

Abstract
Bradykinin stimulation of B2 kinin receptors has been shown to promote matrix metalloproteinase (MMP) secretion from trabecular meshwork cells and to increase conventional outflow facility. Because acute secretion of MMPs can be dependent on the activity of extracellular signal-regulated MAP kinases (ERK1/2), experiments were performed to determine bradykinin effects on ERK1/2 in cultured human trabecular meshwork cells and the relationship of these effects to MMP-9 release. Treatment of cells with bradykinin produced a rapid 4-to 6-fold increase in ERK1/2 phosphorylation. Stimulation of ERK1/2 activity peaked within 2 min and then declined to control levels by 60 min. The response maximum occurred with 100 nM bradykinin and the estimated EC50 was 0.7 nM. Treatment of cells with the B2 kinin receptor agonist, Tyr8- bradykinin, also stimulated ERK1/2 phosphorylation while the B1 agonist, Lys-[Des-Arg9]- bradykinin had no significant effect. In addition, activation of ERK1/2 by bradykinin or Tyr8- bradykinin was blocked by the selective B2 receptor antagonist, Hoe-140. Inhibition of MAP kinase kinase (MEK) with U0126 also blocked bradykinin-induced ERK1/2 phosphorylation. Suppression of protein kinase C activity with the nonselective inhibitor, GF109203X, or by down-regulation with phorbol ester, diminished, but did not eliminate, bradykinin activation of ERK1/2. A similar decrease of ERK1/2 stimulation was observed when Src kinase was inhibited by 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2). Finally, blockade of bradykinin-induced ERK1/2 activation substantially reduced the peptide's action to stimulate MMP-9 release into the extracellular environment. The data demonstrate that bradykinin promotes ERK1/2 activation in human trabecular meshwork cells. The effect is mediated by B2 kinin receptors, involves two different signaling pathways, and results in increased secretion of MMP-9.
Keywords: trabecular meshwork cell, ERK1/2, bradykinin, B2 receptor, MMP-9
1. Introduction
The trabecular meshwork is a specialized region of the ocular anterior chamber consisting of connective tissue beams, trabecular meshwork cells and extracellular matrix (Rohen and Lutjen-Drecoll, 1989). The primary pathway for aqueous outflow from the anterior chamber is through the trabecular meshwork into Schlemm's canal and the venous circulation. The complex matrix and cellular network of the trabecular region provides the principal resistance to the outflow of aqueous humor (Bill and Svedberg, 1972; Ethier et al., 1986) and, consequently, is a major determinant of intraocular pressure. Tissue components for production and inactivation of kinin peptides have been identified within the human anterior segment and ciliary body (Ma et al., 1996; Webb et al., 2009). Tissue kallikrein, a serine protease, is the principal enzyme for kinin generation by local cells and tissues (Bhoola et al., 1992). This enzyme has been shown to be expressed by the non-pigmented epithelium of the ciliary body as well as by ciliary smooth muscle and trabecular meshwork cells. Each of these cell types also express both B1 and B2 kinin receptors and represent targets for the actions of locally generated peptide. In addition, both ciliary muscle and trabecular cells possess kininases for rapid degradation and termination of kinin actions (Webb et al., 2009). Kininogen, the substrate for tissue kallikrein, does not appear to be produced by tissues of the anterior segment but is, nonetheless, present in aqueous humor (Chowdhury et al., 2010). The protein, presumably, is delivered into the angle of the anterior chamber by a documented pathway for diffusional movement of proteins from plasma into the aqueous compartment (Barsotti et al., 1992; Bert et al., 2006). Such data support the concept that kinins produced locally within the anterior segment may act in an autocrine/paracrine fashion to influence multiple cell types and actively participate in the regulation of anterior segment function.
Stimulation of B2 kinin receptors by bradykinin has been shown to activate multiple signaling events in both human and bovine trabecular meshwork cells (Sharif and Xu, 1996; Llobet et al., 1999; Webb et al., 2003), and also to promote secretion of the constitutively-expressed matrix metalloproteinase, MMP-9 (Webb et al., 2006). Further, stimulation of MMP secretion by bradykinin has been associated with an action to enhance conventional outflow facility in perfused anterior segments (Webb et al., 2006). Secretion and activation of constitutively-expressed MMPs such as MMP-2 and MMP-9 is commonly regulated in a variety of cell types through signaling pathways involving mitogen activated protein (MAP) kinases (Shearer and Crosson, 2002; Liu et al., 2002; Husain et al., 2005; Okada et al., 2010). Consequently, experiments were performed in the present study to determine the effects of bradykinin on MAP kinase activities in human trabecular meshwork cells, and to evaluate the relationship of such effects to the secretion of MMP-9. Bradykinin was found to act on B2 kinin receptors in trabecular cells to rapidly and selectively promote ERK1/2 activation. Promotion of ERK1/2 activity by bradykinin utilized at least two different upstream signaling pathways and was a requirement for kinin-induced MMP-9 release.
2. Materials and methods
2.1. Reagents
Dulbecco's modified Eagle's medium (DMEM) was purchased from GIBCO-BRL (Grand Island, NY, USA) and fetal bovine serum from Invitrogen (Carlsbad, CA, USA). Penicillin, streptomycin, and trypsin were acquired from Mediatech, Inc (Manassas, VA, USA). Bradykinin, Hoe-140, GF109203X, and phorbol 12-myristate 13-acetate (PMA) were from Sigma (St. Louis, MO, USA). Lys-[Des-Arg9]-bradykinin and [Tyr8]-bradykinin were obtained from American Peptide (Vista, CA, USA). U0126 and PP2 were purchased from Calbiochem (San Diego, CA, USA). Polyclonal antibodies to ERK 1/2 and phospho ERK 1/2 and p38 MAP kinases were obtained from Cell Signaling Technologies (Danvers, MA). Antibodies to JNK were from Promega (Madison, WI, USA) and polyclonal anti-MMP-9 antibodies were purchased from Fitzgerald (Concord, MA, USA).
2.2. Cell culture
Human tissue was handled in accordance with the Declaration of Helsinki. Experiments were performed with primary cultures of trabecular meshwork cells isolated from normal human eyes. Human eyes were obtained from the National Disease Research Interchange (Philadelphia, PA, USA) and Life-Point Ocular Tissue Division (Storm Eye Institute; MUSC, Charleston, SC, USA). The donor age averaged 57 ± 6 years. Primary cultures of trabecular meshwork cells were isolated as performed previously (Webb et al., 2003). Once established in culture, cells were maintained on polypropylene cell culture plates in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum, 100 U/mL penicillin and 0.1 mg/mL streptomycin in a humidified atmosphere of 5% CO2 in air at 37°C. The medium was routinely changed every 48 hr and cells were subcultured weekly after detachment with 0.05% trypsin in phosphate-buffered saline. Cells from passages 3 through 6 were used for studies. At least 2 different cell populations were evaluated in each series of experiments.
