

Abstract
In Pseudomonas aeruginosa, the chromosomally encoded class C cephalosporinase (AmpC β-lactamase) is often responsible for high-level resistance to β-lactam antibiotics. Despite years of study of these important β-lactamases, knowledge regarding how amino acid sequence dictates function of the AmpC Pseudomonas-derived cephalosporinase (PDC) remains scarce. Insights into structure-function relationships are crucial to the design of both β-lactams and high-affinity inhibitors. In order to understand how PDC recognizes the C3/C4 carboxylate of β-lactams, we first examined a molecular model of a P. aeruginosa AmpC β-lactamase, PDC-3, in complex with a boronate inhibitor that possesses a side chain that mimics the thiazolidine/dihydrothiazine ring and the C3/C4 carboxylate characteristic of β-lactam substrates. We next tested the hypothesis generated by our model, i.e. that more than one amino acid residue is involved in recognition of the C3/C4 β-lactam carboxylate, and engineered alanine variants at three putative carboxylate binding amino acids. Antimicrobial susceptibility testing showed that the PDC-3 β-lactamase maintains a high level of activity despite the substitution of C3/C4 β-lactam carboxylate recognition residues. Enzyme kinetics were determined for a panel of nine penicillin and cephalosporin analog boronates synthesized as active site probes of the PDC-3 enzyme and the Arg349Ala variant. Our examination of the PDC-3 active site revealed that more than one residue could serve to interact with the C3/C4 carboxylate of the β-lactam. This functional versatility has implications for novel drug design, protein evolution, and resistance profile of this enzyme.
Keywords: β-lactamase inhibitor resistance, AmpC cephalosporinases, Pseudomonas aeruginosa
Introduction
Pseudomonas aeruginosa is a Gram-negative bacillus responsible for life-threatening nosocomial infections including pneumonia, blood stream, intra-abdominal, wound, eye, ear, and urinary tract infections.1 The organism, “an opportunistic pathogen,” is often isolated from patients with multiple illnesses, in-dwelling catheters, burns, and surgical devices.2 P. aeruginosa is also one of the most commonly isolated Gram-negative bacteria from patients in intensive care units in the United States, and the incidence has been rising during the past several decades.3 Mortality rates up to 60% are reported for P. aeruginosa infections, particularly in patients with immune compromise or underlying comorbidities.1,4 Treatment guidelines often advise empiric administration of a β-lactam in combination with a fluoroquinolone or aminoglycoside, but despite this “combination approach,” outcomes are poor.5 The prevalence of P. aeruginosa strains resistant to three or more antimicrobial agents can range from 3 to 50%.6,7 Consequently, treatment of P. aeruginosa infections is challenging, and the scarcity of new agents being developed and released into the clinic stresses the need to use currently available agents in a judicious manner.8
The major antibiotic resistance determinants in P. aeruginosa strains are β-lactamase enzymes (e.g., class C of AmpC cephalosporinases and class B verona integron-encoded metallo-β-lactamase (VIM) and β-lactamase active on imipenem (IMP) type metallo-β-lactamases), antibiotic efflux pumps (e.g., MexAB-OprM), and impermeability of the outer membrane.9,10 Production of chromosomal AmpCs mediates resistance to β-lactams, and both genetic mutations and induction from certain β-lactams can significantly increase the expression of these β-lactam-hydrolyzing enzymes.11,12 Protecting the activity of β-lactams by inhibiting β-lactamases is a strategic approach for preserving our current antimicrobial armamentarium. However, the commercially available β-lactamase inhibitors are less effective at inactivating β-lactamases from class C than class A.13–15 Despite the immense clinical significance of this resistance determinant, relatively little research has examined the structure–function relationships of the “Pseudomonal” AmpC (here referred to as Pseudomonas-derived cephalosporinase, PDC) for either substrates or inhibitors [Fig. 1(a)].16,17 A better understanding of the molecular details of catalysis and inhibition of this AmpC can help lead design of more effective inhibitors.
Figure 1.

