

Abstract
As a master transcription factor in cellular responses to external stress, tumor suppressor p53 is tightly regulated. Excessive p53 activity during myocardial ischemia causes irreversible cellular injury and cardiomyocyte death. p53 activation is dependent on lysine acetylation by the lysine acetyltransferase and transcriptional co-activator CBP (CREB-binding protein) and on acetylation-directed CBP recruitment for p53 target gene expression. Here, we report a small molecule ischemin, developed with a structure-guided approach to inhibit the acetyl-lysine binding activity of the bromodomain of CBP. We show that ischemin alters post-translational modifications on p53 and histones, inhibits p53 interaction with CBP and transcriptional activity in cells, and prevents apoptosis in ischemic cardiomyocytes. Our study suggests small molecule modulation of acetylation-mediated interactions in gene transcription as a new approach to therapeutic interventions of human disorders such as myocardial ischemia.
INTRODUCTION
Cardiovascular diseases continue to be an epidemic in the United States and the Western world. Half of all hospital admissions are related to cardiovascular disorders; 80% of those who have fatal consequences are 65 years or older (Gillum et al., 1998; Wei, 1992). The salient feature of cardiac ischemia, which is mainly due to coronary syndromes, includes lack of oxygen and nutrition, which generates stress signals to activate pathways leading to cardiac myocyte death. It has been reported that ischemia-induced myocyte DNA damage results in enhanced transcriptional activity of the tumor suppressor p53 as well as p53-dependent cardiac myocyte apoptosis; the latter is a key feature in the progression of ischemic heart disease (Gudkov and Komarova, 2005; Komarova and Gudkov, 1998). Myocardial ischemia can also induce inflammatory responses and cardiomyocyte necrosis, depending on the intensity and duration of ischemia and reperfusion (Fliss and Gattinger, 1996; Kajstura et al., 1996; Lefer and Granger, 2000). Thus, it appears that attenuation of myocyte apoptosis could potentially prevent myocyte loss, improve myocardial dysfunction, and decrease mortality rate that is associated with myocardial ischemia and reperfusion (Eefting et al., 2004). Previous studies have shown that exposure of myocytes to hypoxia results in increased p53 trans-activating activity and protein accumulation along with the expression of p21/WAF-1/CIP-1, a well-characterized target of p53 transactivation (Long et al., 1997). While p53 activation has been recognized for therapeutic potential in cancer treatments, its hyper-activation could also be detrimental in both normal and ischemic conditions (Komarova and Gudkov, 1998, 2000). Therefore, in a different biological context, modulation of p53 function as a transcriptional regulator, either activation or inhibition, could present valid therapeutic opportunities.
Transcriptional co-activators CREB-binding protein (CBP) and p300 (also known as KAT3B and KAT3A, respectively) (Giordano and Avantaggiati, 1999; Imhof et al., 1997; Ogryzko et al., 1996; Sterner and Berger, 2000) play a central role in regulating p53 stability and its function as a transcription factor in response to genotoxic stress (Ferreon et al., 2009; Ott et al., 1999). The main functions of CBP/p300 in gene transcription are dependent upon activities of both the lysine acetyltransferase (KAT) and the acetyl-lysine binding bromodomain (BRD). Like histones, lysine acetylation of transcription factors facilitates the recruitment of BRD-containing cofactors required for chromatin structural change and transcriptional initiation and elongation (Dhalluin et al., 1999; Mujtaba et al., 2007; Sanchez and Zhou, 2009). The biochemical contribution of acetylation to p53 transcription functions has been attributed to nuclear translocation, alteration of DNA binding ability and enhancement of transcriptional potential (Prives and Manley, 2001). Previous studies from us and other laboratories have supported the role of p53 acetylation in promoting molecular interactions with transcriptional co-regulators leading to target gene activation that ultimately determines cellular responses to stress in the forms of senescence, cell growth arrest or apoptosis (Barlev et al., 2001; Mujtaba et al., 2004; Sakaguchi et al., 1998; Zhao et al., 2006). Specifically, we demonstrated that CBP acetylates p53 C-terminal lysine residues upon DNA damage, and subsequently the BRD of CBP binds to p53 at acetylated lysine 382 (p53K382ac), which is essential for p53-induced transcriptional activation of target genes such as cyclin-dependent kinase inhibitor 1A (CDKN1A/p21) (Mujtaba et al., 2004; Mujtaba et al., 2006).
Activation of p53 in cancer is not restricted to the killing of tumor cells (Rogel et al., 1985). p53-dependent apoptosis also occurs in sensitive tissues shortly after gamma irradiation (Komarova et al., 1997a), and p53-deficient mice survive higher doses of gamma irradiation than do wild-type animals (Westphal et al., 1998; Westphal et al., 1997). These studies highlight the toxic nature of p53 after chemo- and radiation-therapies on normal tissues that ameliorate the disease condition and extend the recovery in cancer patients. Therefore, suppression of p53 functions has been suggested as a therapeutic strategy to prevent damage of normal tissues (Komarova and Gudkov, 2001). Consequently, Pifithrin was identified from cell based chemical library screening, which is probably the first p53 inhibitor reported (Komarov et al., 1999). In addition to inhibiting p53-dependent apoptosis, Pifithrin has been shown to reduce the activation of p53-regulated target genes, including cyclin G, p21/WAF-1/CIP-1, and MDM2 (Komarov et al., 1999). Anti-apoptotic properties of Pifithrin has also been demonstrated in cellular and mouse models of genotoxic stress (Komarov et al., 1999; Liu et al., 2004), dopamine-induced apoptosis in neurons (Culmsee et al., 2003; Culmsee et al., 2001), cisplatin-induced apoptosis in cochlear and vestibular hair cells (Zhang et al., 2003), as well as endotoxin-induced apoptosis in liver tissue (Schafer et al., 2003). Despite these studies, Pifthrin’s mechanism of action, however, is not well understood (Murphy et al., 2004).
Because activated p53 is associated with CBP in an acetylation-sensitive manner, we postulated that small molecules that inhibit the acetyl-lysine binding activity of the BRD of CBP would inhibit p53’s ability to direct its target gene activation. Towards this goal, in this study we have developed small molecules that target the CBP BRD, and characterized their biochemical and functional effects of modulating gene transcription in osteocarcinoma cells and primary cardiomyocytes where DNA damage represents intracellular signaling during ischemia.
RESULTS
Target Structure-Guided Lead Discovery
We conducted NMR-based screening of 3,000 diversity chemical compounds selected from our in-house collection of 115,000 compounds to identify small molecules for the bromodomain of CBP. Ligand binding to the protein was detected by monitoring changes of backbone amide resonances of 15N-labeled CBP BRD in 2D 1H-15N-HSQC NMR spectra upon addition of the chemicals as mixtures of thirty. Subsequently, the positive mixtures with estimated affinity better than Kd of about 300 μM were deconvoluted to identify individual small molecules that bind CBP BRD at the acetyl-lysine binding site (Figure 1A). This process was guided by our NMR resonance assignment of the protein. This target structure-based screening yielded 10 promising hits for the CBP BRD, several of which share the azobenzene scaffold, as exemplified by 4-hydroxyphenylazo-benzenesulfonic acid MS456 (Table 1). We solved the three-dimensional structure of MS456 bound to the CBP BRD with NMR spectroscopy, which reveals that this compound binds at the entrance of the acetyl-lysine binding pocket in the BRD, thereby blocking the protein from interacting with lysine-acetylated binding partners (Figure S1A and Table 2).
