

Abstract
Background and Purpose
Because the potential neuroprotective effect of isoflurane is controversial, we attempted to study whether isoflurane post-treatment provides neuroprotection in a rat model of hyperglycemia-induced ischemic hemorrhagic transformation (HT).
Methods
Rats received an injection of 50% dextrose (6 ml/kg intraperitoneally) and had a middle cerebral artery occlusion (MCAO) 30 minutes later. Four groups were included: sham-operated, ischemia/reperfusion, isoflurane treatment, and vehicle groups. In the treatment group, after 2h of ischemia, 2% isoflurane was administered at the onset of reperfusion. We measured the level of blood glucose at 0, 2.5, 4.5, and 6.5 hrs after dextrose injection. Infarct and hemorrhagic volumes, neurologic scores, oxidative stress (malondialdehyde (MDA), 4-Hydroxy-2-Nonenal (4-HNE) and Nitrotyrosine) and the activities of superoxide dismutase (SOD) and catalase (CAT) were measured at 24h after ischemia.
Results
Isoflurane had no effects on blood glucose, and it failed to reduce infarct, hemorrhage volume, brain edema, and enhanced neurobehavioral deficits when compared with the ischemia/reperfusion group at 24h after MCAO. On contrary, isoflurane exacerbated these parameters compared with the vehicle group. In addition, it increased the expressions of MDA, 4-HNE and Nitrotyrosine, and decreased the activities of SOD and CAT compared to the vehicle group.
Conclusions
Isoflurane post-treatment worsened physiological and neurological outcomes in this ischemia hyperglycemia-induced HT possibly by impairing the antioxidant defense system.
Keywords: hemorrhagic transformation, hyperglycemia, isoflurane, MCAO, oxidative stress
Introduction
Hyperglycemia has been claimed to be associated with hemorrhagic transformation (HT) which is a major factor limiting the use of tissue plasminogen activator (tPA), the only FDA-approved treatment for ischemic stroke.1 Experimental studies have shown that preischemic hyperglycemia dramatically aggravated brain infarct and hemorrhagic conversion in rat middle cerebral artery occlusion (MCAO) with reperfusion model.2 Hyperglycemia-enhanced HT may be linked to increased inflammatory activity and oxidative stress,2-3 which cause the blood–brain barrier (BBB) disruption and neural cell death. There is currently no effective treatment to prevent hemorrhagic transformation after ischemia stroke.
As one of the most widely used volatile anesthetics, isoflurane has been recognized for their potential neuroprotective properties since the 1980s.4 Some evidences indicated that isoflurane, administered before, or after experimental cerebral ischemia, provided neuroprotection in in vivo and in vitro models.5-8 However, an equivalent number of experiments suggested that isoflurane exposure during experimental focal or global stroke offered little or no protection and may even worsen histological and functional outcomes.9-11 In addition, the effect of isoflurane on hyperglycemia-induced HT after stroke had not been tested. Therefore, we attempted to study the effects of isoflurane post-treatment on acute hyperglycemia-induced HT after MCAO in rats.
Materials and Methods
Animal Preparation and Middle Cerebral Artery Occlusion
All experiments were approved by the Institutional Animal Care and Use Committee of Loma Linda University. 131 male Sprague-Dawley rats were purchased from Harlan Laboratories, Indianapolis and randomly divided into the following groups: sham-operated (DX+Sham, n=23), ischemia/reperfusion (DX+MCAO, n=31), isoflurane treatment (DX+MCAO+ISO, n=42), vehicle groups (DX+MCAO+O2, n=28), and additional sham-operated group treated with isoflurane for testing the effects of isoflurane on blood glucose of hyperglycemic rats (DX+Sham+ISO, n=7). All rats were injected with 50% dextrose (DX) (6 ml/kg) i.p. 30 min before MCAO to induce acute hyperglycemia. Anesthesia was induced with ketamine and xylazine (80 mg/kg+10 mg/kg, i.p.) followed by atropine at a dose of 0.1mg/kg s.c. During surgery and postoperative period rectal temperature was maintained at 37.5 °C by means of a feedback-controlled heating pad.
