

Abstract
Alkoxyalkyl esters of cidofovir (CDV) are orally active agents which inhibit the replication of a variety of double stranded DNA (dsDNA) viruses including variola, vaccinia, ectromelia, herpes simplex virus, cytomegalovirus, adenovirus and others. One of these compounds, hexadecyloxypropyl-CDV (HDP-CDV, CMX001) is in clinical development for prevention and treatment of poxvirus infection, vaccination complications, and for infections caused by cytomegalovirus, adenovirus, herpesviruses and other dsDNA viruses. This class of lipid analogs is potentially prone to undergo omega oxidation of the alkyl moiety which can lead to a short chain carboxylic acid lacking antiviral activity. To address this issue, we synthesized a series of alkoxyalkyl or alkyl glycerol esters of CDV and (S)-HPMPA having modifications in the structure of the alkyl residue. Antiviral activity was assessed in cells infected with vaccinia, cowpox or ectromelia viruses. Metabolic stability was determined in S9 membrane fractions from rat, guinea pig, monkey and human liver. All compounds had substantial antiviral activity in cells infected with vaccinia, cowpox or ectromelia. Metabolic stability was lowest in monkey liver S9 incubations where rapid disappearance of HDP-CDV and HDP-(S)-HPMPA was noted. Metabolic stability in monkey preparations increased substantially when a ω-1 methyl group (15-methyl-HDP-CDV) or a terminal cyclopropyl residue (14-cyclopropyl-tetradecyloxypropyl-CDV) was present in the alkyl chain. The most stable compound was 1-O-octadecyl-2-O-benzyl-sn-glycero-3-CDV (ODBG-CDV) which was not metabolized extensively by monkey liver S9. In rat, guinea pig or human liver S9 incubations, most of the modified antiviral compounds were considerably more stable.
1. Introduction
Esterification of 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]cytosine (cidofovir, CDV) or 9-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]adenine (HPMPA) with various alkoxyalkyl or alkylglycerol groups leads to remarkable increases in antiviral activity against herpes virus1 and orthopoxviruses.2 This is due primarily to increased cellular uptake of the alkoxyalkyl esters of CDV and HPMPA and conversion to CDV diphosphate3 or HPMPA diphosphate.4 In mammalian cells cidofovir (CDV) and HPMPA alkoxyalkyl esters are cleaved by phosphatases of the phospholipase C type, releasing CDV and HPMPA which are sequentially phosphorylated to CDV- and HPMPA-diphosphate, the active antivirals.3-5 Alkoxyalkyl esters of CDV are orally active6 against a variety of dsDNA virus infections including vaccinia, ectromelia, cytomegalovirus and adenovirus. 7-10
Long hydrocarbon chains such as those present in hexadecyloxypropyl-CDV (HDP-CDV) or hexadecyloxypropyl-(S)-HPMPA (HDP-HPMPA) may be hydroxylated at the ω or ω-1 carbon atoms by a mixed function oxidase (EC 1.14.14.1) in a process referred to as omega oxidation. The enzymes of omega oxidation are present in the endoplasmic reticulum of liver and kidney where the substrates are fatty acids; preferred substrates are fatty acids having 10 or 12 carbons in length11; longer hydrocarbon chains may also be hydroxylated at the ω carbon or the ω-1 carbon.12 After further oxidations catalyzed by alcohol dehydrogenase (EC 1.1.1.1) and aldehyde dehydrogenase (EC 1.2.1.3) the ω carbon is converted to a carboxylic acid which may then undergo repeated cycles of beta oxidation in mitochondria or peroxisomes leading to a short carboxylic acid metabolite of the alkoxyalkyl nucleotides. This pathway represents a route which does not lead to metabolites with antiviral activity. For example, HDP-CDV was reported to be rapidly degraded by this route in cynomolgus monkeys and levels of active antiviral sufficient to treat viral infections were not achieved.13
To see if the omega oxidation pathway for HDP-CDV or HDP-HPMPA can be slowed, we synthesized a series of alkoxyalkyl or alkylglycerol esters of CDV and HPMPA having modifications in their alkyl structure and assessed their in vitro cytotoxicity and antiviral activity against ectromelia, vaccinia and cowpox. Metabolic stability of the modified alkoxyalkyl esters was compared with their straight chain counterparts in liver S9 assays using liver preparations from guinea pig, rat, monkey and human sources.
2. Results
2.1. Synthesis
Straight chain alkyl groups of HDP-CDV and HDP-(S)-HPMPA were modified by introducing residues near the terminus which might slow the ω oxidation pathway (Figure 1). To introduce methyl groups at the ω-1 carbon of the alkyl chain (e. g. 15M-HDP-CDV and 15M-HDP-(S)-HPMPA), we first synthesized the ω-1 methyl alkanols using Grignard reactions of isoamyl or isobutylmagnesium bromide and ω-bromo-α-alkanols as described previously by Yuasa et al.14 The alkanols were then converted to methansulfonates and treated with 1,3-propanediol or ethylene glycol to give a series of ω-1-methylalkoxyalkanols which were coupled to cyclic cidofovir or cyclic (S)-HPMPA using the Mitsunobu esterification reaction (Scheme 1).15 Basic hydrolysis of the cyclic CDV and (S)-HPMPA diesters gave the target phosphonoesters. Similarly, cyclopropyl16 and double bond17 terminated alkoxyalkanols were prepared and coupled to cCDV and cyclic (S)-HPMPA. Hydrolysis of these diesters gave 14-cyclopropyltetradecyloxypropyl cidofovir (14-cp-TDP-CDV) and ω-hexadecenyloxyethyl-(S)-HPMPA (HDNE-(S)-HPMPA). We also studied a previously reported analog, 1-O-octadecyl-2-O- benzyl sn-3-glyceryl CDV (ODBG-CDV), which has a benzyl substitution at the sn-2 hydroxyl of glycerol, a position remote from the ω carbon of the straight octadecyl chain.15 The antiviral activity and metabolic stability of these compounds was evaluated.
Figure 1.
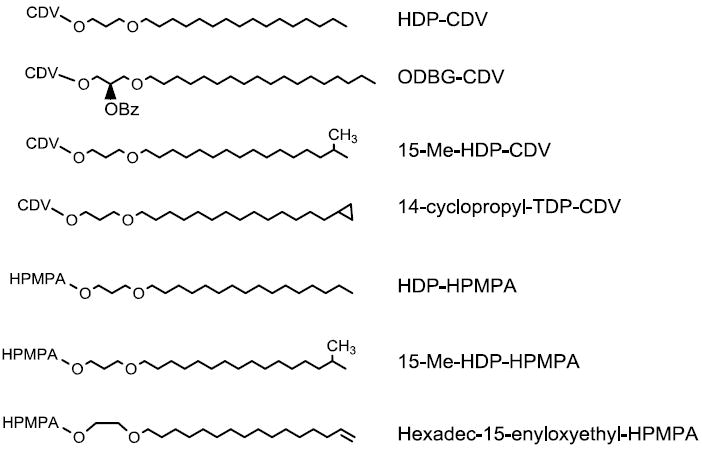
Structures of Modified Alkoxyalkyl Moieties of CDV and (S)-HPMPA
Scheme 1.
