

Abstract
Platelet adhesion to adsorbed plasma proteins, such as fibrinogen (Fg), has been conventionally thought to be mediated by the GPIIb/IIIa receptor binding to Arg-Gly-Asp (RGD)-like motifs in the adsorbed protein. In previous studies, we showed that platelet adhesion response to adsorbed Fg and Alb was strongly influenced by the degree of adsorption-induced protein unfolding and that platelet adhesion was only partially blocked by soluble RGD, with RGD-blocked platelets adhering without activation. Based on these results, we hypothesized that in addition to the RGD-specific GPIIb/IIIa receptor, which mediates both adhesion and activation, a non-RGD-specific receptor set likely also plays a role in platelet adhesion (but not activation) to both Fg and albumin (Alb). To identify and elucidate the role of these receptors, in addition to GPIIb/IIIa, we also examined the GPIb-IX-V receptor complex, which has been shown to mediate platelet adhesion (but not activation) in studies by other groups. The platelet suspension was pretreated with either a GPIIb/IIIa-antagonist drug Aggrastat® or monoclonal antibodies 6B4 or 24G10 against GPIb-IX-V prior to adhesion on Fg- and Alb-coated OH- and CH3-functionalized alkanethiol self-assembled monolayer surfaces. The results revealed that GPIIb/IIIa is the primary receptor set involved in platelet adhesion to adsorbed Fg and Alb irrespective of their degree of adsorption-induced unfolding, while the GPIb-IX-V receptor complex plays an insignificant role. Overall, these studies provide novel insights into the molecular-level mechanisms mediating platelet interactions with adsorbed plasma proteins, thereby assisting the biomaterials field develop potent strategies for inhibiting platelet-protein interactions in the design of more hemocompatible cardiovascular biomaterials and effective anti-thrombotic therapies.
1. Introduction
Platelets respond to minimal stimuli, and adhere and activate upon contact with thrombogenic surfaces such as the exposed endothelium/subendothelium at vascular injury sites [1]. These interactions involve the binding of platelet agonists to receptors on the surface of the platelet plasma membrane [2]. Agonists include plasma proteins (e.g., thrombin), components of the vascular wall (e.g., collagen), as well as molecules released by inflammatory cells and platelets (e.g., ADP and serotonin). In the biomaterials field, thrombus formation is recognized as one of the major problems that generally occur whenever blood comes in contact with synthetic material surfaces, with the thrombotic response being induced by platelet interactions with the layer of plasma proteins that tend to rapidly adsorb over the synthetic material surface. In order to understand the factors underlying platelet interactions with these adsorbed plasma proteins, it is imperative to examine the role of the principal platelet receptors that are involved in platelet adhesion and signaling. These receptors function in positive and negative feedback loops, and play a critical role in mediating platelet responses to material surfaces that come in contact with blood [3]. Two of the most prominent platelet receptors that are involved in platelet adhesion and thrombus formation to regulate hemostasis in the body are the αIIbβ3 integrin, which is also known as GPIIb/IIIa, and the GPIb-IX-V receptor complex.
The αIIbβ3 integrin is the most abundant platelet receptor with 60,000–80,000 copies per platelet [4] plus an additional intracellular pool that is transferred to the platelet’s membrane upon activation [5]. αIIbβ3 mediates the adhesion, aggregation and spreading of platelets at vascular injury sites upon activation, as well as during pathological thrombus formation [5, 6]. It is thus considered to be the main platelet receptor involved in regulating thrombosis and hemostasis [7]. The critical role played by this receptor in mediating platelet response is clearly observed in Glanzmann’s thrombasthenia, which is a blood disorder that is associated with impaired platelet adhesion and aggregation as a result of the lack or dysfunction of the αIIbβ3 platelet integrin [8]. Once activated, αIIbβ3 binds several different ligands, including fibrinogen (Fg) and fibrin, von Willebrand factor (vWf), fibronectin (Fn), and vitronectin (Vn); all which contain the arginine-glycine-aspartic acid (Arg-Gly-Asp or RGD) amino acid sequence. In their resting, non-activated state, however, the αIIbβ3 receptors are maintained in a ‘low-affinity’ conformation with their RGD-binding sites believed to be hidden [4]. Upon agonist-induced platelet activation, the receptor changes to its ‘high-affinity’ state as a result of ‘inside-out’ signaling events, leading to conformational changes in the platelet receptor. This change causes the unmasking of the RGD-binding site, thereby mediating platelet adhesion to RGD motifs in adhesive proteins [4].
The GPIb-IX-V receptor complex (25,000 copies per platelet) mediates the initial adhesion of platelets to sites of vascular injury under conditions of high shear via interactions with the A1 domain of vWf, which becomes exposed when vWf specifically adsorbs from the blood to the exposed extracellular matrix of the damaged blood vessel wall [9, 10]. This interaction, however, is insufficient for stable adhesion of the platelets and is characterized by its rapid dissociation rate, the result of which causes platelets roll or translocate along the vessel wall under flow conditions [11]. The primary role of this receptor is therefore understood to be to reduce the velocity of platelets in flowing blood and recruit them to vascular injury sites, thereby facilitating their interactions with the thrombogenic surface via other platelet receptors, notably αIIbβ3.