MCAO was performed as reported previously.12 The external carotid artery (ECA) was isolated and coagulated, a 4-0 nylon suture with a round tip was inserted into the internal carotid artery through the ECA stump and occluded the MCA for 2h. Blood was obtained from the tail artery for analysis of glucose level before, during, and after the operation.
Isoflurane Post-treatment
In the treatment group, 2% isoflurane carried by 30% O2 and 70% medical air was administered for 1h immediately after ischemia via a chamber. In the vehicle group, rats were placed in the chamber only with 30% O2 and 70% medical air, in which the total concentration of O2 was about 45%. (Supplement 1)
2, 3, 5-triphenyltetrazolium Chloride Staining and Evaluation of Infarction Volume
As studied previously,12 2,3,5-triphenyltetrazolium chloride monohydrate staining was performed to determine the infarct volume at 24 h after MCAO (DX+MCAO+ISO, n=8; other groups, n=6). The possible interference of brain edema with infarct volume was corrected by standard methods (whole contralateral hemisphere volume-nonischemic ipsilateral hemisphere volume) and the infracted volume was expressed as a percentage of the whole contralateral hemisphere.13
Spectrophotometric Assay of Hemoglobin
Rats were anesthetized and subjected to complete transcardial perfusion to remove intravascular blood. The Brain was minced and homogenized in 0.1M PBS to give 10% (w/v) homogenates. The homogenates were then made into aliquots and used for the assessment of hemoglobin and malondialdehyde (MDA) (DX+MCAO+O2, n=5; other groups, n=6).
Cerebral hemorrhage was quantified with a previously described spectrophotometric assay with some modifications.14 A standard curve was obtained using a “virtual” model of hemorrhage. Incremental volumes of homologous blood (0, 2, 4, 8, 16, 32 μl) were added to the perfused brain. After homogenization and centrifugation, Drabkin's reagent (1.6ml, Sigma) was added to 0.4ml supernatant aliquots and optical density was measured at 540nm with a spectrophotometer (Spectronix 3000, Milton-Roy, Rochester, NY, USA). Hemorrhage measurements of the samples were performed and compared with this standard curve to obtain data in terms of hemorrhage volume (μl).
Neurological Deficits
24 hours after MCAO, a neurologic examination was performed by a blinded investigator as previously described with modifications.15 The scores given to each rat at the completion of the evaluation was the summation of all seven individual test scores. (Supplement 2)
Brain Water Content
Animals were anesthetized and decapitated 24 hours after MCAO, as described in our previous study (DX+MCAO+ISO, n=6; other groups, n=5).12 Brain samples were immediately weighed on an electronic balance (model AE 100; Mettler Instrument) to obtain wet weight and then dried in a gravity oven at 120°C for 48 hours to obtain the dry weight. Brain water content was then calculated as (wet weight – dry weight)/ wet weight.
Measurement of Malondialdehyde Content
Malondialdehyde (MDA) content was measured with the thiobarbituric acid (TBA) reaction.16 The method was used to obtain a spectrophotometric measurement of the color produced during the reaction of TBA with MDA at 535nm; estimated MDA level was expressed as nmol/mg-protein.
Western Blot
The brain samples were collected at 24h after MCAO (DX+Sham, n=6; DX+MCAO, n=7; DX+MCAO+ISO, n=8; DX+MCAO+O2, n=7). Proteins of the ipsilateral hemisphere were extracted by homogenizing in 0.1M PBS. Western blotting was performed as described previously using mouse anti-4-Hydroxy-2-Nonenal (4-HNE) (ab48506) and mouse antibodies anti-3-Nitrotyrosine (sc-32757). 12
Activities of Superoxide dismutase (SOD) and Catalase (CAT)
The brain tissue supernatants came from the same for western blot and the blood samples from the heart before killed (n=6 for each group). SOD activity was measured following the reduction of nitrite by a xanthine–xanthine oxidase system (ab65354), and CAT activity was assayed by measuring absorbance at 570 nm according to the protocol (ab83464).