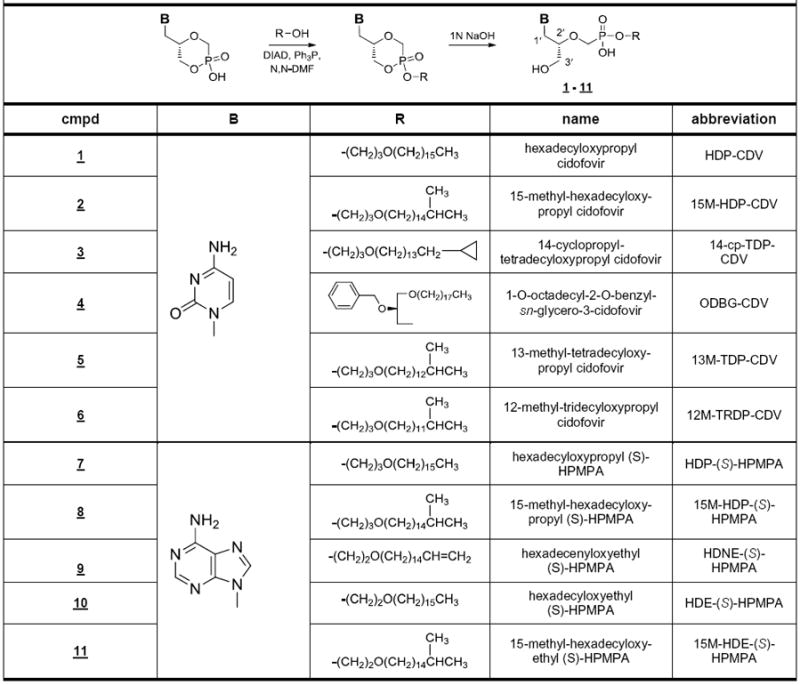
Synthesis of CDV and (S)-HPMPA Alkoxyalkyl Esters
2.2. Antiviral Evaluation
Compounds were screened for antiviral activity in cells infected with ectromelia, cowpox and vaccinia viruses whereas cytotoxicity was determined in uninfected cells (Table 1). In ectromelia infected cells the EC50 of HDP-CDV was 0.125 ± 0.06 μM while 15M-HDP-CDV and ODBG-CDV were more active with EC50s of 0.043 and 0.030 μM, respectively. The terminal cyclopropyl analog (14-cp-TDP-CDV) and shorter ω-1 methyl compounds (13M-TDP-CDV and 12M-TrDP-CDV) were less active than HDP-CDV. HDP-HPMPA was slightly more active than the corresponding CDV analog with an EC50 of 0.066 ± 0.014 μM. Compared with HDP-HPMPA, 15M-HDE-HPMPA was more active, EC50 0.010 ± 0.006 μM, while HDNE-HPMPA was considerably less active and more toxic. In cells infected with cowpox or vaccinia virus, EC50 values for the analogs of CDV ranged from 0.1 to 0.5 μM except for the shortest ω-1 branch methyl compound (12M-TrDP-CDV) which was less active, EC50 0.8 to 1.0 μM. The HPMPA analogs were more active with EC50 values ranging from 0.01 to 0.09 μM. Again, considerable toxicity was noted with HDNE-(S)-HPMPA which had a CC50 of 1.7 μM. Selectivity indexes for the CDV analogs ranged from 75 to 1800; various HPMPA analogs had selectivity index values ranging from 85 to 5900 except for the HDNE-(S)-HPMPA analog with a selectivity index of 7. With the exception of HDNE-(S)-HPMPA, it appears that alkyl substitutions to HDP-CDV or HDP-(S)-HPMPA are generally permissive to maintaining good antiviral activity and selectivity in poxvirus infected cells.
Table 1.
Antiviral Activity of Metabolically Stable Alkoxyalkyl Prodrugs of CDV and (S)-HPMPA against Orthopoxviruses
| Ectromelia | Cowpox | Vaccinia | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | EC50 | CC50 | S.I. | EC50 | CC50 | S.l. | EC50 | CC50 | S.I. |
| Cidofovir compounds | |||||||||
| HDP-CDV | 0.125 ± 0.06 | 25.3 ± 7.5 | 202 | 0.2 ± 0.1 | 25 ± 9.5 | 125 | 0.08 ± 0.03 | 25 ± 9.5 | 313 |
| 15M-HDP-CDV | 0.043 ±0.007 | 78.0 ± 9.9 | 1810 | 0.1 ± 0.04 | 15 ± 4.0 | 150 | 0.2 ± 0.06 | 15 ± 4.0 | 75 |
| 14-cp-TDP-CDV | 0.74 ± 0.22 | 74.0 ± 4.5 | 100 | 0.5 ± 0.05 | 56 ± 13 | 112 | 0.3 ± 0.2 | 56 ± 13 | 187 |
| ODBG-CDV | 0.03 ± 0.007 | 351 | 1100 | 0.3 ± 0.12 | 47 ± 24 | 157 | 0.4 ± 0.12 | 47 ± 24 | 118 |
| 13M-TDP-CDV | 0.39 ± 0.12 | 82 | 210 | 0.3 ± 0.1 | 37 ± 5.6 | 123 | 0.3 ± 0.2 | 37 ± 5.6 | 123 |
| 12M-TRDP-CDV | 0.35 ± 0.07 | 82 | 234 | 0.8 ± 0.2 | 44 ± 4.0 | 55 | 1.0 ± 0.4 | 44 ± 4.0 | 44 |
| (S)-HPMPA compounds | |||||||||
| HDP-(S)-HPMPA | 0.066 ± .014 | 84.7 ± 26.4 | 1280 | 0.02 ± .006 | 2.8 ± 1.3 | 140 | 0.01 ± 0.006 | 2.8 ± 1.3 | 280 |
| 15M-HDP-(S)-HPMPA | 0.044 ± 0.016 | 11.7 ± 5 | 266 | 0.04 ± 0.02 | 11.4 ± 14 | 285 | 0.04 ± 0.03 | 11.4 ± 14 | 285 |
| HDNE-(S)-HPMPA | 0.32 ± 0.12 | 2.32 ± 0.94 | 7.2 | 0.02 ±0.002 | 1.7 ± 0.9 | 85 | 0.02 ± 0.004 | 1.7 ± 0.9 | 85 |
| HDE-(S)-HPMPA | 0.14 ± 0.06 | 40.5 ± 17.5 | 289 | 0.09 ± 0.03 | 14 ± 1.9 | 156 | 0.09 ± 0.05 | 14 ± 1.9 | 156 |
| 15M-HDE-(S)-HPMPA | 0.01 ±0.006 | 59.6 ± 16.7 | 5960 | 0.01 ±0.005 | 2.5 ± 0.1 | 250 | 0.01 ± 0.002 | 2.5 ± 0.1 | 250 |
Data are μM, mean ± SD (n=3). When no SD is given the result is the average of two determinations. Abbreviations: HDP-CDV, hexadecyloxypropyl-cidofovir; 15M-HDP-CDV, 15-methyl-hexadecyloxypropyl-cidofovir; 14-cp-TDP-CDV, 14-cyclopropyl-tetradecyloxypropyl-cidofovir; ODBG-CDV, 1-O-octadecyl-2-O-benzyl-sn-glyceryl-3-cidofovir ; 13M-TDP-CDV, 13-methyl-tetradecyloxypropyl-cidofovir; 12M-TRDP-CDV, 12-methyl-tridecyloxypropyl-cidofovir; HDP-(S)-HPMPA; hexadecyloxypropyl-(S)-HPMPA, 15M-HDP-(S)-HPMPA, 15-methyl-hexadecyloxypropyl-(S)-HPMPA; HDNE-(S)-HPMPA, ω-hexadecenyloxyethyl-(S)-HPMPA, HDE-(S)-HPMPA, hexadecyloxyethyl-(S)-HPMPA; 15M-HDE-(S)-HPMPA, 15-methyl-hexadecyloxyethyl-(S)-HPMPA. SI, selective index; EC50, 50% effective concentration; CC50, 50% cytotoxic concentration as μM. Antiviral assays are plaque reduction assays as noted in Methods.
Hostetler, K. Y. et al. Antiviral Res. 2007, 73, 212.;
Keith, K. A. et al. Antimicrob. Agents Chemother., 2004, 48, 1869.
2.3. Metabolic Stability of CDV analogs
Various alkoxyalkyl esters of CDV were incubated with liver S9 fractions and an NADPH+ generating system for 90 minutes and disappearance was measured by LC-MS. To verify activity of the preparations, 7-ethoxycoumarin was used as a positive control (data not shown). Disappearance curves with rat, guinea pig and human liver S9 incubations show slow metabolism of HDP-CDV at both 1 μM and 10 μM drug concentration (Figure 2). However, monkey S9 degrades HDP-CDV very rapidly with 50% loss in 20 minutes. In contrast, ODBG-CDV is very slowly degraded by monkey S9; 15M-HDP-CDV and 14-cp-TDP-CDV are metabolized at intermediate rates with 50% reduction in approximately 45 to 70 minutes. ODBG-CDV metabolism at 10 μM and 1 μM is minimal with 100% and 88.3% remaining at 90 minutes. Comparing the amount of intact drug remaining in 1 μM monkey and human S9 incubations at 90 minutes, the % remaining of ODBG-CDV, 15M-HDP-CDV and 14-cp-TDP-CDV is 88.3%, 29.0% and 34.7% versus 16.7% for HDP-CDV, a difference which is highly significant at the p<0.001 level (Table 2). The differences in human S9 incubations are much smaller, but there is a statistically significant difference at 90 minutes between HDP-CDV (83.3%) and ODBG-CDV (92.3%) and 15M-HDP-CDV (91.3%) at the p<0.05 level (Table 2).
Figure 2.