Although the exact nature of GPIb-vWf interactions remain to be elucidated, the process has been shown to involve conformational changes in the GPIb-IX-V receptor complex, as well as the vWf [12]. A recent study by Lecut et al. [13] showed that the GPIb-IX-V receptor binding to vWf does not induce platelet integrin activation but primarily only functions to mediate platelet adhesion to collagen, thereby establishing one of its primary roles to be that of an adhesive, non-activation receptor. Studies using inhibitory antibodies have narrowed down the vWf binding epitopes on GPIb to amino acids 1–81 and 201–268 [14, 15]. Vanhoorelbeke et al. [16] recently reviewed the potential of GPIb-IX-V as an antithrombotic target, highlighting its role in binding other ligands such as a-thrombin, factor XII (FXII), factor XI (FXI), high molecular weight kininogen (HMWK), macrophage integrin Mac-1 and P-selectin. These findings suggest that GPIb-IX-V receptors play a multifaceted and more important role in mediating platelet responses beyond its recognized ability to bind to vWf at sites of vascular injury.
While much is understood regarding the role that platelet receptors play in normal physiological processes to mediate thrombotic responses, much less is understood regarding how adsorbed proteins influence the interactions of platelets and subsequent thrombus formation with synthetic biomaterials that are used in medical implants, such as stents, vascular prostheses, and ventricular assist devices, or extracorporeal devices used for hemodialysis and blood oxygenation during heart-lung bypass surgery. For example, while studies have shown that non-activated platelets adhere to adsorbed Fg via interactions between αIIbβ3 platelet receptors and the C-terminus amino acid sequence of the γ-chain of Fg [17, 18], it has remained unclear how this is able to actually occur without prior platelet activation when an activation event is first required before platelets will bind to soluble Fg in the blood stream. This behavior suggests that adsorption induces conformational changes in the Fg, as a result of which αIIbβ3 platelet receptors are somehow able to bind to Fg even when they are in their non-activated, resting state. In an effort to address this issue, we previously investigated how platelet adhesion was influenced by the degree of adsorption-induced unfolding of Fg as measured by adsorbed-state circular dichroism spectropolarimetry (CD) [19]. This study showed that platelet adhesion is strongly correlated with the degree of adsorption-induced unfolding of Fg, thus indicating that adsorption is indeed somehow altering the conformational state of Fg leading to the exposure and/or possibly even the formation of sites in Fg for platelet adhesion that are not present when Fg is in its native state in solution. From these studies, we also observed the surprising result that non-activated platelets are even able to adhere to adsorbed albumin (Alb) once a critical degree of adsorption-induced unfolding is reached, with the platelet response again being strongly correlated with the degree of adsorption-induced unfolding and occurring in a manner that is very similar to the platelet adhesion response to adsorbed Fg [20]. We were then able to prove that platelet adhesion to the adsorbed Alb was a receptor-mediated response by demonstrating that the addition of soluble RGD (but not RGE) reduced platelet adhesion by about 60%. This was further confirmed by showing that the chemical modification of arginine residues in the adsorbed Alb resulted in a similar reduction in the platelet adhesion response. The involvement of RGD-specific receptors in platelet adhesion to adsorbed Alb was particularly surprising given the fact that Alb does not contain any RGD sequences in its primary structure. To explain this behavior, we hypothesized that beyond a critical degree of adsorption-induced unfolding, oppositely-charged residues from non-consecutive regions in Alb (e.g., salt bridges) may become spatially oriented such that they form RGD-like motifs, which then become recognizable to the RGD-specific platelet receptors. Scanning electron microscopy evaluation of the adherent platelets from these previous studies also showed the interesting results that the platelets that were adherent to the adsorbed Alb following the addition of soluble RGD or arginine modification exhibited minimal activation in contrast to the much more highly activated state of platelets that adhered to the adsorbed Alb without either of these blocking treatments. The combined results from these studies suggested that two different platelet receptor sets may be responsible for platelet adhesion to adsorbed proteins, such as Alb; one that involves a receptor that binds to RGD-like motifs that are either exposed or formed by adsorption-induced protein unfolding leading to platelet activation, and another set that bind to a non-RGD-like motif that leads to platelet adhesion without activation.
In an effort to determine which platelet receptors are specifically involved in platelet adhesion to adsorbed Fg and Alb, and to determine how the mechanism of platelet adhesion is influenced by the degree of adsorption-induced unfolding, we planned a new set of studies involving the use of blocking agents targeted against specific platelet receptors. We desired to target the αIIbβ3 platelet receptor because of its known ability to bind RGD-like motifs, leading to platelet activation, and the GPIb-IX-V receptor because of its known function to mediate platelet adhesion without activation [13]. The selection of these receptor targets was reinforced by a recent study by Wu et al., which showed that the joint inhibition of the αIIbβ3 platelet integrin and the GPIb-IX-V receptor complex was effective in preventing arterial thrombosis [21], thus pointing to the synergy of these two platelet receptor sets in mediating a platelet-induced thrombotic response. The objectives of our present study were therefore to determine if an approach similar to Wu et al. [21] would also be effective in completely blocking platelet adhesion to adsorbed Fg and Alb, and whether or not the degree of unfolding of these two adsorbed proteins would influence the relative involvement of these two types of platelet receptors.