2.3. Extracellular signal-regulated kinase (ERK1/2)
Cells were cultured in serum-free medium for 16 hr before the addition of any test agent. Unless otherwise noted, cells were treated with bradykinin or bradykinin analogues for 2 min. In experiments evaluating effects of the B2 kinin receptor antagonist, Hoe-140 (100 nM), the protein kinase C inhibitor, GF109203X (1 μM), the MEK inhibitor, U0126 (1 μM), or the Src tyrosine kinase inhibitor, PP2 (15 μM), cells were pretreated for 30 min with inhibitor prior to the addition of bradykinin. In experiments evaluating the effects of protein kinase C down-regulation, cells were pretreated with phorbol 12-myristate 13-acetate (PMA, 1 μM) for 24 hr before bradykinin treatment. At the end of the incubation periods, cells were rinsed with ice-cold phosphate-buffered saline and lysed by the addition of lysis buffer (50 mM Tris-HCl buffer, pH 8.0, containing 100 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 0.1% SDS, 0.5% deoxycholate, 50 mM NaF, 1 mM Na3VO4, 5 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, and 50 μg/ml aprotinin) for 20 min on ice. To determine the level of ERK1/2 activation (phosphorylation), equivalent amounts of protein (15 μg) were loaded onto 10% SDS-polyacrylamide gels, and proteins were separated according to molecular weight using standard SDS-polyacrylamide gel electrophoresis protocols and transferred to a nitrocellulose membrane. The membranes were then probed with anti-phospho-ERK1/2 antibodies overnight at 4°C. Bands were visualized by the addition of anti-rabbit HRP-conjugated secondary antibodies (at 1:3000) and ECL reagents. Blots were then stripped by incubation in stripping buffer (62.5 mM Tris-HCl, pH 6.7, 100 mM β-mercaptoethanol, and 2% SDS) for 30 min at 50°C, and total ERK levels (phosphorylated and nonphosphorylated forms) were determined by immunoblot techniques using polyclonal anti-ERK1/2 antibodies. Band densities were quantified with a BioRad Versa Doc Imaging System (Bio-Rad Laboratories). To determine effects of bradykinin on other MAP kinases, cell lysates were prepared as described above, subjected to gel electrophoresis and probed with antibodies to JNK or p38 MAP kinase.
2.4. MMP-9
For experiments on acute regulation of MMP-9 secretion, equal numbers of human trabecular meshwork cells were grow to confluence in 100 mm culture dishes. Confluent cultures were then maintained in serum-free medium for 16 hr prior to initiation of the experiments. Cells were treated with bradykinin (100 nM) or U0126 (1 μM), alone and in combination, for 2 hr. Control cells received vehicle only. At the end of the 2-hr experimental period, the media was collected, concentrated to a standard volume (200 μl) using a Centricon concentrator (Millipore, Corp., Bedford, MA), and equal volumes of concentrate analyzed for MMP-9 by Western blotting. In each experiment, culture plates were evaluated to determine if the treatment induced any changes in cell attachment or morphology. Equivalent volumes of concentrate were loaded onto 10% SDS-polyacrylamide gels, and proteins were separated according to molecular weight using standard SDS-polyacrylamide gel electrophoresis protocols and transferred to a nitrocellulose membrane. The membranes were then probed with anti-MMP-9 antibodies overnight at 4°C as performed previously (Webb et al., 2006). Bands were visualized by the addition of anti-mouse HRP-conjugated secondary antibodies (at 1:3000) and ECL reagents. The band intensities were quantified by densitometry as described above. Purified MMP-9 was run in parallel as a positive control to identify the MMP-9 band.
2.5. Data Analysis
Data are presented as means ± S.E. and were analyzed using analysis of variance followed by Neuman-Keuls multiple comparison test for detecting differences, with P < 0.05 considered as significant. The number of observations reported represents the number of individual experiments conducted.
3. Results
3.1. Bradykinin activation of ERK1/2 in cultured human trabecular meshwork cells
Incubation of human trabecular meshwork cells with bradykinin produced a rapid activation of ERK1/2 as measured by increased levels of phosphorylated products (Fig. 1). Erk1/2 phosphorylation was increased four to six-fold relative to basal activity by bradykinin (10 nM) treatment. Activation of ERK1/2 peaked within 2 to 10 min of kinin exposure, and then declined to control levels by 60 min. For most experiments, trabecular meshwork cells were treated with peptide for 2 min. Bradykinin activation of ERK1/2 occurred in a concentration-dependent manner (Fig. 2). The response maximum was achieved with 100 nM bradykinin and the EC50 for the effect was approximately 0.7 nM. To determine the subtype of kinin receptor that mediated stimulation of ERK1/2 activity, cells were treated with the selective B2 receptor agonist, Tyr8-bradykinin, or the B1 agonist, Lys-[Des-Arg9]-bradykinin. Incubation of cells with Tyr8-bradykinin produced a rapid increase in phosphorylated ERK1/2 that was equivalent in magnitude to that observed for bradykinin (Fig. 3). In comparison, Lys-[Des-Arg9]-bradykinin failed to promote ERK1/2 activation. Further, pretreatment of trabecular meshwork cells with the selective B2 receptor antagonist, Hoe-140, inhibited ERK1/2 activation by both bradykinin and Tyr8-bradykinin (Fig. 3). In comparable experiments, treatment of human trabecular cells with bradykinin was found to have no effect on phosphorylation of either JNK or p38 MAP kinase (data not shown).
Fig. 1.
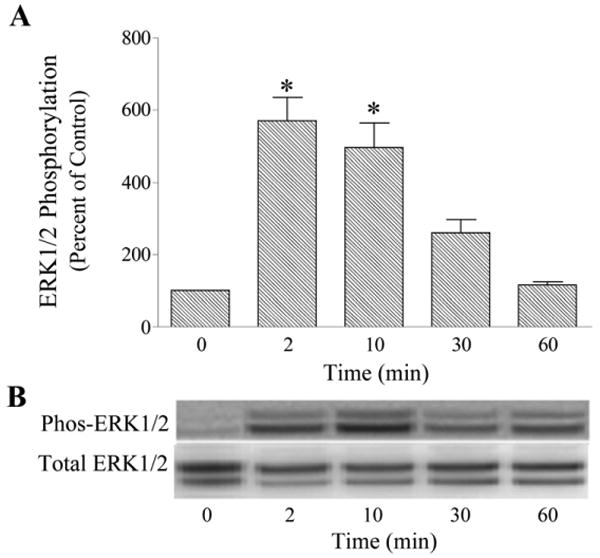
Time-course of bradykinin activation of ERK1/2 in cultured human trabecular meshwork cells. Serum-deprived trabecular meshwork cells were incubated with bradykinin (10 nM) for varying time periods. At the end of incubation, cell lysates were analyzed for phosphorylated ERK1/2 (Phos-ERK1/2) and total ERK1/2 by immunoblot using antibodies to phosphorylated ERK1/2 and total ERK1/2, respectively. A, values represent means ± S. E. of densitometry measurements from phosphorylation data in 5 to 8 experiments. * Differs from 0 time control at P< 0.05. B, representative immunoblot of phosphorylated and total ERK1/2 from trabecular meshwork cells incubated with bradykinin (10 nM) for varying time periods.