Figure 1. Target structure-guided design of chemical probes for the CBP bromodomain.

(A) Superimposition of 2D 1H-15N-HSQC spectra of the 15N-labeled CBP BRD highlighting changes of protein NMR resonances upon binding to a small molecule, ischemin (black, free protein; red, in the presence of ischemin). (B), (C) Ribbon diagram of the lowest energy NMR structures of the CBP BRD bound to ischemin (left) versus to histone H4K20ac peptide (right), respectively. Side chains of the key protein residues at the ligand-binding site are shown and color-coded by atom type. See also Figure S1.
Table 1.
Structure-Activity Relationships of Azobenzene Compounds in p53 Inhibition
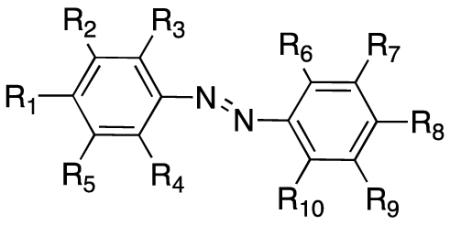
| Compound | R1 | R2 | R3 | R4 | R5 | R6 | R7 | R8 | R9 | R10 |
% Inhibition |
Kd (μM) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MS456 | OH | H | H | H | H | H | H | SO3H | H | H | 4.6 | |
| MS450 | OH | CH3 | H | H | H | H | H | SO3H | H | H | 85.6 | 76 |
| MS113 | OH | CH3CH2 | H | H | H | H | H | SO3H | H | H | 25.7 | |
| MS451 | OH | CH3 | H | H | CH3 | H | H | SO3H | H | H | 87.4 | 44 |
| MS110 | OH | CH2CHCH2 | H | H | CH3 | H | H | SO3H | H | H | 86.2 | 104 |
| MS111 | OH | (CH3)3C | H | H | CH3 | H | H | SO3H | H | H | 22.8 | |
| MS105 | OH | (CH3)2CH | H | H | (CH3)2CH | H | H | SO3H | H | H | 26.4 | |
| MS101 | OH | CH3 | H | CH3 | H | H | H | SO3H | H | H | 82.9 | |
| MS103 | OH | CH3 | H | CH3 | CH3 | H | H | SO3H | H | H | 38.9 | |
| MS100 | OH | H | CH3 | CH3 | H | H | H | SO3H | H | H | 36.4 | |
| Ischemin / MS120 | OH | H | NH2 | H | CH3 | H | SO3H | CH3 | H | CH3 | 104.5 | 19 |
| MS148 | OH | H | CH3CH2(NH2)CH | H | CH3 | H | SO3H | CH3 | H | CH3 | 12.8 | |
| MS155 | OH | H | CO2H | H | CH3 | H | SO3H | CH3 | H | CH3 | 13.1 | |
| MS119 | OH | H | CH3 | CH3 | H | H | SO3H | CH3 | H | CH3 | 54.0 | |
| MS153 | OH | H | CH3 | CH3 | H | H | SO3H | OH | Cl | H | 39.0 | |
| MS131 | OH | Cl | H | H | H | H | SO3H | CH3 | H | CH3 | 49.7 | |
| MS124 | OH | CH3CH2 | H | H | H | H | SO3H | CH3 | H | CH3 | 25.0 | |
| MS126 | OH | CH3CH2CH2 | H | H | H | H | SO3H | CH3 | H | CH3 | 93.5 | 97 |
| MS127 | OH | CH3CH2CO | H | H | H | H | SO3H | CH3 | H | CH3 | 86.8 | 385 |
| MS109 | OH | CH3 | H | H | CH3 | H | SO3H | CH3 | H | CH3 | 60.1 | |
| MS130 | OH | CH2CHCH2 | H | H | CH3 | H | SO3H | CH3 | H | CH3 | 40.4 | |
| MS129 | OH | (CH3)2CH | H | H | CH3 | H | SO3H | CH3 | H | CH3 | 44.6 | |
| MS128 | OH | (CH3)2CH | H | H | (CH3)2CH | H | SO3H | CH3 | H | CH3 | 47.2 | |
| MS154 | OH | Cl | H | CH3 | H | H | SO3H | CH3 | H | CH3 | 6.9 | |
| MS135 | OH | NH2 | H | CH3 | H | H | SO3H | CH3 | H | CH3 | 54.8 | |
| MS118 | OH | CH3 | H | CH3 | H | H | SO3H | CH3 | H | CH3 | 49.7 | |
| MS146 | OH | CH3 | H | CH3 | CH3 | H | SO3H | CH3 | H | CH3 | 30.8 |
Notes:
All compounds were used at 50 μM concentration.
Percent inhibition was calculated by [1-(A/B)]*100, where A is the difference of luciferase activity measured between cells treated with a compound and doxorubicin and the negative control, and B is the difference of luciferase activity between cells treated with and without doxorubicin.
Compounds shown 80%+ inhibition of p53 activity are highlighted in blue.
Kd (μM) was determined in a titration of protein tryptophan fluorescence change as a function of ligand concentration.
Table 2.
NMR Statistics of the Structures of the CBP BRD/Ligand Complexes
| NMR distance and dihedral constraints | CBP BRD/MS456 | CBP BRD/Ischemin |
|---|---|---|
| Distance constraints | ||
| Total NOE | 2,888 | 2,805 |
| Intra-residue | 1,155 | 1,131 |
| Inter-residue | 1,714 | 1,674 |
| Sequential (|i − j| = 1) | 560 | 554 |
| Medium-range (|i − j| < 4) | 654 | 665 |
| Long-range (|i − j| > 5) | 500 | 455 |
| Hydrogen bonds | 50 | 50 |
| Inter-molecular constraints | 53 | 25 |
| Total dihedral angle restraints | ||
| φ | 81 | 81 |
| ψ | 81 | 81 |
| Structure statistics | ||
| Violations (mean ± s.d.) | ||
| Distance constraints (Å) | 0.078 ± 0.0072 | 0.069 ± 0.0064 |
| Dihedral angle constraints (°) | 0.50 ± 0.046 | 0.49 ± 0.19 |
| Max. dihedral angle violation (°) | 0.58 | 0.63 |
| Max. distance constraint violation (Å) | 0.092 | 1.03 |
| Deviations from idealized geometry | ||
| Bond lengths (Å) | 0.0078 ± 0.00014 | 0.0076± 0.00014 |
| Bond angles (°) | 0.86 ± 0.011 | 0.84 ± 0.012 |
| Impropers (°) | 0.47 ± 0.0051 | 0.48 ± 0.0084 |
| Average pairwise RMSD** (Å) | ||
| Heavy – protein | 0.76 ± 0.088 | 0.71 ± 0.12 |
| Backbone - protein | 0.34 ± 0.074 | 0.32 ± 0.093 |
| Ramachandran map analysis ** (%) | ||
| Most favourable regions | 78.6 | 76.2 |
| Additionally allowed regions | 17.1 | 18.7 |
| Generously allowed regions | 2.7 | 3.6 |
| Disallowed regions | 1.6 | 1.5 |
Root-mean-square deviation (RMSD) calculations were performed for 20 final NMR structures of the CBP bromodomain consisting of residues 11-120. Pair-wise root mean square deviation was calculated among the 20 refined structures.