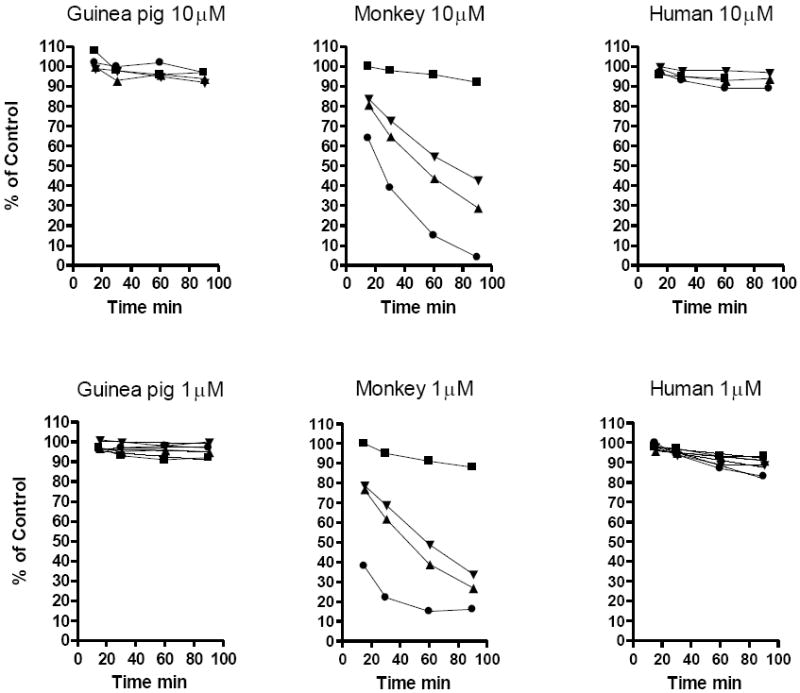
Metabolic stability of various alkoxyalkyl esters of CDV in Liver S9 fractions
Legend: HDP-CDV, circles; ODBG-CDV, squares; 15M-HDP-CDV, triangles; 14-cp-TDP-CDV, inverted triangles
Table 2.
Metabolic stability of CDV analogs in human and monkey liver S9 fractions after a 90 minute exposure
| Monkey S9 | Human S9 | |||
|---|---|---|---|---|
| Compound | Mean ± SD | p value | Mean ± SD | p value |
| HDP-CDV | 16.7 ± 2.1 | - | 83.3 ± 2.3 | - |
| ODBG-CDV | 88.3 ± 2.5 | <0.001 | 92.3 ± 4.0 | <0.05 |
| 15M-HDP-CDV | 29.0 ± 1.0 | <0.001 | 91.3 ± 1.5 | <0.05 |
| 14-cp-TDP-CDV | 34.7 ± 0.58 | <0.001 | 88.0 ± 2.6 | ns |
Result expressed as % compound remaining after 90 minute incubation at 1 μM. Data are % of compound remaining after a 90 minute incubation, n=3. Abbreviations as in Table 1. The p values are vs. HDP-CDV. Statistics by Tukey-Kramer Multiple Comparisons test (Instat, San Diego). Additional statistical comparisons: ODBG-CDV vs. 15M-HDP-CDV: monkey p<0.001; ODBG-CDV vs 14-cp-TDP-CDV: monkey p<0.001. 15M-HDP-CDV vs. 14-cp-TDP-CDV: monkey p<0.05.
2.4. Metabolic Stability of HPMPA analogs
Several alkoxyalkyl esters of (S)-HPMPA were also compared in monkey and human liver S9 incubations (Figure 3). In human liver S9, HDP-HPMPA and 15M-HDP-HPMPA were degraded slowly at both 1 and 10 μM concentrations. However, HDNE-HPMPA is degraded very rapidly. This was especially pronounced in monkey S9 fractions where there was 98% degradation (1 μM) and 80% degradation by 90 min (10 μM). Interestingly, monkey S9 degradation of HDP-CDV (Figure 2) is much more rapid than HDP-HPMPA (Figure 3) suggesting that the nature of the nucleobase may also affect metabolic stability independently of the nature of the alkyl chain.
Figure 3.
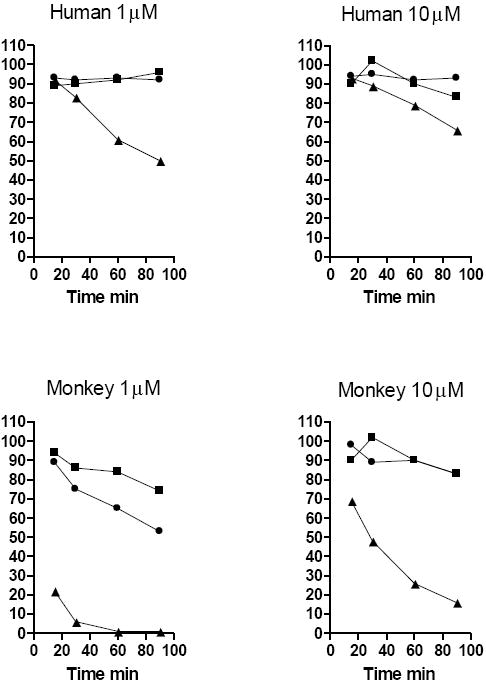
Metabolic stability of alkoxyalkyl esters of (S)-HPMPA in liver S9 fractions
Legend: HDP-(S)-HPMPA, circles; 15M-HDP-(S)-HPMPA, squares; HDNE-(S)-HPMPA, triangles
3. Discussion
Introduction of a ω-1 methyl, a terminal double bond, or a terminal cyclopropyl residue in the alkyl moiety had little effect on antiviral activity of the various compounds compared with the activity of HDP-CDV or HDP-(S)-HPMPA. Esterification of CDV with a 1-O-octadecyl-2-O-benzyl-sn- glycerol moiety also produced a highly active compound. In general all antiviral compounds with modifications in the alkyl moiety retained excellent submicromolar EC50 values although HDNE-(S)-HPMPA had unacceptably low selectivity (Table 1). In contrast, changes to the alkyl function or addition of a 2-O-benzyl group had a pronounced effect on the metabolic stability versus that of straight chain alkyl compounds such as HDP-CDV especially in monkey liver S9 fractions (Figure 2, Table 2). The order of metabolic stability in monkey liver S9 in 1 μM and 10 μM incubations was as follows: ODBG-CDV ≫ 14-cp-TDP-CDV = 15M-HDP-CDV > HDP-CDV. The presence of a ω-1 methyl group or a terminal cyclopropyl residue appears to increase metabolic stability. Unexpectedly, introduction of a 2-O-benzyl group (ODBG-CDV) provides the greatest metabolic stability in spite of the remote position of the benzyl group to the omega end of the octadecyl alkyl chain. The reasons for this observation are not clear, but the 2-O-benzyl group may provide steric hindrance preventing proper binding of the alkyl chain to the enzymes which catalyze omega oxidation. Although HDP-CDV is highly susceptible to metabolic conversion in monkey liver (Figure 2), compound stability was much greater in S9 fractions from rat, guinea pig and human liver. For example, in monkey S9 fractions at 1 μM and 10 μM compound, only 4-16% of HDP-CDV remained after 90 minutes, but in human and guinea pig liver S9 incubations, 83% to 97% of the initial HDP-CDV remained after 90 minutes. This suggests that in vivo testing of HDP-CDV in monkey models of smallpox or monkeypox infection is unlikely to produce levels of intact drug sufficient for successful treatment of these infections. A better choice for treating monkey poxvirus infection models with CDV analogs might be ODBG-CDV, 14-cp-TDP-CDV or 15M-HDP-CDV, in that order, based on the data in Figure 2. Although HDP-CDV is relatively stable in human S9 incubations, ODBG-CDV and 15M-HDP-CDV are somewhat more stable at 90 minutes with 91-92% remaining versus 83% for HDP-CDV (p<0.05) (Table 2).
However, it is important to note that rapid metabolism of HDP-CDV (CMX001) in humans has not been an issue and the drug has been used successfully to treat a variety of infections with double stranded DNA viruses including adenovirus, cytomegalovirus, herpes simplex virus, BK virus, JC virus where it is effective orally when administered once or twice a week.18-20 In an immunosupressed patient with a disseminated vaccinia infection, after 3 weeks of ineffective treatment with ST-246 and vaccinia immune globulin, the infection cleared after CMX001 was added to the regimen.21,22
With analogs of (S)-HPMPA, metabolic stability of straight chain alkoxyalkyl esters was substantially greater in monkey liver S9 fractions with 53 to 83% of the intact compound remaining after 90 minutes (Figure 3). This suggests that the nature of the nucleobase (adenine versus cytosine) may also be important in determining metabolic stability. In human liver S9 the straight alkyl chain HDP-(S)-HPMPA was metabolically stable with 92-93% of intact compound after 90 minutes. Of note was the lack of stability and rapid metabolism of HDNE-(S)-HPMPA, a compound with a double bond at the end of the alkyl chain. Although the antiviral activity of the compound was generally good (0.3-0.74 μM), selectivity was poor and it was rapidly metabolized by monkey and human liver S9 fractions (Figure 3).