Fig. 2.
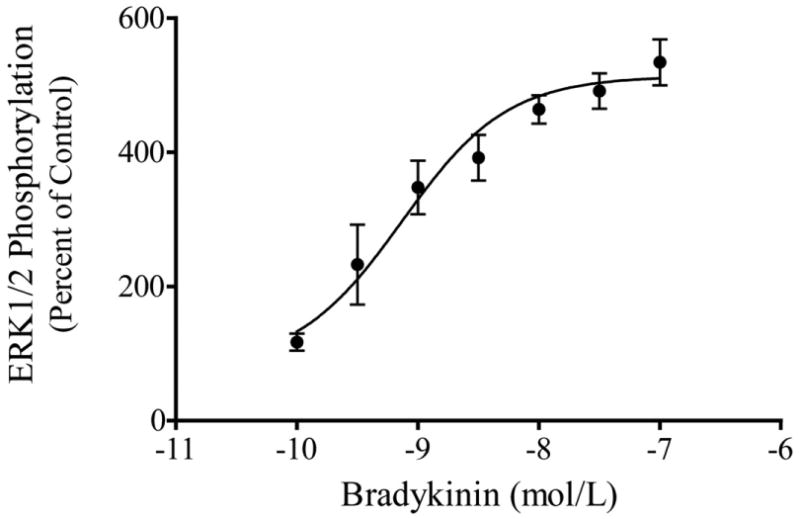
Concentration-dependent activation of ERK1/2 by bradykinin in cultured human trabecular meshwork cells. Serum-deprived trabecular meshwork cells were incubated with varying concentrations of bradykinin for 2 min. At the end of incubation, cell lysates were analyzed for phosphorylated ERK1/2 by immunoblot using antibodies to phosphorylated ERK1/2. Values represent means ± S. E. of densitometry measurements from phosphorylation data in 4 to 7 experiments.
Fig. 3.
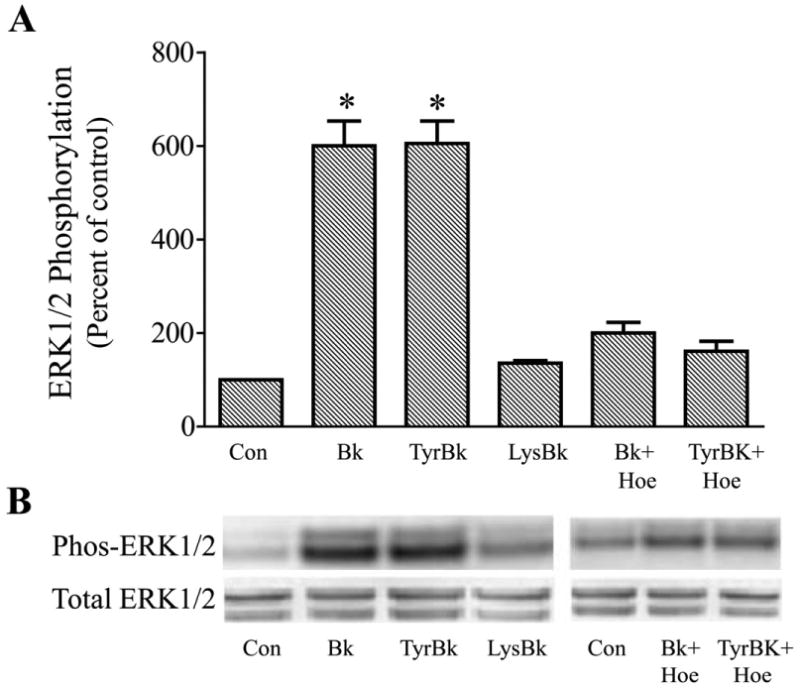
Effects of receptor-selective kinin agonists and antagonist on ERK1/2 activation in cultured human trabecular meshwork cells. Serum-deprived trabecular meshwork cells were incubated for 2 min with 10 nM bradykinin (Bk), Tyr8-bradykinin (TyrBk), or Lys-[Des-Arg9]-bradykinin (LysBk) alone or in combination with 100 nM Hoe-140 (Hoe). In experiments with Hoe-140, cells were pretreated with the antagonist for 30 min prior to addition of kinin agonist. At the end of incubation, cell lysates were analyzed for phosphorylated ERK1/2 (Phos-ERK1/2) and total ERK1/2 by immunoblot using antibodies to phosphorylated ERK1/2 and total ERK1/2, respectively. A, values represent means ± S. E. of densitometry measurements from phosphorylation data in 6 to 8 experiments. * Differs from control at P< 0.05. B, representative immunoblot of phosphorylated and total ERK1/2 from trabecular meshwork cells incubated with test agonist alone or in combination with the antagonist Hoe-140.
3.2. Role of protein kinases in bradykinin activation of ERK1/2
To determine if MAP kinase kinase (MEK) was involved in the signaling pathway for bradykinin activation of ERK1/2, trabecular meshwork cells were treated with the MEK inhibitor, U0126. In this series of experiments, bradykinin treatment increased phosphorylated ERK 1/2 by 380 ± 44 % above control. Pretreatment of cells for 30 min with U0126 (1 μM) reduced basal levels of phosphorylated ERK1/2 by approximately 90 %, and completely blocked activation of ERK1/2 by bradykinin (Fig. 4).
Fig. 4.