Synthesis and Structure-Activity Relationship Analysis of Chemical Analogs
To improve lead compound binding affinity and selectivity for the CBP BRD, we synthesized a series of azobenzene analogs by introducing various chemical functional groups on the aromatic rings, as well as by changing the sulfonate group from para- to meta- position with respect to the azo linkage on the benzensulfonate (Table 1). These compounds were synthesized using a two-step reaction scheme (Scheme S1). Specifically, the synthesis starts with treatment of a substituted sulfanilic acid with concentrated HCL, and resulting amine is diazotized by addition of sodium nitrite to produce diazonium salt. The latter is added to a solution containing a substituted phenol to yield a corresponding diazobenzene compound. Towards the objective of developing small molecules that could be used to study effects of inhibiting the stress-induced p53/CBP interaction in cells, we systematically evaluated structure-activity relationship (SAR) of these azobenzene analogs in their ability to inhibit p53 activation in human osteosarcoma (U2OS) cells. The p53 activation was induced by DNA damage upon treatment of doxorubicin, and effects of compound treatment were measured by p53-dependent p21 luciferase activity (Table 1 and Figure S2A). All compounds were tested at concentration of 50 μM in U2OS cells.
Of the diazobenzene analogs, 5-(2-amino-4-hydroxy-5-methyl-phenylazo)-2,4-dimethyl-benzenesulfonic acid, MS120, exhibited most potent cellular activity in inhibiting p53-dependent p21 luciferase activity by almost 100% as compared to the controls. Using a tryptophan fluorescence binding assay (Sachchidanand et al., 2006), we determined the binding affinity of ischemin to the CBP BRD to be Kd = 19 μM (Figure S2B), which is consistent with its cellular p21 inhibition activity as compared to those analogs that exhibited more than 80% inhibition of doxorubicin-induced p53 activation in the p21 luciferase assay (Table 1). Given its effective cellular protective activity against myocardial ischemia (see below), we named this compound MS120 as ischemin. We further determined IC50 = 5 μM for ischemin in inhibition of p53-induced p21 activation using the luciferase activity assay, which is markedly stronger than an analog MS119 with IC50 = ~100 μM (Figure 2A). This difference agrees with the difference in their inhibition activities observed in the p21 luciferase activity measurement (Table 1 and Figure S2A).
Figure 2. Functional characterization of CBP BRD chemical modulators in transcription.

(A) Dose-dependent inhibition of p21 luciferase activity in U2OS cells upon treatment of ischemin or MS119. The luciferase activity was normalized to renilla luciferase as a control. The IC50 was calculated using PRISM software. (B) Effects of the CBP BRD ligands on BRDU incorporation in U2OS cells upon doxorubicin treatment. The data showing that ischemin or MS119 prevents a doxorubicin-induced decrease of BRDU incorporation. See also Figure S2.
Molecular Basis of Lead Recognition by the CBP BRD
To understand the molecular basis of CBP BRD recognition of the diazobenzenes, we determined the three-dimensional structure of the ischemin/CBP BRD complex by using NMR (Figure 1B; also see Figure S1B and Table 2). The overall position and orientation of ischemin bound to CBP BRD is similar to that of the initial hit MS456 (Figure S1). It is worth noting that binding ischemin caused severe line broadening of several protein residues at the ligand-binding site, which include Pro1110, Phe1111, Ile1122, Tyr1125, Ile1128, and Tyr1167. The ligand binding induced line-broadening resulted in a fewer number of intermolecular NOE-derived distance constraints used for the ischemin-bound structure determination than that for MS456, i.e. 25 versus 53, respectively (see Table 2). Nevertheless, the ischemin/CBP BRD structure is better defined than the latter, consistent with its higher affinity (Figure S1). Ischemin binds across the entrance of the acetyl-lysine binding pocket in an extended conformation with its phenoxyl group forming a hydrogen bond (~2.8 Å) to the amide nitrogen of Asn1168 in CBP. The latter is a highly conserved residue in the BRDs whose amide nitrogen is hydrogen-bonded to the acetyl oxygen of the acetyl-lysine in a biological binding partner as seen with acetylated-lysine 20 of histone H4 recognition by the CBP BRD (Figure 1B vs. 1C). The sulfonate group forms electrostatic interactions with quanidinium group of Arg1173 in the BC loop and possibly also with side chain amide of Gln1113 in the ZA loop.
Ischemin in the acetyl-lysine binding pocket is sandwiched through hydrophobic and aromatic interactions between the diazobenzene and Leu1109, Pro1110 and Val1174 on one side, Leu1120 and Ile1122 in the ZA loop on the other. Since all the diazobenzenes contain a para-phenoxyl group, a hydrogen bond between the phenoxyl with Asn1168 is likely present in all the compounds when bound to the CBP BRD. As such, this structure explains our SAR data (Table 1). For instance, with a para-sulfonate in the diazonbenzene, ortho- but not meta-substitution of methyl groups on the phenol ring results in a marked increase in the lead’s ability to inhibit p53-dependent p21 luciferase activity, e.g. MS450, MS451, and MS101 versus MS110. Ortho-substitution of a larger alkyl group such as ethyl (MS113), isopropyl (MS105), or t-butyl (MS111) showed reduced activity on p21 inhibition as compare to that of ortho-methyl. The preferred small hydrophobic group at ortho-position is due to its possible interaction with a small hydrophobic cavity formed with Ile1122, Tyr1125 and Tyr1167 that is positioned next to the conserved Asn1168 in the acetyl-lysine binding pocket (see below).
When resided at meta-position in diazobenzene, sulfonate establishes electrostatic interactions with quanidinium side chain of Arg1173 (Figure 1B); this alters CBP preference for substitutions on the aromatic ring. For instance, inhibition of p21 expression seems less sensitive to variations of size and position of hydrophobic substituent groups on the phenol. Nevertheless, ortho-propyl (MS126) and ortho-ethyl-keto (MS127) substituted diazobenzenes exhibit 93.5% and 86.8% inhibition activity, respectively. This preferred ortho-substituent likely interacts with side chains of Ile1122, Tyr1125 and Tyr1167, a small hydrophobic pocket embedded in the acetyl-lysine binding site (Figure 1B). With a meta-amino substituent, which electron-donating functionality may aid formation of a hydrogen bond between the phenoxyl in the diazobenzene and side chain amide of Asn1168 of the protein, ischemin nearly completely suppresses the p21 expression. While many ischemin binding residues in the acetyl-lysine binding pocket are conserved among human BRDs (Sanchez and Zhou, 2009), we observed that ischemin exhibits up to five-fold selectivity for the CBP BRD over several other human BRDs including PCAF, BRD41, BAZ1B and BAZ2B as determined by an in vitro tryptophan fluorescence binding assay (Figure S2C). This level of selectivity may attribute to several ischemin binding residues in CBP such as Pro1110, Gln1113 and Arg1173 that are not conserved in other human BRDs (Zeng et al., 2008). Collectively, the new structure provides the detailed molecular basis of ischemin recognition by the CBP BRD.