Statistical Analyses
Data were expressed as the mean±SEM. Statistical differences among groups were analyzed by using one-way analysis of variance followed by the Turkey method. Mortality was analyzed by fisher's exact test. P<0.05 was considered statistically significant.
Results
Physiological Data
The glucose levels in all groups during surgery were significantly higher than the level at the time point of dextrose injection. Isoflurane had no effects on the blood glucose level (Figure 1).
Figure 1.
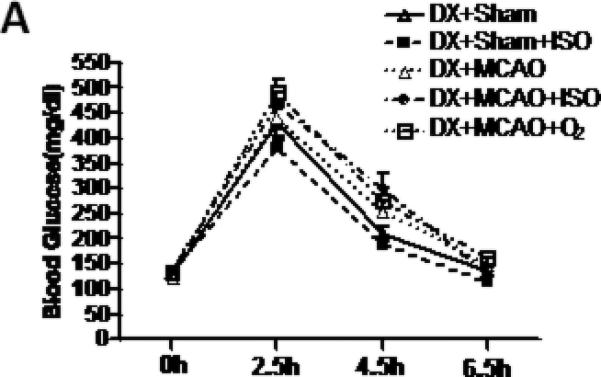
Statistic analysis of blood glucose levels. Isoflurane had no effect on blood glucose at the 4 time points (DX+Sham, n=7; other groups, n=12).
Isoflurane Deteriorated the Outcomes of Hyperglycemia Induced HT 24 hours after MCAO
Hyperglycemia induced extensive HT in ischemic territories in MCAO rats 24 hours after ischemia (Figure 2A). Compared with the vehicle, isoflurane post-treatment for 1h significantly exacerbated the infarction ratio (from 0.33±0.01 to 0.44±0.02, P<0.05) and hemorrhage volume (from 6.29±0.35 to 0.7.44±0.56μl, P<0.05). In addition, in vehicle group, 45% O2 significantly reduced the hemorrhage volume (from 6.29±0.35 to 4.28±0.61μl, P<0.05) when compared with DX+MCAO group (Figure 2).
Figure 2.
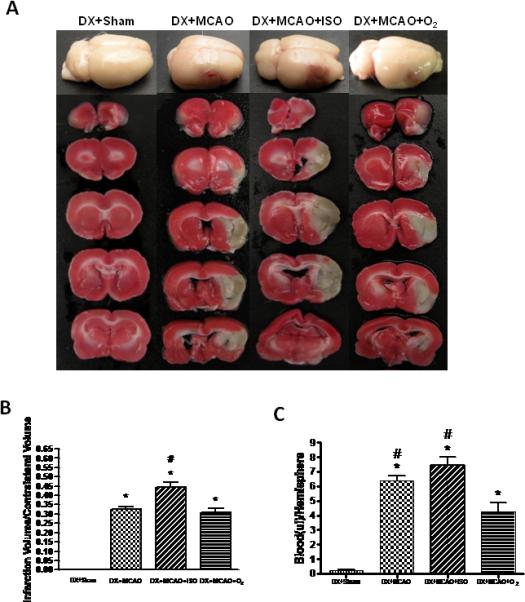
Whole brains and brain slices with TTC staining (A), and quantitative analysis of infarct volume (B) and hemorrhage volume (C) 24h after MCAO in all groups. Isoflurane significantly enlarged the infarct volume and hemorrhage volume compared with the vehicle group. Values are the mean ± SEM. *P<0.05 versus DX+Sham; #P<0.05 versus DX+MCAO+O2.
The mortality was also higher in Isoflurane post-treatment group than in the others, which were showed as follows respectively: DX+Sham, 0% (0/23); DX+MCAO, 22.58% (7/31); DX+MCAO+ISO, 33.33% (14/42) and DX+MCAO+O2, 17.86% (5/28) (Figure 3A). Neurological deficits were worsened 24 hours after MCAO. Animals in the DX+MCAO+ISO group exhibited more severe neurobehavioral deficits compared to the vehicle group (9. 91±0.46 vs. 13.39±0.50) (P<0.05), but not DX+MCAO group (Figure 3B). In addition, isoflurane increased the brain water content in the ischemic hemisphere at 24h after MCAO compared with the vehicle group (0.8284±0.0028 vs. 0.8102±0.0016, P<0.05) (Figure 3C).