(a) Molecular representation of the Pseudomonas-derived cephalosporinase model created to study potential substrate carboxylate binding residues. Ser64 is shown in yellow. (b) Protein sequence alignment of E. coli AmpC and P. aeruginosa PDC-3. Highly conserved class C motifs are underlined including S-V-S-K (first serine is Ser64), Y-X-N, and K-T-G. Amino acids Asn343, Asn346, and Arg349 with potential roles in C3/C4 substrate carboxylate binding are also indicated. Numbers listed are based on the consensus numbering of the mature E. coli AmpC protein, which differs slightly from the PDC-3 protein.17
Using boronate substrate analogs, we explored the role of specific residues in the catalytic and inhibitory profile of the P. aeruginosa AmpC. Our target β-lactamase, Pseudomonas-derived cephalosporinase-3 (PDC-3), has a single amino acid change from the reference strain P. aeruginosa PAO1, and was studied by Rodriguez-Martinez et al. for its “extended-spectrum” properties.17 We examined a particular region of the PDC-3 enzyme active site, which has been implicated in the binding of the C3/C4 carboxylate common to penicillins, cephalosporins, and sulfone inhibitors. Previous studies in class A β-lactamases demonstrated that recognition of this C3/C4 carboxylate contributes directly to substrate and inhibitor affinity through hydrogen bonding with areas of positive charge such as Arg244, Arg220, and Arg276.18–25
While the amino acids that participate in C3/C4 carboxylate binding are described for class A, the molecular and biochemical correlates of this recognition are less well understood in class C enzymes. Comparative studies of the Enterobacter cloacae P99 and Escherichia coli AmpC enzymes suggest that the sites such as Xaa343, Asn346, Arg349, or Thr316 interact with the C3/C4 carboxylate of the β-lactam [Fig. 1(b)].26–33 Our examination of the PDC-3 β-lactamase with a boronate β-lactamase inhibitor docked in the active site lead us to hypothesize that AmpCs may have also have a region or “pocket,” which serves to guide the substrate's catalytically competent conformation.33 A molecular model of PDC-3 in complex with a boronate derivative possessing the C3/C4 carboxylate informed our hypothesis that multiple possible “recognition residues” are present in the active site. We tested this notion by creating single amino acid substitutions in PDC-3 at three active site residues. The effects of these substitutions were examined by β-lactam substrate and β-lactamase inhibitor susceptibility. Detailed steady-state kinetic assays were performed for the PDC-3 Arg349Ala variant, which showed the largest phenotype difference in the antimicrobial susceptibility. To understand further the mechanistic basis for changes in kinetic parameters, we next synthesized a panel of boronate compounds, designed as substrate analogs, allowing us to probe the PDC-3 active site topology. Our comparative analysis reveals the complex relationship between sequence, structure, catalysis and inhibition in class C cephalosporinases and, more importantly, PDC is a uniquely plastic enzyme with a region of positive charge that drives the recognition of the C3/C4 carboxylate.34–36
Results and Discussion
C3/C4 Carboxylate binding in PDC-3 cephalosporinase
To better understand the interactions between the PDC-3 β-lactamase and both β-lactam substrates and inhibitors, we first created a molecular model of the cephalothin analog boronate 5 covalently bound to Ser64 of PDC-3 (Fig. 2). This boronate's meta-carboxylate group is designed to mimic in distance and stereochemistry the conserved C3/C4 carboxylate on β-lactam substrates and β-lactamase inhibitors. Thus, the placement of this group offers insight into possible carboxylate binding residues in PDC-3.
Figure 2.
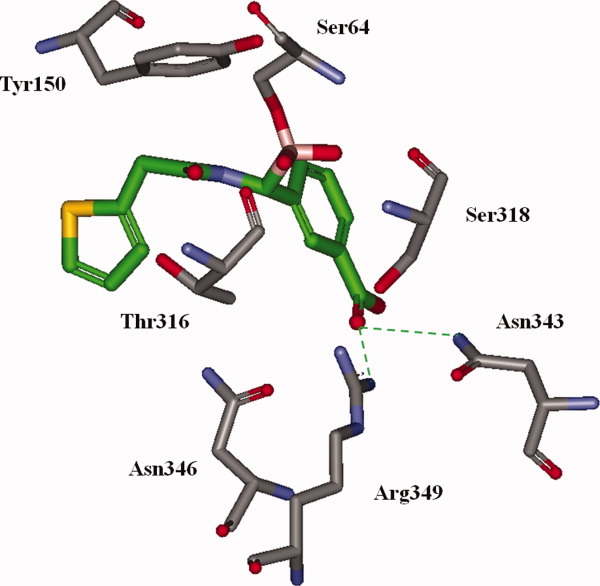
Cephalothin analog 5 docked into the active site of our PDC-3 model to gain insight into the binding relationships of the meta-carboxyphenyl group. In this model, the boronate interacts with Arg349 and Asn343 (represented by dashed green lines), and has possible longer range interactions with Asn346. Active site residues Ser64, Ser318, Thr316, and Tyr150 are labeled.