Ischemin Inhibits p53 Activation upon DNA Damaging Stress
DNA damage induced by doxorubicin leads to p53 stimulated cellular responses including cell cycle arrest, damage repair, and apoptosis (Prives and Hall, 1999; Vogelstein et al., 2000; Woods and Vousden, 2001). To determine the effect of ischemin on dividing U2OS cells, we treated U2OS cells with 5-bromo-2-deoxyuridine (BRDU) and measured the incorporated BRDU during in DNA synthesis using an ELISA assay. The result showed that doxorubicin treatment of U2OS cells resulted in a 45% decrease of BRDU incorporation, indicative of doxorubicin induced cell cycle arrest. However, the presence of ischemin or MS119 (50 μM) almost completely prevented U2OS cells from undergoing doxorubicin-induced cell cycle arrest (Figure 2B). Note that these results also indicate that ischemin is not toxic to the cells at this concentration.
We then examined biochemical effects of ischemin on p53 stability and function as transcription factor. We incubated U2OS cells in the presence of doxorubicin with or without ischemin at concentration of 50 or 100 μM for 24 hours. Subsequently, cellular proteins were subjected to western blot analysis. As shown in Figure 3A, the doxorubicin-induced increased levels of p53 protein, its Ser15-phosphorylated (p53S15p) and Lys382-acetylated (p53K382ac) forms underwent marked reduction in the presence of ischemin as assessed by direct western blots of cell lysate or following immunoprecipitation. Further, we observed that p53 directed expression of its target genes p21, PUMA and 14-3-3s induced by doxorubicin retreatment was significantly decreased in the presence of ischemin whereas the level of actin remained the same.
Figure 3. Effects of ischemin on p53 activation induced by DNA damage.

(A) Immunoblots showing ischemin effects on levels of endogenous p53, p53 phosphorylation on serine 15, p53 acetylation on lysine 382, as well as p53 target genes. (B) Immunoblots showing ischemin effects on levels of correlated H3K9 acetylation and H3S10 phosphorylation, and unaffected upstream kinases CHK1 and ATM upon doxorubicin treatment. (C) Inhibition of over-expressed HA-tagged CBP and flag-tagged p53 interaction in 293T cells by ischemin in a concentration-dependent manner under doxorubicin-induced DNA damaging condition. An arrow indicates the expressed Flag-tagged p53 in the HEK 293T cells.
As a transcription factor, p53 ability to activate gene expression is also dependent upon chromatin modifications. Since CBP acetylates both histones and p53, we evaluated possible changes of epigenetic marks on p53 and global histones in presence of ischemin. The western blot analysis of the nuclear extracts from U2OS cells revealed that p53 inhibition by ischemin is associated with an increase in histone H3 phosphorylation at Ser10 and a decrease in H3 acetylation at Lys9 (Figure 3B). These changes of post-translational modifications on p53 and histone H3 are associated with down-regulation of p21, PUMA and 14-3-3, but not the controls of actin, histone H3 and lamin B. Noticeably, ischemin treatment did not affect the level or functional phosphorylation state of ATM and CHK1, which are the upstream signal transducers of p53 (Figure 3B). Collectively, these results suggest that ischemin inhibits doxorubicin-induced p53 activation and transcriptional functions by altering post-translational modification states on p53 and histones.
We further investigated whether ischemin down-regulates p53 by blocking p53 binding to CBP. We over-expressed haemaglutinin-tagged CBP (HA-CBP) and Flag-tagged p53 (Flag-p53) in human embryonic kidney (HEK) 293T cells. Treatment of the 293T cells with ischemin in the presence or absence of doxorubicin did not affect the expression of HA-CBP or Flag-p53, or acetylation and phosphorylation levels on p53 as assessed by immunoprecipiation with anti-Flag antibody followed by Western blot analysis using specific antibodies (Figure 3C). The results reveal that ischemin was clearly capable of inhibiting in a dose-dependent manner p53 binding to CBP, particularly upon under doxorubicin treatment (Figure 3C, lanes 8 and 9 vs. lane 7). Note that p53 associated with HA-CBP is phosphorylated on Ser15, indicating that p53 is transcriptionally active. These results confirm that ischemin inhibits p53-induced p21 activation upon doxorubicin exposure by blocking p53 recruitment of CBP, which is required for p53 target gene activation (Mujtaba et al., 2004).
Ischemin Inhibits p53 Cellular Signaling Pathways
We next investigated selectivity of ischemin in transcription inhibition of p53 target genes using a RT-PCR array analysis of RNA isolated from biological samples of U2OS cells (Figure 4A). The results show that doxorubicin treatment up-regulated p53 target genes that include CCNB2, CCNH, CDC25C, and CDK4, but did not affect housekeeping genes GAPDH, β-2 microglobulin (B2M) and actin (ACTB) (Figure 4B; also see Figure S3). On the other hand, ischemin can differentially reduce doxorubicin-induced expression of p53 target genes CCNE2, CCNG2, CDC2, CDC25A, CDKN1A, CDKN2A (p21), GADD45A, E2F1, E2F3, PCNA, SESN1 and SESN2 (Figure 4B). These gene products are known to participate in different cellular pathways driven by p53, of which the best known is CDKN1A (p21) that functions as an inhibitor for cell cycle progression. Taken together, these results confirm our hypothesis that small-molecule inhibition of the acetyl-lysine binding activity of the CBP BRD could down-regulate p53 activation and its ability to activate its target genes under stress conditions.
Figure 4. Microarray analysis of effects of ischemin on p53 target gene expression, as assessed by using Ingenuity System.

(A) Heat-map of the microarray data of selected genes in U2OS cells upon doxorubicin treatment with and without ischemin. Changes of gene expression are highlighted in red and green as for up- or down-regulation as compared to the control, respectively. (B) Schematic illustration of selected genes associated with p53 signaling pathways that underwent change of expression levels upon doxorubicin treatment with (left panel) and without (right panel) the presence of ischemin. Gene products are color-coded in red (increase in expression), green (decrease in expression), and white (no significant change in expression). Figure S3 depicts a more complete signaling diagram of the genes subject to the microarray analysis. The abbreviations used in the diagrams are PP, protein-protein interaction; A, activation; M, modification; E, expression; I, inhibition; RE, reaction; and EC, phosphorylation. See also Figure S3.
