

Abstract
Agrin is a large heparin sulphate proteoglycan with multiple domains, which is located in the extracellular matrix. The C-terminal G3 domain of agrin is functionally one of the most important domains. It harbors an α-dystroglycan binding site and carries out acetylcholine receptor clustering activities. In the present study, we have fused the G3 domain of agrin to an IgG Fc domain to produce a G3-Fc fusion protein that we intend to use as a tool to investigate new binding partners of agrin. As a first step of the study, we have characterized the recombinant fusion protein using a multidisciplinary approach using dynamic light scattering, analytical ultracentrifugation and small angle X-ray scattering (SAXS). Interestingly, our SAXS analysis using the high-resolution structures of G3 and Fc domain as models indicates that the G3-Fc protein forms a T-shaped molecule with the G3 domains extruding perpendicularly from the Fc scaffold. To validate our models, we have used the program HYDROPRO to calculate the hydrodynamic properties of the solution models. The calculated values are in excellent agreement with those determined experimentally.
Keywords: agrin, analytical ultracentrifugation, dynamic light scattering, small angle X-ray scattering, splice insert
Introduction
Agrin is a heparin sulphate proteoglycan that is predominantly located in the extracellular matrix.1 It is synthesized by motor neuron cells, transported along their axons and then secreted from their terminals to accumulate in the basal lamina, which occupies the synaptic cleft at the neuromuscular junction (NMJ). Agrin plays a crucial role during the aggregation of the acetylcholine receptor (AChR) and the clustering of other postsynaptic molecules at the NMJ.2–5 It is responsible for the generation of synapses at the NMJ and their maintenance. Agrin is vital for the formation of immunological synapses 6 and synapses in the brain.7 8 It mediates transcytosis of HIV-1 across epithelial cell monolayers during the formation of virological synapses.9 It is essential to generate functional postsynaptic structures in skeletal muscle. Very recently, it has been shown that low-density lipoprotein receptor-related protein 4 (LRP4) acts as a receptor of agrin. LRP4 is required for Muscle Specific Kinase (MuSK) signaling, which in turn induces AChR clustering activity mediated by agrin.10 11 The importance of agrin is underlined by the fact that agrin deficient mice die at birth because of the failure of the respiratory system.12 13
Agrin is a multidomain protein that consists of an N-terminal domain (NtA), followed by a series of follistatin-like domains (FS) 14–17 and three laminin G-like C-terminal domains (G1, G2, and G3).18 19 Different activities of agrin appear to be regulated by the process of alternative mRNA splicing, which gives rise to many different forms that have distinct expression patterns and functions. For example, the G2 domain has a splicing site “A” in chicken (or “y” in rodents) and can accommodate a four amino acids long insert (A4) that encodes a heparin-binding site. The variant without insert (A0) has no heparin binding activity. The G3 domain incorporates a splicing site “B” in chicken (or “z” in rodents) that generates four variants with an insert of either 8, 11, or 19 amino acid residues (B8, B11, and B19).20 21 The C-terminal G2 and G3 domains are mainly responsible for the binding to α-dystroglycan (α-DG), and the presence of an amino acid insert increases the binding of agrin with α-DG by a factor of 1000.22–24 The interaction of α-DG with agrin is calcium dependent. Interestingly, binding of heparin to the C-terminal domains abolishes AChR clustering activity and reduces the affinity of binding to α-DG.20 25 Interaction of agrin with heparin requires two domains: the G2 domain together with either the G1 or the G3 domain.21 This suggests that the interaction of cell surface proteoglycans with agrin could be multivalent. It was suggested that agrin also binds with FGF-2, thrombospondin, and tenascin.26 A role of agrin in T-cell receptor signaling and cell activation has also been proposed.27 An interaction between the G3 domain and Na+, K+-ATPase was recently explored that may regulate functions mediated by Na+, K+-ATPase by displacing its β-subunit.28 29
As discussed above, the G3 domain of agrin is functionally one of the most important domains of this protein. It interacts with various ligands and subsequently influences a number of biological processes, which is the motivation behind selecting the G3 domain for further studies. The fusion of the G3 domain with an Fc portion of IgG was carried out to (1) establish a scaffold for the use of surface plasmon resonance (SPR) to screen for the binding of novel ligands, (2) to study how naturally occurring agrin splice variants of the G3 domain mediate protein-ligand interactions, and (3) to assess conformational changes that occur in the G3 domain upon binding of different ligands. Binding requires two G domains, which is one of the reasons why we chose to introduce an Fc tag. In this way, we can accommodate two G3 domains in a single construct (G3-Fc-G3) to mimic genuine agrin-ligand interactions. Our method of choice to investigate novel ligands for G3 domains of agrin is SPR. This is one of the most widely used methods to study protein-ligand interactions.30 31 Briefly, the Fc region of the G3-Fc protein will be immobilized on an anti-IgG Fc antibody chip. The immobilized protein will then be exposed to cell lysate from different sources. The binding of ligands with the G3-Fc protein immobilized on chip will be studied using SPR. The reaction mixture will be washed to eliminate unbound material and the bound ligands will then be investigated using Mass Spectroscopy.
Scotton et al. have previously shown that the G3-Fc system can be used for affinity assays to detect binding of heparin and α-DG, respectively.19 As a first part of the study, which will underpin a range of future experimentation in the areas indicated, we are presenting in this article the biophysical analysis of the G3-Fc protein complex using a combined approach using the solution techniques dynamic light scattering (DLS), analytical ultracentrifugation (AUC) and small angle X-ray scattering (SAXS).32 SAXS and AUC methods have been widely used for the investigation of large macromolecules and their assemblies. We have built 3D domain models for the G3-Fc protein using SAXS data and show that the recombinant protein adapts a T-shaped conformation in solution. To validate our approach, we have used the program HYDROPRO to calculate the hydrodynamic parameters of the models and have found that they agree with those determined experimentally.
