

Abstract
Hydroxysteroid dehydrogenases (HSDs) represent a major class of NAD(P)(H) dependent steroid hormone oxidoreductases involved in the pre-receptor regulation of hormone action. This is achieved by HSDs working in pairs so that they can interconvert ketosteroids with hydroxysteroids resulting in a change in ligand potency for nuclear receptors. HSDs belong to two protein superfamilies the aldo-keto reductases and the short-chain dehydrogenase/reductases. In humans, many of the important enzymes have been thoroughly characterized including the elucidation of their three-dimensional structures. Because these enzymes play fundamental roles in steroid hormone action they can be considered to be drug targets for a variety of steroid driven diseases: e.g. metabolic syndrome and obesity, inflammation, and hormone dependent malignancies of the endometrium, prostate and breast. This article will review how fundamental knowledge of these enzymes can be exploited in the development of isoform specific HSD inhibitors from both protein superfamilies.
Keywords: aldo-keto reductases, short-chain dehydrogenase/reductases, steroid hormone receptors, selective steroid receptor modulator
1. Introduction
In steroid hormone target tissues pairs of hydroxysteroid dehydrogenases (HSDs) co-exist which interconvert potent steroid hormones with their cognate inactive metabolites and are thus uniquely positioned to regulate the amount of ligand available to bind and trans-activate nuclear receptors [1]. This is achieved by HSDs functioning preferentially as either NADPH-dependent ketosteroid reductases or as NAD+-dependent hydroxysteroid oxidases. The enzymes responsible for these transformations belong to two large gene superfamilies the aldo-keto reductases (AKRs) and the short-chain dehydrogenase reductases (SDRs) [2, 3].
HSDs catalyze positional and stereospecific reactions on ketone or hydroxy- substituents on the steroid nucleus and side-chain. For example starting with the C3 position, type 3 3α-HSD (AKR1C2) in the prostate is responsible for the reduction of 5α-dihydrotestosterone (a potent androgen) to yield 3α-androstanediol (a weak androgen), whereas “RoDH like 3α-HSD” (17β-HSD6) is responsible for the reverse reaction [4, 5]. Thus this enzyme pair acts as a molecular switch to regulate ligand occupancy of the androgen receptor (AR), Scheme 1. At the C11 position, type 1 11β-HSD is responsible for the reduction of cortisone (a weak glucocorticoid) to yield cortisol (a potent glucocorticoid), whereas type 2 11β-HSD will catalyze the reverse reaction. Thus this enzyme pair will act as a molecular switch to regulate the ligand occupancy of the mineralocorticoid receptor (MR) in the kidney, where cortisol has high affinity for the MR. The same enzyme pair can also regulate the ligand occupancy of the glucocorticoid receptor (GR) in peripheral tissues [6–8], where type 1 11β-HSD can act as a local amplifier of glucocorticoid action. At the C17 position, type 1 17β-HSD will catalyze the reduction of estrone (weak estrogen) to yield 17β-estradiol (a potent estrogen) in the breast, whereas type 2 and type 4 17β-HSD will catalyze the reverse reaction [9–11]. Thus these enzymes will act as molecular switches to regulate ligand occupancy of the estrogen receptor (ER). Additionally, type 5 17β-HSD (AKR1C3) will catalyze the reduction of Δ4-androstene-3,17-dione (a weak androgen) to testosterone (a potent androgen) in the prostate, whereas type 2 and type 4 17β-HSD will catalyze the reverse reaction [12, 13]. Thus these enzymes will act as molecular switches to regulate ligand occupancy of the AR. Finally, at the C20 position, 20α(3α-)-HSD (AKR1C1) is responsible for the reduction of progesterone (a potent progestin) to yield 20α-hydroxyprogesterone (a weak progestin) while type 2 17β-HSD will catalyze the reverse reaction [10, 14, 15]. Thus these enzymes will act as a molecular switch to regulate ligand access to the progesterone receptor (PR).
Scheme 1.

Pre-receptor regulation of hormone action by HSDs
Thus pairs of HSDs can regulate ligand occupancy of the AR, MR, GR, ER and PR in a tissue specific manner. Specific inhibitors of these enzymes could benefit the treatment of hormone dependent malignancies driven by androgens and estrogens as well as metabolic syndrome where the underlying cause may be related to excess local glucocorticoid production [7]. During the last ten years tremendous progress has been made in identifying the discrete HSDs involved in the tissue specific control of steroid hormone levels and this has culminated in the elucidation of the crystal structures of many of the relevant enzymes. Thus the field is poised for the development of HSD isoform specific inhibitors for clinical use.
Concurrently, rapid progress has been made in the development of selective steroid receptor modulators (SSRMs) which can act as agonists in some tissues and antagonists in another to gain tissue specific effects of steroid hormones [16, 17]. In this instance, ligand induced conformational changes dictate whether there is recruitment of co-activators or co-repressors to the steroid receptor-ligand complex. It is apparent that rational design of SSRMs is challenging due to the targeting of macromolecule complexes of increasing complexity. By contrast, HSDs represent single protein entities for which structures exist and appear to be more tractable targets to attain tissue specific hormone effects. Because HSDs are involved in the intracrine regulation of steroid hormone action [11, 18], drugs that act in this manner can be referred to as “Selective Intracrine Modulators (SIMs)”. It is predicted that SSRMs and SIMs will have the same pharmacological effect but different modes of action. In the former case, the pharmacology is at the receptor level, but in the latter case the pharmacology is at the enzyme level. For example, RU486 (a PR antagonist) can be used to terminate early pregnancy by depriving the PR of its agonist [19]. By contrast epostane (a type 1 3β-HSD inhibitor) can also be used to terminate pregnancy by blocking the intracrine formation of progesterone at the enzyme level [20]. This article will review some of the principles that should be considered in designing and evaluating tissue specific HSD inhibitors. The reader is also referred to other articles in this special issue on hydroxysteroid inhibitors, [21–27].
2. Consideration of HSD Enzyme Superfamily
HSDs belong to one of two protein superfamilies the AKRs and SDRs, which differ in their protein folds, stereochemistry of hydride transfer, kinetic, and catalytic mechanisms, Schemes 2,3 [3, 28]. An appreciation of these differences is important in inhibitor design and evaluation.
Scheme 2.

Properties of AKRs. Inset shows structure of the AKR1C2●NADP+●Ursodexycholate complex (PDB11H1) (blue = α-helix, green = β-strand, red = loop structures, yellow-stick = cofactor, and red-stick = steroid), where the amino acid numbering is given for rat liver 3α-hydroxysteroid dehydrogenase (AKR1C9).
Scheme 3.

Properties of SDRs. Only some representative SDRs are shown. Inset shows the monomer structure of human 17β-HSD type1●NADP+● 17β-estradiol complex (1FDS) (blue = α-helix, green = β-strand, yellow = loop structures, magenta-stick = cofactor, and red-stick = steroid)
HSDs in the AKR superfamily are NAD(P)(H)-dependent oxidoreductases and work in cells predominately in the reduction direction due to their very high affinity for NADP(H) [5, 29]. They can be thought of as NADPH-dependent ketosteroid reductases. Human HSDs which belong to the AKR superfamily include: 20α(3α)-HSD (AKR1C1); type 3 3α-HSD (AKR1C2); type 5 17β-HSD (AKR1C3); and type 1 3α-HSD (AKR1C4) [15, 28]. They are soluble enzymes and are monomeric. They exhibit a high degree of sequence identity (>86%) and have a characteristic (αβ)8 barrel protein fold. This fold is often referred to as a triose-phosphate isomerase (TIM) barrel. The fold consists of an alternating arrangement of α-helices and β-strands which repeats eight-times, where the β-strands coalesce in the center of the structure to form the staves of a barrel. At the back of the barrel three large loops exist which act as antennae to capture the steroid substrate and cap the steroid once bound. An appreciation of this loop-structure is critical in homology modeling especially when using computer-assisted docking experiments, or in silico-drug screening to aid-drug design.
HSDs in the SDR superfamily are NAD(P) (H)-dependent oxidoreductases and can work either as ketosteroid reductases or hydroxysteroid oxidases based on their preference for NADP(H) or NAD(H). Human HSDs that belong to this superfamily include: type 1 3β-HSD and type 2 3β-HSD ; type 1 11β-HSD and type 2 11β-HSD; and all 17β-HSD isoforms with the exception of type 5 17β-HSD [11, 30–32]. These enzymes are often membrane bound and multimeric. They exhibit a low degree of sequence identity (> 20%) but share a characteristic protein fold in which there are seven β-strands flanked on either side by three α-helices. The cofactor straddles a Rossmann fold [3]. An appreciation of this structure shows that it is hard to predict based on amino sequence alone making homology building challenging.
3. Consideration of Kinetic Mechanism
AKRs and SDRs can catalyze their bi-substrate reactions via different kinetic mechanisms. An appreciation of these mechanisms is important since they identify the number of different enzyme forms available for inhibitor binding.
AKRs catalyze sequential reactions leading to the formation of a central complex (ternary complex) in which chemistry takes place. This sequential reaction is without exception an ordered bi bi reaction. In this sequence, the binding of cofactor is the obligatory first step, steroid then binds, the central complex forms and chemistry occurs. After the chemical step the steroid product and cofactor product are released in that order, see Scheme 4 [33, 34].
Scheme 4.
