

Abstract
Background
Pathologic complete response (pCR) to neoadjuvant chemoradiation (CRT) is an important prognostic factor in locally advanced rectal cancer. However, it is uncertain whether histopathological techniques accurately detect pCR. We tested a novel molecular approach for detecting pCR and compared it to current histopathological approaches.
Study design
Pre-treatment tumor biopsies and surgical specimens were collected from 96 patients with locally advanced rectal cancer treated with neoadjuvant CRT and surgery. Tumor response was categorized by tumor regression grade (TRG). Tumor DNA from pre-CRT tumor biopsies was screened for K-ras and p53 mutations. DNA from paired surgical specimens was then screened for the same mutations using highly sensitive polymerase chain reaction (PCR)-based techniques.
Results
Sixty-eight out of 96 (71%) pre-treatment biopsies harbored K-ras and/or p53 mutation; 36 (38%) had K-ras mutations, 52 (54%) had p53 mutations and 20 (21%) carried both mutations. Of 70 patients with TRG 1–3, 66 (94%) had a concordant K-ras and p53 mutation profile in pre- and post-treatment tissues. Of 26 patients with TRG 0 (pCR), 12 had K-ras or p53 mutations in pre-treatment biopsies. Of these, 2 (17%) patients had the same K-ras (n=1) or p53 (n=1) mutation detected in post-treatment tissue.
Conclusions
Sensitive molecular techniques detect K-ras and p53 mutations in post-CRT surgical specimens in some patients with a pCR. This suggests histopathological techniques may not be completely accurate, and that some patients diagnosed with a pCR to CRT may indeed have occult cancers cells in their surgical specimens with K-ras and p53 mutations serving as reliable surrogates for residual disease.
Keywords: chemoradiation, surgery, rectal cancer, molecular analysis, K-ras, p53
Introduction
Pre-operative neoadjuvant chemoradiation (CRT) has an established role in the management of patients with rectal adenocarcinoma because it provides the opportunity to downstage tumors, increase sphincter preservation, and decrease the risk of local disease recurrence. (1–3) Rectal cancer response to CRT varies from minimal tumor downsizing to an apparent complete disappearance of all viable cancer cells, known as a pathologic complete response (pCR). Rectal cancer patients with a pCR after CRT have lower local recurrence rates and improved overall survival compared to patients with residual cancer cells in their surgical specimens. (4–10) These findings have clinical implications, as surgeons are cautiously proposing less extensive surgical resections, or even no surgery at all in patients who achieve a pCR to CRT. (11) However, before these approaches can be widely accepted, it is important to ensure that a diagnosis of pCR accurately represents the complete eradication of all tumor cells. Otherwise, the oncologic outcomes of these patients could be significantly compromised.
A pCR is currently diagnosed using histopathology. Representative sections of the tumor and lymph nodes are embedded in paraffin. A number of 5–10 μm sections are then stained with hematoxylin and eosin (H&E) and analyzed microscopically for the presence of cancer cells. However, given that a small number of rectal cancer patients with a pCR develop a local relapse after 5 years (10) it is currently unclear whether this method can accurately detect complete disease eradication or whether it can precisely detect the presence of minimal residual disease, information which is critical to predict oncologic control.
Previous studies have proven that molecular techniques able to identify tumor-specific molecular-genetic alterations can be more accurate than conventional histopathology in identifying cancer cells in surgical resection margins and regional lymph nodes of a variety of tumors. (12–16) Using this rationale we considered whether similar molecular techniques could be used to verify pCR or the presence of minimal residual disease in rectal cancer patients who undergo CRT. We used standard polymerase chain reaction (PCR) as well as the highly-sensitive PCR-based techniques, peptide nucleic acid (PNA) clamp PCR and pyrophosphorolysis activated polymerization-allele specific (PAP-A) PCR to determine the mutation status of K-ras and p53, two of the genes more frequently mutated in rectal cancer patients. (17–27) We prospectively analyzed mutation status in pre- and post-treatment samples from 96 rectal cancer patients who received CRT followed by surgery and compared the results of molecular and histopathological analysis of the surgical specimen to determine the accuracy of these techniques for verifying pCR.
Methods
Patients and treatment
Ninety-six rectal cancer patients with Stage II (uT3–4, uN0) or Stage III (any T, uN1–2) tumors were enrolled in the Timing of Rectal Cancer Response to Chemoradiation study, a multi-institutional clinical trial investigating the effect of increasing the CRT-to-surgery interval, and adding chemotherapy, modified FOLFOX-6 (mFOLFOX-6), during the waiting period (ClinicalTrials.org Identifier: NCT00335816). This trial was designed as a series of sequential Phase II trials or study groups (SGs), each with a progressively longer CRT-to-surgery interval and increasing cycles of pre-operative mFOLFOX-6. This study was approved by an Institutional Review Board (IRB) at each participating institution as well as a central IRB, and informed written consent was obtained from each patient prior to enrollment in the trial. Further details of patient eligibility for this trial are presented elsewhere. (28)
Patients in both SGs were treated with CRT; 5-FU 225 mg/m2/day for 7 days in continuous infusion and a total of 50.4 Gy of radiation. Patients in SG1 underwent total mesorectal excision (TME) an average of 6 weeks after completing CRT (standard of care). Following CRT, patients in SG2 with no evidence of stable disease received 2 cycles of additional chemotherapy (mFOLFOX-6); leucovorin 200 mg/m2 or 400 mg/m2 plus oxaliplatin 85 mg/m2 by 2h infusion, followed by bolus of 5-FU 400 mg/m2 and a 46h infusion of 5-FU 2,400 mg/m2. Patients in SG2 underwent TME an average of 11 weeks after completing CRT. The clinical outcomes for these patients are presented elsewhere. (28)
Assessment of response
Pathologic response after CRT was determined according to the AJCC tumor regression grading (TRG) classification. (29) Briefly, TRG 0 - complete response: no viable cancer cells; TRG 1 - moderate response: single cancer cells or small groups of cancer cells; TRG 2 - minimal response: residual cancer outgrown by fibrosis; TRG 3 - poor response: extensive residual cancer. TRG 0 was scored as a pCR. Surgical tissues were evaluated and graded by pathologists using standard histopathological techniques and TRG was determined for each tumor by two independent blinded pathologists.