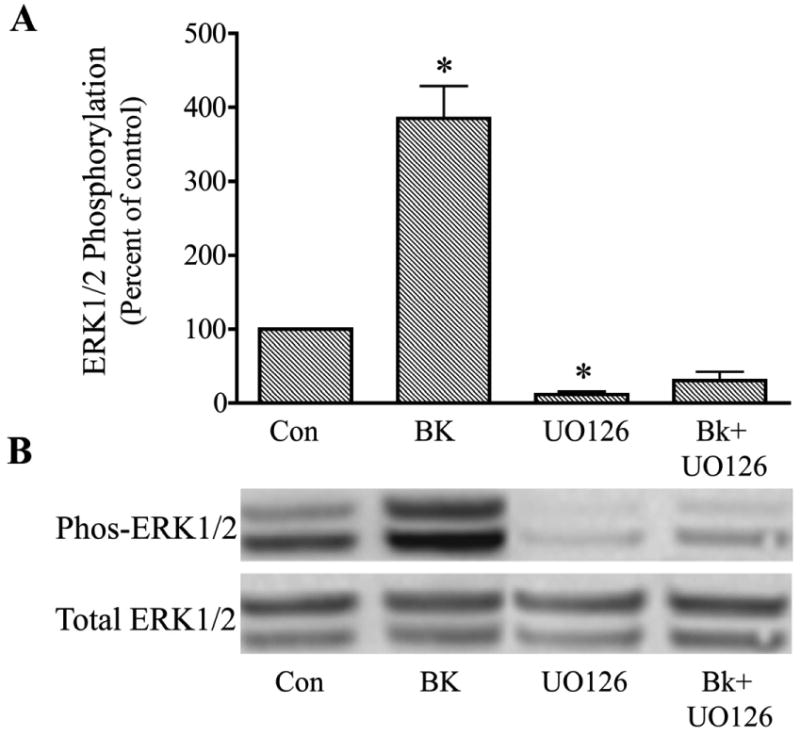
Inhibition of bradykinin (Bk) stimulation of ERK1/2 activation in cultured human trabecular meshwork cells by the MAP kinase kinase inhibitor, U0126. Serum-deprived trabecular meshwork cells were treated with 100 nM Bk, 1 uM U0126, or Bk + U0126. Cells were pretreated with vehicle or U0126 for 30 min prior to incubation with Bk for 2 min. At the end of incubation, cell lysates were analyzed for phosphorylated ERK1/2 (Phos-ERK1/2) and total ERK1/2 by immunoblot using antibodies to phosphorylated ERK1/2 and total ERK1/2, respectively. A, values represent means ± S. E. of densitometry measurements from phosphorylation data in 4 experiments. * Differs from control at P< 0.05. B, representative immunoblot of phosphorylated and total ERK1/2 from trabecular meshwork cells treated with Bk, U0126, or Bk + U0126.
To test the possibility that protein kinase C (PKC) had a role in the pathway for ERK1/2 stimulation, cells were either treated with the PKC inhibitor, GF109203X, or PKC was down-regulated by stimulation of cells for 24 hr with the phorbol ester, PMA (1μM), prior to incubation with bradykinin. With each approach, the effect of bradykinin to promote ERK1/2 activation was diminished, but not completely inhibited, by modification of PKC activity. In experiments in which trabecular cells were pretreated for 30 min with GF 109203X (1 μM), bradykinin alone increased phosphorylated ERK1/2 by 394 ± 8 % while bradykinin in the presence of PKC inhibitor increased phosphorylated ERK1/2 by only 252 ± 36%, a 36% reduction in response (P< 0.05 compared to bradykinin alone) (Fig. 5). Down-regulation of PKC with PMA had a similar effect. In this series, BK increased phosphorylated ERK1/2 by 633 ± 20% in control cells and 256 ± 23% in cells pretreated with PMA, a reduction in effect of 60% (P< 0.05 compared to bradykinin alone)(Fig. 6). Neither GF109203X nor PMA treatment significantly modified basal levels of phosphorylated ERK1/2.
Fig. 5.
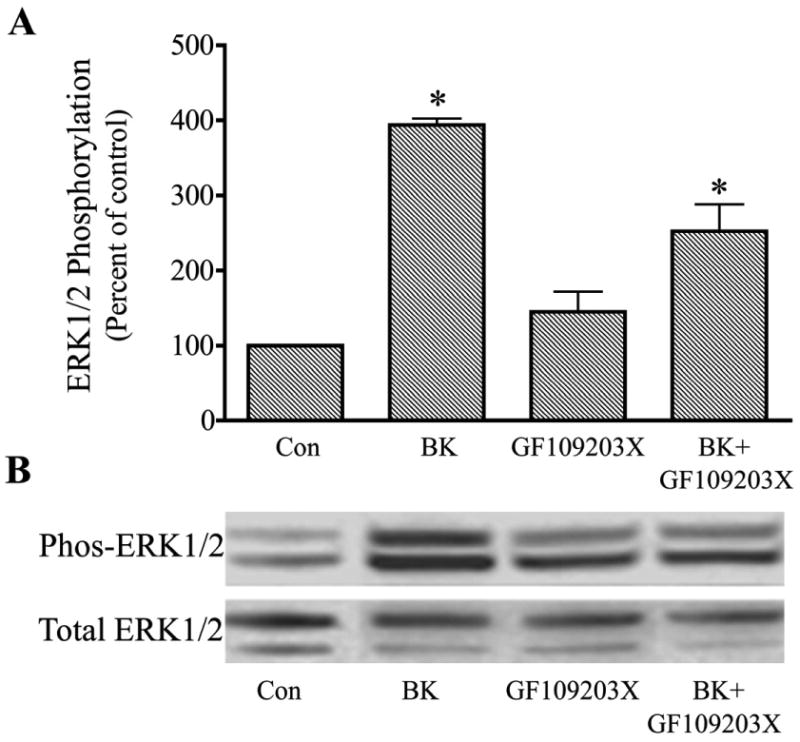
Attenuation of bradykinin (Bk) stimulation of ERK1/2 activation in cultured human trabecular meshwork cells by the protein kinase C inhibitor, GF109203X. Serum-deprived trabecular meshwork cells were treated with 100 nM Bk, 1 μM GF109203X, or Bk + GF109203X. Cells were pretreated with vehicle or GF109203X for 30 min prior to incubation with Bk for 2 min. At the end of incubation, cell lysates were analyzed for phosphorylated ERK1/2 (Phos-ERK1/2) and total ERK1/2 by immunoblot using antibodies to phosphorylated ERK1/2 and total ERK1/2, respectively. A, values represent means ± S. E. of densitometry measurements from phosphorylation data in 4 experiments. * Differs from control at P< 0.05. B, representative immunoblot of phosphorylated and total ERK1/2 from trabecular meshwork cells treated with Bk, GF109203X, or Bk + GF109203X.
Fig. 6.