Figure 3.
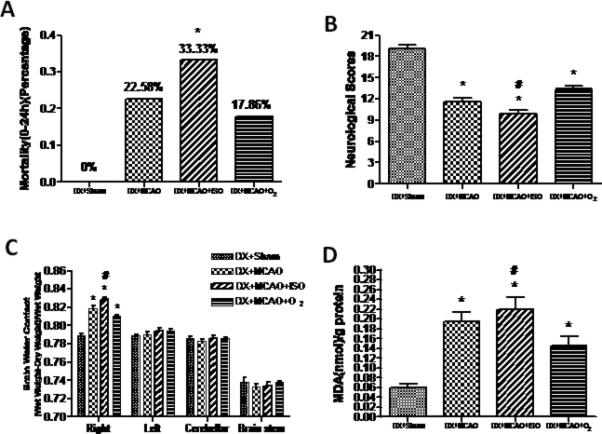
Statistic analysis of mortality (A), neurobehavioral deficits (B), brain water content (C) and MDA content (D). Animals in the DX+MCAO+ISO group exhibited higher mortality, more severe neurobehavioral deficits, increased brain water content and MDA content compared to DX+MCAO+O2 group. Values are the mean ± SEM. *P<0.05 versus DX+Sham; #P<0.05 versus DX+MCAO+O2.
Taken together, isoflurane post-treatment increased the infarct, hemorrhage volume, brain edema and mortality, and worsened the neurobehavioral deficits in the hyperglycemia induced HT model in rats when compared to the vehicle group. And the same results were also observed in the female rats (data not shown).
Isoflurane Increased the Oxidative Stress and Impaired the Activities of SOD and CAT in Hyperglycemia rats after MCAO
The MDA content was greatly increased in the ischemic brain tissue 24 h after MCAO. The content in isoflurane treated group was much higher than that in DX+MCAO+ O2 group (0.219±0.024 vs. 0.145±0.019 nmol/g protein, P<0.05) (Figure 3D). Consistent with the MDA content, the western blot revealed a significant increase of 4-HNE (Figure 4A-B) and Nitrotyrosine (Figure 4C-D) at 24 h after ischemia, this high level of 4-HNE and Nitrotyrosine was enhanced by isoflurane post-treatment (P>0.05 vs. DX+MCAO; P<0.05 vs. DX+MCAO+O2) (Figure 4).
Figure 4.
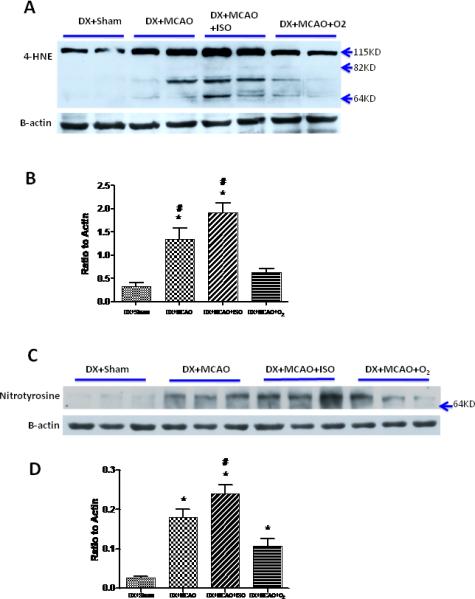
Representative western blots and quantitative analysis of 4-HNE (A and B) and Nitrotyrosine (C and D) in ischemic hemisphere of all groups. Isoflurane post-treatment intensified the expression of both 4-HNE and Nitrotyrosine compared with the vehicle group. Values are the mean ± SEM. *P<0.05 versus DX+Sham; #P<0.05 versus DX+MCAO+O2.