Sample preparation and DNA extraction
Pre-treatment biopsies and post-treatment surgical tissues were collected prospectively and 5–10 μm tissue sections were taken from formalin-fixed, paraffin-embedded (FFPE) blocks, placed on microscope slides, and de-paraffinized. Slides were stained with 0.2% methylene blue and tumor cells were micro-dissected manually from pre- and post treatment samples under inverted microscopy.
For all post-treatment samples (TRG 0–3), representative tissue sections from throughout each tumor block were assessed to ensure sampling of the entire surgical specimen. Slide sections were pooled and used for DNA extraction. Pre- and post-treatment DNA was extracted using the QIAamp DNA FFPE Tissue Kit (Qiagen; Valencia, CA) with the following modifications to the manufacturer’s protocol: an extension of digestion time at 56°C from 1 hour to 48 hours and the addition of three 20 μl aliquots of Proteinase-K at 4, 20, and 28 hours during digestion. DNA was quantified by measuring absorbance (NanoDrop, Thermo Fisher Scientific, Inc.; Wilmington, DE).
Mutation analysis of pre-treatment biopsies and post-treatment surgical specimens
Tumor cells from pre-treatment biopsies and post-treatment surgical specimens were micro-dissected and tumor DNA was extracted as indicated above. Standard PCR analysis was performed to detect mutations in p53 and K-ras. Primers were used to amplify genomic sequences in exons 4–8 of p53 and exons 2–3 of K-ras with the following cycling conditions; 98°C for 10 seconds, 60°C for 20 seconds, and 72°C for 30 seconds, for 45 cycles. Primer sequences are listed in Supplementary Table 1 (online only). All sequencing reactions were performed in both the sense and antisense directions with PCR primers, and all mutations were confirmed by two independently derived PCR products.
Identification of rare mutations in post-treatment surgical specimens
In patients with TRG 0, FFPE tissue sections obtained from the treated tumor bed were examined and DNA was extracted from areas with ulceration or fibrosis. Since these tissues were scored as having no residual cancer cells, we employed more sensitive PCR techniques to detect minimal residual or occult disease.
PNA clamp PCR was performed to identify K-ras mutations at codons 12 and 13 in TRG 0 DNA extracted from post-treatment specimens. This technique was established to identify a single mutant allele in the presence of over 10,000 copies of wild-type DNA. (30–34) PNA clamp PCR reactions were performed using Phusion HS II high fidelity polymerase (Thermo Fisher Scientific, Lafayette, CO). Reactions consisted of 1 X Phusion HF buffer, 0.2 mM dNTP, 0.15 μM primers, 0.25 μM PNA, 1U Phusion HS II DNA polymerase, and 200 ng of template DNA in a final volume of 25 μl. The cycling conditions were 98°C for 10 seconds, 76°C for 10 seconds, 60°C for 20 seconds, and 72°C for 30 seconds, for 60 cycles. PNA clamp information and PCR primer sequences are presented in Supplementary Table 1 (online only). All PCR products were sequenced to verify the presence or absence of K-ras mutations and the reactions were repeated (5 X) for each specimen.
In contrast to K-ras, mutations in p53 can occur at several locations in exons 4–8. (22, 25–27) Therefore, a single PNA clamp PCR-based assay could not be used to detect the multiple p53 mutations that may exist in the hundreds of codons in exons 4–8. Instead, we utilized a PAP-A PCR assay for the detection of known p53 mutations. This technique can detect p53 mutation with high sensitivity in the presence of more than 10,000 copies of wild-type p53 DNA. (35) Post-treatment TRG 0 tissues with p53 mutation in the paired/corresponding pre-treatment biopsies were assessed by PAP-A as follows: PAP-A primers were designed to detect the specific p53 mutation identified in the pre-treatment biopsy (Supplementary Table 1, online only). A 100–200 bp region within the p53 gene was amplified by PAP-A with mutant-specific primers with a 3′ dideoxy block. The blocked primers were synthesized by adding a specific dideoxynucleotide to the oligodeoxynucleotide by terminal transferase. (36) Each reaction contained 50ng of template DNA plus 50 mM Tris-HCl (pH: 7.8, 25uC), 16 mM (NH4)2SO4, 1 mM DTT, 1.5 mM MgCl2, 90 mM PPi, 100 mM blocked primers (P*), 4% DMSO, 25 mM dNTP, and 4 U KlenTaq S in a total volume of 25 μl plus 20 ng fish DNA-carrier in sensitivity assays and negative controls. The cycling conditions were as follows: denaturation at 94°C for 2 minutes, then 94°C for 20 seconds, 58–61°C for 30 seconds, 64°C for 30 seconds, 68°C for 30 seconds, and 72°C for 30 seconds, for 50–55 cycles, and 72°C for 7 minutes for the last extension. 5 μl of PAP-A product was checked by agarose gel electrophoresis (2–4%) and reactions were repeated (5 X) to determine the presence or absence of p53 mutations.
To internally validate the sensitivity of the PNA clamp PCR and PAP-A assays, tissue sections from surgical-specimen FFPE tissue blocks from TRG 1–3 patients were also analyzed by PNA clamp PCR (K-ras mutations) and PAP-A (p53 mutations) as described above, to verify the K-ras and p53 mutation status obtained for TRG 1–3 patients using standard PCR and sequencing.