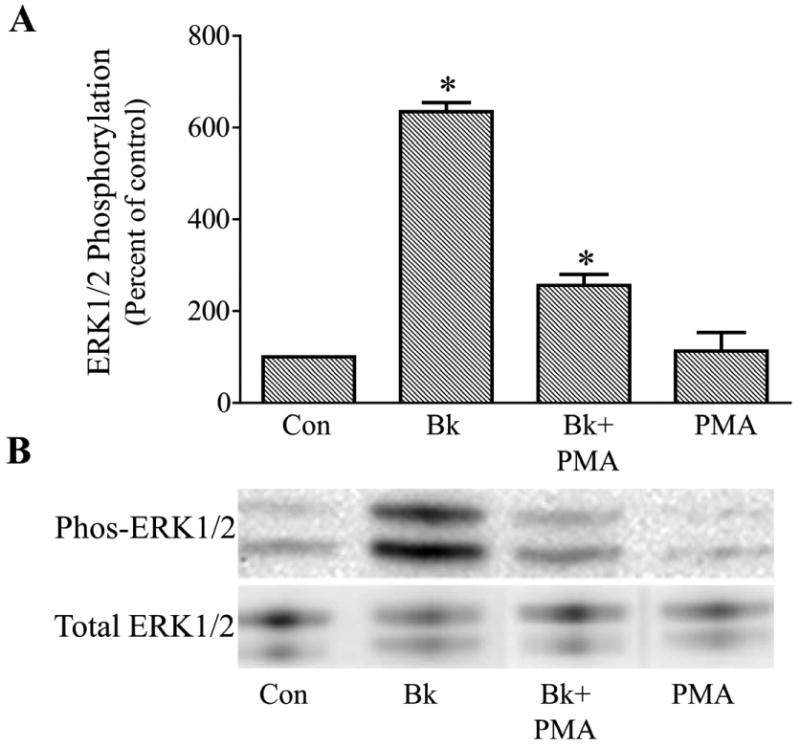
Attenuation of bradykinin (Bk) stimulation of ERK1/2 activation in cultured human trabecular meshwork cells by down-regulation of protein kinase C. Serum-deprived cells were treated for 24 hours with 1 μM PMA or vehicle prior to incubation with 100 nM Bk for 2 min. At the end of incubation, cell lysates were analyzed for phosphorylated ERK1/2 (Phos-ERK1/2) and total ERK1/2 by immunoblot using antibodies to phosphorylated ERK1/2 and total ERK1/2, respectively. A, values represent means ± S. E. of densitometry measurements from phosphorylation data in 6 experiments. * Differs from control at P< 0.05. B, representative immunoblot of phosphorylated and total ERK1/2 from trabecular meshwork cells treated with Bk, Bk following PMA treatment (Bk + PMA), or PMA alone.
An alternative mechanism for ERK1/2 activation through the MEK stimulation pathway is promotion of Src tyrosine kinase activity (Della-Rocca et al., 1997; Goldsmith and Dhanasekaran, 2007). Moreover, activation of Src tyrosine kinase activity has been associated with increased secretion of MMP-9 from some cell types (Cortes-Reynosa et al., 2008). To determine if Src kinase was involved in bradykinin actions in the human trabecular meshwork cell, cells were treated with the Src tyrosine kinase inhibitor, PP2. In these studies, bradykinin alone increased phosphorylated ERK1/2 496 ± 34 % in control cells. Pretreatment of trabecular cells with PP2 (15 μM) for 30 min appeared to lower basal levels of activated ERK1/2 but the effect was not statistically significant. However, bradykinin stimulation of phosphorylated ERK1/2 was diminished approximately 50% following PP2 to a value 263 ± 18% above the control level (P< 0.05 compared to bradykinin alone) (Fig. 7).
Fig. 7.
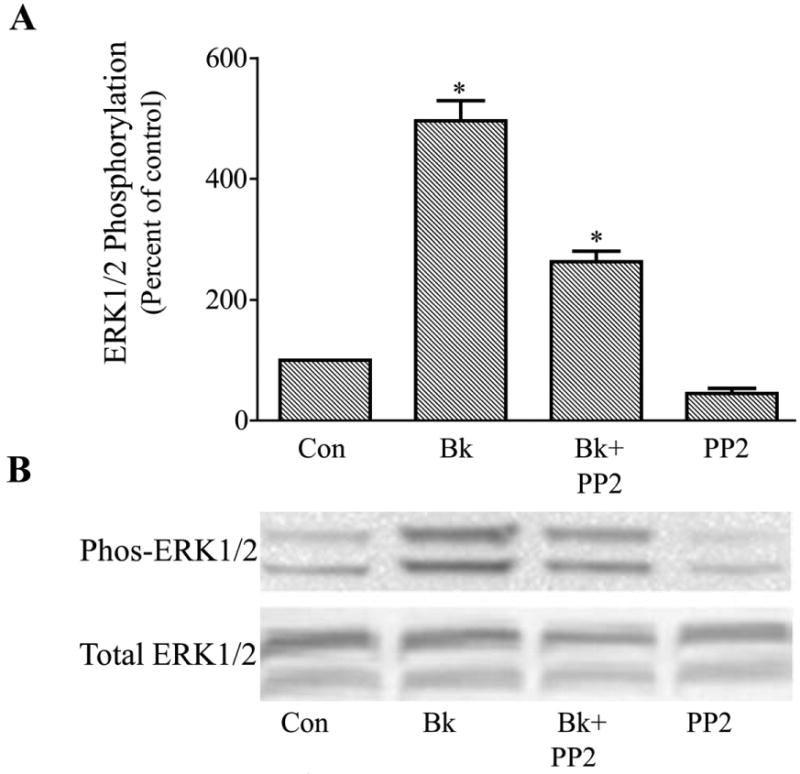
Attenuation of bradykinin (Bk) stimulation of ERK1/2 activation in cultured human trabecular meshwork cells by the Src tyrosine kinase inhibitor, PP2. Serum-deprived trabecular meshwork cells were treated with 100 nM Bk, 15 μM PP2, or BK + PP2. Cells were pretreated with vehicle or PP2 for 30 min prior to incubation with Bk for 2 min. At the end of incubation, cell lysates were analyzed for phosphorylated ERK1/2 (Phos-ERK1/2) and total ERK1/2 by immunoblot using antibodies to phosphorylated ERK1/2 and total ERK1/2, respectively. A, values represent means ± S. E. of densitometry measurements from phosphorylation data in 4 experiments. * Differs from control at P< 0.05. B, representative immunoblot of phosphorylated and total ERK1/2 from trabecular meshwork cells treated with Bk, Bk + PP2, or PP2 alone.