At 24 h after MCAO, SOD activity of the infarct area sharply decreased than that of DX+Sham group (123.4±3.81 vs. 144.8±5.46 U/mg-protein, P<0.05)(Figure 5A). Isoflurane post-treatment strongly decreased the SOD activity compared to the vehicle group in the brain (112.7±3.90 vs. 130.3±3.88 U/mg-protein, P<0.05) (Figure 5A) and in the blood plasma (11.06±0.65 vs. 16.54±0.69 U/mg-protein, P<0.05) (Figure 5B), but showed no statistic difference compared with DX+MCAO group. So was the activity of CAT in the brain tissues (6.79±0.82 vs. 14.31±0.89 U/g-protein, p<0.05) (Figure 5C) and in the blood plasma (5.38±0.63 vs. 10.00±1.15 U/g-protein, P>0.05) (Figure 5D).
Figure 5.
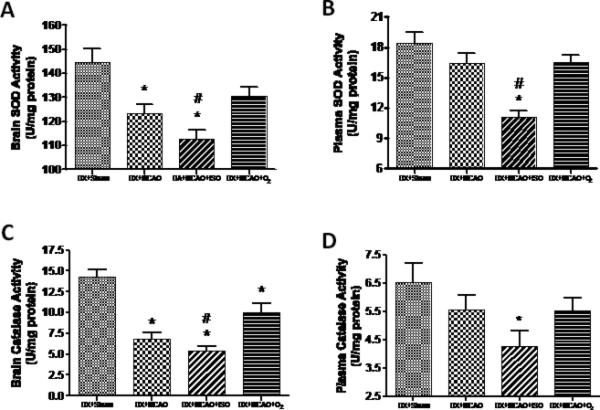
The activities of SOD and CAT in ischemic hemisphere (A and C) and blood plasma (B and D). Isoflurane post-treatment potently inhibited the activities of SOD and CAT both in brain tissue and plasma compared with DX+MCAO+O2 group. Values are the mean ± SEM. *P<0.05 versus DX+Sham; #P<0.05 versus DX+MCAO+O2.
Discussion
We did this study for two main reasons. First, hyperglycemia has been claimed to be strongly associated with HT in patients undergoing tPA therapy, which is the only effective and approved stroke treatment to date, this makes the study on hyperglycemia-induced HT after MCAO in rats is clinically relevant. Second, as one of the most widely used volatile anesthetics in surgical procedures, isoflurane has been shown to be neuroprotective in multiple animal works of ischemic brain injury,17 however, little attention has been given to the possible adverse effects caused by isoflurane or its metabolites. Therefore, before isoflurane is employed for neuroprotective purpose on stroke patients with hyperglycemia, it is imperative that its effects be examined during relevant circumstances.
In the current study, we found that 2% isoflurane, given at the onset of reperfusion for 1h, failed to reduce infarct, hemorrhage volume, or brain edema and failed to improve neurobehavioral deficits when compared with the ischemia/reperfusion group 24h after MCAO. In contrast, isoflurane exacerbated these indicators when compared with the vehicle group. Similar observations were made in female rats that isoflurane failed to reduce infarct and hemorrhage volume but increased mortality when compared with MCAO group (data not shown). These results were consistent with a previous study that post-ischemic isoflurane application after MCAO in baboons was associated with larger infarction, increased risk of hemorrhagic transformation and worse neurobehavioral scores.9 The potential mechanisms for the harmful effect of isoflurane were not studied in that study. Other studies however showed neurotoxicity effect of isoflurane in neonates and young children due to significant neurodegeneration as well as long-lasting post-operative cognitive decline in the elderly.18 In a similar cerebral ischemia/reperfusion injury model, neuroprotective effect of isoflurane was demonstrated in mild to moderate ischemic models, at lower isoflurane concentrations, and after shorter post-injury intervals.17 In these regards, it is plausible that in our experiment, hyperglycemia-induced HT enhanced ischemic injury that post-treatment with isoflurane resulted in adverse effects, although 2% isoflurane for 1h was popularly used in other models mediating neuroprotection.