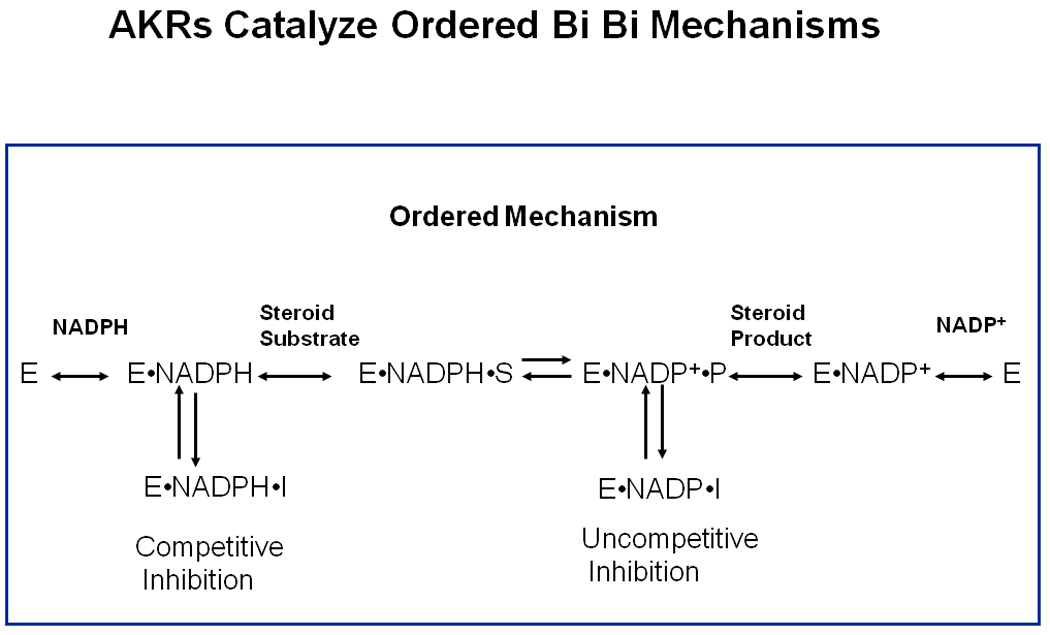
Ordered Bi Bi Kinetic Mechanism used by AKRs. In this scheme: E = enzyme; S = steroid substrate; P = steroid product; and I = inhibitor.
This kinetic mechanism predicts that ligands that bind in the steroid site can bind to either the E●NADPH or E●NADP+ complex, which will give competitive and uncompetitive inhibition patterns, respectively, when the concentration of the ketosteroid substrate is varied. In both instances an abortive ternary complex is formed Scheme 4. The E●NADP+●I complex is a dead-end complex and is responsible for the uncompetitive inhibition pattern. Often investigators may be deterred if uncompetitive inhibition patterns are observed but this is exactly what the kinetic mechanism predicts.
Examination of the rate-determining step in AKRs has been dissected by measuring transient fluorescence changes by stopped flow spectrometry associated with cofactor binding. These studies show that it is often the release of cofactor that is rate-determining since the initial E●NADP(H) complex undergoes isomerization events (associated with loop movement) to form a very tight E**●NADP(H) complex [34, 35]. Thus second ligands that bind tightly to the E**●NADP(H) complex would be desirable since they may form an abortive ternary complex that would be hard to dissociate.
SDRs also catalyze a sequential reaction mechanism. However, depending on the SDR member the mechanism can occur in the absence of a measurable ternary complex (Theorell-Chance) or via a ternary complex (central complex) in which chemistry takes place [36, 37]. In the latter case this sequential reaction can be either an ordered bi bi reaction (see above), or it can be a random mechanism. In the random mechanism either steroid or cofactor can bind first on route to the central complex (Scheme 5). Of the human enzymes of interest type 1 11β-hydroxysteroid dehydrogenase catalyzes an ordered bi bi mechanism [38]. Type 1 17β-hydroxysteroid dehydrogenase was originally thought to catalyze either an ordered mechanism in which steroid bound first or an iso-Theorell Chance mechanism with cofactor binding first [39]. However, crystallographic data supports the presence of a random mechanism since a binary complex of a type1 17β-HSD●NADP+ (1QYV) has been reported and a large number of crystal structures of binary complexes which contain only steroid ligand e.g. type 1 17β-HSD ●17β-estradiol (1FDS); type 1 17β-HSD ●testosterone (1JTV); type 1 17β-HSD●dehydroepiandrosterone (3DHE); and 17β-HSD● 5α-dihydrotestosterone (3KLM) also exist. Importantly, these steroid binary complexes were formed by co-crystallization rather than by crystal soaking. The existence of binary complexes that contain only one ligand supports a random kinetic mechanism. This random kinetic mechanism predicts that inhibitors that bind to the steroid site can bind to one of three enzyme forms. E, E●NAD(P)+, or E●NAD(P)H. Since the ligand can bind to both free enzyme and binary complexes mixed-type inhibition patterns may be observed. These mixed-type inhibition patterns can be simplified if the enzyme is saturated with cofactor first to yield competitive patterns. Often investigators are deterred when mixed-type inhibition patterns emerge but once again they are the predicted ones.
Scheme 5.

Random Bi Bi Kinetic Mechanism used by some SDRs. The binding of substrates (top panel) and the binding of an inhibitor against B (bottom panel) is show. In this scheme: E = enzyme; A = NAD(P)(H); B= steroid substrate; P = steroid product and Q = NAD(P)(H); I = inhibitor
In light of the different kinetic models for AKRs and SDRs, it is important to underscore that comparison of IC50 values for inhibitors is only valid if the inhibitors are binding to the same enzyme complex. Often this is the case for inhibitors from the same structural class. But distinct differences are possible when inhibitors are derived from different structural classes of compounds. It is recommended that for each structural class of inhibitor a complete kinetic profile be performed with the lead compound to determine the pattern of inhibition. Only when patterns of inhibition are the same across different structural classes of inhibitor can the IC50 values be directly compared. When the pattern of inhibition is competitive, the IC50 values can be converted directly to Ki values provided the Km for the steroid substrate is known using the Cheng-Prussof relationship [40]. If only IC50 values are computed it is important to know that all compounds follow the same pattern of inhibition, and to keep the substrate concentration across IC50 value determination constant, and at a value equal to Km. Without these considerations comparison of IC50 values can be meaningless.
4. Consideration of Stereochemistry
HSDs in both families catalyze the reduction of ketones and the oxidation of secondary alcohols. In the reduction direction the ketosteroid is reduced to only one of two stereoisomeric products. In the oxidation direction they are stereospecific for the alcohol utilized. A few exceptions exist in which there is an epimerase activity e.g. a 3α-hydroxy group is converted to a 3β-hydroxy group. But this invariably occurs via the intermediacy of the corresponding ketone and requires build up of the NAD(P)H cofactor [4, 41, 42].
AKRs are A-face dehydrogenases so that during the catalytic mechanism there is 4-pro-R-hydride transfer from the A-face of the cofactor to the recipient carbonyl [43, 44]. To achieve this stereochemical outcome the nicotinamide head group of the cofactor is bound in an extended anti-conformation with respect to the N-glycosidic bond [33, 45]. Hydride transfer is facilitated by protonation of the recipient carbonyl by a highly conserved catalytic tetrad Tyr 55, Lys 84, His 117 and Asp50, where Tyr 55 acts as the general acid-base (numbering with respect to the prototypic rat liver 3α-HSD, AKR1C9) [46–48].
By contrast SDRs are B-face dehydrogenases so that during the catalytic mechanism there is 4-pro-S-hydride transfer from the B-face of the cofactor to the recipient carbonyl [49]. To achieve this stereochemical outcome the nicotinamide head group of the cofactor is bound in a syn-conformation with respect to the N-glycosidic bond. Hydride transfer is facilitated by protonation of the recipient carbonyl by highly conserved catalytic residues Tyr-X-X Ser-(Lys), where the Tyr acts as the general acid-base [50–52]. Thus any bisubstrate analog of AKRs or SDRs must take into account these stereochemical constraints.
5. Consideration of Catalytic Mechanism
Both AKRs and SDRs rely on a catalytic tyrosine to act as the general acid-base in the reduction and oxidation directions, respectively. Spatial overlay of the catalytic residues in AKR1C9 (rat 3α-HSD) with those in the 3α,20α-HSD from Streptomyces hydrogenas (an SDR) revealed that once the nicotinamide head group was flipped to compensate for the differences in the stereochemistry of hydride transfer there was excellent superimposition (1.8 Ǻ rmsd) of the catalytic Tyr, Lys and His from AKRs with the Tyr, Lys, Ser from SDRs [53]. This led to the concept that there had been convergent evolution to a common catalytic mechanism for all HSDs. Further, details of the catalytic mechanisms for the two superfamilies have emerged by detailed site-directed mutagenesis. In the reduction direction in AKRs, the catalytic Tyr partners with the adjacent His residue to generate TyrOH2 + character for proton donation to the recipient carbonyl [46–48]. In the oxidation direction the Tyr partners with the adjacent Lys residue to generate TyrO− (phenolate anion) for proton abstraction from the steroid alcohol, Scheme 6. This mechanism predicts that in the reduction direction an oxyanion hole is generated. It also predicts that the enzyme can exist in two protonation states and a diprotic model has been proposed for enzyme catalysis [48]. In the case of SDRs, the catalytic Tyr is linked to the adjacent lysine so that its pKa is modulated for either reduction or oxidation. In this mechanism the lysine is part of an extensive proton relay that links to the 2’OH group of the nicotinamide ribose[54] that was further elaborated upon by Negri et al for 17β-HSD type 1 [55], Scheme 7.