After energy minimization and equilibration, the PDC-3 model showed us that the meta-carboxylate interacts with Asn343 and Arg349, both of which are implicated in the binding the C3/C4 carboxylate in other AmpC structures.26,32,33 Asn346, another residue responsible for carboxylate recognition, is also found within ∼5 Å of the carboxylate, and potentially plays a role in the overall electrostatic environment of this region. This analysis informed our hypotheses regarding the locations in which C3/C4 carboxylate could bind PDC-3.
Antimicrobial susceptibility testing
Table I summarizes the minimum inhibitory concentration (MIC) values of E. coli DH10B cells expressing PDC-3 alanine variants at residues 343, 346, and 349. Most significantly, the level of resistance to ampicillin, cephalothin, cephaloridine, and cefotaxime conferred by PDC-3 variants in E. coli was lower than the WT enzyme. In contrast, the resistance levels did not decrease more than one dilution for any of the PDC-3 variants for cefoxitin, ceftazidime, and cefepime or the carbapenems (imipenem and meropenem). We note that PDC-3 variants show susceptibilities similar to WT for the substrates with C3 carboxylates, piperacillin and the carbapenems. The exception is ampicillin, which also has a C3 carboxylate but may rely more on recognition of this moiety with its relatively simple R1 and R2 side chains.
Table I.
MIC Values (μg mL−1) of P. aeruginosa 18SH and E. coli DH10B Expressing blaPDC-3 or blaPDC-3 Variants at Possible Carboxylate Binding Sites
| Isolates | Ampicillin | Piperacillin | Cephalothin | Cephaloridine | Cefoxitin | Ceftazidime | Cefotaxime | Cefepime | Aztreonam | Imipenem | Meropenem | Piperacillin/tazobactam |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P. aeruginosa 18SH | 4096 | 512 | >1024 | >1024 | >1024 | 64 | >1024 | 16 | >64 | 2 | 2 | >64 |
| E. coli DH10B | 1 | 2 | 4 | 8 | 8 | <0.06 | <0.06 | <0.06 | 0.125 | 0.5 | <0.06 | 2 |
| E. coli blaPDC-3 | 64 | 16 | 512 | 64 | 16 | 1 | 8 | <0.06 | 0.5–1 | 0.5 | <0.06 | 4 |
| E. coli blaPDC-3 Asn343Ala | 16 | 16 | 256 | 32 | 8 | 1 | 4 | <0.06 | 0.5 | 0.5 | <0.06 | 2 |
| E. coli blaPDC-3 Asn346Ala | 16 | 8 | 256 | 32 | 8 | 1 | 4 | <0.06 | 0.5 | 0.5 | <0.06 | 4 |
| E. coli blaPDC-3 Arg349Ala | 2 | 8 | 64 | 16 | 8 | 1 | 0.5 | <0.06 | 0.25 | 0.5 | <0.06 | 4 |
Tazobactam tested at 4 μg mL−1 and varying concentrations of piperacillin.
Substrate kinetics: PDC-3 Arg349Ala retains catalytic efficiency for cephalosporins
PDC-3 and PDC-3 Arg349Ala β-lactamases were purified using preparative isoelectric focusing, size exclusion, and cation exchange chromatography. The molecular weight of the β-lactamases was confirmed with Electrospray Ionization-Mass Spectrometry (ESI-MS). The observed species were within experimental error of the predicted β-lactamase molecular weights (PDC-3 = 40,775 amu, PDC-3 Arg349Ala = 40,693 amu, all measurements have an error of ±3 amu). Both β-lactamases include an additional Met residue on the N-terminus, as the codon for this residue is introduced with the NdeI restriction site used to clone the gene into the vector.