Ischemin Functions a Cellular Protective Agent against Myocardial Ischemic Stress
We next investigated whether ischemin could inhibit apoptosis in cardiomyocytes under DNA damage stress. Primary neonatal rat cardiomyocytes were isolated and maintained in culture, then, treated with doxorubicin for 24 hours to induce DNA damage in the presence or absence of ischemin. The DNA damage induced by apoptosis was analyzed by the TUNEL (terminal deoxynucleotidyl transferase dUTP nick and end labeling) assay, in which a terminal deoxynucleotidyl transferase was used to identify 3′-OH of DNA generated by DNA fragmentation resulting from apoptosis, and then labels it with biotinylated dUTP. The latter was then detected with avidin-conjugated FITC for specific staining. Using this TUNEL assay we observed that doxorubicin treatment induces apoptosis in the cardiomyocytes (Figure S4), and observed that ischemin, which has no toxicity of its own, can effectively inhibit doxorubicin-induced apoptosis in the cardiomyocytes (Figure 5A). Further, similar to U2OS cells, we confirmed that ischemin was able to inhibit doxorubicin induced p53 activation in the primary neonatal rat cardiomycocytes, but did not alter H2AX phosphorylation at Ser139 (Figure 5B). The latter argues that ATM is active in presence of ischemin, which is consistent with our analysis using Western blots (Figure 3B). We further examined and concluded that ischemin likely blocks apoptosis in cardiomyocytes by inhibiting caspase 3/7 activity in a dose-dependent manner (Figure 5C). Finally, we ruled out that ischemin’s ability to directly inhibit the lysine acetyltransferase activity of CBP/p300 towards a histone H3 peptide substrate in a fluorescence-based assay (data not shown). Taken together, these results strongly demonstrate that ischemin is cell permeable and capable of functioning as a cellular protective agent against myocardial damage by down-regulating p53-induced apoptosis under the stress conditions.
Figure 5. Ischemin functions a cellular protective agent against myocardial ischemic stress.

(A) TUNEL assay showing ischemin inhibition of doxorubicin-induced apoptosis in rat neonatal cardiomyocytes. (B) Evaluation of ischemin effects in U2OS cells and cardiomyocytes. The immunoblots show down-regulation of doxorubicin-induced activated p53 in both cell types in the presence of ischemin, while levels of H2XS139p remained the same. (C) Inhibition of doxorubicin-induced caspase 3/7 activation in cardiomyocytes by ischemin. See also Figure S4.
DISCUSSION
Cardiac dysfunction results from the loss of myocyte mass via necrosis and apoptosis (Joaquin and Gollapudi, 2001; Kajstura et al., 1996; Vanden Hoek et al., 2003). Necrosis is a passive process that causes injury to cellular membranes resulting in the release of intracellular contents and in the marked inflammatory responses. In contrast, apoptosis is an active, gene-directed, and energy-requiring process, in which cells initiate their own death in response to either internal or external stimuli (Coopersmith et al., 1999; Freude et al., 2000; Misao et al., 1996). The p53-dependent apoptosis is a key mechanism by which the host eliminates unwanted or damaged cells thereby ensuring the overall genome integrity (Schafer et al., 2003). Several published studies indicated that apoptosis is the major form of cell loss and that p53-dependent apoptosis plays a critical role in myocardial infarction and reperfusion injury and also in doxorubicin induced cardiomyopathy (Grazette et al., 2004; Komarov et al., 1999). Pifthrin-α, probably the only known p53 inhibitor, has been shown to inactivate p53 function during cardiac ischemia (Gudkov and Komarova, 2005; Komarov et al., 1999; Liu et al., 2004). However, its mechanism of action is not clearly understood. In this study, we have rationally designed and developed a new p53 inhibitor, ischemin, which targets p53 interaction with co-activator CBP that is required for p53 transcriptional activity. We have demonstrated that ischemin can effectively inhibit transcription functions of p53 upon DNA damage and block cardiac myocyte apoptosis during ischemia conditions.
As a master regulator of transcriptional program p53 controls genomic stability by inducing growth arrest or apoptosis in response to either genotoxic stress or activation of oncogenes, thereby protecting normal cell populations from potential tumor progenitors (Vousden, 2000). p53 is frequently mutated in tumors (Soussi and Beroud, 2001). p53-deficient tumors are usually sensitive to ectopically expressed p53 (May and May, 1999) and considered to be targets for p53 gene therapy (Fang and Roth, 2003; Willis and Chen, 2002). However, p53-mediated apoptosis has also been reported to occur in gastrointestinal tract and haemopoeitic cells during radio and/or chemotherapy, which can result in severe and well-known side effects of cancer treatment (Chow et al., 2000; Cui et al., 1995; Komarova et al., 1997a; Komarova et al., 1997b; Song and Lambert, 1999). Therefore, balancing p53 activation in a temporal manner likely holds therapeutic benefits. The primary challenge of this strategy is to block activation of downstream targets of p53 without affecting the upstream pathway of p53 (such as ATR, ATM and ChK kinases). As shown in this study, ischemin can selectively down-regulate p53 downstream target genes without affecting p53 upstream effector proteins and protect the primary rat cardiomyocyte cells from ischemic apoptosis through inhibiting caspase 3/7 activity.
As shown in previously reported studies by us and the others (Barlev et al., 2001; Mujtaba et al., 2004; Mujtaba et al., 2006), bromodomain/acetyl-lysine mediated protein-protein interactions likely play an important role in different steps in stress-induced activation of p53 in gene transcription. These include (1) co-activator CBP/p300 or PCAF binding to p53 leading to acetylation of multiple lysine residues on p53, (2) co-activator recruitment by acetylated p53 to its target gene loci to increase histone lysine acetylation, (3) recruitment of chromatin remodeling complexes to facilitate chromatin opening, and (4) assembly of the active transcriptional machinery for productive gene transcriptional activation and elongation. While the co-activator CBP/p300 has been implicated to participate in all these steps of p53-induced gene activation, the detailed underlying molecular mechanism is yet to be fully elucidated. Other bromodomain-containing proteins could also participate in the process. Given the modest affinity of ischemin to CBP BRD, we cannot rule out ischemin binding to possibly other bromodomain proteins in cells. Nevertheless, additional chemical modifications of the diazobenzene scaffold could be explored, which can be guided by our new insights into the molecular basis of ligand recognition by the target protein. Furthermore, it is conceivable that the diazo moiety could be replaced by carbon-carbon connectivity, which could result in an improvement of chemical stability of lead compounds in cells.
In summary, the new small molecule probes, as described in this study, lay an important foundation to facilitate future functional investigation of endogenous p53 under biologically relevant conditions. Further, we expect that combined use of additional small molecule probes that are high affinity and selective for bromodomains from other chromosomal proteins such as PCAF and BRD4 will further help us dissect detailed mechanistic insights into the complex biological functions of p53 in gene transcriptional control in response to cellular stress in human biology of health and diseases such as myocardial ischemia.
SIGNIFICANCE
Post-translational modifications of proteins are essential in cellular control of physiological processes; their dysregulation has been recognized as the cause of human diseases. Use of small molecules targeting enzymatic activities responsible for such amino acid modifications is an accepted approach in drug discovery as exemplified by the development of kinase inhibitors for cancer treatments (Chico et al., 2009). We argue, however, that small molecule targeting of modification-mediated protein-protein interactions offers unique advantages, particularly in regulation of gene transcription in chromatin. This is because protein functional domains likely have a fewer number of cellular binding partners than that of a given chromatin modifying enzymes. These protein domains have also been shown to bind to modified amino acids of biological partners with high specificity but modest affinity (1-100 μM) (Sanchez and Zhou, 2009; Zeng et al., 2008), which allows necessary dynamic regulation of cellular biology such as gene transcription (Huang et al., 2009). As illustrated in this study, inhibition of acetylation-mediated protein-protein interactions via the bromodomain with selective small molecules can provide a temporal modulation of gene transcription in a chromatin context. Therefore, our study highlights an alternative approach to the future development of new therapeutic strategies to ameliorate stress-induced human cardiovascular disorders such as myocardial ischemia.