Scheme 6.

Catalytic Mechanisms for AKRs
Scheme 7.

Catalytic mechanism for SDRs. From Filling C et al., J. Biol. Chem. 2002, 277, 25677–25684. Reproduced with permission from American Society of Biochemists and Molecular Biologists.
Akhtar et al proposed three possible mechanisms for hydride transfer to a recipient carbonyl [56] and these mechanisms are possible for both AKRs and SDRs, Scheme 8. First, if hydride transfer to the recipient carbonyl occurs before proton donation the intermediate transition state would be an oxyanion. Second, if hydride transfer to the recipient carbonyl occurs after proton donation the intermediate transition state would be a carbocation. Third, if a concerted mechanism occurs with hydride transfer and proton donation occurring simultaneously the transition state would be neutral. Knowledge of this mechanism could have profound consequences on the design and synthesis of transition state analogs for HSDs since it will define whether these should be charged or neutral. Despite the important need to identify the transition state in HSDs, this has yet to be performed with rigor for any HSD isoform. To solve the problem both primary and solvent kinetic isotope effect measurements would have to be performed under conditions in which the chemistry step is isolated kinetically, and this may require the use of transient kinetic approaches (e.g. single turnover experiments). Despite this lack of knowledge many negatively charged inhibitors have been used as HSD inhibitors suggesting a role for the putative oxyanion hole [57–62].
Scheme 8.

Mechanisms of ketosteroid reduction.
6. Thermodynamic Considerations
In vitro, HSDs irrespective of their superfamily can function as fully reversible oxidoreductases where the reaction is governed by an equilibrium constant Keq (equation 1) [63].
| (Equation 1) |
Examination of this equation shows that the direction of the reaction will be governed by cofactor ratio NAD(P)+ : NAD(P)H and by pH. Manipulation of these conditions can thus force the reaction in either the reduction or oxidation direction [5, 29]. Direct measurement of the Keq shows that AKRs have a Keq that favors reduction at physiological pH [29, 34]. Moreover the 200-fold higher affinity for NADPH over NAD+ suggests that they will work as ketosteroid reductases in a cellular environment. Transfection studies of cDNA’s for HSDs into mammalian cells where the HSD is forced to use the prevailing concentration of NAD(P)(H) have been used by investigators to assign the directionality of HSDs [5, 30]. Careful work by Auchus has exposed the danger of this approach since once the enzymes reach their set equilibrium, isotope scrambling demonstrates interconversion of ketosteroids and hydroxysteroids occurs freely in both directions. This work clearly demonstrated that while AKRs had an equilibrium set point that favored reduction this was less clear for 17β-HSD isoforms of the SDR family. However, this approach has its own short-comings since these experiments were performed in HEK-293 cells where the HSD was examined out of context of the steroid metabolic pathway in which it resides [64, 65]. In steroid hormone target tissues the equilibrium end-point many not be reached due to the flux of the keto- or hydroxysteroid down a metabolic pathway.
7. Validating the Target
We and others have made a number of recommendations regarding the validation of an HSD as a drug target [1, 66]. These include: (a) demonstration that the recombinant enzyme (homogeneous or by transfection studies) performs the desired reaction; (b) comparison of the catalytic efficiency of the enzyme (kcat/Km) for the measured reaction versus other isozymes that perform the same reaction; (c) measurement of the HSD (transcript, protein and functionality) in the target organ and cell; (d) demonstration that the HSD is necessary and sufficient to alter trans-activation of a steroid hormone receptor by modulating ligand levels; (e) demonstration that the target shows altered expression in the disease state; (f) conduct proof-of-principle experiments to determine the consequence of enzyme inhibition using si-RNA (cell-based work) or knock-out animals when a rodent ortholog exists; and (g) examine human HSD deficiency syndromes to predict consequences of enzyme inhibition. In this regard, work by the Bunce group has clearly shown that a knock-out mouse approach is not feasible to study the effects of inhibitors on AKRs since the murine homologs of the human enzymes are not possible to assign [67].
8. HSD Inhibitor Classes
On writing this article PubMed listed 1,917 articles for a search of “hydroxysteroid dehydrogenase and inhibitors”. In these articles HSD inhibitors belong to several major classes of compounds: reversible (steroidal and nonsteroidal); bisubstrate analogs; mechanism-based inactivators; as well as natural products and xenobiotics (endocrine disrupting chemicals). Apart from epostane and trilostane which are 3β-HSD inhibitors there are no compounds that have yet been approved for clinical use. However, phase II clinical trials are advanced for type 1 11β-HSD inhibitors [PF(Pfizer)-91575 and INCB(Incyte)-13739] for the treatment of metabolic syndrome and type 2 diabetes [68, 69]. While one of the most promising type 1 17β-HSD inhibitors for the treatment of hormonally dependent breast cancer is STX (Sterix)1040 has undergone extensive preclinical evaluation [66, 70]. In addition a search of the patent literature identified a large number of patents for inhibitors of the different 17β-HSD isoforms [71]. The reader is also referred to a recent comprehensive review of 17β-HSD inhibitors [70].
8. 1. Reversible HSD Inhibitors
Reversible HSD inhibitors can be either steroidal or nonsteroidal [72]. Steroid based ligands have traditionally raised concerns that they could have unintended off-targets e.g. inhibit other steroid hormone transforming enzymes or act as steroid hormone receptor agonists or antagonists. However, with the emergence of SSRMs based on quite different nonsteroidal scaffolds, nonsteroid based reversible inhibitors may also suffer from the same specificity concerns. The advantage of the non-steroidal compounds is that there is enormous structural diversity that can be exploited whereas the rigid steroid ring system limits the size of an inhibitor library. Whichever approach is used lead compounds (sub-micromolar affinity) need to be counter-screened extensively against possible unintended targets.
For HSDs which belong to the AKR superfamily, nonsteroidal based inhibitors have been based on nonsteroidal anti-inflammatory drugs (NSAIDs) following the early observations of Penning & Talalay [61] which showed that these drugs potently inhibited rat liver 3α-HSD (AKR1C9). Human AKR1C isoform specificity can be achieved by using different NSAID scaffolds. Thus N-phenylanthranilic acids are non-discriminatory between AKR1C1 to AKRC14, salicylate inhibitors are specific for AKR1C1 and AKR1C2, while indomethacin and indole acetic acids display specificity for AKR1C3 [57, 58]. Efforts have been made to exploit known structure-activity relationships for COX-1 and COX-2 (accepted NSAID) targets to develop inhibitors that retain AKR1C specificity while eliminating activity for COX-inhibition. Lead compounds, however, still require secondary screening to ensure that this achieved and that inhibition of other human AKRs does not occur. This is particularly important since recent studies show that AKR1B10 is inhibited by N-phenylanthranilates [73]. Recently, 3-(phenylamino)benzoic acids which show nanomolar affinity for AKR1C3 which are 200–300 fold selective versus other AKR1C isoforms have been developed [74]. Salicylate analogs have been pursued as potential specific inhibitors of AKR1C1 since their Ki values are 2-orders of magnitude lower for AKR1C1 than those reported for the reversible inhibition of COX enzymes. The challenge has been to eliminate AKR1C2 inhibition from the salicylates. Lead compounds include 3,5-dichlorosalicylic acid and 3’-bromo-5’-phenylsalicylate where the additional phenyl group provides a 21-fold selectivity for inhibition of AKR1C1 over AKR1C2 [59, 75].
For HSDs that belong to the SDR superfamily, non-steroidal based ligands have been based on a number of different pharmacophores. For type 1 11β-HSD, inhibitor series include diverse scaffolds: triazoles (Merck-544) and azabicyclic sulphonamides developed by both Merck and Eli Lilly [76, 77]; pentanedioic acid diamides developed by Merck-Serono [78]; pyridinyl arylsulfonamides developed by Pfizer (PF-915275) [79]; (phenylsulfonamido-methyl)-nicotine and (phenylsulfonamido-methyl)-thiazole derivatives [80]; arylsulfonylpiperazines, piperdyl- and cyclo-benzamides developed by Amgen (Amgen 2922) [81–83]; thiazolones developed by Biovitrum (BVT-2733) [84]; 1,5-substituted-1H-tetrazoles developed by the group at Edinburgh [85]; and adamantyl ethanones developed by Ipsen/Sterix [86]. Many of these have nM affinity for the target. However, the challenge is to have high selectivity for type 1 11β-HSD over type 2 11β-HSD and to have compounds that may be more efficacious and less toxic than metformin (a biguanide oral anti-diabetic). In this regard, the thiazolones and the adamantyl ethanones have excellent selectivity.