Table II summarizes our kinetic data characterizing PDC-3 and the Arg349Ala variant β-lactamases, which we selected because of its comparatively large phenotypic effect on MICs. In general, the catalytic profile of PDC-3 for the penicillin and cephalosporins tested is consistent with the kinetic behavior described for other AmpC cephalosporinases. Namely, kcat values are in the order of 102 s−1 for narrow-spectrum cephalosporins such as nitrocefin, cephalothin, and cephaloridine, and kcat/Km is typically >1 μM−1 s−1.17,37–39 In contrast, for ampicillin and the extended-spectrum cephalosporins, hydrolysis is poorly measurable, suggesting kcat is <0.5 s−1. However, these β-lactams maintain low μM Kis in competition assays with nitrocefin.
Table II.
PDC-3 and PDC-3 Arg349Ala β-Lactamase Substrate Kineticsa
| PDC-3 | PDC-3 Arg349Ala | |||||
|---|---|---|---|---|---|---|
| Substrates | Km or Ki (μM) | kcat (s−1) | kcat/Km (μM−1s−1) | Km or Ki (μM) | kcat (s−1) | kcat/Km (μM−1s−1) |
| Ampicillin | 0.31 ± 0.02 | 0.96 ± 0.02 | ||||
| Nitrocefin | 20.7 ± 1.8 | 453 ± 45 | 21.9 ± 2.9 | 22.0 ± 2.6 | 310 ± 9 | 14.1 ± 1.7 |
| Cephalothin | 24.3 ± 3.1 | 409 ± 41 | 16.8 ± 2.7 | 1.8 ± 0.1 | 7 ± 1 | 4.0 ± 0.5 |
| Cephaloridine | 33.4 ± 0.7 | 130 ± 13 | 3.9 ± 0.4 | 19.4 ± 1.6 | 27 ± 1 | 1.4 ± 0.1 |
| Ceftazidime | 51.7 ± 2.1 | 36.2 ± 7.6 | ||||
| Cefotaxime | 0.26 ± 0.03 | 2.0 ± 0.2 | ||||
The standard deviation of each value is reported.
Comparison of the PDC-3 Arg349Ala variant to PDC-3 revealed very similar Km values for nitrocefin and cephaloridine. Interestingly, the apparent affinity of PDC-3 Arg349Ala for cephalothin is more than 10-fold higher than PDC-3 (although it should be noted that the relative affinity measurement for cephalothin was based on a competition assay, as the very low kcat value hindered a direct measurement of Km).
The most significant difference between the kinetic parameters of PDC-3 and PDC-3 Arg349Ala is the decrease in kcat observed for nitrocefin, cephalothin, and cephaloridine. The reduction in kcat is largest for cephalothin and cephaloridine, 56-fold and 5-fold lower than PDC-3, respectively. Thus, instead of a predominant effect on the initial substrate affinity, Arg349 appears to be contributing to the turnover of the cephalothin and cephaloridine.
Structural analysis of the reaction pathway of cephalothin with the E. coli AmpC reveals that the position of cephalothin's C4 carboxylate moves more than 7 Å from its original position in the pre-covalent substrate-enzyme complex (Fig. 3).32 In the covalent cephalothin-AmpC complex, the hydrolytic water is observed in the position where the C4 carboxylate was found in the precovalent complex. If PDC-3 and cephalothin engage in a similar reaction pathway as the E. coli AmpC with cephalothin, the decreased kcat values observed for the Arg349Ala substitution could reflect a modification of the local active site topology; the introduction of 349Ala may disrupt the sphere of positive charge around the C4 carboxylate and impair sequestration of the hydrolytic water for the narrow-spectrum cephalosporins.
Figure 3.

Representations of the reaction pathway for cephalothin and the E. coli AmpC: (a) the pre-covalent substrate-enzyme complex; (b) acyl-enzyme complex; and (c) the hydrolyzed product in the active site.32 The position of the C4 carboxylate is circled in dashed lines to show the movement of the substituent over the reaction course. In panel (b), the active site residues are labeled and the red represents the approximate location of the presumed deacylation water. Images created with Discovery Studio Visualizer 2.5 using the deposited crystal structure coordinates 1KVL and 1KVM.
The enzyme–ligand interactions of PDC-3 Arg349Ala were also altered for ampicillin, ceftazidime, and cefotaxime. For both ampicillin and cefotaxime, the apparent Ki of the Arg349Ala variant was increased, 3- and 10-fold, respectively. Such an increase in apparent Ki is consistent with both the increased susceptibility of E. coli cells expressing the Arg349Ala variant and the hypothesis that Arg349 offers a stabilizing hydrogen bond to the conserved C3/C4 carboxylate. The small decrease in the apparent Ki of ceftazidime for PDC-3 Arg349Ala variant is also consistent with the lack of change in MIC susceptibility to this substrate.