Results
Patient demographics and clinical characteristics
Patient demographics and clinical characteristics for the 96 patients included in the analysis are summarized in Table 1. A total of 73 patients (76%) were diagnosed with Stage III rectal cancer. Most patients had T3 disease (92%) and N1 disease (71%). All patients completed the full course of CRT and all patients underwent TME.
Table 1.
Pre-treatment Patient Demographics and Clinical And Pathologic Characteristics
| Characteristics | Patients (n=96) |
|---|---|
| Sex, n (%) | |
| Male | 56 (58) |
| Female | 40 (42) |
| Age, y, median (range) | 57 (32–85) |
| Clinical stage, n (%) | |
| II | 24 (25) |
| III | 73 (76) |
| T Stage, n (%) | |
| T2 | 7 (7) |
| T3 | 88 (92) |
| T4 | 1 (1) |
| Lymph node stage, n (%) | |
| N0 | 24 (25) |
| N1 | 68 (71) |
| N2 | 4 (4) |
Histopathological examination of surgical tissues was performed and response was categorized by TRG. A total of 26 patients (27%) had TRG 0, 21 (22%) had TRG 1, 34 (35%) had TRG 2 and 15 (16%) had TRG 3.
Pre-treatment mutation profile
Standard PCR of the pre-treatment patient biopsies identified K-ras mutation in 36 (38%) and p53 mutation in 52 (54%) out of 96 specimens. The K-ras and p53 mutations stratified by TRG are presented in Table 2. K-ras and p53 mutations were less common in patients with TRG 0 compared to TRG 1–3, but the differences were not statistically significant (p = 0.19). The majority of K-ras mutations (27 out of 36; 82%) were single point mutations in codon 12. The most common mutation was G12D. Other mutations included G12S, G12V, and G13D. In contrast, there was a broad spectrum of p53 mutations including point mutations, base-pair substitutions, insertions, deletions, and nonsense mutations. Point mutations were the predominant p53 aberrancy, identified in 39 out of 52 (75%) patients. The pre-treatment K-ras and p53 genotypes for all patients are presented in Tables 3 and 4.
Table 2.
Frequency of p53 and K-ras Mutations Detected in Pre-Treatment Patient Biopsies
| Gene mutation | Patients screened, n=96* | TRG 0 (n=26) | TRG 1 (n=21) | TRG 2 (n=34) | TRG 3 (n=15) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | n | % | n | % | |
|
p53
|
52 | 54 | 7 | 27 | 12 | 57 | 25 | 74 | 8 | 53 |
| K-ras | 36 | 38 | 5 | 19 | 11 | 52 | 16 | 47 | 4 | 27 |
Of 96 patients screened, 68 carried K-ras or p53 mutations. Percentages for these patients are presented using the total number of patients screened (n=96) as the denominator.
TRG, tumor regression grade.
Table 3.
Pre- and Post-Treatment K-ras and p53 Mutation Analysis in Patients with Residual Disease (TRG 1–3) using Standard Polymerase Chain Reaction
| Patients | Genotype of K-ras | Genotype of p53 | ||
|---|---|---|---|---|
| Pre-T | Post-T | Pre-T | Post-T | |
| TRG-1 | ||||
| 1 | wt | wt | E298Stop | E298Stop |
| 2 | G12D | G12D | R175H | R175H |
| 3 | G12D | G12D | C242F | C242F |
| 4 | wt | wt | wt | wt |
| 5 | wt | wt | Y163N | Y163N |
| 6 | wt | wt | E5p.149InsT | E5p.149InsT |
| 7 | wt | wt | wt | wt |
| 8 | wt | wt | wt | wt |
| 9 | wt | wt | wt | wt |
| 10 | wt | wt | G245S | G245S |
| 11 | G13D | G13D | R181Y | R181Y |
| 12 | G13D | G13D | C176R | C176R |
| 13 * | G12S | G12D | wt | wt |
| 14 | G12D | G12D | wt | wt |
| 15 | G12D | G12D | E285K | E285K |
| 16 | G12D | G12D | wt | wt |
| 17 | G13D | G13D | wt | wt |
| 18 | wt | wt | wt | wt |
| 19 * | G13D | G13D | M237I | wt |
| 20 | G13D | G13D | R306Stop | R306Stop |
| 21 | wt | wt | R175H | R175H |
| TRG-2 | ||||
| 22 | wt | wt | wt | wt |
| 23 | G12D | G12D | wt | wt |
| 24 | G12V | G12V | wt | wt |
| 25 | wt | wt | R282W | R282W |
| 26 | G12V | G12V | wt | wt |
| 27 | G12D | G12D | wt | wt |
| 28 | wt | wt | R282W | R282W |
| 29 | G12D | G12D | E298Stop | E298Stop |
| 30 | G12D | G12D | wt | wt |
| 31 | G13D | G13D | M237I | M237I |
| 32 | wt | wt | wt | wt |
| 33 | G12D | G12D | R175H | R175H |
| 34 | wt | wt | R175H | R175H |
| 35 | wt | wt | R196Stop | R196Stop |
| 36 | wt | wt | R282W | R282W |
| 37 * | G13D | G13D | R273C | wt |
| 38 | wt | wt | R273P | R273P |
| 39 | wt | wt | E286K | E286K |
| 40 | G12D | G12D | R175H | R175H |
| 41 | wt | wt | R181C | R181C |
| 42 | wt | wt | E8p.269delGCT | E8p.269delGCT |
| 43 | G12D | G12D | R273H | R273H |
| 44 | wt | wt | R248W | R248W |
| 45 | G12V | G12V | Y205H | Y205H |
| 46 | wt | wt | wt | wt |
| 47 | wt | wt | R213Q | R213Q |
| 48 | G12D | G12D | Q167Stop | Q167Stop |
| 49 | wt | wt | R273C | R273C |
| 50 | wt | wt | R273C | R273C |
| 51 | G13D | G13D | R213L | R213L |
| 52 | G12D | G12D | R248W | R248W |
| 53 | wt | wt | Q144Stop | Q144Stop |
| 54 | wt | wt | wt | wt |
| 55 | G12V | G12V | E6p.768-769delTTT211-F212 | E6p.768-769delTTT211-F212 |
| TRG-3 | ||||
| 56 * | wt | wt | wt | L130H |
| 57 | G12D | G12D | wt | wt |
| 58 | wt | wt | R248Q | R248Q |
| 59 | wt | wt | R175H | R175H |
| 60 | G12V | G12V | M237I | M237I |
| 61 | wt | wt | R306Stop | R306Stop |
| 62 | G12D | G12D | G244D | G244D |
| 63 | wt | wt | R209Stop | R209Stop |
| 64 | wt | wt | E285K | E285K |
| 65 | wt | wt | wt | wt |
| 66 | wt | wt | wt | wt |
| 67 | wt | wt | wt | wt |
| 68 | G13D | G13D | wt | wt |
| 69 | wt | wt | A138T | A138T |
| 70 | wt | wt | wt | wt |
Patient had a discordant mutation profile between pre-treatment biopsy and post-treatment surgical specimen.