4. Conclusion
A series of alkoxyalkyl esters of CDV and (S)-HPMPA designed to resist omega oxidation were synthesized placing functional groups at the end or at the ω-1 position of the alkyl chain or adding a 2-O-benzyl residue as in ODBG-CDV. The antiviral activity of the modified compounds was not greatly affected by these changes and all compounds retained excellent submicromolar EC50 values. However, there were profound changes in the metabolic stability of the compounds versus the straight alkyl chain compounds, particularly in monkey liver S9 preparations where straight chain HDP-CDV was rapidly metabolized and the modified compounds were substantially more stable. ODBG-CDV, which has a straight alkyl chain at the sn-1 position of glycerol and a 2-O-benzyl group, was the most stable even though the added functional group is not close to the ω end of the alkyl chain. In human liver S9 fractions, HDP-CDV was substantially more stable than in monkey S9 fractions. OBDG-CDV and 15M-HDP-CDV were slightly more stable than HDP-CDV in human liver S9 (p<0.05). Our findings may have significance which could affect the design of new alkoxyalkyl esters of acyclic nucleoside phosphonates for treatment of human viral infections.
5. Experimental
5.1 Chemistry
1H NMR spectra were recorded on a Varian HG spectrophotometer operating at 400 MHz and are reported in units of ppm relative to internal tetramethylsilane at 0.00 ppm and the numbering system used for peak assignments is as shown in Scheme 1. Low resolution electrospray ionization mass spectra (ESI-MS) were recorded on a Finnigan LCQDECA spectrometer and high resolution mass spectra (HRMS) were obtained on an Agilent 6230 Accurate-Mass TOFMS spectrometer, both at the small molecule facility, Department of Chemistry, University of California, San Diego. Chromatographic purification was done using the flash method with silica gel 60 (EMD Chemicals, Inc., 230–400 mesh). Cyclic cidofovir (cCDV) was provided by Gilead Sciences, Inc. and cyclic (S)-HPMPA (cHPMPA) was prepared as described previously.23 Purity of the new, tested compounds was assessed by HPLC analysis at the Department of Chemistry, University of California, San Diego using an Agilent 1260 Infinity Binary HPLC, a Phenomenex (Torrance, CA) Synergi™ Polar-RP (4μm,150 × 4.6 mm) column equipped with SecurityGuard™ (pentafluorophenylpropyl) protection column and UV detection at 274 nm. Solutions were prepared in MeOH (1 mg/mL). Mobile phase A was HPLC grade water with 0.1% formic acid and mobile Phase B was HPLC grade methanol with 0.1% formic acid. The gradient (1.0 mL/min.) was run as follows: 0-15 minutes: 10%B to 95% B; 15-18 minutes: 95% B; 18-20 minutes: 95%B to 10% B, and 20-25 minutes: 10% B. Analytical thin layer chromatography (TLC) was performed using Analtech silica gel-GF (250 μm) plates. The products were visualized with UV light, phospray (Supelco, Bellefonte, PA, USA) and charring. Syntheses and characterization of compounds 1, 4, 7, and 8 (Scheme 1) were previously reported by Kern et al.2, Wan et al.15, Beadle et al.24, and Morrey et al.25, respectively. Grignard coupling reactions between isoamyl or isobutyl magnesium bromide and ω-bromo-α-alkanols were carried out as described by Yuasa and Tsuruta14 to provide 15-methyl-hexadecanol, 14-methyl-pentadecanol, 13-methyl-tetradecanol and 12-methyl-tridecanol.
5.1.1. General procedure. Synthesis of (ω-1)-methyl-alkylmethanesulfonates
The (ω-1)-methyl alkanol (1 mol equiv) and triethylamine (1.2 mol equiv) were dissolved in CH2Cl2 and cooled to 0 °C. Methanesulfonyl chloride (1.2 mol equiv in CH2Cl2) was added dropwise to the stirred solution over 10 min. The reaction was stirred overnight, quenched with 1 N aq HCl and extracted three times with diethyl ether. The organic layer was then washed with brine, dried over magnesium sulfate, filtered and concentrated under reduced pressure to give the corresponding (ω-1)-methylalkylmethanesulfonates which were used in the next step without further purification.
5.1.1.1 15-Methyl-hexadecylmethanesulfonate
Yield 98%. 1H NMR (300 MHz, CDCl3): δH 4.20 (t, 2H, J=6.9Hz), 3.00 (s, 3H), 1.1-1.4 (m, 27H), 0.86 (d, 6H, J=6.6Hz).
5.1.1.2. 14-Methyl-pentadecylmethanesulfonate
Yield 87%. 1H NMR (300 MHz, CDCl3): δ 4.22 (t, 2H, J=6.9 Hz), 3.03 (s, 3H), 1.1-1.4 (m, 25H), 0.87 (d, 6H, J=6.6 Hz).
5.1.1.3. 13-Methyl-tetradecylmethanesulfonate
Yield 88%. 1H NMR (300 MHz, CDCl3): δ 4.23 (t, 2H, J=6.9Hz), 2.99 (s, 3H) 1.1-1.8 (m, 23H), 0.86 (d, 6H, J=6.6Hz).
5.1.1.4. 12-Methyl-tridecylmethanesulfonate
Yield 88%. 1H NMR (300MHz, CDCl3): δH 4.21 (t, 2H, 6.9Hz), 2.99 (s, 3H), 1.1-1.8 (m, 21H), 0.86 (d, 6H, 6.6Hz).
5.1.2. 14-Cyclopropyl-tetradecyl bromide
A solution of trifluoroacetic acid (2.96 g, 26 mmol) in CH2Cl2 (25 mL) was slowly added to a solution of diethyl zinc (26 mL of 1M solution in hexanes) and freshly distilled CH2Cl2 (25 mL) being stirred at 0 °C in an ice bath. After stirring the mixture for 20 minutes, a solution of CH2I2 (6.94 g, 26 mmol) in CH2Cl2 (10 mL) was added followed by 20 min. of stirring. Finally, a solution of 16-bromohexadECene (3.19 g, 13 mmol, prepared according to Balachander and Sukenik17) in 10 mL of dichloromethane was added, the ice bath was removed and the reaction was stirred for an additional 30 min. The reaction was quenched by addition of aq HCl followed by aq NH4Cl solution. The mixture was extracted with hexanes and the organic layer washed with saturated aq NaHCO3 and dried with brine and anhydrous MgSO4. The residue was purified by chromatography on silica gel. Elution with hexanes gave 3.34 g (79%) of product. 1H NMR (400 MHz, CDCl3): δ 3.38 (t, 2H,J=7.2Hz), 1.8 (m, 2H), 1.12-1.4 (m, 24H), 0.63 (m, 1H), 0.36 (m, 2H), 0.037 (m, 2H). ESI-MS [M+H]+ = 316.
5.1.3. General procedure. Synthesis of (ω-1)-methyl-alkoxyalkan-1-ols
To a solution of 1,3-propanediol or ethylene glycol (5 mol equiv) in anhydrous N,N-DMF (20 mL/mmol) was added NaH (1.2 mol equiv) and the mixture was stirred for 30 min. To this solution was added the (ω-1)-methylalkylmethanesulfonate (1 mol equiv) all at once. The reaction mixture was heated to 80 °C and kept overnight. The solvents were evaporated under vacuum and the residue was added to water and extracted three times with CH2Cl2. The organic phase was dried over MgSO4 and concentrated under vacuum. The residue was adsorbed on silica gel and purified by flash chromatography. Elution with Hexanes/EtOAc (80%:20%) gave the pure products as waxy solids.
5.1.3.1. 15-Methyl-hexadecyloxypropanol
Yield 74%. 1H NMR (400 MHz, CDCl3) δ 3.77 (t, 2H), 3.61 (t, 2H), 3.42 (t, 2H), 1.83 (pentet, 2H), 1.56 (m, 2H), 1.53 (septet, 1H), 1.25 (m, 20H), 1.15 (m, 2H), 0.86 (d, 6H).