To accomplish our objectives, we selected Aggrastat® (trade name for tirofiban), which is a peptidomimetic αIIbβ3-antagonist drug, as a suitable blocking agent for the αIIbβ3 platelet receptor [22]. We then selected monoclonal antibodies 24G10 (targeting residues 1–81 of GPIb) and 6B4 (targeting residues 201–268 of GPIb) to block the GPIb-IX-V receptor complex [15]. Fg and Alb were then adsorbed on CH3- and OH-functionalized surfaces to induce substantially different degrees of adsorption-induced unfolding, with the degree of unfolding characterized by adsorbed-state CD. Platelet adhesion studies were then performed on each surface-protein combination with the blocking agents applied to assess their influence on the platelet adhesion response.
2. Materials and Methods
2.1 Gold substrates
Functionalized 1.0 cm × 1.0 cm silicon wafers (Silicon Quest, Inc.) were used as substrates for the platelet adhesion experiments, while functionalized fused silica slides (0.375” × 1.625” × 0.0625”, Chemglass) were used for CD experiments. These substrates were cleaned by immersing them in a piranha solution (7:3 v/v H2SO4:H2O2) at 50°C for 30 min, followed by an RCA basic wash (1:1:5 v/v NH4OH:H2O2:H2O), and this cleaning procedure was repeated twice, as described previously [19, 20]. The cleaned substrates were rinsed with 100% ethanol (Pharmco-Aaper; Catalog No. 111000200) and nanopure water, and then dried using a stream of nitrogen gas.
The cleaned silicon wafers were coated with a 50 Å chromium adhesion layer and 1,000 Å of gold, while the quartz slides for CD were coated with 30 Å of chromium and 100 Å of gold, via a thermal vapor deposition process. The thicknesses of the gold and chromium layers were verified using a DekTak profilometer and a GES5 ellipsometer (Sopra, Inc., Palo Alto, CA).
2.2 Formation of self-assembled monolayers (SAMs) of alkanethiols
The gold-coated substrates were cleaned by dipping them in a modified piranha wash (4:1 v/v H2SO4:H2O2), followed by an RCA basic wash, for 1 minute each and then rinsed with 100% ethanol, prior to incubation in 1.0 mM alkanethiol solutions for 24 h, as per the standard protocols [23, 24].
The following alkanethiols were used for creating the SAM surfaces, and were prepared in 100% ethanol, as described previously [19, 20]:
1-Dodecanethiol (SH-(CH2)11CH3; Aldrich; CH3), and
11-Mercapto-1-undecanol (SH-(CH2)11OH; Aldrich; OH).
The SAM surfaces were cleaned to remove any traces of hydrophobic contaminants on their surface prior to surface characterization and protein adsorption [25]. The CH3-SAM surfaces were cleaned by sonication in ethanol, hexane and ethanol, and then rinsed with nanopure water. The OH-SAMs were sonicated in ethanol, and then incubated in a 25 mM potassium phosphate buffer containing 0.005 volume % Triton-X-100 (Sigma; Catalog No. T-9284), in order to block hydrophobic defect sites (e.g. grain boundaries), and then rinsed thoroughly with acetone, ethanol and nanopure water to remove loosely-bound Triton.
2.3 Buffers
Protein adsorption was carried out in a 25 mM potassium phosphate buffer (pH 7.4), which was made by combining suitable amounts of the mono- and dibasic salts (Sigma-Aldrich), in order to maintain the pH at 7.4. This buffer is recommended for CD experiments for determining the structure of native/adsorbed proteins [26–28] as it permits measurement of CD spectra with minimal noise below 200 nm, especially the positive CD peaks at 193 and 195 nm, which are critical for the accurate determination of the α-helix and β-sheet content of the proteins, respectively.
The platelet suspension buffer (PSB, pH 7.4) contained 137 mM NaCl, 2.7 mM KCl, 5.5 mM dextrose, 0.4 mM sodium phosphate monobasic, 10 mM HEPES and 0.1 U/mL apyrase [29]. 2.5 mM CaCl2 and 1.0 mM MgCl2 were added to the PSB to provide a platelet suspension buffer with metal ions (PSB+MI).
2.4 Protein adsorption
Human Fg (FIB3; plasminogen-, vWf- and Fn-depleted, Enzyme Research Laboratories) and Alb (Sigma, Catalog No. A9511) were dissolved in 25 mM phosphate buffer solution (pH 7.4), to prepare the protein stock solutions, and protein adsorption was carried out as described previously [19, 20], at a protein concentration of 0.1 mg/mL. To briefly summarize, the cleaned SAM surfaces were incubated in 25 mM potassium phosphate buffer (pH 7.4) in a Pyrex petridish and a suitable amount of protein stock solution was then added to give a 0.1 mg/mL bulk protein solution concentration, taking care to ensure that the tip of the pipette was held below the air-water interface to avoid denaturation of the protein at this interface. The surfaces were exposed to the protein solution to allow the protein to adsorb and equilibrate on the surface for 2 h, after which an infinite dilution step was carried out to wash away the bulk protein solution in addition to any loosely adherent protein on the SAM surfaces. The SAM surfaces with preadsorbed protein were then incubated with the platelet suspension in a well-plate.