Results
To characterize the recombinant protein G3-Fc (see Fig. 1) in terms of its solution conformation, we have performed a combination of DLS, AUC, and SAXS. The fusion protein construct starts with the G3 domain comprising amino acid residues 1752-1944 from chicken agrin at the N-terminus. It is followed by two immunoglobulin heavy chain constant domains CH2-CH3 of the Fc portion of human IgG (Fig. 1A). A 10-residue linker connects G3 with the Fc-fragment, equivalent to the natural hinge region between the CH1 and CH2 domains in antibodies. The G3-Fc recombinant protein was purified using affinity column chromatography and purified protein fractions were assayed for the presence of protein. The fractions were then combined and concentrated for further studies. The purity of the concentrated fractions was studied using tricine SDS-PAGE, and the results are presented in Figure 1. Lane 1 shows the G3-Fc protein in the presence of β-mercaptoethanol, and Lane 3 contains the protein without reducing agent. The protein is highly pure, and there is no evidence of degradation.
Figure 1.
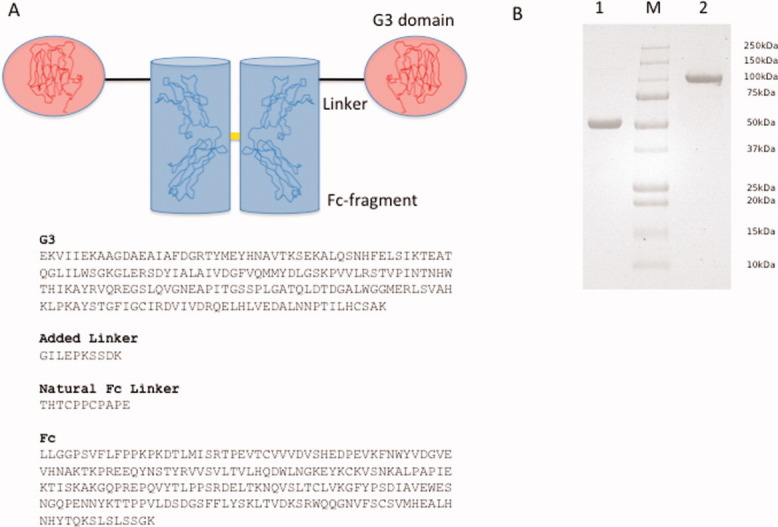
The recombinant agrin G3-human IgG Fc protein. (A) Topology scheme of the recombinant G3-Fc protein with the N-terminal G3 domains highlighted in red and the Fc fragment highlighted in blue. The individual structural coordinates are underlined as ribbon models. The yellow bar symbolizes the disulfide linkage within the Fc fragment (top). Sequence pattern of the 428 residue encompassing G3-Fc fusion protein. Individual segments are highlighted in bold (bottom). (B) SDS-PAGE analysis of the G3-Fc fusion protein. SDS-PAGE of the G3-Fc in reducing (lane 1) and oxidizing conditions (lane 2) confirm the formation of a disulfide-linked chimera between human IgG Fc (Mw 32 kDa) and chicken agrin G3 (Mw 18 kDa). Lane “M” contains the standard molecular weight markers. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
DLS
We have studied the G3-Fc protein at various concentrations to gain information about its purity and diffusion coefficient at 20.0°C, and the results are presented in Figure 2. From the DLS data measured at different concentrations, we have concluded that G3-Fc is homogenous and highly pure with ≥ 95% of the protein present as a single species (Fig. 2A). We have experimentally determined a value of 4.60 ± 0.10 nm for the hydrodynamic radius (Rh, Fig. 2B). For the diffusion coefficient (D020,w) we have obtained a value of 4.70 ± 0.20 × 10−7 cm2/s (corrected to standard conditions of water) that is related to Rh via the Stokes-Einstein equation (Eq. 1) (Table I). We have found from the DLS data that the G3-Fc has almost no concentration dependence and that the protein exists as a monodisperse solution over the wide range of concentrations.
Figure 2.

Hydrodynamic properties of the G3-Fc fusion protein. (A) DLS profile of G3-Fc suggesting the presence of a single component at 3 mg/mL. (B) Plot of concentration (g/mL) versus Rh (nm) for G3-Fc. The hydrodynamic radius Rh has been determined by DLS with concentrations in the range of 1 to 4.0 mg/mL and extrapolating the values to zero concentration. (C) SEDFIT analysis fit and residuals for the 1 mg/mL sample. (D) The c(s) fits for absorbance data (0.48, 0.72, and 1.00 mg/mL concentrations). The X-axis in the bottom panel represents the sedimentation coefficient and the Y-axis shows c(s) distribution. (E) Plot of concentration (g/mL) versus 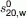 (S) for G3-Fc. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
(S) for G3-Fc. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Table I.