5.1.3.2. 14-Methyl-pentadecyloxypropanol
Yield 80%. 1H NMR (400 MHz, CDCl3) δ 3.79 (q, 2H), 3.61 (t, 2H), 3.42 (t,2H), 1.83 (pentet, 2H), 1.56 (m, 2H), 1.53 (septet, 1H), 1.25 (m, 20H), 1.15 m,2H), 0.86 (d, 6H).
5.1.3.3. 13-Methyl-tetradecyloxypropanol
Yield 66%. 1H NMR (400 MHz, CDCl3) δ 3.77 (q, 2H), 3.62 (t, 2H), 3.43 (t, 2H), 1.82 (pentet, 2H), 1.56 (m, 2H), 1.53 (septet, 1H), 1.25 (m, 18H), 0.86 (d, 6H).
5.1.3.4. 12-Methyl-tridecyloxypropanol
Yield 65%. 1H NMR (400 MHz, CDCl3) δ 3.77 (t, 2H), 3.61 (t, 2H), 3.42 (t, 2H), 1.83 (pentet, 2H), 1.56 (m, 2H), 1.54 (septet, 1H), 1.25 (m, 16H), 1.15 (m, 2H), 0.86 (d, 6H).
5.1.3.5. 15-methyl-hexadecyloxyethanol
Yield 58%. 1H NMR (400 MHz, CDCl3) δ 3.72 (q, 2H), 3.53 (t, 2H), 3.47 (t, 2H), 1.59 (pentet, 2H), 1.51 (septet, 1H), 1.26 (m, 22H), 1.15 (m, 2H), 0.86 (d, 6H).
5.1.3.6. ω-Hexadecenyloxyethanol
Yield 61%. 1H NMR (400 MHz, CDCl3) δ 5.81 (m, 1H), 4.97 (m, 1H), 4.91 (m, 1H), 3.73 (q, 2H), 3.53 (t, 2H), 3.47 (t, 2H), 2.04 (q, 2H), 1.58 (m, 2H), 1.26 (m, 22H)
5.1.4. 14-Cyclopropyl-tetradecyloxypropanol
To a solution of 1,3-propanediol (6.08 g, 80 mmol) in anhydrous N,N-DMF (20 mL) was carefully added NaH (1.2 g, 30 mmol as 60% oil suspension) and the mixture was stirred for 30 min. This solution was slowly added to a stirred solution of 14-cyclopropyltetradecyl bromide (2.5 g, 7.9 mmol) in N,N-DMF (19 mL). The reaction mixture was kept overnight at 80 °C. Solvents were evaporated and the residue was treated with water and extracted with CHCl3. The organic phase was dried over MgSO4. Purification on silica gel (hexanes/EtOAc 80:20) gave 1.56 g (65% yield) of 14-cyclopropyl-tetradecyloxypropanol as a white solid. 1H NMR δ (400 MHz, CDCl3): 3.78 (q, 2H; J= 6 Hz), 3.61 (t, 2H, J=6Hz), 3.42 (t, 2H, J=6.8Hz), 2.53 (t,1H, J=5.6Hz), 1.8 (m, 2H), 1.5 (m, 2H), 1.14-1.04 (m, 24H), 0.64 (m, 1H), 0.38 (m, 2H), 0.013 (m, 2H). APCI-MS: 313.23 [M+H]+.
5.1.5. General Procedure. Esterification of cyclic cidofovir
Anhydrous cyclic cidofovir (cCDV, 1 mol equiv), the appropriate alkoxyalkanol (2 mol equiv) and triphenylphosphine (2 mol equiv) were added to anhydrous N,N-dimethylformamide (6.5 mL per mmol cCDV) and stirred vigorously under a nitrogen atmosphere. Diisopropyl azodicarboxylate (DIAD, 2 mol equiv) was added in three portions over 15 minutes and then the mixture was allowed to stir overnight. The solvent was evaporated under reduced pressure and the residue adsorbed onto silica gel and purified by flash column chromatography. Gradient elution from 100% CH2Cl2 to 15% EtOH/85% CH2Cl2 yielded the cyclic diesters which were also recrystallized from p-dioxane. Each cCDV ester was isolated as an approx. equimolar mixture of the axial and equatorial diastereomers and yields are reported based on cCDV.
5.1.5.1. 15-Methyl-hexadecyloxypropyl cyclic cidofovir
Synthesized from cCDV (835 mg, 3.2 mmol), 3-(15-methyl-hexadecyloxy)propan-1-ol (2.00 g, 6.4 mmol), triphenylphosphine (838 mg, 6.4 mmol) and DIAD (1.3 g, 6.4 mmol). Yield 950 mg (53%) as a white solid. 1H NMR (400 MHz, CD3OD) δ 7.27, 7.24 (pair d, 1H), 5.78, 5.76 (pair d, 1H), 4.42-4.34 (m, 1H), 3.89-3.84 (m, 1H), 4.25-4.05 (m, 5H), 3.48, 3.51 (pair t, 2H), 3.40, 3.49 (pair t, 2H), 1.98, 1.92 (pair q, 2H), 1.53 (m, 2H), 1.51 (m, 1H), 1.26 (m, 22H), 1.15 (m, 2H), 0.86 (d, 6H). ESI-MS 558.54 [M+H]+, 580.52 [M+Na]+.
5.1.5.2. 13-Methyl-tetradecyloxypropyl cyclic cidofovir
Synthesized from cCDV (335 mg, 1.28 mmol), 3-(13-methyltetradecyloxy)propan-1-ol (680 mg, 2.57 mmol), triphenylphosphine (675 mg, 2.57 mmol) and DIAD (480 mg, 2.37 mmol). Obtained 311 mg (46%) as a white solid. 1H NMR (400 MHz, CD3OD) δ 7.51, 7.49 (pair d, 1H), 5.71, 5.66 (pair d, 1H), 4.47-3.93 (m, 7H), 3.84-3.69 (m, 2H), 3.51, 3.49 (pair t, 2H), 3.43, 3.41 (pair t, 2H), 1.93 (m, 2H), 1.55 (m, 2H), 1.52 (m, 1H), 1.29 (m, 18H), 1.16 (m, 2H), 0.88 (d, 6H). ESI-MS 530.27 [M+H]+, 552.25 [M+Na]+.
5.1.5.3. 12-Methyl tridecyloxypropyl cyclic cidofovir
Synthesized from cCDV (335 mg, 1.28 mmol), 3-(12-methyltridecyloxy)propan-1-ol (700 mg, 2.57 mmol), triphenylphosphine (675 mg, 2.57 mmol) and DIAD (480 mg, 2.57 mmol). Yield 265 mg (40%) as an off-white solid. 1H NMR (400 MHz, CD3OD) δ 7.53, 7.44 (pair d, 1H), 5.62, 5.66 (pair d, 1H), 4.42-3.85 (m, 7H), 3.82-3.82 (m, 2H), 3.43, 3.41 (pair t, 2H), 3.46, 3.33 (pair t, 2H), 1.83 (m, 2H), 1.49 (m, 2H), 1.47 (m, 1H), 1.24 (m, 16H), 1.14 (m, 2H), 0.84 (d, 6H). ESI-MS 516.26 [M+H]+, 538.26 [M+Na]+.
5.1.5.4. 14-cyclopropyl-tetradecyloxypropyl cyclic cidofovir
Synthesized from cCDV (130 mg, 0.5 mmol), 3-(14-cyclopropyltetradecyloxy)propan-1-ol (312 mg, 1 mmol), triphenylphosphine (524 mg, 2 mmol), DIAD (400 mg, 2.0 mmol). Obtained 160 mg (30%) as an off-white solid. 1H NMR (300MHz, CDCl3): δH 7.2(broad s,3H), 5.67 (d, 1H, J=7.2Hz), 4.23 (m,m, 6H), 3.85 (m, 1H), 3.4 (m, 4H), 1.92 (m, 2H), 1.76 (m, 2h), 1.53 (m, 2H), 1.25 (m, 24H), 0.66 (m, 1H), 0.37 (m ,2H), -0.009 (m, 2H). 31P NMR (300 MHz, CDCl3) δ 13.75 (s),12.115 (s). ESI-MS: 556.43 [M+H]+.
5.1.6. General Procedure. Synthesis of cidofovir esters
The cyclic cidofovir diesters were suspended in 1 N NaOH (10 mL/ 1 mmol) and stirred while heating at 60°C for 1 h, during which time the solution became clear. The solution was then cooled to 25°C and acidified with acetic acid to a pH of approximately 5. The crude product was collected by vacuum filtration and then purified by flash column chromatography on silica gel. The CDV monoesters were eluted from silica using CH2Cl2/MeOH 80%:20%. Evaporation of the product-containing fractions gave the final products as white to off white powders.