3.3. Bradykinin activation of ERK1/2 and MMP-9 release from trabecular meshwork cells
Acute secretion of constitutively expressed MMPs from trabecular meshwork cells has been observed with agonists that promote ERK1/2 activation (Shearer and Crosson, 2002; Liu et al., 2002; Husain et al., 2005; Okada et al., 2010) and bradykinin has been previously shown to stimulate MMP-9 release from bovine trabecular cells (Webb et al., 2006). Therefore, experiments were conducted to determine if bradykinin promotes MMP-9 secretion in association with ERK1/2 activation in human trabecular meshwork cells. For these studies, bradykinin effects on MMP-9 release into the extracellular media over 2 hr was examined in control cells and in cells pretreated for 30 min with the U0126 (1μM), which blocks ERK1/2 activation (Fig. 4). Treatment of cells with bradykinin produced almost a 6-fold increase in MMP-9 release into the extracellular environment in control human trabecular cells, and this increase in MMP-9 secretion was substantially reduced in cells treated with U0126 (Fig. 8). While there was a trend toward increased MMP-9 release in cells treated with U0126 alone, the value did not differ significantly from control levels in the 6 experiments performed. Most importantly, MMP-9 protein released in response to bradykinin in cells treated with U0126 did not differ from that released by cells treated with U0126 alone (Fig. 8).
Fig. 8.
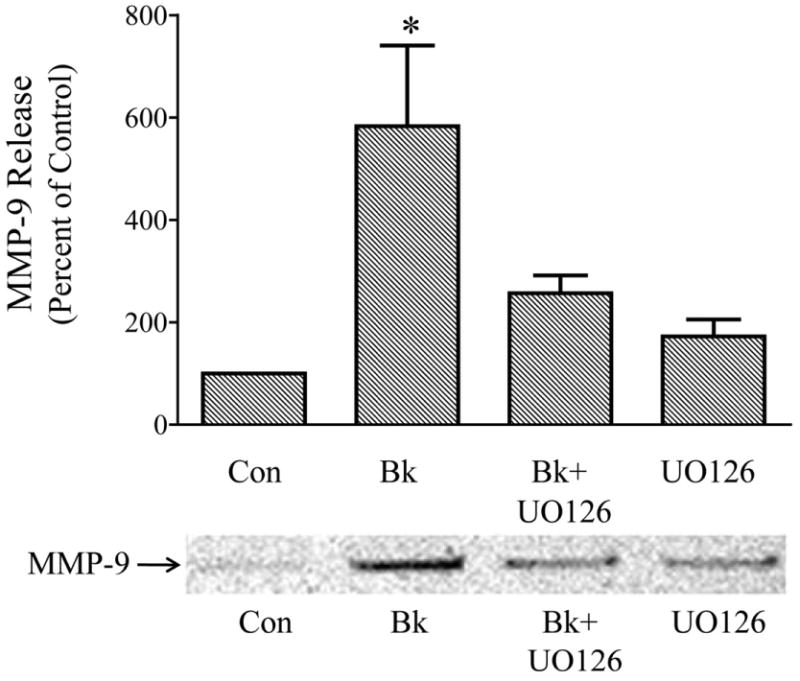
Inhibition of bradykinin (Bk)-induced release of matrix metalloproteinase 9 (MMP-9) from cultured human trabecular meshwork cells by the MAP kinase kinase inhibitor, U0126. Serum-deprived trabecular meshwork cells were treated for 2 hr with 100 nM Bk, 1 μM U0126, or Bk+ U0126. Cells incubated with U0126 were pretreated 30 min prior to the addition of Bk and U0126 remained in the media for the duration of the experiment. At the end of incubation, the media were collected, concentrated and analyzed for MMP-9 by immunoblot. A, values represent means ± S. E. of densitometry measurements of MMP-9 data in 6 experiments. * Differs from control at P< 0.05. B, representative immunoblot of MMP-9 detected in media from vehicle-treated control cells, and from cells treated with Bk, Bk + U0126, or U0126 alone.
4. Discussion
This study examined effects of bradykinin on the activity of extracellular-regulated MAP kinases (ERK1/2) in trabecular meshwork cells cultured from human anterior segment. Incubation of trabecular meshwork cells with bradykinin produced a rapid increase in the levels of phosphorylated ERK1/2 with peak effect observed within 2 min. The EC50 for the bradykinin response was approximately 0.7 nM, a concentration consistent with a physiological role for kinins in the modulation of ERK1/2 activity (Campbell, 2000). Interestingly, the effect of bradykinin to promote ERK1/2 activation in the human trabecular meshwork cell was relatively selective as bradykinin treatment did not stimulate either the JNK or p38 kinase pathways in the present study.
Generally, the principal physiological actions of kinins are mediated by the B2 subtype of kinin receptor, while the B1 receptor is believed to be of importance only when induced during certain pathophysiological states and responds preferentially to the desArg9 metabolites of kinin peptides (Bhoola et al., 1992; Blais et al., 2000). In evaluating kinin actions in the present study, the B2 kinin receptor agonist, Tyr8- bradykinin, was found to be equivalent to bradykinin in stimulating ERK1/2 phosphorylation in trabecular meshwork cells while the selective B1 receptor agonist, Lys-Des-Arg9- bradykinin, failed to promote ERK1/2 activation. In addition, bradykinin stimulation of ERK1/2 activity was blocked by pretreatment of cells with the selective B2 receptor antagonist, Hoe-140. These results clearly establish a role for the B2 kinin receptor in mediating bradykinin activation of ERK1/2 in the human trabecular meshwork cell. Such a conclusion is consistent with prior studies of bradykinin actions in ocular tissues where effects to stimulate phospoinosotide metabolism and increase Ca2+ mobilization in trabecular meshwork cells have been attributed to B2 receptor stimulation (Sharif and Xu, 1996; Llobet et al., 1999; Webb et al., 2003) as have the effects of bradykinin to promote outflow facility in perfused anterior segments (Webb et al., 2006).