Hydrodynamic Results for the G3-Fc
| Hydrodynamic properties | G3-Fc (Experimental) | G3-Fc best fit model (HYDROPRO) | G3-Fc - lower and upper limit (HYDROPRO) | G3-Fc - Mean (HYDROPRO)e |
|---|---|---|---|---|
| Rh (nm)a | 4.60 ± 0.10 | 4.77 | 4.75–4.90 | 4.82 (5.35 × 10−02) |
| D020,w (10−7 cm2/s)a | 4.70 ± 0.20 | 4.50 | 4.37–4.51 | 4.45 (5.16 × 10−09) |
| s020,w (S)b | 4.95 ± 0.10 | 5.00 | 4.84–5.00 | 4.92 (5.73 × 10−02) |
| P | 1.30 ± 0.04 | 1.34 | 1.33–1.37 | 1.35 (1.51 × 10−02) |
| Mwd (kDa) from s20,w and D020,w | 98.50 | 103.00 | 97–103 | 102 (1.72 × 10−02) |
| f/f0 | 1.50 ± 0.04 | 1.50 | 1.49–1.54 | 1.51 (1.73 × 10−02) |
| Rg (nm)c | 5.00 ± 0.40 | 4.90 | 4.78–5.00 | 4.94 (7.91 × 10−02) |
| G | 2.53 ± 0.06 | 2.53 | 2.53–2.67 | 2.57 (8.03 × 10−02) |
| Dmax (nm)c | 17.50 | 17.60 | 17.40–18.00 | 17.70 (3.06 × 10−01) |
| Rg/Rh | 1.06 | 1.03 | 1.00–1.04 | 1.02 (1.36 × 10−02) |
Dynamic light scattering.
Analytical ultracentrifugation.
Small-angle X-ray scattering.
The sequence-based molecular weight for G3-Fc is 99.50kDa.
Standard deviations are in brackets.
The partial specific volume ( ) and molecular weight (Mw) have been calculated from the amino acid sequence using the SEDNTERP algorithm.33
) and molecular weight (Mw) have been calculated from the amino acid sequence using the SEDNTERP algorithm.33
Rh, hydrodynamic radius; D020,w, diffusion coefficient; s020,w, sedimentation coefficient; P, Perrin function (For the Perrin function we have considered a 3% error since it has been derived from sedimentation data that contain 3% error); f/f0, frictional ratio; Rg, radius of gyration; G, reduced radius of gyration function; and Dmax, maximum dimension of the scattering particle. The Perrin function and reduced radius of gyration have been calculated from sedimentation and SAXS data, respectively, using the Universal_Param utility by Harding et al.35
| (1) |
where D is the diffusion coefficient, k is the Boltzmann constant (1.38 × 10−16 erg K−1), T is the temperature (K) and η is the solvent viscosity.
AUC
We have used AUC in sedimentation velocity mode at 20.0°C to investigate the purity of G3-Fc protein and to analyse its “in solution” behavior at multiple concentrations. The sedimentation velocity experiment indicates an aggregation-free preparation and the presence of only one significant component, demonstrating that G3-Fc is highly pure and monodisperse in solution (Figs. 2C,E). Regarding to the purity of the fusion protein, these results are consistent with observations made from the DLS. The c(s) profile from SEDFIT for G3-Fc at three concentrations is shown in Figure 2C. We have determined a value of 4.95 ± 0.10 S for the sedimentation coefficient (s020,w, see Table I). The s020,w (S) value was obtained from the s20,w (S) for each concentration that was corrected for viscosity and density of water using SEDNTERP 33 followed by their extrapolation to infinite dilution.
In summary, SDS-PAGE, DLS, and AUC methods indicate that the protein sample is highly pure and monodisperse in solution, which is a prerequisite for the SAXS studies. Further, as discussed by Solanki et al., 34 any interparticulate or aggregation effects are generally absent at low concentration of antibodies. Therefore, we have selected low concentration ranges to study this protein where no such effects are observed.
Biophysical analysis
We have calculated an f/f0 value of 1.50 ± 0.04 for G3-Fc from the sedimentation data using the “Universal_Param” utility by Harding et al.35 The f/f0 is a hydration dependent parameter of macromolecules that can give us an indication of its shape in solution. Interestingly, Carrasco et al.36 have reported f/f0 values of different subclasses of IgG molecules in the range from 1.50 to 1.80 (Mw 150 – 160 kDa), which agrees with our observation for an antibody chimera-like structure of G3-Fc (Mw 99.5 kDa). Furthermore, we also have obtained an f/f0 value of ˜1.50 from SEDFIT analysis. In general, frictional ratios of 1.0–1.3 are observed for globular molecules, 1.5–1.8 are observed for asymmetric or glycosylated proteins and values higher than 1.8 are obtained for elongated molecules.37–39
We have further combined the results from DLS and AUC to calculate the molecular weight of the G3-Fc protein using the Svedberg equation (Eq. 2).40
| (2) |
where M is the molecular weight, s0 is the sedimentation coefficient, R is the gas constant (8.31 × 107 erg K−1 mol−1), T is the temperature, D0 is the diffusion coefficient,  is the partial specific volume and ρ is the density. We have calculated a partial specific volume of 0.739 mL/g for the G3-Fc protein using the program SEDNTERP. The superscript zero indicates that the values of the sedimentation and diffusion coefficients, which were measured at several different concentrations, have been extrapolated to zero concentration to remove any effects of interactions between particles on their movement. From the results presented in Table I and using Eq. 2, we have calculated a molecular weight of 98.5 kDa for G3-Fc that is in excellent agreement with the sequence-based Mw of 99.5 kDa.
is the partial specific volume and ρ is the density. We have calculated a partial specific volume of 0.739 mL/g for the G3-Fc protein using the program SEDNTERP. The superscript zero indicates that the values of the sedimentation and diffusion coefficients, which were measured at several different concentrations, have been extrapolated to zero concentration to remove any effects of interactions between particles on their movement. From the results presented in Table I and using Eq. 2, we have calculated a molecular weight of 98.5 kDa for G3-Fc that is in excellent agreement with the sequence-based Mw of 99.5 kDa.