5.1.6.1. 15-methyl-hexadecyloxypropyl cidofovir, sodium salt
Yield 76%. 1H NMR (methanol-d4): δ 7.72 (d, 1H, J = 7.3 Hz, H-6); 5.88 (d, 1H, J = 7.3 Hz, H-5); 4.09 (dd, 1H, J1’a,2’ = 3.3 Hz, Jgem = 14.0 Hz, H-1’a); 3.96-3.91 (m, 2H, -P-O-CH2); 3.80 (dd, 1H, J1’b,2’ = 7.6 Hz, Jgem = 13.9 Hz, -CHa-P-); 3.74-3.71 (m, 2H, H-3’); 3.66-3.63 (m, 1H, H-2’); 3.59 (dd, 1H, JCHb,P = 9.3 Hz, Jgem = 12.8 Hz, -CHb-P-); 3.51 (t, 2H, J = 6.7 Hz, -CH2-O-CH2-); 3.41 (t, 2H, J = 6.6 Hz, - CH2-O-CH2-); 1.84 (pentet, 2H, -OCH2CH2CH2O-); 1.58-1.49 (m, 2H, CH2-O-CH2CH2-); 1.28 (m, 24H, -(CH2)12-); 1.19-1.14 (m, 1H, -CH(CH3)2); 0.88 (d, 6H, -CH(CH3)2). 31P NMR (160 MHz) δ 17.12, singlet. HRMS (ESI-) calcd. for C28H53N3O7P [M-H]- 574.3627, found 574.3625 (E = -0.4 ppm). HPLC analysis: retention time 16.87 min., purity 99.6 %.
5.1.6.2. 13-methyl-tetradecyloxypropyl cidofovir, sodium salt
Yield 57%. 1H NMR (methanol-d4): δ 7.90 (d, 1H, J = 8.0 Hz, H-6); 5.97 (d, 1H, J = 7.2 Hz, H-5); 4.17 (dd, 1H, J1’a,2’ = 2.8 Hz, Jgem = 13.6 Hz, H-1’a); 3.94-3.89 (m, 2H, -P-O-CH2); 3.81 (dd, 1H, J1’b,2’ = 8.0 Hz, Jgem = 14.0 Hz, H-1’b); 3.79 (dd, 1H, JCHa,P = 2.8 Hz, Jgem = 9.0 Hz, -CHa-P-); 3.75 (dd, 1H, JCHb,P = 2.4 Hz, Jgem = 11.6 Hz, -CHb-P-); 3.69 (m, 1H, H-2′); 3.59; 3.57;3.52 (t, 2H, J = 6.4 Hz, -CH2-O-CH2-); 3.41 (t, 2H, J = 6.6 Hz, -CH2-O-CH2-); 1.84 (pentet, 2H, -OCH2CH2CH2O-); 1.57-1.48 (m, 2H, CH2-O-CH2CH2-); 1.28 (m, 20H, -(CH2)10-); 1.19-1.14 (m, 1H, -CH(CH3)2); 0.87 (d, 6H, J = 6.8 Hz, -CH(CH3)2). 31P NMR (160 MHz) δ 17.18, singlet. HRMS (ESI-) calcd. for C26H49N3O7P [M-H]- 546.3314, found 546.3312 (E = -0.4 ppm). HPLC analysis: retention time 16.36 min., purity 95.5 %.
5.1.6.3. 12-methyl-tridecyloxypropyl cidofovir, sodium salt
Yield 77%. 1H NMR (methanol-d4): δ 7.65 (d, 1H, J = 7.3 Hz, H-6); 5.84 (d, 1H, J = 7.2 Hz, H-5); 4.05 (dd, 1H, J1’a,2’ = 3.6 Hz, Jgem = 13.9 Hz, H-1’a); 3.95-3.90 (m, 2H, -P-O-CH2); 3.82 (dd, 1H, J1’b,2’ = 7.2 Hz, Jgem = 13.9 Hz, H-1’b); 3.75-3.62 (m, 3H, H-3’ + -CHa-P-); 3.60 (dd, 1H, JCHb,P = 9.2 Hz, Jgem = 12.8 Hz, -CHb-P-); 3.51 (t, 2H, J = 6.4 Hz, -CH2-O-CH2-); 3.41 (t, 2H, J = 6.6 Hz, -CH2-O-CH2-); 1.84 (pentet, 2H, -OCH2CH2CH2O-); 1.59-1.48 (m, 2H, CH2-O-CH2CH2-); 1.29 (m, 18H, -(CH2)-); 1.20-1.14 (m, 1H, -CH(CH3)2); 0.88 (d, 6H, J = 6.6 Hz, -CH(CH3)2). 31P NMR (160 MHz) δ 17.35, singlet. HRMS (ESI-) calcd. for C25H47N3O7P [M-H]- 532.3157, found 532.3155 (E = - 0.4 ppm). HPLC analysis: retention time 16.03 min., purity 92.0 %.
5.1.6.4. 14-cyclopropyltetradecyloxypropyl cidofovir, sodium salt
Yield 80%. 1H NMR (methanol-d4): δ 7.68 (d, 1H, J = 7.6 Hz, H-6); 5.94 (d, 1H, J = 7.6 Hz, H-5); 4.15 (dd, 1H, J1’a,2’ = 2.8 Hz, Jgem = 13.6 Hz, H-1’a); 3.96-3.91 (m, 2H, -P-O-CH2); 3.81 (dd, 1H, J1’b,2’ = 7.6 Hz, Jgem = 14.0 Hz, H-1’b); 3.76 (dd, 1H, JCHb,P = 4.8 Hz, Jgem = 9.2 Hz, -CHb-P-); 3.73;3.66;3.59;3.54;3.52 (t, 2H, J = 6.4 Hz, -CH2-O-CH2-); 3.41 (t, 2H, J = 6.8 Hz, -CH2-O-CH2-); 1.84 (pentet, 2H, -OCH2CH2CH2O-); 1.57-1.49 (m, 2H, CH2-O-CH2CH2-); 1.29 (m, 20H, - (CH2)10-); 1.20-1.16 (m, 2H, -CH2-cyclopropyl); 0.65 (m, 1H, cyclopropyl); 0.39 (ddd, 2H, J = 1.6 Hz, 8 Hz, 8 Hz, cyclopropyl); -0.003 (ddd, 2H, J = 5.5 Hz, 10 Hz, 10 Hz). 31P NMR (160 MHz) δ 17.03, singlet. HRMS (ESI-) calcd. for C28H51N3O7P [M-H]- 572.3470, found 572.3471 (E = 0.2 ppm). HPLC analysis: retention time 16.73 min., purity 99.5 %.
5.1.7. General procedure. Esterification of cyclic HPMPA
Diethylazodicarboxylate (DIAD, 1.4 mol equiv) was added to a mixture of cyclic HPMPA (cHPMPA, 1 mol equiv), an alkoxyalkanol (1 mol equiv), triphenylphosphine (1.4 mol equiv), and dry N,N-dimethylformamide (10 mL/mmol cHPMPA); and the mixture was stirred overnight at room temperature. The solvent was then evaporated under reduced pressure, the residue adsorbed onto silica gel and purified by flash column chromatography. Gradient elution from 100% CH2Cl2 to 15% EtOH/85% CH2Cl2 yielded the cyclic diesters which were also recrystallized from p-dioxane. Each cHPMPA ester was isolated as an approx. equimolar mixture of the axial and equatorial diastereomers and yields are reported based on cHPMA.
5.1.7.1. Hexadecyloxyethyl (S)-cyclic HPMPA
Synthesized from cHPMPA (200 mg, 0.7 mmol), 2-(hexadecyloxy)ethan-1-ol (mg, 0.7 mmol), triphenylphosphine (262 mg, 1.0 mmol) and DIAD (202 mg, 1.0 mmol). Obtained 182 mg (47%) as an off-white solid. 1H NMR (400 MHz, CD3OD) δ 8.24, 8.22 (pair s, 1H), 8.04, 8.01 (pair s, 1H), 4.59-4.15 (m, 9H), 3.51 (t, 2H), 3.41 (t, 2H), 1.54 (m, 2H), 1.27 (m, 26 H), 0.87 (t, 3H); ESI-MS: m/z 552.38 [M-H]-.