B2 kinin receptors couple through the heterotrimeric G protein, Gq, to activate phospholipase C. Consequently, bradykinin stimulation of trabecular meshwork cells yields diacylglycerol (DAG) and also inositol trisphosphate (IP3), which then promotes the mobilization of intracellular free Ca2+. With this combination of second messengers, ERK1/2 activation can then proceed through a DAG-PKC (Ueda et al., 1996; Schonwasser et al., 1998) mechanism or, alternatively, through an IP3-Ca2+- dependent mechanism, which commonly involves Src kinase as an intermediate (Lev et al., 1995; Dikic et al., 1996). The dominant mechanism for ERK1/2 activation in this setting of signaling pathways varies with cell type (Hawes et al., 1995; Dikic et al., 1996), though, in some cells, both pathways have been shown to contribute significantly (Della-Rocca et al., 1997). This latter possibility appears to be the case for bradykinin activation of ERK1/2 in the human trabecular meshwork cell. In the present study, inhibition of PKC with the nonselective inhibitor, GF109203X, or down-regulation of PKC activity by persistent stimulation with phorbol ester blunted bradykinin stimulation of ERK1/2, but neither intervention abolished the response. Similarly, inhibition of Src kinase activity with PP2 reduced bradykinin activation of ERK1/2, but by only 50 %. These results indicate that at least two signaling pathways can contribute to bradykinin -induced ERK1/2 phosphorylation in trabecular meshwork cells, and it seems likely that the contribution of a particular pathway will vary with physiological or pharmacological conditions. Regardless of the upstream events, however, a common final step in ERK1/2 activation by a variety of signaling cascades is phosphorylation of ERK1/2 by MAP kinase kinase (MEK) (Goldsmith and Dhanasekaran, 2007). This was also determined to be a key component in the action of bradykinin in trabecular meshwork cells as inhibition of MEK with U0126 completely blocked bradykinin stimulation of ERK1/2 activity.
Trabecular meshwork cells have been shown to express tissue kallikrein, the primary enzyme for kinin production within tissues, and also to possess kininase activity for termination of peptide effects (Webb et al., 2009). Consequently, kinins generated locally within the trabecular meshwork may participate in the regulation of anterior segment function, an idea supported by the finding that activation of B2 kinin receptors by bradykinin results in increased outflow facility in perfused bovine anterior segments (Webb et al., 2006). However, while stimulation of signaling by bradykinin in trabecular meshwork cells occurs in minutes as observed for ERK1/2 activation in the present study, functional effects of the peptide on outflow facility are much slower to develop. Indeed, bradykinin's effect to increase outflow facility in perfused anterior segments was only evident after one hr of treatment and required 4 hr for the peak response to be achieved. This suggests that to alter facility in the conventional pathway bradykinin stimulation of signaling in trabecular cells must initiate a cellular response with the potential to only gradually diminish resistance to aqueous flow through the trabecular meshwork. An action to increase MMP levels is one potential mechanism as elevated MMPs in the extracellular environment are known to promote matrix degradation and have been shown to enhance outflow through the conventional pathway (Bradley et al., 1998; Bradley et al., 2001). Moreover, this possibility is supported by the finding that bradykinin's action to stimulate outflow facility in perfused anterior segments was blocked when MMP activity was inhibited (Webb et al., 2006).
Trabecular meshwork cells are a source of multiple MMP isozymes (Alexander et al., 1998; Zhou et al., 1998; 2001 Shearer and Crosson, 2002). Expression and activation of these enzymes is generally regulated at a transcriptional level, which is a process that occurs over days. However, MMP-2 and MMP-9 are constitutively expressed by trabecular meshwork cells (Shearer and Crosson, 2002; Webb et al., 2006) as well as other cell types, and the level of these MMPs in the extracellular environment is regulated primarily by acute changes in secretion from the cell. Moreover, activation of ERK1/2 is a common signaling mechanism for regulating MMP secretion (Shearer and Crosson, 2002; Liu et al., 2002; Husain et al., 2005; Okada et al., 2010). Since we had previously demonstrated that bradykinin stimulation of B2 kinin receptors increased MMP-9 secretion from cultured bovine trabecular meshwork cells (Webb et al., 2006), it was of interest in the present study to determine if bradykinin promoted MMP-9 release from the human cell, and if this effect was linked to the activation of ERK1/2. Bradykinin stimulation was observed to increase MMP-9 release from human trabecular cells into the extracellular media over a 2-hr treatment period, consistent with the prior finding in bovine cells. Further, pretreatment of cells with the MEK inhibitor, U1026, to block ERK1/2 activation substantially blunted bradykinin stimulation of MMP-9 secretion. There was no attempt in these experiments to assess MMP activity. The data from this group of experiments establish an action of bradykinin to promote MMP-9 release from human trabecular meshwork cells and suggest that the effect results from stimulation of ERK1/2 activity.
In conclusion, bradykinin was observed to act on human trabecular meshwork cells to rapidly activate the extracellular signal-regulated MAP kinases, ERK1/2. This effect was mediated by B2 kinin receptors and involved 2 different signaling pathways, each of which utilized MEK activity to enhance ERK1/2 phosphorylation. ERK1/2 activation, in turn, promoted release of MMP-9 from the human trabecular cell. These events, viewed together, provide a cellular basis for bradykinin's functional effect to increase outflow facility in the anterior segment by a MMP-dependent mechanism. In this context, bradykinin stimulation of B2 receptors on the trabecular meshwork cell leads to ERK1/2 activation and subsequent secretion of constitutively-expressed MMP-9, which, over a period of hours, alters the extracellular matrix and decreases resistance to aqueous flow through the conventional pathway.