SAXS
Solution X-ray scattering experiments have been performed on protein solutions at a concentration range between 2 and 4 mg/mL (Fig. 3A,B). We have carried out a Guinier analysis using the ATSAS suite 41 and have obtained an Rg value of 4.80 ± 1.00 nm for the G3-Fc fusion protein with a s•Rg limit of 1.30 (see Table I). This finding is confirmed by the program GNOM, which has yielded an Rg value of 5.00 ± 0.40 nm that is in agreement with the Rg from Guinier analysis. The maximum dimension (Dmax) determined from GNOM analysis is 17.50 nm.
Figure 3.

SAXS and 3D domain structure. (A) Pair (or distance) distribution function [p(r)] and maximum dimension (Dmax) of the SAXS data of G3-Fc. Values were obatained using the program GNOM. (B) Plot of scattering intensity I(s) versus momentum transfer [s(Å−1)] for G3-Fc. Red colored dots represent raw data and superimposed blue coloured lines represent BUNCH fit data. The satisfactory value of 1.6 demonstrates the excellent agreement between raw data and BUNCH fit data. (C) Solution conformation of the G3-Fc in “surface” representation. N-G3 and C-G3 represent the N-terminus and the C-terminus of the G3 domain. N-Fc and C-Fc indicate the N-terminus and C-terminus of the Fc domain. (D) Schematic representation of the maximum displacement of individual BUNCH models. (Left) the 10-amino acid residue linker segments shows a transversal dislocation of 6.8Å (blue box) causing a maximum displacement of 14.5Å for the outer edge of the recognition surface of the G3 domain. (Right) The overall radius of gyration of the center of gravity for the recognition surface area is 58°. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
After using the program CRYSOL to calculate the scattering profiles, we have used the program BUNCH to determine the solution structure of G3-Fc (Fig. 3B,C). During the BUNCH calculations, we have fixed the structure of the Fc region and allowed the G3 domains to move and rotate freely within the P2 point symmetry restraints. The 12 G3-Fc models obtained from the program BUNCH have χ values (goodness-of-fit of the models with experimental data) of about 1.6. The normalized spatial discrepancy (NSD) factor of 0.8 was obtained from program DAMAVER which indicates close agreement of the independent BUNCH models.
In addition to the frictional ratio f/fo, the ratio of Rg and Rh can provide an indication about the solution conformation of macromolecules. It has been reported previously that the ratio of Rg/Rh is ˜ 0.7 for globular proteins and > 2.0 for elongated structures.42 For G3-Fc, the ratio is 1.06, which further demonstrates that this protein is neither globular nor very elongated in solution.
Discussion
G3-Fc protein structure evaluation
SAXS has emerged as a powerful technique to study the solution conformation of macromolecules and their assemblies as discussed in several reviews over the last few years. It can provide information about size, shape, and domain organization of macromolecules in solution. The Universal shape parameters (or size-independent parameters) called Perrin function (P, frictional ratio due to shape) and the reduced radius of gyration (G) have been previously used to study shape information of antibodies in solution.
The G3-Fc fusion protein has a T-shaped form where both G3 domains extrude almost perpendicularly from longitudinal axis of the Fc fragment (Fig. 3C). The spatial distance of 33Å between the G3 domain and the Fc tail allows for a large degree of structural flexibility and excludes any steric hindrance in their relative quaternary assembly. The recognition surfaces (equivalent to the splice inserts at the splicing site B of the G3 domain) are located at the outer edge of the recombinant molecule and are freely accessible for binding small molecule ligands (e.g., Heparin) or other proteins (e.g., Dystroglycan). This fulfils our goal to construct a G3-Fc fusion protein that can be attached to an SPR chip and will enable us to investigate novel ligands from cell lysates from different origins.
To validate the G3-Fc models, we have used the program HYDROPRO to calculate the hydrodynamic properties (Rh, Rg, G, s20,w, P, D20,w, and Dmax) for each structure that we have obtained from the program BUNCH. Thus, we can compare the theoretical values with the experimental data (see Table I). Close agreement between experimentally determined parameters via DLS, AUC, and SAXS and the calculated parameters from HYDROPRO strongly supports our modeling approach. An overall comparison of all obtained BUNCH models have revealed that the linker segment undergoes only small-scale motions (Fig. 3D). Based on the experimental and HYDROPRO results, we have selected the single best-fitting model out of the 12 models calculated and presented in Figure 3(C). It should be noted that the NSD value of 0.8 suggests that all the models are significantly similar to each other. With a Cα-atom displacement of maximum 14.5 Å at the outer edge of the recognition surface area of G3 and a maximum outer radius of rotation of ˜58°, the G3-Fc fusion protein has revealed a well defined G3-Fc structure with limited spatial flexibility between both covalently linked subfragments.
Because the G3 domains are located at the same position as “Fab” domains on human IgG Fc, we have expected a similarity of shape between the G3-Fc protein and human IgG Fc. Remarkably, while investigating the solution conformation for the G3-Fc protein, we have discovered a “T”-shaped arrangement of the G3 domains attached to the Fc domain as compared to the “Y”-shaped arrangement of “Fab” domains (Fig. 3C). Both globular G3 domains of agrin are almost perpendicularly oriented relative to the Fc fragment. This is in contrast to the ˜110° elbow angle of the hinge region of antibody structures. All of our attempts in BUNCH to model the G3 domain in elbow angles according to hinge regions of immunoglobulins have failed. Deviations in the kink angle of +/− 10° caused already NSD values of 2.8 and the obtained models were not interpretable in BUNCH/DAMAVER (data not shown).