EXPERIMENTAL PROCEDURES
Chemical Synthesis
All compounds were synthesized using commercially available starting materials without further purification unless otherwise stated. See Supplemental Experimental Procedures for details.
Cell Lines, Plasmids and Reagents
U2OS cells were grown in DMEM (Eagle’s minimal essential medium) (Mediatech) supplemented with 10% fetal bovine serum (Invitrogen) and antibiotics (Invitrogen). For p53 activation, doxorubicin (Sigma) was used. The compounds were dissolved in DMSO (Sigma). The antibodies used for immunoprecipitation and western blot are p53 (sc-6243), p21 (sc-397), 14-3-3 (sc-7683), lamin B (sc-6215) from Santa Cruz Biotech; p53Ser15p (9282), p53K382ac (2525), ATM (2873), ATMp1981 (4526), CHK (2345), CHKp (2341) and PUMA (4976) from Cell Signaling Tech; H3 (ab1791), H3KS10p (ab14955), H3K9ac (ab4441) from ABCAM; and Actin A4700) from Sigma.
Western Blotting
U2OS cells were harvested cells and lysed in lysis buffer (20 mM Tris (pH 8.0), 150 mM NaCl, 1 mM EGTA, 1% Triton X-100, and 50 mM NaF) containing protease inhibitor cocktail (Sigma). The cells were sonicated and spun down at 14,000 rpm for 30 min at 4°C. After protein estimation, 30-50 micrograms of lysates were subjected to SDS-PAGE, transferred onto nitrocellulose membranes, blocked with 5% milk/PBS and blotted with a primary antibody. Horse radish peroxidase-labeled secondary antibodies (goat anti-Mouse or anti-Rabbit) were added for 60 min at room temperature, and the blots were washed with TBS (20mM Tris, 150 mM Nacl, and .05% tween −20) and subjected to autoradiography after development of reaction by ECL (GE health care).
Luciferase Assay
U2OS Cells were transfected with p21 luciferase (1 μg) and renilla luciferase (100 ng) vectors in 6 well plate format using Fugene 6 (Roche). Briefly, total of 1.1 micrograms of vector was incubated with 3 mL of Fugene 6 reagent for 30 min. After 3-4 hours of transfection, cell were treated with compounds for overnight, and then exposed to 300 nanogram of doxorubicin for next 24 hours. In these experiments, DMSO, transfected cells with empty vector and cell without doxorubicin were used as controls. DMSO concentration is maintained at 0.01%. Transfected cells with doxorubicin treatment were used as positive control. The luciferase activity was estimated by following the manufacturer’s instruction (Promega) in a luminometer. Both active and passive lysis of cells yielded consistent results. The inhibitory activity (IC50) of a small molecule on p21 luciferase activity was obtained from the average of three biological replicates using PRISM software.
BRDU Cell Cycle Analysis
BRDU incorporation assay for cell cycle evaluation was performed in 96 well plates using calorimetric based kit from Calbiochem (Cat# QiA58). Hundred microliter of 1×105/ml cells were plated in DMEM media (Mediatech) with 10 % fetal bovine serum (FBS). After 12 hours cells were treated with compounds ischemin and MS119 (50 μM) with or without doxorubicin treatment (5 μM). The controls were DMSO and untreated cells. BRDU was added for 24 hours treatment. After 24 hours cells were fixed and treated with anti-BRDU antibody. After washings, the wells were incubated with peroxidase. After final wash, the color was developed using TMB as substrate and the reaction was stopped with stop solution and optical density was estimated at 450 nm.
HA-CBP and Flag-p53 Pull-down Assay
HA-CBP and Flag-p53 were transfected into human embryonic kidney (HEK) 293T cells with recommended amount of Fugene 6 (Roche). After transfection, the HA-CBP and Flag-p53 co-transfected cells were treated with ischemin in the presence or absence of doxorubicin. To test the inhibitory potential of ischemin against CBP and p53 association, CBP was first immunoprecipitated by pulling-down with HA-agarose beads (Sigma) and its association with p53 was then determined with western blot using anti-Flag antibody (Sigma).
Microarray Analysis
Effects of ischemin on transcription inhibition of p53 target genes were assessed using a RT-PCR array study. The array was performed on RNA isolated from three different biological repeats in U2OS cells using a set of primers selected for a group of genes that are known to be associated within p53 signaling pathways. The differentially expressed genes in treated related to untreated groups, i.e. doxorubicin treated versus untreated, or doxorubicin plus ischemin versus doxorubicin alone, were subjected to pathway analysis by using the Ingenuity System software (http://www.ingenuity.com/). The fold changes of these genes were converted to log2Ratio and then imported into IPA tool along with gene symbols. The enriched pathways in the gene list were identified by Fisher exact test at p value of 0.05 and visualized in Canonical pathway explorer.
Cardiac Myocyte Isolation
Neonatal rat ventricular myocytes (NRVMs) were isolated by enzymatic dissociation of cardiac ventricle from 1-to-2-day-old Sprague-Dawley pups using the Worthington neonatal cardiomyocyte isolation system (Worthington). Briefly, the pups were anesthetized and their hearts were excised. The ventricular tissues were minced in ice cold HBSS and then digested with trypsin overnight at 4°C followed by collagenase treatment for 45 min at 37°C. Cells were collected by centrifugation at 800 rpm for 5 min and subsequently underwent two rounds of preplating on culture dishes to minimize nonmyocyte contamination. The enriched cardiomyocytes were cultured in DMEM/F12 nutrient mixture (Invitrogen) with 10% horse serum and 5% fetal calf serum (Invitrogen). After 48 hours, the medium was changed to DMEM/F12 containing 1% insulin, transferrin, and selenium media supplement (ITS; Invitrogen) and 0.1% BSA.
Apoptosis Assays in Cardiomyocytes
Caspase 3/7 and TUNEL assays were performed to assess inhibition of apoptosis by ischemin. Caspase assay and TUNEL assays were performed using Caspase-Glo 3/7 and DeadEnd kits from Promega. Caspase assay was performed on live cardiomyocytes in 96 wells plate on three different days. Similarly, TUNEL assay was performed in triplicate on three different days. For caspase assay 7500 cardiomyocytes were plated in 96 well plates. After treatment with compounds overnight and then doxorubicin for 24 hours, the intensities of luminecnce were read. Similarly, the TUNEL assay was performed on cardiomyocytes attached on coverslips. Briefly, cells were fixed with 4% paraformaldehyde in phosphate buffer saline and permeablized with 0.5% Tween 20 for 10 minutes. The TUNEL reaction was performed on cells with nucleotide labeled with FITC by following manufacturer’s instruction.
Supplementary Material
ACKNOWLEDGEMENT
We acknowledge the use of the NMR facility at the New York Structural Biology Center. G.G.-N. is recipient of the Research Supplements to Promote Diversity in Health-Related Research Program from the National Cancer Institute. This work was support by research grants from the National Institutes of Health (to M.-M.Z., S.M. and R.J.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information including four figures and a table can be found with this article online.
Accession Numbers
Coordinates for the three-dimensional NMR solution structures of CBP bromodomain bound to ischemin and MS456 are deposited in the Protein Data Bank with accession numbers of PDB ID 2L84 and 2L85, and RCSB ID RCSB102075 and RCSB102076.