2.5 Preparation of washed platelet suspension
Platelet adhesion was carried out, using a suspension of washed platelets from human blood, as described in our earlier work [19, 20], in a well plate (Corning Costar, Catalog No. 3527). Briefly, 25.0 mL of blood was collected from healthy volunteers as per research protocols approved by the Institutional Review Board (IRB) and Institutional Biosafety Committee (IBC) at Clemson University. The first few mL of blood was discarded, as it is rich in clotting factors, and then 25 mL of blood was collected in BD Vacutainer tubes (Becton-Dickinson, Catalog No. 364606) containing an acid-citrated dextrose (ACD) anti-coagulant.
The ACD-anticoagulated blood was centrifuged (225g, 15 min, 25°C) to generate platelet rich plasma (PRP), and platelets were separated from PRP via a gel separation method [29], using a liquid chromatography column (Sigma-Aldrich, Catalog No. C4169) containing Sepharose 2B (Sigma-Aldrich, Catalog No. 2B-300). The Sepharose column was first equilibrated with PSB, after which the PRP was carefully pipetted on to the column and allowed to pass into the column. PSB was then added to the column from a reservoir, while keeping the column running. Fractions were collected from the bottom of the column, with the platelet-rich fractions being identified by increased effluent turbidity. These fractions were pooled, and platelet concentration was measured using a Beckman Coulter Z2 Coulter Particle Count and Size Analyzer (Beckman Coulter, Fullerton, CA). The platelet count was then adjusted to 108 platelets/mL with PSB, and CaCl2 and MgCl2 were added to give 2.5 mM and 1.0 mM concentrations of these salts, respectively. The platelet suspension was allowed to rest for 30 min, and then the platelet suspension was placed over each of the protein-coated SAMs for 1 h at 37°C to allow the platelets to interact with the adsorbed proteins.
At the end of the platelet adhesion step, the platelet suspension was aspirated from each well, and the non-adherent platelets were rinsed away by filling and aspirating the wells five times with PBS. The substrates were then transferred to a fresh well-plate, and the adhesion levels were quantified using a lactate dehydrogenase (LDH) assay.
2.6 Measurement of platelet adhesion using lactate dehydrogenase (LDH) assay
The platelets adherent on the Fg- and Alb-coated SAMs were lysed with a Triton-PSB buffer, (2% v/v Triton-X-100 in PSB), and the platelet adhesion levels on the SAMs was quantified by measuring the lactate dehydrogenase (LDH) released from these lysed, adherent platelets using a CytoTox96® Non-Radioactive Cytotoxicity Assay (Promega Corporation, Madison, WI), as described in previous studies by our group [19, 20]. A calibration curve was constructed using a known number of platelets, counted using a Beckman Coulter Z2 Coulter Particle Count and Size Analyzer (Beckman Coulter, Fullerton, CA), and the platelet adhesion on the SAM surfaces was determined from this calibration curve.
2.7 Inhibition of platelet-protein interactions using anti-GPIb antibodies 6B4 and 24G10
Monoclonal antibodies 6B4 and 24G10 against the GPIb platelet receptor were generously provided by Dr. Hans Deckmyn (Laboratory for Thrombosis Research, Katholieke Universiteit, Leuven, Belgium). The platelet suspension was incubated with 5 μg/mL of each anti-GPIb antibody for 30 min at room temperature in order to inhibit any potential interactions between the GPIb platelet receptors and the adsorbed Fg/Alb. These platelets were then allowed to adhere on the Fg- and Alb-coated SAM surfaces for 1 h at 37°C, after which the platelet adhesion was quantified using an LDH assay and the morphology of the adherent platelets was examined using SEM. A platelet suspension without antibody pre-treatment was used as a control.
2.8 Aggrastat treatment of platelets
Aggrastat is a potent αIIbβ3 antagonist, and has been shown to inhibit platelet adhesion and activation on collagen [30]. The washed platelet suspension was treated with 5 μg/mL of Aggrastat (Baxter Healthcare, Product Code 2J1400) for 30 min at room temperature prior to the platelet adhesion step. The Aggrastat-treated platelets were then allowed to adhere to the Fg- and Alb-coated SAMs for 1 h at 37°C, with the platelet adhesion levels being quantified using the LDH assay. A platelet suspension without Aggrastat pre-treatment was used as a control.
2.9 CD studies to quantify the adsorption-induced conformational changes and total surface coverage of Fg and Alb on SAM surfaces
The secondary structures of Fg and Alb in the solution and adsorbed states, along with the surface coverage of adsorbed protein, was determined using a Jasco J-810 spectropolarimeter (Jasco, Inc., Easton, MD), as described in a previous paper [31]. The solution structure was determined using a special high-transparency quartz cuvette (Starna Cells, Inc., Atascadero, CA), while the adsorbed structure of Fg and Alb on the SAM surfaces was determined using a special cuvette, custom-designed by our group, which maximizes the signal-to-noise ratio [31]. The ellipticity of the samples (θ, in mdeg) was converted to molar ellipticity (designated as [θ], with standard units of deg·cm2/dmol) using the following equation [28, 32]:
| (1) |
where L is the path length of the cuvette in cm, Csoln is the solution concentration of the protein in g/mL, and M0 is the mean residue molecular weight of 118 g/mol.