Finally, we would like to point out that the obtained G3-Fc fusion protein structure corresponds to the best representative of the experimentally measured parameters and is thus a time-averaged model. We consider that the G3-Fc is a semiflexible molecule based on the following arguments. First, it has been suggested that for IgG and IgA, two clear peaks in the distance distribution function profile are indicative of a nonflexible nature.34 43 Here, the distance distribution plot shows mainly one maxima but there is clearly second minor peak present for the G3-Fc (see, Fig. 3A). Second, the low NSD value of 0.8 indicates that the individual BUNCH models agree with each other, meaning that they are not highly flexible (Fig. 3D). The upper and lower limits of solution properties calculated from HYDROPRO for all 12 models also vary only slightly, which further gives an indication of close agreement of the models with each other. Finally, the simulated annealing protocols for the individual 3D domain structures revealed only a very limited spatial grid window of 100 Å3 for the center of gravity of the G3 domain (Fig. 3D). Despite the fact that the G3 domains and Fc-fragments do not show any steric clashes and are only connected via the linker segment, the spatial flexibility seems likely to be limited in solution.
Conclusions
The data presented here show for the first time a solution structure of the agrin G3-Fc fusion protein at nanoscale resolution. Our experiments indicate a “T”-shaped molecule with a limited flexible linker connecting both G3 domains to their Fc scaffold and free accessibility of both binding regions. Our data also support strategies in which tandem domains of G2 and G3 can be linked to the Fc-tag, allowing free access to the binding partners. We aim to continue in this direction, and our future studies will focus on screening of novel ligands to study protein-ligand complexes using our integrated approach.
Materials and Methods
Expression and purification
The pCEP-Pu plasmid containing the G3 domain of chicken (Gallus gallus) agrin fused to the Fc region of human IgG was used for eukaryotic expression of the G3-Fc fusion protein. We have established a stably transfected HEK 293 cell line to obtain the G3-Fc fusion protein using the nonliposomal lipid transfection reagent Effectene™ (Qiagen, CA), using the protocol described by the manufacturer. Dulbecco's modified Eagle's medium containing1% glutamine, 10-mM sodium pyruvate, 10% fetal bovine serum (FBS), 100 μg/mL of penicillin and 100 μg/mL of streptomycin was used as growth medium. The transfected cells were selected for puromycin resistance, using puromycin at a concentration of 2 μg/mL. Within 3 weeks, colonies of transfected cells started to appear. The stably transfected cells were then allowed to grow at 37°C in growth medium until about 80% confluence level was reached. Then the cells were transferred to an expression medium (growth medium without 10% FBS) that was collected every 48 h (as these proteins are being secreted) followed by an exchange with fresh expression media. The collected medium was centrifuged at 2000 × g for 5 min to pellet the cells before storing at −20°C. After thawing, it was first dialyzed overnight against dialysis buffer 1 (PBS, pH 7.5) at room temperature and then concentrated using a membrane filter with a molecular weight cutoff of 30 kDa. The C-terminal Fc domain allowed the purification of the fusion protein to homogeneity by affinity chromatography using a Protein A column (GE Healthcare). Fractions of 1 mL were collected from the column and then analyzed by tricine SDS-PAGE. The protein was then dialysed at room temperature against dialysis buffer 2 (50-mM Tris, 200-mM NaCl, 10-mM EDTA, pH 7.5). The EDTA was removed by a third dialysis step at room temperature against buffer 3 (50-mM Tris, 200-mM NaCl, pH 7.5). The concentration of the purified protein was calculated from the measured absorbance at 280 nm, using a molar extinction coefficient of 66850 M−1 cm−1. The value of the extinction coefficient was obtained from the ProtParam tool available on ExPASy server. The purified proteins were stored at 4°C.
DLS
The G3-Fc fusion protein was filtered using a 0.1 μm centrifugal filter (Millipore) in a buffer containing 50-mM Tris at pH 7.5 and 200-mM NaCl before DLS analysis at concentrations up to 4 mg/mL. Samples were allowed to equilibrate for 4 min at 20.0°C before collecting data in the “automatic mode”. For better reproducibility, four measurements were made at each protein concentration, and the average value was used in the subsequent calculations. The resulting data were analysed using DTS software (Version 5.10.2, Malvern Instruments, Malvern, UK). The hydrodynamic radius (Rh) was measured at different concentrations before extrapolating to infinite dilution.
AUC
Sedimentation velocity experiments were performed using a Beckman (Palo Alto) Optima XL-I analytical ultracentrifuge equipped with absorption and interference optics and an automatic online data capture system. Standard 12-mM double sector cells were loaded with 0.4 mL of samples at 0.48, 0.72, and 1.00 mg/mL concentrations (for absorbance optics) and 2.90 mg/mL for interference optics with reference solvent (50-mM Tris, 200-mM NaCl, pH 7.5) in the appropriate channels. The balanced cells were placed in an analytical four-hole rotor (An60-Ti). After allowing time for vacuum formation and temperature equilibration (20.0°C), the rotor was accelerated to 45,000 rpm. Using the UV/Interference optical system, scans of relative concentration versus radial displacement r from the axis of rotation were taken at 4 min intervals throughout the duration of the experiment. The weight average sedimentation coefficient s20,b (S) (measured in seconds or Svedberg units S = 10−13 sec), was obtained from SEDFIT [c(s)] analysis 44 45 and was subsequently corrected to standard solvent conditions (density and viscosity of water at 20°C) to yield s20,w using SEDNTERP. To account for hydrodynamic nonideality, the apparent sedimentation coefficients (s20,w) were calculated at each concentration and extrapolated to infinite dilution to obtain s020,w (S).