5.1.7.2. ω-Hexadecenyloxyethyl (S)-cyclic HPMPA
Synthesized from cHPMPA (100 mg, 0.35 mmol), 2-(hexadecenyloxy)ethan-1-ol (100 mg, 0.35 mmol), triphenylphosphine (138 mg, 0.52 mmol) and DIAD (106 mg, 0.52 mmol). Obtained 75 mg (39%) as an off-white solid. 1H NMR (400 MHz, CD3OD) δ 8.27, 8.28 (pair s, 1H), 7.93, 7.90 (pair s, 1H), 5.81 (m, 1H), 4.97 (m, 1H), 4.92 (m, 1H), 4.56-4.06 (m, 7H), 3.66 (t, 2H), 3.60 (t, 2H), 3.47 (m, 2H), 2.04 (pentet, 2H), 1.57 (m, 2H), 1.26 (broad s, 22 H). ESI-MS: m/z 550.44 [M-H]-.
5.1.7.3. 15-Methylhexadecyloxyethyl (S)-cyclic HPMPA
Synthesized from cHPMPA (190 mg, 0.67 mmol), 2-(15-methylhexadecyloxy)ethan-1-ol (201 mg, 0.67 mmol), triphenylphosphine (245 mg, 0.94 mmol) and DIAD (190mg, 0.94 mmol). Obtained 130 mg (34%) as an off-white solid. 1H NMR (400 Mz, CD3OD) δ 8.26, 8.24 (pair s, 1H), 8.04, 8.02 (pair s, 1H), 4.59-4.15 (m, 9H), 3.51 (t, 2H), 3.41 (t, 2H), 1.54 (m, 2H), 1.52 (m, 1H), 1.27 (broad s, 22 H), 1.16 (m, 2H), 0.87 (d, 6H). ESI-MS: m/z 566.26 [M-H]-.
5.1.8. General procedure. Synthesis of (S)-HPMPA Esters
The cyclic (S)-HPMPA diesters were suspended in 1 N NaOH (10 mL/ 1 mmol) and stirred while heating at 60°C for 1 h, during which time the solution became clear. The solution was cooled to 25°C and acidified with acetic acid to a final pH of approximately 5. The precipitated crude product was collected by vacuum filtration and then purified by flash column chromatography on silica gel. The alkoxyalkyl monoesters were eluted from silica using 20% methanol-dichloromethane. Evaporation of product fractions gave the monoesters as white to off-white powders.
5.1.8.1. Hexadecyloxyethyl (S)-HPMPA, sodium salt
Yield 73%. 1H NMR (400 MHz, CD3OD) δ 8.29 (s, 1H, H-8), 8.22 (s, 1H, H-2), 4.47 (dd, 1H, J1’a,2’ = 3.8 Hz, Jgem = 14.5 Hz, H-1’a); 4.39 (dd, 1H, J1’b,2’ = 6.8 Hz, Jgem = 13.7 Hz, H-1’b); 3.95 (m, 2H, -P-O-CH2-); 3.80 (m, 1H, H-2’); 3.73 – 3.62 (m, 3H, H-3′ + -CHa-P-); 3.53 – 3.49 (m, 3H, -CH2-O-CH2- + -CHb-P-); 3.40 (t, 2H, -CH2-O-CH2-); 1.50 (m, 2H, -O-CH2CH2(CH2)13-); 1.26 (m, 26H, -(CH2)13-); 0.87 (t, 3H, -CH3). ESI-MS: m/z 570.32 [M-H]-. . HRMS (ESI-) calcd. for C27H49N5O6P [M-H]- 570.3426, found 570.3428 (E = 0.4 ppm). HPLC analysis: retention time 16.43 min., purity 90.8 %.
5.1.8.2. ω-Hexadecenyloxyethyl (S)-HPMPA, sodium salt
Yield 80%. 1H NMR (400 MHz, CD3OD) δ 8.38 (s, 1H, H-8), 8.25 (s, 1H, H-2), 5.79 (m, 1H, -CH=CH2); 4.97 (d, 1H, -CH=CH2); 4.91 (d, 1H, -CH=CH2); 4.55 (dd, 1H, J1’a,2’ = 2.8 Hz, Jgem = 14.4 Hz, H-1’a); 4.43 (dd, 1H, J1’b,2’ = 7.2 Hz, Jgem = 14.4 Hz, H-1’b); 4.05 (m, 2H, -P-O-CH2-); 3.91 (m, 1H, H-2’); 3.87 (dd, 1H, JCHa,P = 9.2 Hz, Jgem = 13.2 Hz, -CHa-P-); 3.76 (dd, 1H, JCHb,P = 8.8 Hz, Jgem = 13.2 Hz, -CHb-P-); 3.68 (dd, 1H, J3’a,2’ = 4.6 Hz, Jgem = 12.6 Hz, H-3’a); 3.60 (t, 2H, J = 6.7 Hz, - CH2-O-CH2-); 3.54 (dd, 1H, J3’b,2’ = 4.4 Hz, Jgem = 12.8 Hz, H-3’b); 3.47 (t, 2H, J = 6.6 Hz, -CH2-O-CH2-); 1.55 (m, 2H, -CH2(CH2)11-); 1.37 (m, 2H, -CH2-CH=CH2); 1.26 (m, 22H, -CH2)11-). ESI-MS: m/z 568.32 [M-H]-. . HRMS (ESI-) calcd. for C27H47N5O6P [M-H]- 568.3269, found 568.3268 (E = -0.2 ppm). HPLC analysis: retention time 16.13 min., purity 94.3 %.
5.1.8.3. 15-methylhexadecyloxyethyl (S)-HPMPA, sodium salt
Yield 66%. 1H NMR (400 MHz, CD3OD) δ 8.29 (s, 1H, H-8), 8.22 (s, 1H, H-2), 4.47 (dd, 1H, J1′a,2′ = 3.9 Hz, Jgem = 14.5 Hz, H-1′a); 4.39 (dd, 1H, J1′b,2′ = 6.8 Hz, Jgem = 14.6 Hz, H-1′b); 3.94 (m, 2H, -P-O-CH2-); 3.80 (m, 1H, H-2′); 3.78-3.60 (m, 4H, -CH2-P- + -CH2-OH); 3.51 (t, 2H, -CH2-O-CH2-), 3.40 (t, 2H, -CH2-O-CH2-), 1.49 (m, 2H, -CH2-O-CH2CH2-); 1.27 (m, 24 H, -(CH2)12-), 1.17 (m, 1H, - CH(CH3)2), 0.87 (d, 6H, -CH(CH3)2). ESI-MS: m/z 586.25 [M+H]+ . HRMS (ESI-) calcd. for C28H51N5O6P [M-H]- 584.3582, found 584.3586 (E = 0.7 ppm). HPLC analysis: retention time 16.48 min., purity 94.7 %.
5.2. Antiviral Methods
5.2.1. Ectromelia inhibition assay
ECTV-Moscow strain was used to infect BSC-1 cells at 50-75 plaques per well for one hour in DMEM (Lonza, Basel) supplemented with 2% Fetal Clone II (Hyclone, ThermoFisher, Pittsburgh). Serial dilutions of the compounds were made in DMEM-2% FCII and added to the infected cells along with DMEM-5% FCII-1% Carboxymethylcellulose (CMC Overlay media Lonza, Hyclone, Sigma, St. Louis, MO). The cultures were incubated 4-5 days and stained with crystal violet (0.13% crystal violet, 5% ethanol, 10% formaldehyde) to visualize plaque formation. The percent plaque reduction was determined by comparing the number of plaques in the treated wells to a group of untreated wells. The final concentration of compound in the culture was plotted versus the % plaque reduction to determine the concentration at which the plaque reduction is 50%.
5.2.2. Cytotoxicity assay BSC-1 Cells
Serial dilutions of the compounds were made in DMEM (Lonza) supplemented with 2% Fetal Clone II (Hyclone). The compound dilutions were mixed with BSC-1 cells at 5000 cells per reaction in 100μL total volume. At the given time point, 20 μL of MTS solution (CellTiter 96 Aqueous One Solution Cell Proliferation Assay, Promega, Madison, WI) was added to each reaction and incubated 2-4 hours. The OD490 was read and the % cell viability for each reaction was calculated based on untreated cells. The compound concentration was plotted versus the % cell viability to determine the concentration at which the cell viability was 50%.
5.2.3. Vaccinia and cowpox inhibition assays
Vaccinia virus (Copenhagen) and cowpox virus (Brighton), were obtained from Dr. John Huggins of the U.S. Army Medical Research Institute for Infectious Diseases, Frederick, MD. Monolayers of human foreskin fibroblast (HFF) cells in 6-well plates were infected with either vaccinia virus or cowpox virus to yield 20 to 30 plaques per well. Following a 1 h adsorption, the medium was aspirated from the wells, solutions containing different concentrations of test compounds were added in an agarose overlay, and plates were incubated for 3 d. Infected monolayers were then stained with a 0.02% solution of neutral red in PBS, plaques were enumerated, and EC50 values were calculated as described above.