Since proteins exhibit an absorbance peak at 195 nm [33], we used the height of this absorbance peak (A195) for constructing a calibration curve of A195 vs. Csoln for various known concentrations of Fg and Alb [31]. The slope of this plot is “εprotein·L” from Beer’s Law, which can be written as:
| (2) |
where εprotein is the extinction coefficient of the protein in mL · g−1 · cm−1 (or cm2/g) and L is the path length of the cuvette.
The term “Csoln · L” in eq. 2 (above) has units of g/cm2, which is equivalent to the amount of protein per unit area. Assuming that the absorbance is dependent on the total amount of protein present per unit area through which the beam of light passes, irrespective of whether the protein is in the solution or the adsorbed state, the calibration curve of A195 vs. Csoln can also be used for calculating the amount of Fg or Alb adsorbed per unit area on the SAMs (i.e., Qads). The validity of this method for measuring the surface coverage of adsorbed protein has been confirmed by independent measurement of Qads from the thickness of the adsorbed protein film obtained by ellipsometry [31] using de Feijter’s formula [34].
Hence, while calculating the molar ellipticity for the adsorbed Fg or Alb layer on the SAMs, the term “Csoln · L” in eq. 1 can be replaced by the term Qads to give the following equation:
| (3) |
The CD spectra (molar ellipticity vs. wavelength) thus obtained were deconvoluted using the SP-22X algorithm and analyzed using the CONTIN/LL software package to quantify the percentage of α-helix and β-sheet content of the Fg and Alb in the solution and adsorbed states [35].
2.10 Statistical analysis
The results we present are the mean values with 95% confidence intervals (CI). Statistical significance of differences between mean values for different samples and conditions was evaluated using a Student’s t-test, with p ≤ 0.05 considered as statistically significant.
3. Results and Discussion
3.1 Effect of surface chemistry on the conformation of adsorbed Fg and Alb
The secondary structural content (percentage α-helix and β-sheet) of Fg and Alb adsorbed on the OH and CH3 SAMs, measured using CD spectropolarimetry is presented in Table 1, with the native/solution structure included for purposes of comparison.
Table 1.
Comparison of secondary structural content Fg and Alb in solution (native) and adsorbed on the CH3 and OH SAMs from 0.1 mg/mL bulk solution concentration. (n=6, mean ± 95% CI).
| Sample | α-helix [%] | β-sheet [%] |
|---|---|---|
| Native Fg | 40.0 ± 1.1 | 9.6 ± 1.1 |
| OH, 0.1 mg/mL Fg | 40.6 ± 0.5 | 12.2 ± 0.8 |
| CH3, 0.1 mg/mL Fg | 11.3 ± 0.7 | 33.3 ± 1.7 |
|
| ||
| Native Alb | 65.1 ± 1.3 | 2.6 ± 1.1 |
| OH, 0.1 mg/mL Alb | 43.1 ± 2.1 | 13.4 ± 1.1 |
| CH3, 0.1 mg/mL Alb | 14.4 ± 0.7 | 28.3 ± 0.7 |
As indicated, the average conformational states of the adsorbed Fg and Alb were very different on the OH SAMs compared to the CH3 SAMs. The proteins underwent greater adsorption-induced conformational changes (i.e., loss of α-helix content, accompanied by an increase in β-sheet content) on the hydrophobic CH3 SAMs compared to the hydrophilic OH SAMs. This was consistent with observations made by other groups [36, 37], and is attributed to the stronger thermodynamic driving force on these surfaces to cause the protein molecules to unfold on hydrophobic surfaces [31], thereby reducing the overall solvent-accessible surface area of the system [38].
3.2 Effect of anti-GPIb antibodies on platelet adhesion to adsorbed Fg and Alb
Our previous results indicated that platelet adhesion to adsorbed Fg [19] and Alb [20] was potentially mediated by a non-RGD-specific receptor set, in addition to an RGD-specific receptor set. RGDS-treated platelets were found to be moderately spread with little or no filopodia, suggesting that the non-RGD-specific receptor set mediated adhesion without activation [19, 20]. Since the GPIb-IX-V receptor complex is the second most abundant receptor complex on platelets [4], and has been shown to mediate adhesion without activation on collagen [13], we examined its role as the non-RGD-specific platelet receptor set mediating adhesion to adsorbed Fg and Alb. Since studies [14, 15] have implicated two vWf-binding domains in platelet GPIba spanning amino acid residues 1–81 and 201–268, we used monoclonal antibodies 24G10 and 6B4 against these domains, respectively, in delineating the interactions of this receptor complex with adsorbed Fg and Alb.
As shown in Fig. 1, the addition of monoclonal antibodies 24G10 and 6B4 did not lead to a significant decrease in platelet adhesion levels for either the adsorbed Fg or Alb protein layers. Fig. 1 in combination with the data presented in Table 1, also indicate that blocking this receptor did not significantly reduce platelet adhesion irrespective of the degree of adsorption-induced unfolding for either protein, which was vastly different on the OH and CH3 SAMs (~0% Fg unfolding on OH and ~72% Fg unfolding on CH3; ~34% Alb unfolding on OH and ~78% Alb unfolding on CH3). These combined results indicate that the GPIb-IX-V complex does not play a significant role in mediating platelet adhesion to adsorbed Fg and Alb irrespective of its degree of adsorption-induced unfolding.
Figure 1.