SAXS
The SAXS experiments were performed at a protein concentration range of 2–4 mg/mL using a Rigaku 3-pinhole camera (S-MAX3000) equipped with a Rigaku MicroMax+002 microfocus sealed tube (Cu Kα radiation at 1.54 Å) and a Confocal Max-Flux optics system operating at 40 W. The system had a 3-m fully evacuated camera length. Data were recorded using a 200-mM multiwire 2D detector, which was calibrated with gold particles (NIST Standard Reference Material 8012, NIST, MD). SAXS data for the G3-Fc protein were collected within the range of 0.008 ≤ s ≤ 0.27 Å−1 and an exposure time of 2 to 4 h. The momentum transfer, s is defined as 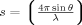 , where θ is the scattering angle and λ the wavelength of the X-ray radiation. The data reduction was performed using Rigaku's SAXGUI data processing software. The data were normalized by the scattering cross section per unit sample volume. Scattering data collected from buffer (50-mM Tris, 200-mM NaCl, pH 7.5) were subtracted from the sample data. Datasets from all concentrations were then carefully merged using the primary data analysis package- PRIMUS 46 to obtain a single data set for further analysis as described previously. 47 Radiation damage was not detected.
, where θ is the scattering angle and λ the wavelength of the X-ray radiation. The data reduction was performed using Rigaku's SAXGUI data processing software. The data were normalized by the scattering cross section per unit sample volume. Scattering data collected from buffer (50-mM Tris, 200-mM NaCl, pH 7.5) were subtracted from the sample data. Datasets from all concentrations were then carefully merged using the primary data analysis package- PRIMUS 46 to obtain a single data set for further analysis as described previously. 47 Radiation damage was not detected.
3D domain structure
The raw data were processed with PRIMUS. The distance distribution function p(r), radii of gyration (Rg) and the maximum dimension of particle (Dmax) were obtained from the program GNOM. This program provides the radius of gyration and maximum particle dimension for monodisperse samples by evaluating distance distribution function.48 An additional value for the Rg was obtained using the Guinier approximation 49 from data from the low angle region. Since the high-resolution structures of the G3 and Fc fragment of human IgG are known, we followed the modeling approach where we could use the high-resolution structure information for most of the part of the protein except that for the linker. First, the atomic coordinates of the solution NMR model of G3 (PDB-code: 1Q56) and the X-ray crystal structure of IgG Fc (PDB-code: 2DTQ) were obtained from the RCSB Protein Data Dank and their solution scattering profiles was calculated using the program CRYSOL.50 In the second step, the program BUNCH 51 was used to determine the optimal positions and orientations of the high-resolution structural models and to place the dummy residues for the part of the protein for which the high-resolution information was missing. The 10 amino acid linker segment between the G3 and Fc domains was modelled using MODLOOP and the connection between both domains was manually adjusted. 52 Twelve different conformations were generated for the G3-Fc fusion protein in the program BUNCH. We kept the Fc domain fixed whereas the G3 domains were allowed to rotate and translate freely within the symmetry restraints. Finally, all the models we calculated using BUNCH were compared using the DAMAVER package 53 that uses the program SUPCOMB. 54 The goodness of the superimposition of these models was estimated by the overlap function –NSD.
To analyze the flexibility in the relative orientation of both individual domains, we used a combined simulated annealing—Powel minimization protocol, treating Fc-fragment and both G3 domains as rigid body system. The starting temperature for the slow-cooling approach was 2500K with a drop in temperature of 25K per dynamic cycle. For each individual subset of the obtained 3D domain structures from BUNCH we performed 500 cycles of conjugate gradient minimization using the force field parameters from CNS.55 During refinement, strict constraints were applied on the torsion angles of the 10 amino acid residue linker segment. Harmonic restraints were imposed on both individual domains with increased weight (25kcal/mol/Å2) for the Cα atoms.
Comparison of the experimental and calculated hydrodynamic parameters
We used the program HYDROPRO 56 to calculate the hydrodynamic parameters such as the sedimentation coefficient, diffusion coefficient, hydrodynamic radius and the radius of gyration for each model generated by BUNCH. We set the atomic element radius to 3.3 Å, the temperature to 293 K, the partial specific volume to 0.739 mL/g and the molecular weight of protein corresponding to an Fc domain (consisting of two disulfide-linked chains) and two G3 domains (see Table I).
Acknowledgments
T.R. Patel has received a Manitoba Health Research Council/Manitoba Institute of Child Health Postdoctoral Fellowship. We would like to thank Prof. Klaus Wrogemann and Ms. Nehal Patel for helping us with stable transfection. J. Stetefeld holds a Canada Research Chair in Structural Biology.