For type 1 17β-HSD inhibition, inhibitors have been developed based on bis-(hydroxyphenyl)azoles [87], 6-phenyl-2-naphthols [88, 89] and substituted, thiophenes, benzenes and aza benzenes [90, 91]. The most promising compounds had selectivity for type 1 17β-HSD over type 2 17β-HSD and have low binding affinity for ERα. Several of the lead non-steroidal compounds mimic the substrate estrone. They contain a phenolic ring that will bind to the site occupied by the phenolic A ring of estrone and a bridge to another ring that will occupy the site that binds the D-ring of estrone, which is in proximity to the catalytic residues of the enzyme [92]. A similar approach was used to develop biphenyl ethanone-based molecules which showed promising inhibition of type 1 17β-HSD, where the best compounds had an IC50 = 3.7 µM [93]. For type 3 17β-HSD, inhibitors with pM-nM affinity based on tetrahydrodibenzazocines have been developed for prostate cancer [94]. Using these compounds as leads, an homology model of type 3 17β-HSD was built for structure-aided drug design which led to 1-(4-[2-(4-chloro-phenoxy)phenylamino]-piperidin-1-yl)ethanone which inhibited the enzyme with an IC50 770 nM in transfected cells [95].
8.2. Bisubstrate Analogs
Bisubstrate analogs refer to compounds that contain both a portion of the cofactor and steroid substrate or product. This approach has been used extensively to develop 17β-HSD inhibitors by Poirier and co-workers [96–98] and by the Reed group [66, 99, 100]. While these compounds may have the potential to be higher affinity ligands than steroid-based reversible inhibitors, there are several challenges. First, if the HSD target has an ordered bi bi kinetic mechanism it is uncertain that these compounds will even bind since they may have insufficient cofactor character. Second, the synthesis of these compounds can be challenging. Third, bisubstrate analogs for AKRs may by necessity require to be linked to a AMP analog with a 2’phosphate group. This phosphate group is essential for the anchoring of the cofactor where the tight binding of the cofactor precedes steroid binding in the ordered bi bi mechanism [33–35]. Additionally, phosphate analogs and their mimics have difficulty crossing the plasma membrane. Despite these concerns a bi-substrate analog of type 1 17β-HSD which contains a 16β-methylene-benzamide substituent (to mimic the nicotinamide head group) of the cofactor yields an IC50 value of 42 nM for the inhibition of the conversion of estrone to 17β-estradiol in T47D cells [96–98]. A similar inhibitor, 2-ethyl-16β-m-pyridylmethylamidomethyl-estrone (STX1040) inhibited the conversion of estrone to 17β-estradiol in T47D cells with an IC50 value of 27 nM [66].
8.3. Mechanism-based Inactivators
Mechanism-based inactivators of HSDs refer to steroidal and nonsteroidal ligands that act as pseudo-substrates so that upon turn over by their target enzyme they form a potent electrophile that can alkylate an active site residue leading to irreversible enzyme inhibition [101]. Early proof-of-principle experiments showed that using acetylenic alcohols as substrates, HSDs could be tricked into oxidizing these substrates to the corresponding α,β-unsaturated acetylenic ketones which would then cause enzyme inactivation [102, 103]. This approach also poses several challenges. First, if the kcat/kinact (partition ratio is high) the enzyme generated electrophile may react with other unintended macromolecules in the cell. Second, many of the HSDs that need to be targeted work in the reduction direction and thus far it has been difficult to develop mechanism-based inactivators in which the reduction step produces an enzyme generated electrophile. Generally the introduction of a carbonyl group rather than its removal increases electrophilicity.
8.4. Natural Products as HSD Inhibitors
Natural products also can suffer from specificity problems. For example, licorice derivatives (carbenoxolone and glycyrrhetinic acid) were shown to be prototypic inhibitors of type 2 11β-HSD but also inhibit many related SDRs [60, 104]. 18β-Glycyrrhetinic (GA) a metabolite of the natural product glycchizin inhibits both type 1 11β-HSD and type 2 11β-HSD. Interestingly, the diastereomer 18α-GA inhibits only type 1 11β-HSD, and this has been explained by docking experiments into the type 1 11β-HSD crystal structure [105].
9. Structure-Based Drug Design and HTS
HSDs which appear to be the most promising drug targets are as follows: type 1 11β-HSD (for metabolic syndrome, obesity and type 2 diabetes); type 1 17β-HSD for hormonally dependent breast cancer; and type 3 17β-HSD and type 5 17β-HSD for hormonally dependent and castrate resistant prostate cancer, respectively. The PDB lists 14 structures for human type 1 11β-HSD and its complexes; 18 structures for human type 1 17β-HSD and its complexes; and 11 structures for human type 5 17β-HSD and its complexes. Many of these structures are of abortive complexes of E●NADP+●Inhibitor. In each of these cases atomic details of how the inhibitor is bound are clearly discerned. These structures were initially used to explain how an inhibitor binds to the complex. More recently, many of these structures have been used for in silico screening of large compound libraries to identify potential leads. This approach has been used for AKR1C1 [106, 107], type 1 11β-HSD [108] and type 1 17β-HSD [109].
Simultaneously there has been a revolution in screening compounds for HSD inhibitor activity because of their potential as therapeutics. This has been facilitated by high-through put screening (HTS) assays and the availability of large compound libraries. HTS assays can now be performed using human recombinant enzymes in vitro, and in cell-based assays. Counter screens can also be performed efficiently for AR, ER, PR and GR trans-activation using appropriate luciferase reporter gene assays.
10. Future Directions
For many of the HSD isoforms inhibitors with nanomolar affinity now exist that have been developed by diverse approaches. The challenge will be to identify compounds that are truly selective in vitro in counterscreens and which have favorable absorption, disposition, metabolism, excretion and toxicological properties and move them into preclinical testing in animals. In vivo screening requires animal models of disease to test their efficacy. For example xenograft models in which AKR1C3 or type 1 17β-HSD are overexpressed in prostate and breast cancer cells can be used to test the efficacy of inhibitors for prostate and breast cancer treatment [110, 111]. Similarly, 11β-HSD transgenic mice which are models for obesity and metabolic syndrome can be used to screen inhibitors of this enzyme [112]. Much of this work is ongoing and 11β-HSD inhibitors are in clinical trial. The future is bright since continued progress will result in new clinical trials that will bring SIMs into the clinic, where they can ultimately share a place with SSRMs.
Acknowledgements
Dr. Yi Jin is thanked for her critical reading of the manuscript. This work was supported in part by grants P30- ES013508-05 and R01-DK47015 and R01CA90744 (awarded to TMP).
Abbreviations
- AKR
aldo-keto reductases
- AR
androgen receptor
- ER
estrogen receptor
- GA
glycyrrhetinic acid
- GR
glucocorticoid receptor
- HSD
hydroxysteroid dehydrogenase
- HTS
high throughput screening
- MR
mineralocorticoid receptor
- NSAID
non-steroidal anti-inflammatory drug
- rmsd
root mean square deviation
- SDR
short chain dehydrogenase reductase
- SIM
selective intracrine modulator
- SSRM
selective steroid receptor modulator
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Penning TM. Hydroxysteroid dehydrogenases and pre-receptor regulation of steroid hormone action. Human Reproduction Update. 2003;9:193–205. doi: 10.1093/humupd/dmg022. [DOI] [PubMed] [Google Scholar]
- 2.Bauman DR, Steckelbroeck S, Penning TM. The roles of aldo-keto reductases in steroid hormone action. Drugs News Perspect. 2004;17:563–578. doi: 10.1358/dnp.2004.17.9.872570. [DOI] [PubMed] [Google Scholar]