REFERENCES
- Barlev NA, Liu L, Chehab NH, Mansfield K, Harris KG, Halazonetis TD, Berger SL. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell. 2001;8:1243–1254. doi: 10.1016/s1097-2765(01)00414-2. [DOI] [PubMed] [Google Scholar]
- Chico LK, Van Eldik LJ, Watterson DM. Targeting protein kinases in central nervous system disorders. Nat Rev Drug Discov. 2009;8:892–909. doi: 10.1038/nrd2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow BM, Li YQ, Wong CS. Radiation-induced apoptosis in the adult central nervous system is p53-dependent. Cell Death Differ. 2000;7:712–720. doi: 10.1038/sj.cdd.4400704. [DOI] [PubMed] [Google Scholar]
- Coopersmith CM, O’Donnell D, Gordon JI. Bcl-2 inhibits ischemia-reperfusion-induced apoptosis in the intestinal epithelium of transgenic mice. Am J Physiol. 1999;276:G677–686. doi: 10.1152/ajpgi.1999.276.3.G677. [DOI] [PubMed] [Google Scholar]
- Cui YF, Zhou PK, Woolford LB, Lord BI, Hendry JH, Wang DW. Apoptosis in bone marrow cells of mice with different p53 genotypes after gamma-rays irradiation in vitro. J Environ Pathol Toxicol Oncol. 1995;14:159–163. [PubMed] [Google Scholar]
- Culmsee C, Siewe J, Junker V, Retiounskaia M, Schwarz S, Camandola S, El-Metainy S, Behnke H, Mattson MP, Krieglstein J. Reciprocal inhibition of p53 and nuclear factor-kappaB transcriptional activities determines cell survival or death in neurons. J Neurosci. 2003;23:8586–8595. doi: 10.1523/JNEUROSCI.23-24-08586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, Greig NH, Mattson MP. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J Neurochem. 2001;77:220–228. doi: 10.1046/j.1471-4159.2001.t01-1-00220.x. [DOI] [PubMed] [Google Scholar]
- Dhalluin C, Carlson J, Zeng L, He C, Aggarwal K, Zhou M. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- Eefting F, Rensing B, Wigman J, Pannekoek WJ, Liu WM, Cramer MJ, Lips DJ, Doevendans PA. Role of apoptosis in reperfusion injury. Cardiovasc Res. 2004;61:414–426. doi: 10.1016/j.cardiores.2003.12.023. [DOI] [PubMed] [Google Scholar]
- Fang B, Roth JA. Tumor-suppressing gene therapy. Cancer Biol Ther. 2003;2:S115–121. [PubMed] [Google Scholar]
- Ferreon JC, Martinez-Yamout MA, Dyson HJ, Wright PE. Structural basis for subversion of cellular control mechanisms by the adenoviral E1A oncoprotein. Proc Natl Acad Sci U S A. 2009;106:13260–13265. doi: 10.1073/pnas.0906770106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliss H, Gattinger D. Apoptosis in ischemic and reperfused rat myocardium. Circ Res. 1996;79:949–956. doi: 10.1161/01.res.79.5.949. [DOI] [PubMed] [Google Scholar]
- Freude B, Masters TN, Robicsek F, Fokin A, Kostin S, Zimmermann R, Ullmann C, Lorenz-Meyer S, Schaper J. Apoptosis is initiated by myocardial ischemia and executed during reperfusion. J Mol Cell Cardiol. 2000;32:197–208. doi: 10.1006/jmcc.1999.1066. [DOI] [PubMed] [Google Scholar]
- Gillum BS, Graves EJ, Wood E. National hospital discharge survey. Vital Health Stat. 1998;13:i–v. 1–51. [PubMed] [Google Scholar]
- Giordano A, Avantaggiati ML. p300 and CBP: partners for life and death. J Cell Physiol. 1999;181:218–230. doi: 10.1002/(SICI)1097-4652(199911)181:2<218::AID-JCP4>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Grazette LP, Boecker W, Matsui T, Semigran M, Force TL, Hajjar RJ, Rosenzweig A. Inhibition of ErbB2 causes mitochondrial dysfunction in cardiomyocytes: implications for herceptin-induced cardiomyopathy. J Am Coll Cardiol. 2004;44:2231–2238. doi: 10.1016/j.jacc.2004.08.066. [DOI] [PubMed] [Google Scholar]
- Gudkov AV, Komarova EA. Prospective therapeutic applications of p53 inhibitors. Biochem Biophys Res Commun. 2005;331:726–736. doi: 10.1016/j.bbrc.2005.03.153. [DOI] [PubMed] [Google Scholar]
- Huang B, Yang X-D, Zhou M-M, Ozato K, Chen L-F. Brd4 Coactivates Transcriptional Activation of NFkB via Specific Binding to Acetylated RelA. Molecular and Cellular Biology. 2009;29:1375–1387. doi: 10.1128/MCB.01365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imhof A, Yang XJ, Ogryzko VV, Nakatani Y, Wolffe AP, Ge H. Acetylation of general transcription factors by histone acetyltransferases. Curr Biol. 1997;7:689–692. doi: 10.1016/s0960-9822(06)00296-x. [DOI] [PubMed] [Google Scholar]
- Joaquin AM, Gollapudi S. Functional decline in aging and disease: a role for apoptosis. J Am Geriatr Soc. 2001;49:1234–1240. doi: 10.1046/j.1532-5415.2001.04990.x. [DOI] [PubMed] [Google Scholar]
- Kajstura J, Cheng W, Reiss K, Clark WA, Sonnenblick EH, Krajewski S, Reed JC, Olivetti G, Anversa P. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab Invest. 1996;74:86–107. [PubMed] [Google Scholar]
- Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–1737. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- Komarova EA, Chernov MV, Franks R, Wang K, Armin G, Zelnick CR, Chin DM, Bacus SS, Stark GR, Gudkov AV. Transgenic mice with p53-responsive lacZ: p53 activity varies dramatically during normal development and determines radiation and drug sensitivity in vivo. EMBO J. 1997a;16:1391–1400. doi: 10.1093/emboj/16.6.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova EA, Gudkov AV. Could p53 be a target for therapeutic suppression? Semin Cancer Biol. 1998;8:389–400. doi: 10.1006/scbi.1998.0101. [DOI] [PubMed] [Google Scholar]
- Komarova EA, Gudkov AV. Suppression of p53: a new approach to overcome side effects of antitumor therapy. Biochemistry (Mosc) 2000;65:41–48. [PubMed] [Google Scholar]
- Komarova EA, Gudkov AV. Chemoprotection from p53-dependent apoptosis: potential clinical applications of the p53 inhibitors. Biochem Pharmacol. 2001;62:657–667. doi: 10.1016/s0006-2952(01)00733-x. [DOI] [PubMed] [Google Scholar]
- Komarova EA, Zelnick CR, Chin D, Zeremski M, Gleiberman AS, Bacus SS, Gudkov AV. Intracellular localization of p53 tumor suppressor protein in gamma-irradiated cells is cell cycle regulated and determined by the nucleus. Cancer Res. 1997b;57:5217–5220. [PubMed] [Google Scholar]
- Lefer DJ, Granger DN. Oxidative stress and cardiac disease. Am J Med. 2000;109:315–323. doi: 10.1016/s0002-9343(00)00467-8. [DOI] [PubMed] [Google Scholar]