References
- 1.Tsen G, Halfter W, Kroger S, Cole GJ. Agrin is a heparan sulfate proteoglycan. J Biol Chem. 1995;270:3392–3399. doi: 10.1074/jbc.270.7.3392. [DOI] [PubMed] [Google Scholar]
- 2.Bowe MA, Fallon JR. The role of agrin in synapse formation. Ann Rev Neuro. 1995;18:443–462. doi: 10.1146/annurev.ne.18.030195.002303. [DOI] [PubMed] [Google Scholar]
- 3.Fallon JR, Gelfman CE. Agrin-related molecules are concentrated at acetylcholine receptor clusters in normal and aneural developing muscle. J Cell Biol. 1989;108:1527–1535. doi: 10.1083/jcb.108.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nitkin RM, Smith MA, Magill C, Fallon JR, Yao YM, Wallace BG, McMahan UJ. Identification of agrin, a synaptic organizing protein from Torpedo electric organ. J Cell Biol. 1987;105:2471–2478. doi: 10.1083/jcb.105.6.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wallace BG. Agrin-induced specializations contain cytoplasmic, membrane, and extracellular matrix-associated components of the postsynaptic apparatus. J Neurosci. 1989;9:1294–1302. doi: 10.1523/JNEUROSCI.09-04-01294.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khan AA, Bose C, Yam LS, Soloski MJ, Rupp F. Physiological regulation of the immunological synapse by agrin. Science. 2001;292:1681–1686. doi: 10.1126/science.1056594. [DOI] [PubMed] [Google Scholar]
- 7.Hilgenberg LGW, Su H, Gu H, O'Dowd DK, Smith MA. α3Na+/K+-ATPase is a neuronal receptor for agrin. Cell. 2006;125:359–369. doi: 10.1016/j.cell.2006.01.052. [DOI] [PubMed] [Google Scholar]
- 8.Hoover CL, Hilgenberg LGW, Smith MA. The COOH-terminal domain of agrin signals via a synaptic receptor in central nervous system neurons. J Cell Biol. 2003;161:923–932. doi: 10.1083/jcb.200301013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alfsen A, Yu H, Magerus-Chatinet A, Schmitt A, Bomsel M. HIV-1-infected blood mononuclear cells form an integrin- and agrin-dependent viral synapse to induce efficient HIV-1 transcytosis across epithelial cell monolayer. Mol Biol Cell. 2005;16:4267–4279. doi: 10.1091/mbc.E05-03-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML, Burden SJ. Lrp4 is a receptor for agrin and forms a complex with MuSK. Cell. 2008;135:334–342. doi: 10.1016/j.cell.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008;60:285–297. doi: 10.1016/j.neuron.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sane JR. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- 13.Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee K-F. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 2001;410:1057–1064. doi: 10.1038/35074025. [DOI] [PubMed] [Google Scholar]
- 14.Stetefeld J, Jenny M, Schulthess T, Landwehr R, Schumacher B, Frank S, Ruegg MA, Engel J, Kammerer RA. The laminin-binding domain of agrin is structurally related to N-TIMP-1. Nat Struct Biol. 2001;8:705–709. doi: 10.1038/90422. [DOI] [PubMed] [Google Scholar]
- 15.Mascarenhas JB, Ruegg MA, Sasaki T, Eble JA, Engel J, Stetefeld J. Structure and laminin-binding specificity of the NtA domain expressed in eukaryotic cells. Matrix Biol. 2005;23:507–513. doi: 10.1016/j.matbio.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Mascarenhas JB, Ruegg MA, Winzen U, Halfter W, Engel J, Stetefeld J. Mapping of the laminin-binding site of the N-terminal agrin domain (NtA) EMBO J. 2003;22:529–536. doi: 10.1093/emboj/cdg041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McFarlane AA, Stetefeld J. An interdomain disulfide bridge links the NtA and first FS domain in agrin. Protein Sci. 2009;18:2421–2428. doi: 10.1002/pro.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stetefeld J, Alexandrescu AT, Maciejewski MW, Jenny M, Rathgeb-Szabo K, Schulthess T, Landwehr R, Frank S, Ruegg MA, Kammerer RA. Modulation of agrin function by alternative splicing and Ca2+ binding. Structure. 2004;12:503–515. doi: 10.1016/j.str.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Scotton P, Bleckmann D, Stebler M, Sciandra F, Brancaccio A, Meier T, Stetefeld J, Ruegg MA. Activation of muscle-specific receptor tyrosine kinase and binding to dystroglycan are regulated by alternative mRNA splicing of agrin. J Biol Chem. 2006;281:36835–36845. doi: 10.1074/jbc.M607887200. [DOI] [PubMed] [Google Scholar]
- 20.Gesemann M, Cavalli V, Denzer AJ, Brancaccio A, Schumacher B, Ruegg MA. Alternative splicing of agrin alters its binding to heparin, dystroglycan, and the putative agrin receptor. Neuron. 1996;16:755–767. doi: 10.1016/s0896-6273(00)80096-3. [DOI] [PubMed] [Google Scholar]
- 21.Campanelli JT, Gayer GG, Scheller RH. Alternative RNA splicing that determines agrin activity regulates binding to heparin and alpha-dystroglycan. Development. 1996;122:1663–1672. doi: 10.1242/dev.122.5.1663. [DOI] [PubMed] [Google Scholar]
- 22.Ferns MJ, Campanelli JT, Hoch W, Scheller RH, Hall Z. The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface proteoglycans. Neuron. 1993;11:491–502. doi: 10.1016/0896-6273(93)90153-i. [DOI] [PubMed] [Google Scholar]
- 23.Gesemann M, Denzer AJ, Ruegg MA. Acetylcholine receptor-aggregating activity of agrin isoforms and mapping of the active site. J Cell Biol. 1995;128:625–636. doi: 10.1083/jcb.128.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hopf C, Hoch W. Agrin binding to alpha-dystroglycan. J Biol Chem. 1996;271:5231–5236. doi: 10.1074/jbc.271.9.5231. [DOI] [PubMed] [Google Scholar]
- 25.O'Toole JJ, Deyst KA, Bowe MA, Nastuk MA, McKechnie BA, Fallon JR. Alternative splicing of agrin regulates its binding to heparin, alpha -dystroglycan, and the cell surface. Proc Natl Acad Sci USA. 1996;93:7369–7374. doi: 10.1073/pnas.93.14.7369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cotman SL, Halfter W, Cole GJ. Identification of extracellular matrix ligands for the heparan sulfate proteoglycan agrin. Exp Cell Res. 1999;249:54–64. doi: 10.1006/excr.1999.4463. [DOI] [PubMed] [Google Scholar]