- 3.Jornvall H, Persson B, Krook M, Atrian S, Gonzalez-Duarte R, Jeffery J, Ghosh D. Short-chain dehydrogenases/reductases (SDR) Biochemistry. 1995;34:6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- 4.Bauman DR, Steckelbroeck S, Williams MV, Peehl DM, Penning TM. Identification of the major oxidative 3α-hydroxysteroid dehydrogenase in human prostate that converts 5α-androstane-3α,17β-diol to 5α-dihydrotestosterone: A potential therapeutic target for androgen dependent disease. Mol. Endocrinol. 2006;20:444–458. doi: 10.1210/me.2005-0287. [DOI] [PubMed] [Google Scholar]
- 5.Rizner T, Lin H-K, Peehl DM, Steckelbroeck S, Bauman DR, Penning TM. Human type 3 3α-hydroxysteroid dehydrogenase (AKR1C2) and androgen metabolism in prostate cells. Endocrinology. 2003;144:2922–2932. doi: 10.1210/en.2002-0032. [DOI] [PubMed] [Google Scholar]
- 6.Funder JW, Pearce PT, Smith R, Smith AI. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science. 1988;242(4878):583–585. doi: 10.1126/science.2845584. [DOI] [PubMed] [Google Scholar]
- 7.Seckl JR, Walker BR. Minireview: 11β-Hydroxysteroid dehydrogenase type 1 a tissue-specific amplifier of glucocorticoid action. Endocrinology. 2001;142:1371–1376. doi: 10.1210/endo.142.4.8114. [DOI] [PubMed] [Google Scholar]
- 8.Stewart PM, Whorwood CB. 11β-Hydroxysteroid dehydrogenase activity and corticosteroid hormone action. Steroids. 1994;59(2):90–95. doi: 10.1016/0039-128x(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 9.Adamski J, Normand T, Leenders F, Monte D, Begue A, Stehelin S, Jungblut PW, de Launoit Y. Molecular cloning of a novel widely expressed human 80 kDa 17β-hydroxysteroid dehydrogenase IV. Biochem J. 1995;311(Pt 2):437–443. doi: 10.1042/bj3110437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andersson S. 17β-hydroxysteroid dehydrogenase: isozymes and mutations. J Endocrinol. 1995;146:197–200. doi: 10.1677/joe.0.1460197. [DOI] [PubMed] [Google Scholar]
- 11.Labrie F, Luu-The V, Lin SX, Simard J, Labrie C, El-Alfy M, Pelletier G, Belanger A. Intracrinology: role of the family of 17β-hydroxysteroid dehydrogenases in human physiology and disease. J. Mol. Endocrinol. 2000;25:1–16. doi: 10.1677/jme.0.0250001. [DOI] [PubMed] [Google Scholar]
- 12.Fung K-M, Shea-Samara EH, Wong C, Krin R, Jones AM, Bane B, Liu CZ, Yang JT, Pitha JV, Culkin DJ, Koop BP, Penning TM, Lin H-K. Increased expression of type 2 3α-hydroxysteroid dehydrogenase/type 5 17β-hydroxysteroid dehydrogenase (AKR1C3) and its relationship with the androgen receptor in prostate carcinoma. Endocr. Related Cancer. 2006;13:169–180. doi: 10.1677/erc.1.01048. [DOI] [PubMed] [Google Scholar]
- 13.Penning TM, Burczynski ME, Jez JM, Lin H-K, Ma H, Moore M, Ratnam K, Palackal N. Structure-function aspects and inhibitor design of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3) Mol. Cell. Endcorinol. 2001;171:137–149. doi: 10.1016/s0303-7207(00)00426-3. [DOI] [PubMed] [Google Scholar]
- 14.Casey ML, MacDonald PC, Andersson S. 17β-Hydroxysteroid dehydrogenase type 2: chromosomal assignment and progestin regulation of gene expression in human endometrium. J Clin Invest. 1994;94(5):2135–2141. doi: 10.1172/JCI117569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Penning TM, Burczynski ME, Jez JM, Hung C-F, Lin H-K, Ma H, Moore M, Palackal N, Ratnam K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem. J. 2000;351:67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ibrahim N, Hortobagyi G. The evolving role of specific estrogen receptor modulators (SERMs) Surg. Oncol. 1999;8:103–122. doi: 10.1016/s0960-7404(99)00047-x. [DOI] [PubMed] [Google Scholar]
- 17.McDonnell D. Selective estrogen receptor modulators (SERMs): A first step in the development of a perfect hormone replacement therapy regimen. J Soc Gynecol. 2000;7:S10–S15. doi: 10.1016/s1071-5576(99)00055-6. [DOI] [PubMed] [Google Scholar]
- 18.Labrie F, Belanger A, Simard J. Intracrinology. Autonomy and freedom of peripheral tissues. Annuals Endocrinology. 1995;56:23–29. [PubMed] [Google Scholar]
- 19.Cadepond F, Ulmann A, Baulieu EE. RU486 (mifepristone): mechanisms of action and clinical uses. Annu Rev Med. 1997;48:129–156. doi: 10.1146/annurev.med.48.1.129. [DOI] [PubMed] [Google Scholar]
- 20.Crooij MJ, de Nooyer CC, Rao BR, Berends GT, Gooren LJ, Janssens J. Termination of early pregnancy by the 3β-hydroxysteroid dehydrogenase inhibitor epostane. N Engl J Med. 1988;319(13):813–817. doi: 10.1056/NEJM198809293191301. [DOI] [PubMed] [Google Scholar]
- 21.Byrns MC, Jin Y, Penning TM. Inhibitors of type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3): Overview and structural insights. J Steroid Biochem Mol Biol. 2010 doi: 10.1016/j.jsbmb.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El-Kabbani O, Dhagat U. A. Hara Inhibitors of human 20α-hydroxysteroid dehydrogenase (AKR1C1) Journal Steroid Biochemistry and Mol. Biol. 2011 doi: 10.1016/j.jsbmb.2010.10.006. In Press. [DOI] [PubMed] [Google Scholar]
- 23.Kratschmar DV, Vuorinen A, Cunha TA, Wolber G, Classen-Houben D, Doblhoff O, Schuster D, Odermatt A. Characterization of activity and binding mode of glycyrrhetinic acid derivatives inhibiting 11β-hydroxysteroid dehydrogenase type 2. Journal Steroid Biochemistry and Mol. Biol. 2011 doi: 10.1016/j.jsbmb.2010.12.019. In Press. [DOI] [PubMed] [Google Scholar]
- 24.Marchais-Oberwinkler S, Henn C, Möller G, Klein T, Negri M, Oster A, Spadaro A, Werth R, Wetzel M, Xu K, Frotscher M, Hartmann RW, Adamski J. 17β-Hydroxysteroid dehydrogenases (17β-HSDs) as therapeutic targets: protein structures, functions, and recent progress in inhibitor development. Journal Steroid Biochemistry and Mol. Biol. 2011 doi: 10.1016/j.jsbmb.2010.12.013. In Press. [DOI] [PubMed] [Google Scholar]
- 25.Poirier D. Contribution to the development of inhibitors of 17β hydroxysteroid dehydrogenase types 1 and 7: key tools for studying and treating estrogen-dependent diseases. Journal Steroid Biochemistry and Mol. Biol. 2011 doi: 10.1016/j.jsbmb.2010.12.007. In press. [DOI] [PubMed] [Google Scholar]
- 26.Schuster D, Kowalik D, Kirchmair J, Laggner C, Markt P, Aebischer-Gumy C, Ströhle F, Möller G, Wolber G, Wilckens T, Langer, Odermatt A, Adamski J. Identification of chemically diverse, novel inhibitors of 17β–hydroxysteroid dehydrogenase type 3 and 5 by pharmacophore-based virtual screening. Journal Steroid Biochemistry and Mol. Biol. 2011 doi: 10.1016/j.jsbmb.2011.01.016. In Press. [DOI] [PubMed] [Google Scholar]
- 27.Thomas JL, Bucholtz KM, Kacsoh B. Selective inhibition of human 3β-hydroxysteroid dehydrogenase type 1 as a potential treatment for breast cancer. J Steroid Biochem Mol Biol. 2010 doi: 10.1016/j.jsbmb.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jez JM, Bennett MJ, Schlegel BP, Lewis M, Penning TM. Comparative anatomy of the aldo-keto reductase superfamily. Biochem. J. 1997;325:625–636. doi: 10.1042/bj3260625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin Y, Penning T. Multiple steps determine the overall rate of the reduction of 5α-dihydrotestosterone catalyzed by human type 3 3α-hydroxysteroid dehydrogenase: Implications for the elimination of androgens. Biochemistry. 2006:13054–13063. doi: 10.1021/bi060591r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Labrie F, Luu-The V, Lin S-X, Labrie C, Simard J, Breton R, Belanger A. The key role of 17β-hydroxysteroid dehydrogenases in sex steroid biology. Steroids. 1997;62:148–158. doi: 10.1016/s0039-128x(96)00174-2. [DOI] [PubMed] [Google Scholar]
- 31.Lachance Y, Luu-The V, Verreault H, Dumont M, Rheaume E, Leblanc G, Labrie F. Structure of the human type II 3β-hydroxysteroid dehydrogenase/Δ5−Δ4 isomerase (3β-HSD) gene: adrenal and gonadal specificity. DNA and Cell Biology. 1991;10:701–711. doi: 10.1089/dna.1991.10.701. [DOI] [PubMed] [Google Scholar]
- 32.Simard J, de Launoit Y, Labrie F. Characterization of the structure-activity relationships of rat types I and II 3β-hydroxysteroid dehydrogenase/Δ5−Δ4 isomerase by site-directed mutagenesis and expression in HeLa cells. J Biol Chem. 1991;266(23):14842–14845. [PubMed] [Google Scholar]