Acknowledgments
This study was supported by National Eye Institute grants EY014653 (J.G.W.), EY09741(C.E.C.), EY01479 (C.E.C.) and Research to Prevent Blindness.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander JP, Samples JR, Acott TS. Growth factor and cytokine modulation of trabecular meshwork matrix metalloproteinase and TIMP expression. Curr Eye Res. 1998;17:276–285. doi: 10.1076/ceyr.17.3.276.5219. [DOI] [PubMed] [Google Scholar]
- Barsotti MF, Bartels SP, Freddo TF, Kamm RD. The source of protein in the aqueous humor of the normal monkey eye. Invest Ophthalmol Vis Sci. 1992;33:581–595. [PubMed] [Google Scholar]
- Bert RJ, Caruthers SD, Jara H, Krejza J, Melhem ER, Kolodny NH, Patz S, Freddo TF. Demonstration of an anterior diffusional pathway for solutes in the normal human eye with high spatial resolution contrast-enhanced dynamic MR imaging. Invest Ophthalmol Vis Sci. 2006;47:5153–5162. doi: 10.1167/iovs.05-0372. [DOI] [PubMed] [Google Scholar]
- Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens and kininases. Pharmacol Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- Bill A, Svedbergh B. Scanning electron microscopic studies of the trabecular meshwork and the canal of Schlemm – an attempt to localize the main resistance to outflow of aqueous humor in man. Acta Ophthalmol. 1972;50:295–320. doi: 10.1111/j.1755-3768.1972.tb05954.x. [DOI] [PubMed] [Google Scholar]
- Blais C, Jr, Marceau F, Rouleau JL, Adam A. The kallikrein-kininogen-kinin system: lessons from the quantification of enodgenous kinins. Peptides. 2000;21:1903–1940. doi: 10.1016/s0196-9781(00)00348-x. [DOI] [PubMed] [Google Scholar]
- Bradley JM, Vranka J, Colvis CM, Conger DM, Alexander JP, Fisk AS, Samples JR, Acott TS. Effect of matrix metalloproteinases activity on outflow in perfused human organ culture. Invest Ophthalmol Vis Sci. 1998;39:2649–2658. [PubMed] [Google Scholar]
- Bradley JM, Kelley MJ, Zhu X, Anderssohn AM, Alexander JP, Acott TS. Effects of mechanical stretching on trabecular matrix metalloproteinases. Invest Ophthalmol Vis Sci. 2001;42:1505–1513. [PubMed] [Google Scholar]
- Campbell DJ. Towards understanding the kallikrein-kinin system: insights from measurements of kinin peptides. Braz J Med Biol Res. 2000;33:665–677. doi: 10.1590/s0100-879x2000000600008. [DOI] [PubMed] [Google Scholar]
- Chowdhury UR, Madden BJ, Charlesworth MC, Fautsch MP. Proteome analysis of human aqueous humor. Invest Ophthalmol Vis Sci. 2010;51:4921–4931. doi: 10.1167/iovs.10-5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Reynosa P, Robledo T, Macias-Silva M, Wu SV, Salazar EP. Src kinase regulates metalloproteinase-9 secretion induced by type IV collagen in MCF-7 human breast cancer cells. Matrix Biol. 2008;27:220–231. doi: 10.1016/j.matbio.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Della Rocca GJ, van Biesen T, Daaka Y, Luttrell DK, Luttrell LM, Lefkowitz RJ. Ras-dependent mitogen-activated protein kinase activation by G protein coupled receptors. Convergence of Gi and Gq-mediated pathways on calcium/calmodulin, Pyk2 and Src kinase. J Biol Chem. 1997;272:19125–19132. doi: 10.1074/jbc.272.31.19125. [DOI] [PubMed] [Google Scholar]
- Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383:547–550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- Ethier CR, Kamm RD, Palaszewski BA, Johnson MC, Richardson TM. Calculations of flow resistance in the juxtacanalicular meshwork. Invest Ophthalmol Vis Sci. 1986;27:1741–1750. [PubMed] [Google Scholar]
- Goldsmith ZG, Dhanasekaran DN. G protein regulation of MAPK networks. Oncogene. 2007;26:3122–3142. doi: 10.1038/sj.onc.1210407. [DOI] [PubMed] [Google Scholar]
- Hawes BE, van Biesen T, Koch WJ, Luttrell LM, Lefkowitz RJ. Distinct pathways of Gi- and Gq-mediated mitogen-activated protein kinase activation. J Biol Chem. 1995;270:17148–17153. doi: 10.1074/jbc.270.29.17148. [DOI] [PubMed] [Google Scholar]
- Husain S, Jafri F, Crosson CE. Acute effects of PGF2α on MMP-2 secretion from human ciliary muscle cells: a PKC- and ERK-dependent process. Invest Ophthalmol Vis Sci. 2005;46:1706–1713. doi: 10.1167/iovs.04-0993. [DOI] [PubMed] [Google Scholar]
- Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J. Protein tyrosine kinase PYK2 involved in Ca(2+)-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376:737–745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- Liu JF, Crepin M, Liu JM, Barritault D, Ledoux D. FGF-2 and TPA induce matrix metalloproteinase-9 secretion in MCF-7 cells through PICC activation of the Ras/ERK pathway. Biochem Biophys Res Commun. 2002;293:1174–1182. doi: 10.1016/S0006-291X(02)00350-9. [DOI] [PubMed] [Google Scholar]
- Llobet A, Gual A, Pales J, Barraquer R, Tobias E, Nicolas JM. Bradykinin decreases outflow facility in perfused anterior segments and induces shape changes in passaged BTM cells in vitro. Invest Ophthalmol Vis Sci. 1999;40:113–125. [PubMed] [Google Scholar]
- Ma JX, Song Q, Hatcher HC, Crouch RK, Chao L, Chao J. Expression and cellular localization of the kallikrein/kinin system in human ocular tissues. Exp Eye Res. 1996;63:19–26. doi: 10.1006/exer.1996.0087. [DOI] [PubMed] [Google Scholar]
- Okada M, Yamawaki HJ, Hara Y. Angiotensin II enhances interleukin-1β- induced MMP-9 secretion in adult rat cardiac fibroblasts. J Vet Med Sci. 2010;72:735–739. doi: 10.1292/jvms.09-0582. [DOI] [PubMed] [Google Scholar]
- Rohen J, Lutjen-Drecoll E. Morphology of aqueous outflow pathways in normal and glaucomatous eyes. In: Ritch R, Shields MB, Krupin T, editors. The Glaucomas. Vol. 1. Mosby; St. Louis, MO: 1989. pp. 41–74. [Google Scholar]
- Schonwasser DC, Marais RM, Marshall CJ, Parker PJ. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel and atypical protein kinase C isotypes. Mol Cell Biol. 1998;18:790–798. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif NA, Xu SX. Pharmacological characterization of bradykinin receptors coupled to phosphoinositide turnover in SV40-immortalized human trabecular meshwork cells. Exp Eye Res. 1996;63:631–637. doi: 10.1006/exer.1996.0157. [DOI] [PubMed] [Google Scholar]
- Shearer TW, Crosson CE. Adenosine A1 receptor modulation of MMP-2 secretion by trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2002;43:3016–3020. [PubMed] [Google Scholar]
- Ueda Y, Hirai S, Osada S, Suzuki A, Mizuno K, Ohno S. Protein kinase C activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. J Biol Chem. 1996;271:23512–23519. doi: 10.1074/jbc.271.38.23512. [DOI] [PubMed] [Google Scholar]
- Webb JG, Husain S, Yates PW, Mukhin YV, Crosson CE. Kinin modulation of conventional outflow facility in the bovine eye. J Ocul Pharmacol Ther. 2006;22:310–316. doi: 10.1089/jop.2006.22.310. [DOI] [PubMed] [Google Scholar]
- Webb JG, Shearer TW, Yates PW, Mukhin YV, Crosson CE. Bradykinin enhancement of PGE2 signaling in bovine trabecular meshwork cells. Exp Eye Res. 2003;76:283–289. doi: 10.1016/s0014-4835(02)00313-5. [DOI] [PubMed] [Google Scholar]
- Webb JG, Yang X, Crosson CE. Expression of the kallikrein/kinin system in human anterior segment. Exp Eye Res. 2009;89:126–132. doi: 10.1016/j.exer.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Li Y, Yue BY. Glucocorticoid effects on extracellular matrix proteins and integrins in bovine trabecular meshwork cells in relation to glaucoma. Int J Mol Med. 1998;1:339–346. [PubMed] [Google Scholar]