Platelet adhesion on OH and CH3 SAM surfaces preadsorbed with Alb and Fg from 0.1 mg/mL solutions blocked with anti-GPIb antibodies 24G10 and 6B4, compared to untreated platelets. (n=6, mean ± 95% CI). * denotes no statistically significant difference, p > 0.05.
3.2 Effect of Aggrastat-pretreatment on platelet adhesion to adsorbed Fg and Alb
Platelet adhesion and activation on collagen has been shown to be inhibited by the αIIbβ3 antagonist Aggrastat [30]. This is a potent, small molecule RGD-mimetic drug, which utilizes the separation distance between the Arg and Asp residues as a key determinant of its potency [39]. We therefore examined the ability of Aggrastat to inhibit interactions with adsorbed Fg and Alb, which, based on literature reports [30], should occur by blocking the αIIbβ3 platelet receptor.
The addition of Aggrastat to the washed platelet suspension prior to the platelet adhesion step led to a strong inhibition of platelet adhesion to both adsorbed Fg and Alb on both the OH- and CH3-SAM surfaces, as shown in Fig. 2. These results thus reveal that the αIIbβ3 integrin is the dominant receptor that mediates platelet adhesion to both of these proteins irrespective of their degree of unfolding on the surface. All but the OH-SAM surface preadsorbed with Alb, however, still exhibited a detectable, non-negligible level of platelet adhesion despite Aggrastat-pretreatment. This small remaining degree of platelet adhesion indicates that either Aggrastat does not completely inhibit the αIIbβ3 receptor or that other receptor(s), such as the aVβ3 integrin, may still be involved. Competitive binding studies are necessary with different concentrations of Aggrastat in solution to determine if the residual degree of platelet adhesion is still being mediated by αIIbβ3 integrin receptors. On the other hand, the αVβ3 receptor, which belongs to the β3 family of integrins, is not inhibited by Aggrastat [40] and is present on platelets with several hundred copies per platelet [3], thus making it a possible candidate as the mediator of this residual platelet adhesion response. Despite this small remaining level of a platelet adhesion, Aggrastat is clearly a potent inhibitor of platelet interactions with adsorbed Fg and Alb under each of the applied conditions, thus implicating the role of αIIbβ3 as the primary receptor responsible for platelet adhesion to adsorbed Fg and Alb irrespective of the degree of adsorption-induced protein unfolding..
Figure 2.
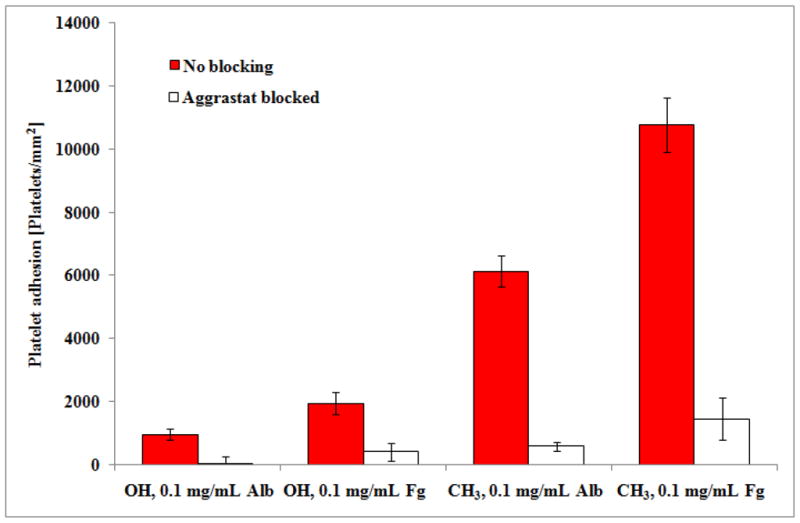
Adhesion of Aggrastat-treated platelets on OH and CH3 SAM surfaces preadsorbed with Alb and Fg from 0.1 mg/mL solutions, compared to untreated platelets. (n=6, mean ± 95% CI). * denotes no statistically significant difference, p > 0.05.
4. Conclusions
These studies were aimed at delineating the roles of the GPIb-IX-V and αIIbβ3 platelet receptors in mediating platelet adhesion to adsorbed Fg and Alb and to determine how adsorption-induced unfolding of these proteins may influence the platelet adhesion response. Although we had initially hypothesized that the former receptor set likely mediated adhesion with little/no activation as the non-RGD-specific receptor, our studies showed that anti-GPIb antibodies 6B4 and 24G10 were unable to inhibit the adhesion of untreated platelets to the adsorbed Fg and Alb layers. Furthermore, the fact that the anti-αIIbβ3 platelet antagonist drug Aggrastat nearly completely inhibited platelet adhesion indicates that the αIIbβ3 platelet integrin is the primary receptor involved in platelet binding to adsorbed Fg and Alb and that the effectiveness of this platelet adhesion inhibitor is not substantially influenced by the degree of adsorption-induced unfolding of these proteins.