- 33.Askonas LJ, Ricigliano JW, Penning TM. The kinetic mechanism catalysed by homogeneous rat liver 3α-hydroxysteroid dehydrogenase. Evidence for binary and ternary dead-end complexes containing non-steroidal anti-inflammatory drugs. Biochem J. 1991;278(Pt 3):835–841. doi: 10.1042/bj2780835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cooper WC, Jin Y, Penning TM. Elucidation of a complete kinetic mechanisms for a mammalian hydroxysteroid dehydrogenase (HSD) and identification of all enzyme forms on the reaction coordinate: The example of rat liver 3α-HSD (AKR1C9) J. Biol. Chem. 2007;282:33484–33493. doi: 10.1074/jbc.M703414200. [DOI] [PubMed] [Google Scholar]
- 35.Ratnam K, Ma H, Penning TM. The arginine 276 anchor for NADP(H) dictates fluorescence kinetic transients in 3α-hydroxysteroid dehydrogenase, a representative aldo-keto reductase. Biochemistry. 1999;38:7856–7864. doi: 10.1021/bi982838t. [DOI] [PubMed] [Google Scholar]
- 36.Rizner TL, Adamski J, Stojan J. 17β-Hydroxysteroid dehydrogenase from Cocliobolus lunatus: Model structure and substrate specificity. Archives Biochem & Biophys. 2000;384:255–262. doi: 10.1006/abbi.2000.2064. [DOI] [PubMed] [Google Scholar]
- 37.Ueda S, Oda M, Imamura S, Ohnishi M. Transient-phase kinetic studies on the nucleotide binding to 3α-hydroxystreoid dehydrogenase from Pseudomonas sp B-831 using fluorescence stopped-flow procedures. Eur. J. Biochem. 2004;271:1774–1780. doi: 10.1111/j.1432-1033.2004.04089.x. [DOI] [PubMed] [Google Scholar]
- 38.Castro A, Zhu J, Alton G, Rejto P, Emolieff J. Assay optimization and kinetic profile of the human and the rabbit isoforms of 11β-HSD1. Biochem. Biophys. Res. Commun. 2007;357:561–566. doi: 10.1016/j.bbrc.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 39.Warren J, Crist R. Site-specificity and mechanism of human placental 17β-hydroxysteroid dehydrogenase. Archives Biochem & Biophys. 1967;118:577–584. doi: 10.1016/0003-9861(67)90392-x. [DOI] [PubMed] [Google Scholar]
- 40.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22(23):3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 41.Biswas MG, Russell DW. Expression cloning and characterization of oxidative 17β-and 3α-hydroxysteroid dehydrogenases from rat and human prostate. J Biol Chem. 1997;272(25):15959–15966. doi: 10.1074/jbc.272.25.15959. [DOI] [PubMed] [Google Scholar]
- 42.Steckelbroeck S, Jin Y, Gopishetty S, Oyesanmi B, Penning TM. Human cytosolic 3α-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3β-hydroxysteroid dehydrogenase activity: implications for steroid hormone metabolism and action. J Biol Chem. 2004;279(11):10784–10795. doi: 10.1074/jbc.M313308200. [DOI] [PubMed] [Google Scholar]
- 43.Fisher HF, Conn EE, Vennesland B, Westheimer FH. The enzymatic transfer of hydrogen. I. The reaction catalyzed by alcohol dehydrogenase. J Biol Chem. 1953;202(2):687–697. [PubMed] [Google Scholar]
- 44.Loewus FA, Ofner P, Fisher HF, Westheimer FH, Vennesland B. The enzymatic transfer of hydrogen. II. The reaction catalyzed by lactic dehydrogenase. J Biol Chem. 1953;202(2):699–704. [PubMed] [Google Scholar]
- 45.Kersey WH, Wilcox RB. Stereochemistry of hydrogen transfer by rat ovary 20α-hydroxysteroid dehydrogenase. Biochemistry. 1970;9(5):1284–1286. doi: 10.1021/bi00807a033. [DOI] [PubMed] [Google Scholar]
- 46.Bohren KM, Girmshaw CE, Lai C-J, Harrison D, Ringe D, Petsko GA, Gabbay KH. Tyrosine-48 is the proton donor and histidine-110 directs substrate stereochemical selectivity in the reduction reaction of human aldose reductase: Enzyme kinetics and crystal structure of the Y48H mutant enzyme. Biochemistry. 1994;33:2021–2032. doi: 10.1021/bi00174a007. [DOI] [PubMed] [Google Scholar]
- 47.Grimshaw CE, Bohren KM, Lai CJ, Gabbay KH. Human aldose reductase: pK of tyrosine 48 reveals the preferred ionization state for catalysis and inhibition. Biochemistry. 1995;34(44):14374–14384. doi: 10.1021/bi00044a014. [DOI] [PubMed] [Google Scholar]
- 48.Schlegel BP, Jez JM, Penning TM. Mutagenesis of 3α-hydroxysteroid dehydrogenase reveals a "push-pull" mechanism for proton transfer in aldo-keto reductases. Biochemistry. 1998;37:3538–3548. doi: 10.1021/bi9723055. [DOI] [PubMed] [Google Scholar]
- 49.Vogan E, Belamacina C, He X, Liu HW, Ringe D, Petsko G. Crystal structure of 1.8 A resolution of CDP-D-glucose 4,6-dehydratase from Yersinia pseudotuberculosis. Biochemistry. 2004;43:3057–3067. doi: 10.1021/bi035547f. [DOI] [PubMed] [Google Scholar]
- 50.Ghosh D, Weeks CM, Grochulski P, Duax WL, Erman M, Rimsay RL, Orr JC. Three-dimensional structure of holo 3α,20β-hydroxysteroid dehydrogenase: a member of a short-chain dehydrogenase family. Proc Natl Acad Sci U S A. 1991;88:10064–10068. doi: 10.1073/pnas.88.22.10064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghosh D, Wawrzak Z, Weeks CM, Duax WL, Erman M. The refined three-dimensional structure of 3α,20β-hydroxysteroid dehydrogenase and possible roles of the residues conserved in short-chain dehydrogenases. Structure. 1994;2:629–640. doi: 10.1016/s0969-2126(00)00064-2. [DOI] [PubMed] [Google Scholar]
- 52.Ghosh D, Pletnev VZ, Zhu DW, Wawrzak Z, Duax WL, Pangborn W, Labrie F, Lin SX. Structure of human estrogenic 17β-hydroxysteroid dehydrogenase at 2.20 A resolution. Structure. 1995;3(5):503–513. doi: 10.1016/s0969-2126(01)00183-6. [DOI] [PubMed] [Google Scholar]
- 53.Bennett MJ, Schlegel BP, Jez JM, Penning TM, Lewis M. Structure of α–hydroxysteroid/dihydrodiol dehydrogenase complexed with NADP+ Biochemistry. 1996;35:10702–10711. doi: 10.1021/bi9604688. [DOI] [PubMed] [Google Scholar]
- 54.Filling C, Berndt KD, Benach J, Knapp S, Prozoorovski T, Nordling E, Ladenstein R, Jornvall H, Oppermann U. Critical residues for structure and catalysis in short-chain dehydrogenases/reductases. J. Biol. Chem. 2002;277:25677–25684. doi: 10.1074/jbc.M202160200. [DOI] [PubMed] [Google Scholar]
- 55.Negri M, Recanatini M, Hartmann RW. Insights in 17β-HSD1 enzyme kinetics and ligand binding by dynamic motion investigation. PLoS One. 2010;5(8):e12026. doi: 10.1371/journal.pone.0012026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Akhtar M, Wilton DC, Watkinson IA, Rahimtula AD. Substrate activation in pyridine nucleotide-linked reactions: illustrations from the steroid field. Proc R Soc Lond B Biol Sci. 1972;180(59):167–177. doi: 10.1098/rspb.1972.0012. [DOI] [PubMed] [Google Scholar]
- 57.Bauman DR, Rudnick S, Szewczuk L, Sridhar G, Penning TM. Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: potential antineoplastic agents that work independently of cyclooxygenase isozymes. Mol. Pharmacol. 2005;67:60–68. doi: 10.1124/mol.104.006569. [DOI] [PubMed] [Google Scholar]
- 58.Byrns MC, Steckelbroeck S, Penning TM. An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3α-HSD, type 5 17β-HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem Pharmacol. 2008;75(2):484–493. doi: 10.1016/j.bcp.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.El-Kabbani O, Scammells PJ, Gosling J, Dhagat U, Endo S, Mastunnaga T, Soda M, Hara A. Structure-guided design, synthesis, and evaluation of salicyclic acid-based inhibitors targeting a selectivity pocket in the active site of human 20α-hydroxystreoid dehydrogenase. J, Med. Chem. 2009;52:3259–3264. doi: 10.1021/jm9001633. [DOI] [PubMed] [Google Scholar]
- 60.Ghosh D, Erman M, Wawrzak Z, Duax WL, Pangborn W. Mechanism of inhibition of 3α,20β-hydroxysteroid dehydrogenase by a licorice-derived steroidal inhibitor. Structure. 1994;2(10):973–980. doi: 10.1016/s0969-2126(94)00099-9. [DOI] [PubMed] [Google Scholar]
- 61.Penning TM, Talalay P. Inhibition of a major NAD(P)+-linked oxidoreductase from rat liver cytosol by steroidal and nonsteroidal anti-inflammatory agents and by prostaglandins. Proc. Natl. Acad. Sci. USA. 1983;80:4504–4508. doi: 10.1073/pnas.80.14.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Penning TM, Mukharji I, Barrows S, Talalay P. Purification and properties of a 3α-hydroxysteroid dehydrogenase of rat liver cytosol and its inhibition by anti-inflammatory drugs. Biochem J. 1984;222(3):601–611. doi: 10.1042/bj2220601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Talalay P. Hydroxysteroid dehydrogenases. In: Boyer PD, Lardy M, Myrback K, editors. Enzymes. New York: NY Acad Press; 1963. pp. 177–202. [Google Scholar]