- Liu X, Chua CC, Gao J, Chen Z, Landy CL, Hamdy R, Chua BH. Pifithrin-alpha protects against doxorubicin-induced apoptosis and acute cardiotoxicity in mice. Am J Physiol Heart Circ Physiol. 2004;286:H933–939. doi: 10.1152/ajpheart.00759.2003. [DOI] [PubMed] [Google Scholar]
- Long X, Boluyt MO, Hipolito ML, Lundberg MS, Zheng JS, O’Neill L, Cirielli C, Lakatta EG, Crow MT. p53 and the hypoxia-induced apoptosis of cultured neonatal rat cardiac myocytes. J Clin Invest. 1997;99:2635–2643. doi: 10.1172/JCI119452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May P, May E. Twenty years of p53 research: structural and functional aspects of the p53 protein. Oncogene. 1999;18:7621–7636. doi: 10.1038/sj.onc.1203285. [DOI] [PubMed] [Google Scholar]
- Misao J, Hayakawa Y, Ohno M, Kato S, Fujiwara T, Fujiwara H. Expression of bcl-2 protein, an inhibitor of apoptosis, and Bax, an accelerator of apoptosis, in ventricular myocytes of human hearts with myocardial infarction. Circulation. 1996;94:1506–1512. doi: 10.1161/01.cir.94.7.1506. [DOI] [PubMed] [Google Scholar]
- Mujtaba S, He Y, Zeng L, Yan S, Plotnikova O, Sachchidanand, Sanchez R, Zeleznik-Le NJ, Ronai Z, Zhou MM. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol Cell. 2004;13:251–263. doi: 10.1016/s1097-2765(03)00528-8. [DOI] [PubMed] [Google Scholar]
- Mujtaba S, Zeng L, Zhou MM. Modulating molecular functions of p53 with small molecules. Cell Cycle. 2006;5:2575–2578. doi: 10.4161/cc.5.22.3464. [DOI] [PubMed] [Google Scholar]
- Mujtaba S, Zeng L, Zhou MM. Structure and acetyl-lysine recognition of the bromodomain. Oncogene. 2007;26:5521–5527. doi: 10.1038/sj.onc.1210618. [DOI] [PubMed] [Google Scholar]
- Murphy PJ, Galigniana MD, Morishima Y, Harrell JM, Kwok RP, Ljungman M, Pratt WB. Pifithrin-alpha inhibits p53 signaling after interaction of the tumor suppressor protein with hsp90 and its nuclear translocation. J Biol Chem. 2004;279:30195–30201. doi: 10.1074/jbc.M403539200. [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- Ott M, Schnolzer M, Garnica J, Fischle W, Emiliani S, Rackwitz H, Verdin E. Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr Biol. 1999;9:1489–1492. doi: 10.1016/s0960-9822(00)80120-7. [DOI] [PubMed] [Google Scholar]
- Prives C, Hall PA. The p53 pathway. J Pathol. 1999;187:112–126. doi: 10.1002/(SICI)1096-9896(199901)187:1<112::AID-PATH250>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Prives C, Manley JL. Why is p53 acetylated? Cell. 2001;107:815–818. doi: 10.1016/s0092-8674(01)00619-5. [DOI] [PubMed] [Google Scholar]
- Rogel A, Popliker M, Webb CG, Oren M. p53 cellular tumor antigen: analysis of mRNA levels in normal adult tissues, embryos, and tumors. Mol Cell Biol. 1985;5:2851–2855. doi: 10.1128/mcb.5.10.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachchidanand, Resnick-Silverman L, Yan S, Mutjaba S, Liu WJ, Zeng L, Manfredi JJ, Zhou MM. Target structure-based discovery of small molecules that block human p53 and CREB binding protein association. Chem Biol. 2006;13:81–90. doi: 10.1016/j.chembiol.2005.10.014. [DOI] [PubMed] [Google Scholar]
- Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998;12:2831–2841. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez R, Zhou MM. The role of human bromodomains in chromatin biology and gene transcription. Curr Opin Drug Discov Devel. 2009;12:659–665. [PMC free article] [PubMed] [Google Scholar]
- Schafer T, Scheuer C, Roemer K, Menger MD, Vollmar B. Inhibition of p53 protects liver tissue against endotoxin-induced apoptotic and necrotic cell death. FASEB J. 2003;17:660–667. doi: 10.1096/fj.02-0774com. [DOI] [PubMed] [Google Scholar]
- Song S, Lambert PF. Different responses of epidermal and hair follicular cells to radiation correlate with distinct patterns of p53 and p21 induction. Am J Pathol. 1999;155:1121–1127. doi: 10.1016/S0002-9440(10)65215-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Hoek TL, Qin Y, Wojcik K, Li CQ, Shao ZH, Anderson T, Becker LB, Hamann KJ. Reperfusion, not simulated ischemia, initiates intrinsic apoptosis injury in chick cardiomyocytes. Am J Physiol Heart Circ Physiol. 2003;284:H141–150. doi: 10.1152/ajpheart.00132.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Vousden KH. p53: death star. Cell. 2000;103:691–694. doi: 10.1016/s0092-8674(00)00171-9. [DOI] [PubMed] [Google Scholar]
- Wei JY. Age and the cardiovascular system. N Engl J Med. 1992;327:1735–1739. doi: 10.1056/NEJM199212103272408. [DOI] [PubMed] [Google Scholar]
- Westphal CH, Hoyes KP, Canman CE, Huang X, Kastan MB, Hendry JH, Leder P. Loss of atm radiosensitizes multiple p53 null tissues. Cancer Res. 1998;58:5637–5639. [PubMed] [Google Scholar]
- Westphal CH, Rowan S, Schmaltz C, Elson A, Fisher DE, Leder P. atm and p53 cooperate in apoptosis and suppression of tumorigenesis, but not in resistance to acute radiation toxicity. Nat Genet. 1997;16:397–401. doi: 10.1038/ng0897-397. [DOI] [PubMed] [Google Scholar]
- Willis AC, Chen X. The promise and obstacle of p53 as a cancer therapeutic agent. Curr Mol Med. 2002;2:329–345. doi: 10.2174/1566524023362474. [DOI] [PubMed] [Google Scholar]
- Woods DB, Vousden KH. Regulation of p53 function. Exp Cell Res. 2001;264:56–66. doi: 10.1006/excr.2000.5141. [DOI] [PubMed] [Google Scholar]
- Zeng L, Zhang Q, Gerona-Navarro G, Moshkina N, Zhou MM. Structural basis of site-specific histone recognition by the bromodomains of human coactivators PCAF and CBP/p300. Structure. 2008;16:643–652. doi: 10.1016/j.str.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Liu W, Ding D, Salvi R. Pifithrin-alpha suppresses p53 and protects cochlear and vestibular hair cells from cisplatin-induced apoptosis. Neuroscience. 2003;120:191–205. doi: 10.1016/s0306-4522(03)00286-0. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Lu S, Wu L, Chai G, Wang H, Chen Y, Sun J, Yu Y, Zhou W, Zheng Q, et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1) Mol Cell Biol. 2006;26:2782–2790. doi: 10.1128/MCB.26.7.2782-2790.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.