Acknowledgments
The authors would like to thank the volunteers at Clemson University who donated blood for this project, as well as the staff at the Redfern Health Center at Clemson University for their assistance with blood-drawing process. We are particularly grateful to Dr. Hans Deckmyn (Katholieke Universiteit, Leuven, Belgium) for kindly providing us with the anti-GPIb antibodies 6B4 and 24G10. We are also would like to acknowledge Dr. James Harriss at Clemson University, for fabrication of the gold-coated surfaces used in our studies. This project was partially supported by NIH Grant Number P20 RR-016461 from the National Center for Research Resources.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gorbet MB, Sefton MV. Biomaterial-associated thrombosis: roles of coagulation factors, complement, platelets and leukocytes. Biomaterials. 2004;25:5681–703. doi: 10.1016/j.biomaterials.2004.01.023. [DOI] [PubMed] [Google Scholar]
- 2.Blockmans D, Deckmyn H, Vermylen J. Platelet activation. Blood Rev. 1995;9:143–56. doi: 10.1016/0268-960x(95)90020-9. [DOI] [PubMed] [Google Scholar]
- 3.Clemetson KJ, Clemetson JM. Platelet receptors. In: Michelson AD, editor. Platelets. 2. Elsevier Science; 2007. pp. 117–44. [Google Scholar]
- 4.Varga-Szabo D, Pleines I, Nieswandt B. Cell adhesion mechanisms in platelets. Arterioscler Thromb Vasc Biol. 2008;28:403–12. doi: 10.1161/ATVBAHA.107.150474. [DOI] [PubMed] [Google Scholar]
- 5.Shattil SJ, Kashiwagi H, Pampori N. Integrin signaling: the platelet paradigm. Blood. 1998;91:2645–57. [PubMed] [Google Scholar]
- 6.Ruggeri ZM. Platelets in atherothrombosis. Nat Med. 2002;8:1227–34. doi: 10.1038/nm1102-1227. [DOI] [PubMed] [Google Scholar]
- 7.Springer TA, Zhu JH, Xiao T. Structural basis for distinctive recognition of fibrinogen gamma C peptide by the platelet integrin alpha(IIb)beta(3) J Cell Biol. 2008;182:791–800. doi: 10.1083/jcb.200801146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nurden AT. Glanzmann thrombasthenia. Orphanet J Rare Dis. 2006;1:1–10. doi: 10.1186/1750-1172-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruggeri ZM. Structure and function of von Willebrand factor. Thromb Haemost. 1999;82:576–84. [PubMed] [Google Scholar]
- 10.Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 1998;94:657–66. doi: 10.1016/s0092-8674(00)81607-4. [DOI] [PubMed] [Google Scholar]
- 11.Ruggeri ZM. von Willebrand factor. J Clin Invest. 1997;99:559–64. doi: 10.1172/JCI119195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrews RK, Berndt MC. Platelet physiology and thrombosis. Thromb Res. 2004;114:447–53. doi: 10.1016/j.thromres.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 13.Lecut C, Schoolmeester A, Kuijpers MJE, Broers JLV, van Zandvoort M, Vanhoorelbeke K, et al. Principal role of glycoprotein VI in alpha 2 beta 1 and alpha IIb beta 3 activation during collagen-induced thrombus formation. Arterioscler Thromb Vasc Biol. 2004;24:1727–33. doi: 10.1161/01.ATV.0000137974.85068.93. [DOI] [PubMed] [Google Scholar]
- 14.Cauwenberghs N, Meiring M, Vauterin S, van Wyk V, Lamprecht S, Roodt JP, et al. Antithrombotic effect of platelet glycoprotein Ib-blocking monoclonal antibody Fab fragments in nonhuman primates. Arterioscler Thromb Vasc Biol. 2000;20:1347–53. doi: 10.1161/01.atv.20.5.1347. [DOI] [PubMed] [Google Scholar]
- 15.Cauwenberghs N, Vanhoorelbeke K, Vauterin S, Westra DF, Rome G, Huizinga EG, et al. Epitope mapping of inhibitory antibodies against platelet glycoprotein Ib alpha reveals interaction between the leucine-rich repeat N-terminal and C-terminal flanking domains of glycoprotein Ib alpha. Blood. 2001;98:652–60. doi: 10.1182/blood.v98.3.652. [DOI] [PubMed] [Google Scholar]
- 16.Vanhoorelbeke K, Ulrichts H, Van de Walle G, Fontayne A, Deckmyn H. Inhibition of platelet glycoprotein Ib and its antithrombotic potential. Curr Pharm Des. 2007;13:2684–97. doi: 10.2174/138161207781662867. [DOI] [PubMed] [Google Scholar]
- 17.Farrell DH, Thiagarajan P, Chung DW, Davie EW. Role of fibrinogen alpha and gamma chain sites in platelet aggregation. Proc Natl Acad Sci USA. 1992;89:10729–32. doi: 10.1073/pnas.89.22.10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horbett TA, Lew KR. Residence time effects on monoclonal antibody binding to adsorbed fibrinogen. J Biomater Sci Polym Ed. 1994;6:15–33. doi: 10.1163/156856295x00724. [DOI] [PubMed] [Google Scholar]