How isoflurane deteriorated the outcomes of hyperglycemia-induced HT after focal cerebral ischemia? We have found that isoflurane had no effects on the level of blood glucose (Figure 2A), the inducer of hemorrhagic transformation in our model, which indicated that other perpetrators might play a major role. It had been shown that isoflurane induced the oxidative stress either by reducing the antioxidant defense mechanism19 or by generating toxic free radicals such as reactive oxygen species (ROS)20. Thereby, we sought to examine whether there was a role for oxidative stress in mediating the neurotoxicity of isoflurane post-treatment after hyperglycemia-induced HT. In this model, oxidative stress had been massively generated by hyperglycemia21 and ischemia/reperfusion injury22, which seriously disrupted the BBB and developed pronounced cerebral edema.2 It was predictable that further weakened cellular antioxidant defense systems would accelerate and widespread the damage caused by this overloaded oxidative stress. Oxidative stress usually comes from ROS and reactive nitrogen species (RNS), including hydroxyl radical (·OH), superoxide anion (O2-), hydrogen peroxide (H2O2), nitric oxide (NO), and peroxynitrite (ONOO-), leading to subsequent cell death by promoting DNA fragmentation, lipid oxidation, and protein nitrification.22
SOD and CAT are two representative endogenous antioxidant enzymes present in essentially every cell. SOD is considered fundamental in the process of eliminating ROS by reducing (adding an electron to) O2- to form H2O2, and CAT is responsible for decomposing H2O2 to H2O and O2, consequently, remove the deleterious free radicals. It has been demonstrated that reducing activities of SOD and CAT exacerbated neuronal cell injury and/or edema formation after transient focal cerebral ischemia.23-24 In this study, we found that isoflurane increased the expressions of MDA, HNE and Nitrotyrosine, which are markers of lipid peroxidation (MDA and 4-HNE) and protein nitrification (Nitrotyrosine), when compared with the vehicle group. We further observed that the activities of SOD and CAT enzymes in ischemic brain tissue were decreased by isoflurane post-treatment. Therefore, we conceptualize that isoflurane deteriorated the outcomes of hyperglycemia-induced HT after MCAO, possibly by enhancing oxidative stress via impaired activities of SOD and CAT. In 1999, Durak et al. found that exposure to 2% isoflurane for 30 min at three consecutive days could induce renal toxicity in guinea pig by reducing the activity of SOD and CAT. The authors suggested that the increased fluoride concentration, which was one of the metabolites of isoflurane and one of the most electronegative elements, might inhibit the activity of antioxidant enzymes through making complexes with their cofactor metals such as copper (Cu) and zinc (Zn).25 Kenna and Jones also noted that inorganic fluoride from inhaled fluorinated anesthetics might impair antioxidant enzymes by binding tightly to metal cations.26 A recent study verified that 2% isoflurane anesthesia in patients decreased the plasma zinc levels, erythrocyte SOD and glutathione peroxidase (GSH-Px) activities and trace element levels at 24h.19 Therefore it is highly likely that increased fluoride concentration due to isoflurane post-treatment might impair antioxidant defense system by making powerful metal complexes.
One unexpected result from this study was that the vehicle group, in which the oxygen concentration was about 45% and delivered at 2h after MCAO, ameliorated outcomes, including mild infarct and hemorrhage volume, lowered brain edema and mortality, as well as improved neurobehavioral deficits when compared with the ischemia/reperfusion group, consistent with the view that normobaric oxygen (NBO) has neuroprotective effects in acute ischemia stroke and is useful in extending the narrow time window for patients in clinical settings.27 There was one weakness in our experiment that we didn't monitor the physiological parameters such as arterial partial pressure of oxygen (PaO2), arterial partial pressure of carbon dioxide (PaCO2) and mean arterial blood pressure (MABP) for the technical constraints (rats were treated in a sealed chamber). In isoflurane treated group and the vehicle group, the hyperoxic could increase PaO2 and MABP through vasoconstriction,28 which may have an acute influence on the effects of hyperglycemia.
Overall, we concluded that isoflurane post-treatment in hyperglycemia-induced HT after ischemia stroke failed to demonstrate any protective value, but exacerbated the outcomes by impairing the antioxidant defense system. The current findings implied that isoflurane may not an appropriate anesthetic for the stroke patients with hyperglycemia.
Supplementary Material
Acknowledgments
Sources of Funding
This study was supported partially by grants from NIH NS053407 and NS054685 to J.H.Zhang, and NS060936 to J.Tang.