- 64.Agarwal AK, Auchus RJ. Minireview: cellular redox state regulates hydroxysteroid dehydrogenase activity and intracellular hormone potency. Endocrinology. 2005;146(6):2531–2538. doi: 10.1210/en.2005-0061. [DOI] [PubMed] [Google Scholar]
- 65.Papari-Zareei M, Brandmaier A, Auchus RJ. Arginine 276 controls the directional preference of AKR1C9 (rat liver 3α-hydroxysteroid dehydrogenase) in human embryonic kidney 293 cells. Endocrinology. 2006;147(4):1591–1597. doi: 10.1210/en.2005-1141. [DOI] [PubMed] [Google Scholar]
- 66.Day JM, Foster PA, Tutill HJ, Parsons MF, Newman SP, Chander SK, Allan GM, Lawrence HR, Vicker N, Potter BV, Reed MJ, Purohit A. 17β-hydroxysteroid dehydrogenase Type 1, and not Type 12, is a target for endocrine therapy of hormone-dependent breast cancer. Int J Cancer. 2008;122(9):1931–1940. doi: 10.1002/ijc.23350. [DOI] [PubMed] [Google Scholar]
- 67.Veliça P, Davies N, Rocha P, Schrewe H, Ride J, Bunce C. Lack of functional and expression homology between human and mouse aldo-keto reductase 1C enzymes: implications for modelling human cancers. Mol Cancer. 2009;8:121. doi: 10.1186/1476-4598-8-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gathercole LL, Stewart PM. Targeting the pre-receptor metabolism of cortisol as a novel therapy in obesity and diabetes. J. Steroid Biochem. & Mol. Biol. 2010;122:21–27. doi: 10.1016/j.jsbmb.2010.03.060. [DOI] [PubMed] [Google Scholar]
- 69.Ge R, Huang Y, Liang G, Li X. 11β-hydroxysteroid dehydrogenase type 1 inhibitors as promising therapeutic drugs for diabetes: status and development. Curr. Med. Chem. 2010:412–422. doi: 10.2174/092986710790226147. [DOI] [PubMed] [Google Scholar]
- 70.Day JM, Tutill HT, Purohit A, Reed MJ. Design and validation of specific inhibitors of 17β-hydroxysteroid dehydrogenases for therapeutic application in breast and prostate cancer and endometriosis. Endocrine-Related Cancer. 2008;15:665–692. doi: 10.1677/ERC-08-0042. [DOI] [PubMed] [Google Scholar]
- 71.Poirier D. 17β-Hydroxysteroid dehydrogenase inhibitors: a patent review. Expert Opin. Ther. Pat. 2010;20:1123–1145. doi: 10.1517/13543776.2010.505604. [DOI] [PubMed] [Google Scholar]
- 72.Penning TM, Ricigliano JW. Nonsteroidal and nonprostanoid inhibitors of steroid and prostaglandin transforming enzymes. 5,258,296. U.S. Patent. 1993 Nov 2nd;
- 73.Endo S, Matsunaga T, Soda M, Tajima K, Zhao H, El-Kabbani O, Hara A. Selective inhibition of the tumor marker AKR1B10 by anti-inflammatory N-phenylanthraniic acids and glycyrrhetic acid. Biol Pharm. Bull. 2010;33:886–890. doi: 10.1248/bpb.33.886. [DOI] [PubMed] [Google Scholar]
- 74.Adeniji AO, Twenter BM, Byrns MC, Jin Y, Winkler JD, Penning TM. Discovery of substituted 3-(phenylamino)benzoic acids as potent and selective inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3) Biorganic & Medicinal Chemistry Letts. 2011 doi: 10.1016/j.bmcl.2011.01.010. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dhagat U, Endo S, Sumi R, Hara A, El-Kabbani O. Selectivity determinants of inhibitor binding to human 20α-hydroxysteroid dehydrogenase: Crystal structure of the enzyme in ternary complex with coenzyme and the potent inhibitor 3,5-dichlorosalicylic acid. J. Med. Chem. 2008;51:4844–4848. doi: 10.1021/jm8003575. [DOI] [PubMed] [Google Scholar]
- 76.Shah U, Boyle CD, Chackalamannil S, Baker H, Kowalski T, Lee J, Terracina G, Zhang L. Azabicyclic sulfonamides as potent 11β-HSD1 inhibitors. Bioorganic & Medicinal Chemistry Letts. 2010;20:1551–1554. doi: 10.1016/j.bmcl.2010.01.082. [DOI] [PubMed] [Google Scholar]
- 77.Zhu Y, Olson S, Hermanowski-Vostak A, Mundt S, Shah K, Springer M, Thieringer R, Wright S, Xioa J, Zokian H, Balkovec JM. 4-Methyl-5-phenyl trizaoles as selective inhibitors of 11β-hydroxysteroid dehydrogenase type 1. Bioorganic & Medcinal Chemistry Letts. 2008;18:3405–3411. doi: 10.1016/j.bmcl.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 78.Roche D, Carniato D, Leriche C, Lepifre F, Christmann-Franck S, Graedler U, Charon C, Bozec S, Doare L, Schmidlin F, Lecomte M, Valuer E. Discovery and structure-activity relationships of pentanedioic acid diamides as potent inhibitors of 11β-hydroxysteroid dehydrogenase type 1. Bioorganic & Medcinal Chemistry Letts. 2009;19:2674–2678. doi: 10.1016/j.bmcl.2009.03.140. [DOI] [PubMed] [Google Scholar]
- 79.Siu M, Johnson TO, Wang Y, Nair SK, Taylor WD, Cripps SJ, Matthews JJ, Edwards MP, Pauly TA, Ermolieff J, Castro A, Hosea NA, LaPaglia A, Fanjul AN, Vogel JE. N-(pyridin-2-yl)aryl sulfonamide inhibitors of 11β-hydroxysteroid dehydrogenase type 1: Discovery of PF-915275. Bioorganic & Medicinal Chemistry Letts. 2009;19:3493–3497. doi: 10.1016/j.bmcl.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 80.Zhang X, Zhu-ou Y, Du L-i, Chen J-h, Leng Y, Chen J-h. Derivatives of (phenylsulfonoamido-methyl)nicotine and (phenylsulfonomido-methyl)thiazole as novel 11β-hydroxysteroid dehydrogenase type 1 inhibitors: synthesis and biological activities in vitro. Acta Pharmacologica Sinica. 2009;30:1344–1350. doi: 10.1038/aps.2009.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McMinn DL, Rew Y, Sudom A, Caille S, DeGraffenreid M, He X, Hungate R, Jiang B, Jaen J, Julian LD, Kaizerman J, Novak P, Sun D, Tu H, Ursu S, Walker N, Yan X, Ye Q, Wang Z, Powers JP. Optimization of novel di-substituted cyclohexylbenzamide derivatives as potent 11β-HSD1 inhibitors. Bioorganic & Medicinal Chemistry Letts. 2009;19:1446–1450. doi: 10.1016/j.bmcl.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 82.Rew Y, McMinn DL, Wang Z, He X, Hungate RW, Jaen JC, Sudom A, Sun D, Tu H, Ursu S, Villemure E, Walker NPC, Yan X, Ye Q, Powers JP. Discovery and optimization of piperidyl benzamide derivatives as a novel class of 11β-HSD1 inhibitors. Bioorganic & Medcinal Chemistry Letts. 2009;19:1797–1801. doi: 10.1016/j.bmcl.2009.01.058. [DOI] [PubMed] [Google Scholar]
- 83.Sun D, Wang Z, Cardozo M, Choi R, DeGarffenreid M, Di Y, He X, Jen JC, Labelle M, Liu J, Ma J, Miao S, Sudom A, Tang L, Tu H, Ursu S, Walker N, Yan X, Y Q, Powers JP. Synthesis and optimization of arylsulfonylpiperazines as a novel class of inhibitors of 11β-hydroxysteroid dehydrogenase type1. Bioorganic & Medicinal Chemistry Letts. 2009;19:1522–1527. doi: 10.1016/j.bmcl.2008.12.114. [DOI] [PubMed] [Google Scholar]
- 84.Johansson L, Fotsch C, Bartberger MD, Castro VM, Chen M, Emery M, Gustafsson S, Hale C, Hickman D, Homan E, Jordan SR, Komorowski R, Li A, McRae K, Mioniz G, Matsumoto G, Oriheuela C, Palm G, Veniant M, Wang M, Williams M, Zhang J. 2-Amino-1,3-thizaol 4(5H)-ones as potent and selective 11β-hydroxysteroid dehydrogenase type 1 inhibitors: enzyme-ligand co-crystal structure and demonstration of pharrmacodynamic effects in C57B1/6 mice. J. Med. Chem. 2008;51:2933–2943. doi: 10.1021/jm701551j. [DOI] [PubMed] [Google Scholar]
- 85.Webster SP, Binnie M, McConnell KMM, Sooy K, Ward P, Greaney MF, Vinter A, Pallin TD, Dyke HJ, Gill MIA, Warner I, Seckl JR, Walker BR. Modulation of 11β-hydroxysteroid dehydrogenase type 1 activity by 1,5-substituted 1H-tetrazoles. Bioorganic & Medcinal Chemistry Letts. 2010;20:3265–3271. doi: 10.1016/j.bmcl.2010.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Su X, Pradaux-Caggiano F, Thomas MP, Szeto MWY, Halem HA, Culler MD, Vicker N, Potter BVL. Discovery of admantyl ethanone derivatives as potent 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitors. Chem Med. Chem. 2010;5:1–20. doi: 10.1002/cmdc.201000081. [DOI] [PubMed] [Google Scholar]