TRG, tumor regression grade; wt, wild-type; Pre-T, pre-treatment; Post-T, post-treatment.
Table 4.
Molecular Analysis of K-ras and p53 in Patients with a pCR (TRG 0) using PAP-A and PNA Clamp Polymerase Chain Reaction
| Patients | Genotype of K-ras | Genotype of p53 | ||
|---|---|---|---|---|
| Pre-T | Post-T PNA Clamp |
Pre-T | Post-T PAP-A |
|
| TRG 0 | ||||
| 1 | wt | wt | wt | wt |
| 2 | wt | wt | wt | wt |
| 3 | G12D | wt | wt | wt |
| 4 | wt | wt | R175H | wt |
| 5 | wt | wt | R175H | wt |
| 6 | wt | wt | wt | wt |
| 7 | wt | wt | wt | wt |
| 8 | wt | wt | wt | wt |
| 9 | wt | wt | wt | wt |
| 10 | wt | wt | wt | wt |
| 11 | G12D | wt | wt | wt |
| 12 | wt | wt | wt | wt |
| 13 * | wt | wt | R213L | R213L |
| 14 | wt | wt | Exon8/Intron G>A Splice Site | wt |
| 15 | wt | wt | E7p.252del CTCACCATC | wt |
| 16 | wt | wt | wt | wt |
| 17 | G12D | wt | wt | wt |
| 18 * | G12D | G12D | wt | wt |
| 19 | wt | wt | C176R | wt |
| 20 | wt | wt | wt | wt |
| 21 | wt | wt | R175H | wt |
| 22 | wt | wt | wt | wt |
| 23 | wt | wt | wt | wt |
| 24 | G12D | wt | wt | wt |
| 25 | wt | wt | wt | wt |
| 26 | wt | wt | wt | wt |
Patient had mutation in pre-treatment biopsy and post-treatment surgical specimen.
TRG, tumor regression grade; wt, wild-type; Pre-T, pre-treatment; Post-T, post-treatment.
Mutation profile for pre- and post-treatment tissues (TRG 1–3)
Standard PCR of the post-treatment specimens in patients with residual disease (TRG 1–3) revealed that all patients with pre-treatment K-ras mutation (n = 31) had K-ras mutation in the paired post-treatment surgical tissue. K-ras mutation was concordant in 30 out of 31 (97%) patients (Table 3). The one discordant patient (TRG 1) had G12S in the pre-treatment biopsy and G12D in the post-treatment tissue (Table 3, Figure 1a). In the remaining 39 patients with no pre-treatment biopsy K-ras mutation, no K-ras mutation was detected in the paired surgical tissues.
Figure 1.
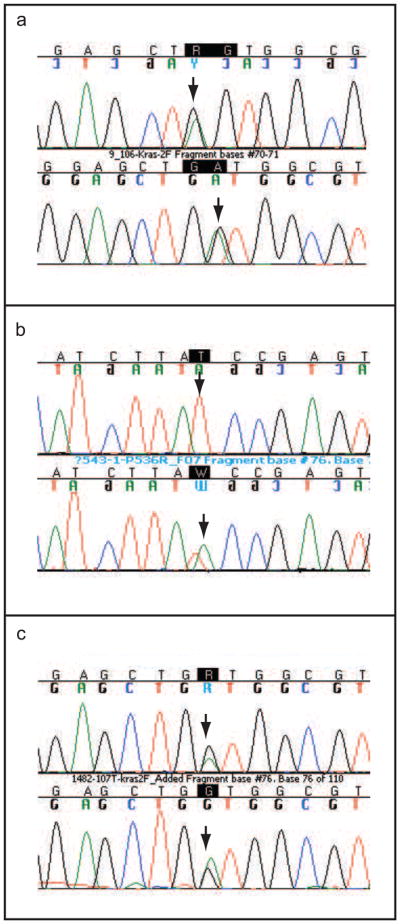
Mutation analysis of pre-and post treatment patient specimens. (a): One patient (TRG 1) had a discordant K-ras mutation profile pre-and post treatment. The mutation is within codon 12. The upper panel shows K-ras mutation in the first position of codon 12 (GGT > AGT) in the pre-treatment biopsy specimen. The lower panel shows K-ras mutation in the second position of codon 12 (GGT>GAT) in the surgical specimen following treatment. (b): One patient (TRG 3) harbored a new p53 mutation in the surgical specimen that was not present in the pre-treatment biopsy. The upper panel shows a wild-type genotype following sequencing exon 6 of the p53 gene in the pre-treatment biopsy. The lower panel shows a new mutation in codon 195 of exon 6 (ATC > AAC) in the surgical specimen following treatment. (c): One patient who was TRG 0 after treatment had a mutation in the K-ras gene. The upper panel shows mutation in codon 12 GGC > GAC of the K-ras gene in the pre-treatment biopsy specimen. The lower panel shows the same mutation in codon 12 GGC > GAC in the surgical sample detected by PNA clamp PCR.