Footnotes
These authors contributed equally to this work.
Disclosures
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Paciaroni M, Agnelli G, Caso V, Corea F, Ageno W, Alberti A, Lanari A, Micheli S, Bertolani L, Venti M, Palmerini F, Billeci AM, Comi G, Previdi P, Silvestrelli G. Acute hyperglycemia and early hemorrhagic transformation in ischemic stroke. Cerebrovasc Dis. 2009;28:119–123. doi: 10.1159/000223436. [DOI] [PubMed] [Google Scholar]
- 2.Kamada H, Yu F, Nito C, Chan PH. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats: Relation to blood-brain barrier dysfunction. Stroke. 2007;38:1044–1049. doi: 10.1161/01.STR.0000258041.75739.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garg R, Chaudhuri A, Munschauer F, Dandona P. Hyperglycemia, insulin, and acute ischemic stroke: A mechanistic justification for a trial of insulin infusion therapy. Stroke. 2006;37:267–273. doi: 10.1161/01.STR.0000195175.29487.30. [DOI] [PubMed] [Google Scholar]
- 4.Newberg LA, Michenfelder JD. Cerebral protection by isoflurane during hypoxemia or ischemia. Anesthesiology. 1983;59:29–35. doi: 10.1097/00000542-198307000-00006. [DOI] [PubMed] [Google Scholar]
- 5.Lee JJ, Li L, Jung HH, Zuo Z. Postconditioning with isoflurane reduced ischemia-induced brain injury in rats. Anesthesiology. 2008;108:1055–1062. doi: 10.1097/ALN.0b013e3181730257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu W, Wang L, Zhang L, Palmateer JM, Libal NL, Hurn PD, Herson PS, Murphy SJ. Isoflurane preconditioning neuroprotection in experimental focal stroke is androgen-dependent in male mice. Neuroscience. 2010;169:758–769. doi: 10.1016/j.neuroscience.2010.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou Y, Lekic T, Fathali N, Ostrowski RP, Martin RD, Tang J, Zhang JH. Isoflurane posttreatment reduces neonatal hypoxic-ischemic brain injury in rats by the sphingosine-1-phosphate/phosphatidylinositol-3-kinase/akt pathway. Stroke. 2010;41:1521–1527. doi: 10.1161/STROKEAHA.110.583757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li L, Zuo Z. Isoflurane preconditioning improves short-term and long-term neurological outcome after focal brain ischemia in adult rats. Neuroscience. 2009;164:497–506. doi: 10.1016/j.neuroscience.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nehls DG, Todd MM, Spetzler RF, Drummond JC, Thompson RA, Johnson PC. A comparison of the cerebral protective effects of isoflurane and barbiturates during temporary focal ischemia in primates. Anesthesiology. 1987;66:453–464. doi: 10.1097/00000542-198704000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Warner DS, Deshpande JK, Wieloch T. The effect of isoflurane on neuronal necrosis following near-complete forebrain ischemia in the rat. Anesthesiology. 1986;64:19–23. doi: 10.1097/00000542-198601000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Warner DS, Zhou JG, Ramani R, Todd MM. Reversible focal ischemia in the rat: Effects of halothane, isoflurane, and methohexital anesthesia. J Cereb Blood Flow Metab. 1991;11:794–802. doi: 10.1038/jcbfm.1991.137. [DOI] [PubMed] [Google Scholar]
- 12.Hu Q, Chen C, Yan J, Yang X, Shi X, Zhao J, Lei J, Yang L, Wang K, Chen L, Huang H, Han J, Zhang JH, Zhou C. Therapeutic application of gene silencing mmp-9 in a middle cerebral artery occlusion-induced focal ischemia rat model. Exp Neurol. 2009;216:35–46. doi: 10.1016/j.expneurol.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 13.Swanson RA, Sharp FR. Infarct measurement methodology. J Cereb Blood Flow Metab. 1994;14:697–698. doi: 10.1038/jcbfm.1994.88. [DOI] [PubMed] [Google Scholar]
