DA, Dhir R, Yin X, Singhal NS, Graham MJ, Crooke RM, Ahima RS. Inhibition of ADRP prevents diet-induced insulin resistance. Am J Physiol Gastrointest Liver Physiol. 2008;295:G621–G628. doi: 10.1152/ajpgi.90204.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohjima M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, Fujino T, Yada M, Yada R, Harada N, Enjoji M, et al. SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int J Mol Med. 2008;21:507–511. [PubMed] [Google Scholar]
- 36.Ahmed MH, Byrne CD. Modulation of sterol regulatory element binding proteins (SREBPs) as potential treatments for non-alcoholic fatty liver disease (NAFLD) Drug Discov Today. 2007;12:740–747. doi: 10.1016/j.drudis.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 37.Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest. 1997;99:846–854. doi: 10.1172/JCI119248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimano H, Yahagi N, Amemiya-Kudo M, Hasty AH, Osuga J, Tamura Y, Shionoiri F, Iizuka Y, Ohashi K, Harada K, et al. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J Biol Chem. 1999;274:35832–35839. doi: 10.1074/jbc.274.50.35832. [DOI] [PubMed] [Google Scholar]
- 39.Yamashita T, Eto K, Okazaki Y, Yamashita S, Yamauchi T, Sekine N, Nagai R, Noda M, Kadowaki T. Role of uncoupling protein-2 up-regulation and triglyceride accumulation in impaired glucose-stimulated insulin secretion in a beta-cell lipotoxicity model overexpressing sterol regulatory element-binding protein-1c. Endocrinology. 2004;145:3566–3577. doi: 10.1210/en.2003-1602. [DOI] [PubMed] [Google Scholar]
- 40.Yahagi N, Shimano H, Hasty AH, Matsuzaka T, Ide T, Yoshikawa T, Amemiya-Kudo M, Tomita S, Okazaki H, Tamura Y, et al. Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice. J Biol Chem. 2002;277:19353–19357. doi: 10.1074/jbc.M201584200. [DOI] [PubMed] [Google Scholar]
- 41.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang GL, Fu YC, Xu WC, Feng YQ, Fang SR, Zhou XH. Resveratrol inhibits the expression of SREBP1 in cell model of steatosis via Sirt1-FOXO1 signaling pathway. Biochem Biophys Res Commun. 2009;380:644–649. doi: 10.1016/j.bbrc.2009.01.163. [DOI] [PubMed] [Google Scholar]