There were 45 patients with p53 mutation in pre-treatment biopsies (TRG 1–3) and 43 of them (96%) had p53 mutation in the corresponding post-treatment surgical tissues. Discordance occurred in 2 patients who each carried a p53 mutation in the pre-treatment biopsy and no p53 mutation in the post-treatment surgical tissues (Table 3). Interestingly, both of these patients had K-ras mutation in both pre- and post-treatment tissues. In the remaining patients with p53 mutation, p53 genotype was unchanged between pre- and post-treatment tissues. One of the 25 patients with no p53 mutation in the pre-treatment biopsy harbored a new p53 mutation in the surgical specimen (Table 3, Figure 1b).
To ensure that mutations did not arise secondary to radiation and that there were no field defects in normal mucosa outside the tumor which had these mutations, we extracted DNA from normal proximal resection margin tissue and screened it for K-ras and p53 mutations. No K-ras or p53 mutations were detected in any TRG 1–3 patients (data not shown).
Internal validation of PAP-A and PNA clamp PCR assays
To assess the sensitivity of our PCR assays, we first tested PAP-A and PNA clamp PCR in FFPE tissue sections from patients with gross or microscopic residual disease (TRG 1–3). For PAP-A, we correctly detected all p53 mutations in post-treatment tissues corresponding to the standard PCR results. Similarly, we detected the same K-ras mutations in both pre- and post-treatment tissues with the PNA clamp PCR assay. No new mutations were detected in tissues from TRG 1–3 specimens using the PNA clamp PCR technique (data not shown).
Detection of K-ras mutation in TRG 0 patients using PNA clamp PCR
In the TRG 0 patient cohort, K-ras mutation was detected in pre-treatment biopsies in 5 out of 26 (19%) patients (Table 4). Despite the expectation that no mutation would be detected in any post-treatment tissues, K-ras mutation was detected in the post-treatment tissue of 1 patient by PNA clamp PCR (Table 4, Figure 1c). The genotype of the mutation was the same as the pre-treatment biopsy. No new mutations were detected by PNA clamp PCR. None of the remaining patients who harbored K-ras gene mutation in the pre-treatment biopsies (n = 4) had mutant K-ras detected in the post-treatment tissues, and no K-ras mutations were detected in patient DNA from proximal resection margin tissue (data not shown).
Detection of p53 mutation in TRG 0 patients using PAP-A
In the TRG 0 patient group, p53 mutation was detected in pre-treatment biopsies in 7 out of 26 (27%) patients (Table 4). PAP-A analysis detected a p53 mutation in 1 post-treatment specimen, matching the p53 genotype of the paired pre-treatment biopsy (Table 4). No new p53 mutations were detected in the post-treatment surgical specimens. None of the remaining patients who harbored p53 mutation in the pre-treatment biopsies (n=6), had p53 mutation detected in the post-treatment tissues and no p53 mutations were detected in patient DNA from proximal resection margin tissue (data not shown).
Discussion
Our study shows that overall p53 and K-ras mutations in rectal cancer rarely change as a result of CRT. We found that for nearly all patients analyzed, the genotype of p53 or K-ras mutation remained consistent between pre-and post-treatment tissues despite CRT. Importantly, using the highly sensitive PCR-based PAP-A and PNA clamp techniques, we were able to detect gene mutations in two post-treatment patient specimens that were determined to be TRG 0 by histopathology. Given that detection of these mutations may be a surrogate for residual or occult cancer cells this is a significant finding as it suggests that current histopathological techniques may miss occult cancer cells present in the surgical specimen and that the application of sensitive molecular-genetic techniques may improve the accuracy for diagnosing pCR in response to CRT in rectal cancer.
We focused on mutations in K-ras and p53, as these are two of the most common genetic aberrations found in colorectal cancer. (17–27, 37–41) Nearly 35–55% of colorectal cancer patients harbor a mutation in the p53 gene and 35–45% of patients harbor a mutation in the K-ras gene. Overall, the prevalence of these mutations in our patient population is consistent with what has been reported previously, (22, 25–27, 38). However, it is important to note that these mutations were only present in sixty-eight (71%) patients in our study. The remaining twenty-eight (29%) patients could therefore not be assessed using molecular approaches. To fully determine the effectiveness of using mutation-screening-based molecular techniques to assess pCR, it will be important to screen a larger panel of colorectal genes for mutations so that all patients can be assessed. Imperiale, et al. adopted this approach in their study to identify abnormal DNA in stool samples from colorectal cancer patients (42). Their fecal DNA panel consisted of 21 mutations present in the K-ras, p53, and APC genes as well as the micro-satellite instability (MSI) marker BAT-26. Screening for a similar panel of mutations may detect additional mutations in our patient cohort, which may be informative for assessing pCR.
With the exception of three patients, p53 and K-ras mutations detected in pre-treatment biopsies could also be detected by conventional PCR in paired post-treatment surgical tissues. Although a small number of studies have compared the mutation profiles between primary tumors and their corresponding metastasis after chemotherapy, ours is the first study comparing mutation profiles pre- and post-treatment in rectal cancer using neoadjuvant CRT and additional chemotherapy (mFOLFOX-6) before surgery. In general, our study is in agreement with prior published data and demonstrates concordance in the mutation profiles of the primary tumor pre- and post-treatment. (41, 43, 44) However, we found discordance in the p53 genotype between pre- and post-treatment tissues in three patients and in one patient we found a change in the K-ras genotype. Of the three patients with a genotype change in p53 from pre- to post-treatment, two had loss of p53 mutation and one patient had a gain of p53 mutation. The two patients with loss of p53 mutation initially harbored both K-ras and p53 mutations in their pre-treatment biopsies; and only mutant p53, not K-ras, was lost in the post-treatment tissues, suggesting that the loss of p53 mutation is probably real and not likely a sequencing artifact. There are no other reported cases of a genotypic change in the same mutant K-ras codon following chemotherapy or radiotherapy. These discordances have some plausible explanations. Solid tumors, and in particular colorectal tumors, are intrinsically heterogeneous and different regions have cell populations with different gene mutations. Therefore sampling different regions may yield a different mutation profile.(15, 45, 46) It is also possible that the genotoxic stress of chemotherapy and radiation may either select clones of cells with a genotype that might contribute to CRT resistance or introduce new mutations in oncogenes and tumor suppressor genes.