- 27.Jury E, Kabouridis P. New role for Agrin in T cells and its potential importance in immune system regulation. Arthritis Res Therapy. 2010;12:205–211. doi: 10.1186/ar2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hilgenberg LGW, Su H, Gu H, O'Dowd DK, Smith MA. α3Na+/K+-ATPase is a neuronal receptor for agrin. Cell. 2006;125:359–369. doi: 10.1016/j.cell.2006.01.052. [DOI] [PubMed] [Google Scholar]
- 29.Tidow H, Aperia A, Nissen P. How are ion pumps and agrin signaling integrated? Trends Biochem Sci. 2010;35:653–659. doi: 10.1016/j.tibs.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 30.Winzor DJ. Surface plasmon resonance as a probe of protein isomerization. Anal Biochem. 2003;318:1–12. doi: 10.1016/s0003-2697(03)00133-7. [DOI] [PubMed] [Google Scholar]
- 31.Karlsson R. SPR for molecular interaction analysis: a review of emerging application areas. J Mol Recog. 2004;17:151–161. doi: 10.1002/jmr.660. [DOI] [PubMed] [Google Scholar]
- 32.Patel TR, Morris GA, Zwolanek D, Keene DR, Li J, Harding SE, Koch M, Stetefeld J. Nano-structure of the laminin γ-1 short arm reveals an extended and curved multidomain assembly. Matrix Biol. 2010;29:565–572. doi: 10.1016/j.matbio.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 33.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding SE, Rowe AJ, Horton JC, editors. Analytical ultracentrifugation in biochemistry and polymer science. Cambridge, United Kingdom: Royal Society of Chemistry; 1992. pp. 90–125. [Google Scholar]
- 34.Solanki AK, Boone CD, Krueger JK. Global structure of HIV-1 neutralizing antibody IgG1 b12 is asymmetric. Biochem Biophys Res Comm. 2010;391:947–951. doi: 10.1016/j.bbrc.2009.11.170. [DOI] [PubMed] [Google Scholar]
- 35.Harding SE, Colfen H, Aziz Z. The ELLIPS suit of whole-body protein conformation algorithms for Microsoft Windowns. In: Scoot DJ, Harding SE, Rowe AJ, editors. Analytical ultracentrifugation techniques and methoda. Cambdrige: Royal Society of Chemistry; 2005. pp. 468–483. [Google Scholar]
- 36.Carrasco B, Garcia de la Torre J, Davis KG, Jones S, Athwal D, Walters C, Burton DR, Harding SE. Crystallohydrodynamics for solving the hydration problem for multi-domain proteins: open physiological conformations for human IgG. Biophys Chem. 2001;93:181–196. doi: 10.1016/s0301-4622(01)00220-4. [DOI] [PubMed] [Google Scholar]
- 37.Tanford C. Physical chemistry of macromolecules. New York: Wiley; 1961. [Google Scholar]
- 38.Schuck P. Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown PH, Balbo A, Schuck P. Characterizing protein-protein interactions by sedimentation velocity analytical ultracentrifugation. Curr Protoc Immunol. 2008:15. doi: 10.1002/0471142735.im1815s81. Chapter 18, Unit 18. [DOI] [PubMed] [Google Scholar]
- 40.van Holde KE. Physical biochemistry. Englewood Cliffs, NJ: Prentice Hall; 1985. [Google Scholar]
- 41.Konarev PV, Petoukhov MV, Volkov VV, Svergun DI. ATSAS 2.1, a program package for small-angle scattering data analysis. J Appl Cryst. 2006;39:277–286. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burchard W. Static and dynamic light scattering approaches to structure determination of biopolymers. In: Harding SE, Sattelle DB, Bloomfield VA, editors. Laser light scattering in biochemistry (.) Cambdrige: Royal Society of Chemistry; 1992. pp. 3–22. [Google Scholar]
- 43.Boehm MK, Woof JM, Kerr MA, Perkins SJ. The Fab and Fc fragments of IgA1 exhibit a different arrangement from that in IgG: a study by X-ray and neutron solution scattering and homology modelling. J Mol Biol. 1999;286:1421–1447. doi: 10.1006/jmbi.1998.2556. [DOI] [PubMed] [Google Scholar]
- 44.Dam J, Schuck P. Calculating sedimentation coefficient distributions by direct modeling of sedimentation velocity concentration profiles. Methods in Enzymology. In: Brand L, Johnson ML, editors. Academic Press; 2004. pp. 185–212. Numerical computer methods, Part E Vol. 384 edit. [DOI] [PubMed] [Google Scholar]
- 45.Schuck P. Sedimentation analysis of noninteracting and self-associating solutes using numerical solutions to the Lamm equation. Biophys J. 1998;75:1503–1512. doi: 10.1016/S0006-3495(98)74069-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Cryst. 2003;36:1277–1282. [Google Scholar]
- 47.Patel TR, Morris GA, Zwolanek D, Keene DR, Li J, Harding SE, Koch M, Stetefeld J. Nano-structure of the laminin gamma-1 short arm reveals an extended and curved multidomain assembly. Matrix Biol. 2010;29:565–572. doi: 10.1016/j.matbio.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 48.Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Cryst. 1992;25:495–503. [Google Scholar]
- 49.Guinier A, Fourner G. Small angle scattering of X-rays. New York: Wiley; 1955. [Google Scholar]
- 50.Svergun D, Barberato C, Koch MHJ. CRYSOL - a program to evaluate X-ray solution scattering of biological macromolecules from attomic coordinates. J Appl Cryst. 1995;28:768–773. [Google Scholar]
- 51.Petoukhov MV, Svergun DI. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fiser A, Sali A. ModLoop: automated modeling of loops in protein structures. Bioinformatics. 2003;19:2500–2501. doi: 10.1093/bioinformatics/btg362. [DOI] [PubMed] [Google Scholar]
- 53.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Cryst. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. J Appl Cryst. 2001;34:33–41. [Google Scholar]
- 55.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 56.GarcÌa de la Torre J, Huertas ML, Carrasco B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys J. 2000;78:719–730. doi: 10.1016/S0006-3495(00)76630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]