- 14.Choudhri TF, Hoh BL, Solomon RA, Connolly ES, Jr., Pinsky DJ. Use of a spectrophotometric hemoglobin assay to objectively quantify intracerebral hemorrhage in mice. Stroke. 1997;28:2296–2302. doi: 10.1161/01.str.28.11.2296. [DOI] [PubMed] [Google Scholar]
- 15.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26:627–634. doi: 10.1161/01.str.26.4.627. discussion 635. [DOI] [PubMed] [Google Scholar]
- 16.Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 17.Kitano H, Kirsch JR, Hurn PD, Murphy SJ. Inhalational anesthetics as neuroprotectants or chemical preconditioning agents in ischemic brain. J Cereb Blood Flow Metab. 2007;27:1108–1128. doi: 10.1038/sj.jcbfm.9600410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei H, Xie Z. Anesthesia, calcium homeostasis and alzheimer's disease. Curr Alzheimer Res. 2009;6:30–35. doi: 10.2174/156720509787313934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turkan H, Bukan N, Sayal A, Aydin A, Bukan MH. Effects of halothane, enflurane, and isoflurane on plasma and erythrocyte antioxidant enzymes and trace elements. Biol Trace Elem Res. 2004;102:105–112. doi: 10.1385/BTER:102:1-3:105. [DOI] [PubMed] [Google Scholar]
- 20.Kim H, Oh E, Im H, Mun J, Yang M, Khim JY, Lee E, Lim SH, Kong MH, Lee M, Sul D. Oxidative damages in the DNA, lipids, and proteins of rats exposed to isofluranes and alcohols. Toxicology. 2006;220:169–178. doi: 10.1016/j.tox.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 21.Li PA, Liu GJ, He QP, Floyd RA, Siesjo BK. Production of hydroxyl free radical by brain tissues in hyperglycemic rats subjected to transient forebrain ischemia. Free Radic Biol Med. 1999;27:1033–1040. doi: 10.1016/s0891-5849(99)00152-5. [DOI] [PubMed] [Google Scholar]
- 22.Sugawara T, Chan PH. Reactive oxygen radicals and pathogenesis of neuronal death after cerebral ischemia. Antioxid Redox Signal. 2003;5:597–607. doi: 10.1089/152308603770310266. [DOI] [PubMed] [Google Scholar]
- 23.Kondo T, Reaume AG, Huang TT, Carlson E, Murakami K, Chen SF, Hoffman EK, Scott RW, Epstein CJ, Chan PH. Reduction of cuzn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J Neurosci. 1997;17:4180–4189. doi: 10.1523/JNEUROSCI.17-11-04180.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gu W, Zhao H, Yenari MA, Sapolsky RM, Steinberg GK. Catalase over-expression protects striatal neurons from transient focal cerebral ischemia. Neuroreport. 2004;15:413–416. doi: 10.1097/00001756-200403010-00006. [DOI] [PubMed] [Google Scholar]
- 25.Durak I, Ozturk HS, Dikmen B, Guven C, Cimen MY, Buyukkocak S, Kacmaz M, Avci A. Isoflurane impairs antioxidant defence system in guinea pig kidney. Can J Anaesth. 1999;46:797–802. doi: 10.1007/BF03013919. [DOI] [PubMed] [Google Scholar]
- 26.Kenna JG, Jones RM. The organ toxicity of inhaled anesthetics. Anesth Analg. 1995;81:S51–66. doi: 10.1097/00000539-199512001-00008. [DOI] [PubMed] [Google Scholar]
- 27.Liu W, Hendren J, Qin XJ, Shen J, Liu KJ. Normobaric hyperoxia attenuates early blood-brain barrier disruption by inhibiting mmp-9-mediated occludin degradation in focal cerebral ischemia. J Neurochem. 2009;108:811–820. doi: 10.1111/j.1471-4159.2008.05821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waring WS, Thomson AJ, Adwani SH, Rosseel AJ, Potter JF, Webb DJ, Maxwell SR. Cardiovascular effects of acute oxygen administration in healthy adults. J Cardiovasc Pharmacol. 2003;42:245–250. doi: 10.1097/00005344-200308000-00014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.