We found that patients with a pCR overall had fewer mutations in p53 and K-ras compared to non-pCR patients. This is an important finding because for the current approach to be successful, it is critical that these mutations are detected in pCR patients. The concordance in mutation profiles in tumors before and after CRT also has important clinical implications. Detection of specific mutation profiles in plasma has been used to monitor tumor response and diagnose early relapse after treatment. (45, 46) However, the accuracy of this method requires that the mutation profile remain consistent throughout treatment. Our data suggest that mutations in two of the genes more commonly mutated in rectal cancer do remain largely stable after treatment with CRT and that for the most part the detection of K-ras and p53 can be used to screen patients for residual or recurrent disease after treatment.
The development of local tumor recurrence after apparent complete surgical resection with negative microscopic surgical margins, and systemic disease in patients with node negative disease, has questioned the sensitivity of standard histopathological techniques to detect occult cancer cells or minimal residual disease. (14) Over the past decade, a number of sensitive molecular-genetic techniques have been developed for the purpose of detecting tumor-specific genetic alterations as indicators of the presence of occult cancer cells in the tissue, body fluids, or peripheral blood of cancer patients. (12–16, 30, 33, 47, 48) PAP-A and PNA clamp PCR are two molecular techniques that have the sensitivity to detect single copies of mutant DNA in a background of abundant wild-type DNA. The utility of PNA clamp PCR in detecting single copies of mutant K-ras alleles has been well established (30–33) however, its use is impractical to detect p53 mutations because of the diverse locations of these mutations. To detect p53 mutations, we utilized the sensitive PAP-A technique. (35, 36) Using these sensitive PCR assays, we detected tumor-specific genetic alterations in 2 out of 12 (17%) of the TRG 0 surgical specimens. This suggests that, similar to what occurs in histologically negative resection margins and lymph nodes, there may be occult cancer cells in some rectal cancer specimens diagnosed with pCR based on histopathological techniques. Our finding may provide a potential explanation as to why some rectal cancer patients develop local recurrence after an apparent complete response after CRT. (10) However, the clinical implications of finding tumor DNA in a surgical specimen with a histopathological diagnosis of pCR are still unknown. To prove that patients with a pCR and mutant DNA behave in a similar fashion to non-pCR patients with respect to local recurrence and oncologic outcomes, a long follow-up and a larger series of patients will be required.
In summary, we have demonstrated that sensitive molecular-genetic techniques can detect tumor-specific gene mutations in rectal cancer specimens diagnosed by standard histopathology as having a pCR in response to CRT. Given the high concordance of K-ras and p53 genes mutations in pre- and post-treatment specimens, the detection of these gene mutation signatures may act as a surrogate of residual or occult disease. This has significant clinical implications for diagnosis of pCR in transrectal biopsies of the tumor bed and when considering a non-operative approach for patients with clinical complete response. Our studies suggest that molecular diagnosis may provide further verification beyond the resolution of histopathology that complete response has in fact been achieved. Future studies are planned to improve the accuracy of pre-operative biopsy such that it may be used to help select therapy after CRT, to follow these patients and look at their long term outcomes, and to extend our study to include additional patients to further strengthen and validate our findings.
Supplementary Material
Acknowledgments
The authors thank Nicola Solomon, PhD, City of Hope, for assistance in writing and editing the manuscript, and Karin Avila, for specimen collection and project management.
This study was supported by the National Institutes of Health (NIH), National Cancer Institute (NCI) R01 Grant CA090559 (JGA). ClinicalTrials.org Identifier: NCT00335816.
Footnotes
Abstract presented at the American College of Surgeons 96th Annual Clinical Congress, Surgical Forum, Washington, DC, October 2010.