5.2.4. Cytotoxicity Assays HFF Cells
HFF cells were seeded into 96-well plates containing 2.5 × 104 cells per well and incubated for 24 h to allow monolayers to form. Growth media was replaced with MEM containing 2% FBS and solutions containing different concentrations of test compounds. The plates were incubated for 7 d, the media aspirated, and the cells stained with 0.01% neutral red in PBS. Absorbance was measured at 540 nm to determine the number of viable cells. Cytotoxicity is expressed as the concentration of drug that reduced cell viability by 50% (CC50), and was interpolated from the experimental data.
5.3 Metabolic Stability in Liver S9 Fractions
5.3.1. Test Method
Test articles were incubated in Liver S9 fractions at 37 °C. Samples were pulled from the incubation mixture at specified time points (0 to 90 minutes) and added to a solution of 1% ammonium hydroxide in N-propyl alcohol. The amount of test article in each sample was quantified by HPLC/MS/MS.
5.3.2. Reaction Mixture Preparations
Test article solutions were prepared in DMSO:ACN:deionized water (5:5:190 v/v/v). A single species liver S9 suspension was prepared by diluting commercial pooled S9 fractions (CellzDirect, Carlsbad, CA for rat, monkey, human; XenoTECh, Lenexa, KS for guinea pig) with phosphate buffer. The test article solutions were diluted 10 fold into the S9 suspension. Reaction mixtures were allowed to equilibrate at 37 °C for approximately two minutes before adding a solution of NADPH regeneration system (BD Biosciences) to start the reaction. The reaction solution was mixed by multiple aspiration/dispense actions. The final reaction mixture contained S9 at 3 mg/mL protein concentration, NADPH regeneration solution at 1x concentration, and test article at either 1 μM or 10 μM.
5.3.3. Sample Analysis
Immediately after the addition, and mixing of NADPH, a Time zero sample (20 μL) was withdrawn from the reaction mixture. The sample was placed in a microtiter plate containing 80 μL of n-propyl alcohol with 1% ammonium hydroxide. The microtiter plate was then sealed and placed on an orbital mixer for 10 minutes. At 15, 30, 60 and 90 minutes after the Time 0 sample was withdrawn, subsequent samples were withdrawn and given the same treatment. The microtiter plates were placed in a refrigerator for at least 48 hours. The plates were then centrifuged for 10 minutes at 2254 RCF; after which, the supernatant was transferred to the final plate. The components in the final plate were eluted from a Zorbax Eclipse XDB C8 HPLC column with a linear gradient (Solvent A=water with 0.1% Formic Acid, Solvent B= isopropyl alcohol with 0.1% Formic Acid). The test article was detected by electrospray ionization-mass spectrometry on an Agilent 1200SL/6410 system.
5.3.4. Special Considerations
Typically, test articles are extracted from S9 incubations by acetonitrile protein precipitation. That method produces inconsistent results with the test articles described here. Extractions of these test articles at basic pH using n-propyl alcohol containing ammonium ions were more efficient and reproducible. Furthermore, the test articles were not immediately extracted from the precipitated protein. It is necessary to wait at least 24 hours at room temperature or 48 hours if refrigerated for extracted concentration to stabilize in the samples. The test articles are very stable in these conditions and no degradation was observed even if the samples were left on the bench for up to 7 days.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the National Institute for Allergy and infectious Disease, AI071803 and AI074057 (KYH); NIH/NIAID/DMID contract NO1-AI-30063 Task C24 (RMB) and NIAID NO1-AI-30049 (MNP). Dr. Hostetler is a consultant and equity holder in Chimerix Inc.
Footnotes
SUPPLEMENTARY DATA Supplementary data (HPLC analysis of compounds 2, 3, 5, 6, 9, 10 and 11 and 1H NMR spectra of 2, 6 and 11) associated with this article can be found in the online version.
The terms of this relationship have been reviewed and approved by the University of California, San Diego in accordance with their conflict of interest policies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beadle JR, Hartline C, Aldern KA, Rodriguez N, Harden E, Kern ER, Hostetler KY. Antimicrob Agents Chemother. 2002;46:2381. doi: 10.1128/AAC.46.8.2381-2386.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kern ER, Hartline C, Harden E, Keith K, Rodriguez N, Beadle JR, Hostetler KY. Antimicrob Agents Chemother. 2002;46:991. doi: 10.1128/AAC.46.4.991-995.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aldern K, Ciesla SL, Winegarden K, Hostetler KY. Mol Pharm. 2003;63:678. doi: 10.1124/mol.63.3.678. [DOI] [PubMed] [Google Scholar]
- 4.Magee WC, Aldern KA, Hostetler KY, Evans DH. Antimicrob Agents Chemother. 2008;52:586. doi: 10.1128/AAC.01172-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Magee WC, Hostetler KY, Evans DH. Antimicrob Agents Chemother. 2005;49:3153. doi: 10.1128/AAC.49.8.3153-3162.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciesla SL, Trahan J, Wan WB, Beadle JR, Aldern KA, Painter GR, Hostetler KY. Antiviral Res. 2003;59:163. doi: 10.1016/s0166-3542(03)00110-4. [DOI] [PubMed] [Google Scholar]
- 7.Quenelle DC, Collins DJ, Wan WB, Beadle JR, Hostetler KY, Kern ER. Antimicrob Agents Chemother. 2004;48:404. doi: 10.1128/AAC.48.2.404-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buller RM, Owens G, Schriewer J, Melman L, Beadle JR, Hostetler KY. Virology. 2004;318:474. doi: 10.1016/j.virol.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 9.Bidanset DJ, Beadle JR, Wan WB, Hostetler KY, Kern ER. J infect Dis. 2004;190:499. doi: 10.1086/421912. [DOI] [PubMed] [Google Scholar]
- 10.Toth K, Spencer JF, Dhar D, Sagartz JE, Buller RML, Painter GR, Wold WSM. Proc of the Natl Acad Sci U S A. 2008;105:7293. doi: 10.1073/pnas.0800200105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lehninger . In: Lehninger’s Principle’s of Biochemistry. Nelson DL, Cox MM, editors. WH Freeman & Co; New York: 2008. p. 664. [Google Scholar]
- 12.Johnston JB, Oueliet H, Podust LM, Ortiz de Montellano PR. Arch Biochem Biophys. 2011;507:86. doi: 10.1016/j.abb.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Painter GR. Presented at the 18th International Conference on Antiviral Research; Barcelona, Spain. April 12, 2005. [Google Scholar]
- 14.Yuasa Y, Tsuruta H. Flavour and Fragrance Journal. 2004;19:199. [Google Scholar]
- 15.Wan W, Beadle JR, Aldern KA, Trahan J, Hostetler KY. Antimicrob Agents Chemother. 2005;49:656. doi: 10.1128/AAC.49.2.656-662.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lorenz JC, Long J, Yang Z, Xue S, Xie Y, Shi Y. Org Chem. 2004;69:327. doi: 10.1021/jo030312v. [DOI] [PubMed] [Google Scholar]
- 17.Balachander N, Sukenik CN. Langmuir. 1990;6:1621. [Google Scholar]
- 18.Trost LC, Tippin TA, Anderson MT, Painter WP. Abstract of Papers 50th ICAAC; Boston, MA. American Society for Microbiology; 2010. Abstract A1-2017a. [Google Scholar]
- 19.Painter W. personal communication. 2010 [Google Scholar]
- 20.Painter W, Mossad S, Siemionow M, Eghtesad B, Djohan R, Papay R, Avery R. Presented at the American Transplant Congress; San Diego, CA. May 10, 2010. [Google Scholar]
- 21.Chong J. Los Angeles Times. 2009 June 7; [Google Scholar]
- 22.Anonymous. Morb Mortal Weekly Rep. 2009;58:532. [PubMed] [Google Scholar]
- 23.Rosenberg I, Hol A. Collect Czech Chem Commun. 1987;52:2792. [Google Scholar]
- 24.Beadle JR, Wan WB, Ciesla SL, Keith KA, Hartline CB, Kern ER, Hostetler KY. J Med Chem. 2006;49:2010. doi: 10.1021/jm050473m. [DOI] [PubMed] [Google Scholar]
- 25.Morrey JD, Korba BE, Beadle JR, Wyles DL, Hostetler KY. Antimicrob Agents Chemother. 2009;53:2865. doi: 10.1128/AAC.00114-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.