- 19.Sivaraman B, Latour RA. The relationship between platelet adhesion on surfaces and the structure versus the amount of adsorbed fibrinogen. Biomaterials. 2010;31:832–9. doi: 10.1016/j.biomaterials.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sivaraman B, Latour RA. The adherence of platelets to adsorbed albumin by receptor-mediated recognition of binding sites exposed by adsorption-induced unfolding. Biomaterials. 2010;31:1036–44. doi: 10.1016/j.biomaterials.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu DM, Meiring M, Kotze HF, Deckmyn H, Cauwenberghs N. Inhibition of platelet glycoprotein Ib, glycoprotein IIb/IIIa, or both by monoclonal antibodies prevents arterial thrombosis in baboons. Arterioscler Thromb Vasc Biol. 2002;22:323–8. doi: 10.1161/hq0202.102321. [DOI] [PubMed] [Google Scholar]
- 22.Honda S, Tomiyama Y, Aoki T, Shiraga M, Kurata Y, Seki J, et al. Association between ligand-induced conformational changes of integrin alpha(IIb)beta(3) and alpha(IIb)beta(3)-mediated intracellular Ca2+ signaling. Blood. 1998;92:3675–83. [PubMed] [Google Scholar]
- 23.Bain CD, Troughton EB, Tao YT, Evall J, Whitesides GM, Nuzzo RG. Formation of monolayer films by the spontaneous assembly of organic thiols from solution onto gold. J Am Chem Soc. 1989;111:321–35. [Google Scholar]
- 24.Gooding JJ, Mearns F, Yang WR, Liu JQ. Self-assembled monolayers into the 21(st) century: recent advances and applications. Electroanalysis. 2003;15:81–96. [Google Scholar]
- 25.Fears KP, Sivaraman B, Powell GL, Wu Y, Latour RA. Probing the conformation and orientation of adsorbed enzymes using side-chain modification. Langmuir. 2009;25:9319–27. doi: 10.1021/la901885d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coelho MAN, Vieira EP, Motschmann H, Mohwald H, Thunemann AF. Human serum albumin on fluorinated surfaces. Langmuir. 2003;19:7544–50. [Google Scholar]
- 27.Damodaran S. In situ measurement of conformational changes in proteins at liquid interfaces by circular dichroism spectroscopy. Anal Bioanal Chem. 2003;376:182–8. doi: 10.1007/s00216-003-1873-6. [DOI] [PubMed] [Google Scholar]
- 28.Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. 2006;1:2876–90. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grunkemeier JM, Tsai WB, Horbett TA. Co-adsorbed fibrinogen and von Willebrand factor augment platelet procoagulant activity and spreading. J Biomater Sci Polym Ed. 2001;12:1–20. doi: 10.1163/156856201744416. [DOI] [PubMed] [Google Scholar]
- 30.Van de Walle GR, Schoolmeester A, Iserbyt BF, Cosemans J, Heemskerk JWM, Hoylaerts MF, et al. Activation of alpha(IIb)beta(3) is a sufficient but also an imperative prerequisite for activation of alpha(2)beta(1) on platelets. Blood. 2007;109:595–602. doi: 10.1182/blood-2005-11-011775. [DOI] [PubMed] [Google Scholar]
- 31.Sivaraman B, Fears KP, Latour RA. Investigation of the effects of surface chemistry and solution concentration on the conformation of adsorbed proteins using an improved circular dichroism method. Langmuir. 2009;25:3050–6. doi: 10.1021/la8036814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akaike T, Sakurai Y, Kosuge K, Senba Y, Kuwana K, Miyata S, et al. Study on the interaction between plasma-proteins and polyion complex by circular-dichroism and ultraviolet spectroscopy. Kobunshi Ronbunshu. 1979;36:217–22. [Google Scholar]
- 33.Qiu J, Lee H, Zhou C. Analysis of guanidine in high salt and protein matrices by cation-exchange chromatography and UV detection. J Chromatogr A. 2005;1073:263–7. doi: 10.1016/j.chroma.2004.10.045. [DOI] [PubMed] [Google Scholar]
- 34.de Feijter JA, Benjamins J, Veer FA. Ellipsometry as a tool to study the adsorption behavior of synthetic and biopolymers at the air–water interface. Biopolymers. 1978;17:1759–72. [Google Scholar]
- 35.Sreerama N, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem. 2000;287:252–60. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 36.Andrade JD, Hlady V. Protein adsorption and materials biocompatibility: a tutorial review and suggested hypotheses. Adv Polym Sci. 1986;79:1–63. [Google Scholar]
- 37.Roach P, Farrar D, Perry CC. Interpretation of protein adsorption: surface-induced conformational changes. J Am Chem Soc. 2005;127:8168–73. doi: 10.1021/ja042898o. [DOI] [PubMed] [Google Scholar]
- 38.Latour RA. Biomaterials: protein-surface interactions. In: Wnek GE, Bowlin GL, editors. The Encyclopedia of Biomaterials and Bioengineering. Informa Healthcare; 2008. pp. 270–84. [Google Scholar]
- 39.Cook JJ, Bednar B, Lynch JJ, Gould RJ, Egbertson MS, Halczenko W, et al. Tirofiban (Aggrastat (R)) Cardiovasc Drug Rev. 1999;17:199–224. [Google Scholar]
- 40.Lele M, Sajid M, Wajih N, Stouffer GA. Eptifibatide and 7E3, but not tirofiban, inhibit alpha(v)beta(3) integrin-mediated binding of smooth muscle cells to thrombospondin and prothrombin. Circulation. 2001;104:582–7. doi: 10.1161/hc3101.092199. [DOI] [PubMed] [Google Scholar]