Disclosure Information: Nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sauer R, Becker H, Hohenberger W, et al. Preoperative versus postoperative chemoradiotherapy for rectal cancer. N Engl J Med. 2004;351:1731–1740. doi: 10.1056/NEJMoa040694. [DOI] [PubMed] [Google Scholar]
- 2.Janjan NA, Khoo VS, Abbruzzese J, et al. Tumor downstaging and sphincter preservation with preoperative chemoradiation in locally advanced rectal cancer: The M.D. Anderson Cancer Center experience. Int J Radiat Oncol Biol Phys. 1999;44:1027–1038. doi: 10.1016/s0360-3016(99)00099-1. [DOI] [PubMed] [Google Scholar]
- 3.Pucciarelli S, Toppan P, Friso ML, et al. Complete pathologic response following preoperative chemoradiation therapy for middle to lower rectal cancer is not a prognostic factor for a better outcome. Dis Colon Rectum. 2004;47:1798–1807. doi: 10.1007/s10350-004-0681-1. [DOI] [PubMed] [Google Scholar]
- 4.Chari RS, Tyler DS, Anscher MS, et al. Preoperative radiation and chemotherapy in the treatment of adenocarcinoma of the rectum. Ann Surg. 1995;221:778–786. doi: 10.1097/00000658-199506000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diaz-Gonzalez JA, Calvo FA, Cortes J, et al. Prognostic factors for disease-free survival in patients with T3–4 or N+ rectal cancer treated with preoperative chemoradiation therapy, surgery, and intraoperative irradiation. Int J Radiat Oncol Biol Phys. 2006;64:1122–1128. doi: 10.1016/j.ijrobp.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 6.Rodel C, Martus P, Papadoupolos T, et al. Prognostic significance of tumor regression after preoperative chemoradiotherapy for rectal cancer. J Clin Oncol. 2005;23:8688–8696. doi: 10.1200/JCO.2005.02.1329. [DOI] [PubMed] [Google Scholar]
- 7.Valentini V, Coco C, Picciocchi A, et al. Does downstaging predict improved outcome after preoperative chemoradiation for extraperitoneal locally advanced rectal cancer? A long-term analysis of 165 patients. Int J Radiat Oncol Biol Phys. 2002;53:664–674. doi: 10.1016/s0360-3016(02)02764-5. [DOI] [PubMed] [Google Scholar]
- 8.Vecchio FM, Valentini V, Minsky BD, et al. The relationship of pathologic tumor regression grade (TRG) and outcomes after preoperative therapy in rectal cancer. Int J Radiat Oncol Biol Phys. 2005;62:752–760. doi: 10.1016/j.ijrobp.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 9.Borschitz T, Wachtlin D, Mohler M, et al. Neoadjuvant chemoradiation and local excision for T2–3 rectal cancer. Ann Surg Oncol. 2008;15:712–720. doi: 10.1245/s10434-007-9732-x. [DOI] [PubMed] [Google Scholar]
- 10.Maas M, Nelemans PJ, Valentini V, et al. Long-term outcome in patients with a pathologic complete response after chemoradiation for rectal cancer: A pooled analysis of individual patient data. Lancet Oncol. 2010;11:835–844. doi: 10.1016/S1470-2045(10)70172-8. [DOI] [PubMed] [Google Scholar]
- 11.Habr-Gama A, Perez RO, Nadalin W, et al. Operative versus nonoperative treatment for stage 0 distal rectal cancer following chemoradiation therapy: Long-term results. Ann Surg. 2004;240:711–717. doi: 10.1097/01.sla.0000141194.27992.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brennan JA, Mao L, Hruban RH, et al. Molecular assessment of histopathological staging in squamous-cell carcinoma of the head and neck. N Engl J Med. 1995;332:429–435. doi: 10.1056/NEJM199502163320704. [DOI] [PubMed] [Google Scholar]
- 13.Kim J, Reber HA, Dry SM, et al. Unfavorable prognosis associated with K-ras gene mutation in pancreatic cancer surgical margins. Gut. 2006;55:1598–1605. doi: 10.1136/gut.2005.083063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Houten VM, Leemans CR, Kummer JA, et al. Molecular diagnosis of surgical margins and local recurrence in head and neck cancer patients: A prospective study. Clin Cancer Res. 2004;10:3614–3620. doi: 10.1158/1078-0432.CCR-03-0631. [DOI] [PubMed] [Google Scholar]
- 15.Braakhuis BJ, Tabor MP, Kummer JA, et al. A genetic explanation of Slaughter’s concept of field cancerization: Evidence and clinical implications. Cancer Res. 2003;63:1727–1730. [PubMed] [Google Scholar]
- 16.Tabor MP, Brakenhoff RH, Ruijter Schippers HJ, et al. Genetically altered fields as origin of locally recurrent head and neck cancer: A retrospective study. Clin Cancer Res. 2004;10:3607–3613. doi: 10.1158/1078-0432.CCR-03-0632. [DOI] [PubMed] [Google Scholar]
- 17.Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 18.Andreyev HJ, Norman AR, Cunningham D, et al. Kirsten ras mutations in patients with colorectal cancer: The multicenter “RASCAL” study. J Natl Cancer Inst. 1998;90:675–684. doi: 10.1093/jnci/90.9.675. [DOI] [PubMed] [Google Scholar]
- 19.Andreyev HJN, Norman AR, Cunningham D, et al. Kirsten ras mutations in patients with colorectal cancer: The ‘RASCAL II’ study. Br J Cancer. 2001;85:692–696. doi: 10.1054/bjoc.2001.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di-Fiore F, Blanchard F, Charbonnier F, et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by cetuximab plus chemotherapy. Br J Cancer. 2007;96:1166–1169. doi: 10.1038/sj.bjc.6603685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Esteller M, Gonzalez S, Risques RA, et al. K-ras and p16 aberrations confer poor prognosis in human colorectal cancer. J Clin Oncol. 2001;19:299–304. doi: 10.1200/JCO.2001.19.2.299. [DOI] [PubMed] [Google Scholar]
- 22.Goh HS, Elnatan J, Low CH, Smith DR. p53 point mutation and survival in colorectal cancer patients: Effect of disease dissemination and tumour location. Int J Oncol. 1999;15:491–498. doi: 10.3892/ijo.15.3.491. [DOI] [PubMed] [Google Scholar]
- 23.Lievre A, Bachet JB, Le-Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 24.Luna-Perez P, Arriola EL, Cuadra Y, et al. p53 protein overexpression and response to induction chemoradiation therapy in patients with locally advanced rectal adenocarcinoma. Ann Surg Oncol. 1998;5:203–208. doi: 10.1007/BF02303772. [DOI] [PubMed] [Google Scholar]