- 87.Al-Soud YA, Bey E, Oster A, Marchais-Oberwinkler S, Werth R, Kruchten P, Forstshcer M, Hartmann RW. The role of the heterocycle in bis(hydroxyphenyl)triazoles for inhibition of 17β-hydroxysteroid dehydrogenase (17β-HSD) type 1 and type 2. Molecular & Cellular Endocrinology. 2009;301:212–215. doi: 10.1016/j.mce.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 88.Frotscher M, Ziegler E, Marchais-Oberwinkler S, Kruchten P, Neugebauer A, Fetzer L, Scherer C, Muller-Vieira U, Messinger J, Thole H, Hartmann RW. Design, synthesis, and biological evaluation of (hydroxyphenyl)naphthalene and -quinoline derivatives: potent and selective nonsteroidal inhibitors of 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1) for the treatment of estrogen-dependent diseases. J Med Chem. 2008;51:2158–2169. doi: 10.1021/jm701447v. [DOI] [PubMed] [Google Scholar]
- 89.Marchais-Oberwinkler S, Kruchten P, Frostcher M, Ziegler E, Negebauer A, Bhoga U, Bey E, Muller-Vieria U, Messinger J, Thole H, Hartmann RW. Substituted 6-phenyl-2-naphthols. Potent and selective nonsteroidal inhibitors of 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1): Design, synthesis, biological evaluation and pharmacokinetics. J. Med. Chem. 2008;51:4685–4698. doi: 10.1021/jm800367k. [DOI] [PubMed] [Google Scholar]
- 90.Bey E, Marchais-Oberwinkler S, Werth R, Negri M, Al-Soud YA, Kruchten P, Oster A, Frostcher M, Birk B, Hartmann RW. Design, synthesis, biological evaluation and pharmacokinetics of bis(hydroxyphenyl)substituted azoles, thiophenes, benzenes, and aza-benzenes as potent and selective nonsteroidal uinhibitors of 17β-hydroxysteroid dehydrogenaase type 1 (17β-HSD1) J. Med. Chem. 2008;51:6725–6739. doi: 10.1021/jm8006917. [DOI] [PubMed] [Google Scholar]
- 91.Bey E, Marchais-Oberwinkler S, Negri M, Kruchten P, Oster A, Klein T, Spadaro A, Werth R, Frotscher M, Birk B, Hartmann RW. New insights into the SAR and binding modes of bis(hydroxyphenyl)thiophenes and -benzenes: influence of additional substituents on 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1) inhibitory activity and selectivity. J Med Chem. 2009;52:6724–6743. doi: 10.1021/jm901195w. [DOI] [PubMed] [Google Scholar]
- 92.Oster A, Klein T, Werth R, Kruchten P, Bey E, Negri M, Marchais-Oberwinkler S, Frotscher M, Hartmann RW. Novel estrone mimetics with high 17β-HSD1 inhibitory activity. Bioorg Med Chem. 2010;18:3494–3505. doi: 10.1016/j.bmc.2010.03.065. [DOI] [PubMed] [Google Scholar]
- 93.Allan GM, Vicker N, Lawrence HR, Tutill HJ, Day JM, Huchet M, Ferrandis E, Reed MJ, Purohit A, Potter BV. Novel inhibitors of 17β-hydroxysteroid dehydrogenase type 1: templates for design. Bioorg Med Chem. 2008;16(8):4438–4456. doi: 10.1016/j.bmc.2008.02.059. [DOI] [PubMed] [Google Scholar]
- 94.Fink BE, Gava AV, Tokarski JS, Goyal B, Misra R, Xiao H-Y, Kimball SD, Han W-C, Norris D, Spires TE, You D, Gottardis MM, Lorenzi MV, Vite GD. Identification of a novel series of tetrahydrodibenzazocines as inhibitors of 17β-hydroxysteroid dehydrogenase type 3. Bioorganic & Medcinal Chemistry Letts. 2006;16:1532–1536. doi: 10.1016/j.bmcl.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 95.Vicker N, Sharland CM, Heaton WB, Gonzalez AM, Bailey HV, Smith A, Springall JS, Day JM, Tutill HJ, Reed MJ, Purohit A, Potter BV. The design of novel 17β-hydroxysteroid dehydrogenase type 3 inhibitors. Mol Cell Endocrinol. 2009;301:259–265. doi: 10.1016/j.mce.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 96.Fournier D, Poirier D, Mazumdar M, Lin S-X. Design and synthesis of bisubstrate inhibitors of type 1 17β-hydroxysteroid dehydrogenase: Overview and perspectives. European J. Med. Chem. 2008;43:2298–2306. doi: 10.1016/j.ejmech.2008.01.044. [DOI] [PubMed] [Google Scholar]
- 97.Mazumdar M, Fornier D, Zhu D-W, Cadot C, Poirier D, Lin S-X. Binary and ternary crystal structure analyses of a novel inhibitor with 17β-HSD type 1:a lead compound for breast cancer therapy. Biochem. J. 2002;424:357–366. doi: 10.1042/BJ20091020. [DOI] [PubMed] [Google Scholar]
- 98.Poirier D, Boivin RP, Tremblay MR, Berube M, Qiu W, Lin S-X. Estradiol-adenosine hybrid compounds designed to inhibit type 1 17β-hydroxysteroiddehydrogenase. J. Med. Chem. 2005;48:8134–8147. doi: 10.1021/jm058235e. [DOI] [PubMed] [Google Scholar]
- 99.Lawrence HR, Vicker N, Allan GM, Smith A, Mahon MF, Tutill HJ, Purohit A, Reed MJ, Potter BV. Novel and potent 17β-hydroxysteroid dehydrogenase type 1 inhibitors. J Med Chem. 2005;48(8):2759–2762. doi: 10.1021/jm049045r. [DOI] [PubMed] [Google Scholar]
- 100.Vicker N, Lawrence HR, Allan GM, Bubert C, Smith A, Tutill HJ, Purohit A, Day JM, Mahon MF, Reed MJ, Potter BV. Focused libraries of 16-substituted estrone derivatives and modified e-ring steroids: inhibitors of 17β-hydroxysteroid dehydrogenase type 1. Chem. Med. Chem. 2006;1:464–481. doi: 10.1002/cmdc.200500087. [DOI] [PubMed] [Google Scholar]
- 101.Penning TM, Thronton R, Ricigliano JW. Clues to the development of mechanism-based inactivators of 3α-hydroxysteroid dehydrogenase comparison of steroidal and nonsteroidal Michael acceptors and epoxides. Steroids. 1991;56:420–427. doi: 10.1016/0039-128x(91)90030-y. [DOI] [PubMed] [Google Scholar]
- 102.Auchus RJ, Covey DF. Mechanism-based inactivation of 17β,20α-hydroxysteroid dehydrogenase by an acetylenic secoestradiol. Biochemistry. 1986;25(23):7295–7300. doi: 10.1021/bi00371a008. [DOI] [PubMed] [Google Scholar]
- 103.Schlegel BP, Pawlowski JE, Hu Y, Scolnick DM, Covey DF, Penning TM. Secosteroid mechanism-based inactivators and site-directed mutagenesis as probes for steroid hormone recognition by 3α-hydroxysteroid dehydrogenase. Biochemistry. 1994;33(34):10367–10374. doi: 10.1021/bi00200a017. [DOI] [PubMed] [Google Scholar]
- 104.Stewart PM, Wallace AM, Valentino R, Burt D, Shackleton CH, Edwards CR. Mineralocorticoid activity of liquorice: 11β-hydroxysteroid dehydrogenase deficiency comes of age. Lancet. 1987;2(8563):821–824. doi: 10.1016/s0140-6736(87)91014-2. [DOI] [PubMed] [Google Scholar]
- 105.Classen-Houben D, Schutser D, Da Cunha T, Odermatt A, Wolber G, Jordis U, Kueenburg B. Selective inhibition of 11β-hydroxysteroid dehydrogenase 1 by 18α-glycyrrhetinic acid but not 18β-glycyrhetinic acid. J. Steroid Biochem. & Mol. Biol. 2009;113:248–252. doi: 10.1016/j.jsbmb.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 106.Brozic P, Turk S, Rizner TL, Gobec S. Discovery of new inhibitors of aldo-keto reductase 1C1 by structure-based virtual screening. Mol. Cell. Endocrinol. 2009;301:245–250. doi: 10.1016/j.mce.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 107.Dhagat U, Carbone V, Chung RP, Matsunaga T, Endo S, Hara A, El-Kabbani O. A salicylic acid-based analogue discovered from virtual screening as a potent inhibitor of human 20α-hydroxysteroid dehydrogenase. Med Chem. 2007;3(6):546–550. doi: 10.2174/157340607782360399. [DOI] [PubMed] [Google Scholar]
- 108.Yang H, Shen Y, Chen J, Jiang Q, Leng Y, Shen J. Structure-based virtual screening for identification of novel 11β-HSD 1 inhibitors. European J. Med. Chem. 2009;44:1167–1171. doi: 10.1016/j.ejmech.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 109.Schuster D, Nashev LC, Kirchmair J, Laggner C, Wolber G, Langer T, Odermatt A. Discovery of nonsteroidal 17β-hydroxysteroid dehydrogenase 1 inhibitors by pharmacophore-based screening of virtual compound libraries. J. Med. Chem. 2008;2008:4188–4199. doi: 10.1021/jm800054h. [DOI] [PubMed] [Google Scholar]
- 110.Hofland J, van Weerden W, Dits N, Steenbergen J, van Leenders G, Jenster G, Schröder F, de Jong F. Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res. 2010;70:1256–1264. doi: 10.1158/0008-5472.CAN-09-2092. [DOI] [PubMed] [Google Scholar]
- 111.Husen B, Huhtinen K, Saloniemi T, Messinger J, Thole HH, Poutanen M. Human hydroxysteroid (17-beta) dehydrogenase 1 expression enhances estrogen sensitivity of MCF-7 breast cancer cell xenografts. Endocrinology. 2006;147(11):5333–5339. doi: 10.1210/en.2006-0778. [DOI] [PubMed] [Google Scholar]
- 112.Wake DJ, Walker BR. 11β-hydroxysteroid dehydrogenase type 1 in obesity and the metabolic syndrome. Mol Cell Endocrinol. 2004;215:45–54. doi: 10.1016/j.mce.2003.11.015. [DOI] [PubMed] [Google Scholar]