- 25.Rebischung C, Gerard JP, Gayet J, et al. Prognostic value of P53 mutations in rectal cancer. Int J Cancer. 2002;100:131–135. doi: 10.1002/ijc.10480. [DOI] [PubMed] [Google Scholar]
- 26.Russo A, Bazan V, Iacopetta B, et al. The TP53 Colorect al Cancer International Collaborative Study on the prognostic and predictive significance of p53 mutation: Influence of tumor site, type of mutation, and adjuvant treatment. J Clin Oncol. 2005;23:7518–7528. doi: 10.1200/JCO.2005.00.471. [DOI] [PubMed] [Google Scholar]
- 27.Soong R, Powell B, Elsaleh H, et al. Prognostic significance of TP53 gene mutation in 995 cases of colorectal carcinoma: Influence of tumour site, stage, adjuvant chemotherapy and type of mutation. Eur J Cancer. 2000;36:2053–2060. doi: 10.1016/s0959-8049(00)00285-9. [DOI] [PubMed] [Google Scholar]
- 28.Garcia-Aguilar J, Smith DD, Avila K, et al. Optimal timing of surgery after chemoradiation for advanced rectal cancer: preliminary results of a prospective trial. Ann Surg. 2010 doi: 10.1097/SLA.0b013e3182196e1f. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edge SB, Compton CC, Fritz AG, et al. American Joint Committee on Cancer (AJCC) cancer staging manual. 7. Springer, Inc; Chicago: 2010. [DOI] [PubMed] [Google Scholar]
- 30.Dabritz J, Hanfler J, Preston R, et al. Detection of K-ras mutations in tissue and plasma samples of patients with pancreatic cancer using PNA-mediated PCR clamping and hybridization probes. Br J Cancer. 2005;92:405–412. doi: 10.1038/sj.bjc.6602319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilje B, Heikkila R, Oltedal S, et al. High-fidelity DNA polymerase enhances the sensitivity of a peptide nucleic acid clamp PCR assay for K-ras mutations. J Mol Diagn. 2008;10:325–331. doi: 10.2353/jmoldx.2008.070183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo JD, Chan EC, Shih CL, et al. Detection of rare mutant K-ras DNA in a single-tube reaction using peptide nucleic acid as both PCR clamp and sensor probe. Nucleic Acids Res. 2006;34:e12. doi: 10.1093/nar/gnj008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taback B, Bilchik AJ, Saha S, et al. Peptide nucleic acid clamp PCR: A novel K-ras mutation detection assay for colorectal cancer micrometastases in lymph nodes. Int J Cancer. 2004;111:409–414. doi: 10.1002/ijc.20268. [DOI] [PubMed] [Google Scholar]
- 34.Thiede C, Bayerdorffer E, Blasczyk R, et al. Simple and sensitive detection of mutations in the Ras proto-oncogenes using PNA-mediated PCR clamping. Nucleic Acids Res. 1996;24:983–984. doi: 10.1093/nar/24.5.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi J, Liu Q, Sommer SS. Detection of ultrarare somatic mutation in the human TP53 gene by bidirectional pyrophosphorolysis-activated polymerization allele-specific amplification. Hum Mutat. 2007;28:131–136. doi: 10.1002/humu.20423. [DOI] [PubMed] [Google Scholar]
- 36.Liu Q, Sommer SS. Pyrophosphorolysis-activatable oligonucleotides may facilitate detection of rare alleles, mutation scanning and analysis of chromatin structures. Nucleic Acids Res. 2002;30:598–604. doi: 10.1093/nar/30.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forrester K, Almoguera C, Han K, et al. Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature. 1987;327:298–303. doi: 10.1038/327298a0. [DOI] [PubMed] [Google Scholar]
- 38.Spitz FR, Giacco GG, Hess K, et al. p53 immunohistochemical staining predicts residual disease after chemoradiation in patients with high-risk rectal cancer. Clin Cancer Res. 1997;3:1685–1690. [PubMed] [Google Scholar]
- 39.Yashiro M, Inoue T, Nishioka N, et al. Allelic imbalance at p53 and microsatellite instability are predictive markers for resistance to chemotherapy in gastric carcinoma. Ann Surg Oncol. 2009;16:2926–2935. doi: 10.1245/s10434-009-0590-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Markowitz SD, Bertagnolli MM. Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med. 2009;361:2449–2460. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Artale S, Sartore-Bianchi A, Veronese SM, et al. Mutations of KRAS and BRAF in primary and matched metastatic sites of colorectal cancer. J Clin Oncol. 2008;26:4217–4219. doi: 10.1200/JCO.2008.18.7286. [DOI] [PubMed] [Google Scholar]
- 42.Imperiale TF, Ransohoff DF, Itzkowitz SH, et al. Fecal DNA versus fecal occult blood for colorectal-cancer screening in an average-risk population. N Engl J Med. 2004;351:2704–2714. doi: 10.1056/NEJMoa033403. [DOI] [PubMed] [Google Scholar]
- 43.Loupakis F, Pollina L, Stasi I, et al. PTEN Expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol. 2009;27:2622–2629. doi: 10.1200/JCO.2008.20.2796. [DOI] [PubMed] [Google Scholar]
- 44.Vincent-Salomon A, Jouve M, Genin P, et al. HER2 status in patients with breast carcinoma is not modified selectively by preoperative chemotherapy and is stable during the metastatic process. Cancer. 2002;94:2169–2173. doi: 10.1002/cncr.10456. [DOI] [PubMed] [Google Scholar]
- 45.Klein CA, Blankenstein TJ, Schmidt-Kittler O, et al. Genetic heterogeneity of single disseminated tumour cells in minimal residual cancer. Lancet. 2002;360:683–689. doi: 10.1016/S0140-6736(02)09838-0. [DOI] [PubMed] [Google Scholar]
- 46.Lips EH, van-Eijk R, de-Graaf EJR, et al. Progression and tumor heterogeneity analysis in early rectal cancer. Clin Cancer Res. 2008;14:772–781. doi: 10.1158/1078-0432.CCR-07-2052. [DOI] [PubMed] [Google Scholar]
- 47.Cohen SJ, Punt CJ, Iannotti N, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2009;26:3213–3221. doi: 10.1200/JCO.2007.15.8923. [DOI] [PubMed] [Google Scholar]
- 48.Smerage JB, Hayes DF. The measurement and therapeutic implications of circulating tumour cells in breast cancer. Br J Cancer. 2006;94:8–12. doi: 10.1038/sj.bjc.6602871. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.