How do cefotaxime and ceftazidime interact differently with PDC-3? Upon inspection of the chemical structures, we see that the R1 side chains of these two extended-spectrum cephalosporins are similar with the replacement of a methyl group with a dimethyl acetate group in ceftazidime. The comparable apparent affinity of ceftazidime for the WT enzyme and Arg349Ala variant suggests that the dimethyl acetate group is engaged in productive enzyme–ligand interactions, an observation consistent with previous work on ceftazidime hydrolysis in the AmpCs from other organisms.40 The crystal structure of the E. coli AmpC complexed with ceftazidime shows that both the R1 dimethyl acetate group and the C4 carboxylate are oriented towards residues 343, 346, and 349. Furthermore, the electron density of the dimethyl acetate group suggests this part of the ligand may be particularly flexible and exist in multiple conformations.40 The flexibility of ceftazidime in the acyl-enzyme form may allow for residues 343, 346, and 349 to interact with both R1 and C4 carboxylate groups during the reaction coordinates. Thus, loss of the Arg residue has minimal impact on the recognition of ceftazidime.
β-Lactamase inhibitor kinetics: Arg349 as part of a carboxylate recognition region
To explore further the mechanism underlying the changes in substrate turnover of PDC-3 and the effects of the substitution on the potential carboxylate-binding region, we tested boronic acid compounds with penicillin- and cephalosporin-like R1 and R2 groups. We compared the inhibition data obtained with these boronate substrate analogs to that for the currently available class A β-lactamase inhibitors clavulanate and tazobactam and the monobactam aztreonam. The results are presented in Table III.
Table III.
Inhibitor Kinetics for PDC-3 and PDC-3 Arg349Alaa
| Inhibitors | PDC-3, Ki (μM) | PDC-3 Arg349Ala, Ki (μM) | PDC-3/PDC-3 Arg349Ala, ΔΔGb (kcal mol−1) |
|---|---|---|---|
| Commercially available inhibitors | |||
| Clavulanate | >10,000 | >10,000 | NA |
| Tazobactam | 24.1 ± 4.1 | 138 ± 10 | +1.03 |
| Aztreonam | 1.31 ± 0.13 | 15.6 ± 1.1 | +1.47 |
| Boronate substrate analogs | |||
| Compound R | 37 ± 6 | 55 ± 4 | +0.23 |
| Compound 1 | 0.62 ± 0.10 | 3.1 ± 0.4 | +0.95 |
| Compound 2 | 0.42 ± 0.03 | 0.77 ± 0.03 | +0.36 |
| Compound 3 | 0.48 ± 0.05 | 0.41 ± 0.03 | −0.09 |
| Compound 4 | 0.010 ± 0.001 | 0.012 ± 0.002 | +0.11 |
| Compound 5 | 0.004 ± 0.001 | 0.015 ± 0.003 | +0.78 |
| Compound 6 | 0.22 ± 0.03 | 0.89 ± 0.10 | +0.83 |
| Compound 7 | 0.17 ± 0.04 | 0.34 ± 0.03 | +0.41 |
| Compound 8 | 0.004 ± 0.001 | 0.21 ± 0.06 | +2.34 |
| Compound 9 | 0.040 ± 0.006 | 0.17 ± 0.04 | +0.86 |
All boronate compounds were incubated with the enzyme for 5 min before addition of nitrocefin.a
The standard deviation of each Ki value is reported.
Differential (Gibbs) free energy of binding for PDC-3 Arg349Ala compared to PDC-3, calculated at 298 K. Values are calculated using ΔΔG = −RT ln [(Ki PDC-3)/(Ki PDC-3 Arg349Ala)]. Positive values indicate decreased affinity.
Commercially available inhibitors
Of the inhibitors examined, the commercially available β-lactamase inhibitors (clavulanate, sulbactam, and tazobactam) demonstrate the highest apparent Kis. We could not achieve reduction of nitrocefin hydrolysis for PDC-3 and the Arg349Ala variant with up to 10 mM of the oxapenam clavulanate, illustrating the enzymes are not inhibited by the compound. The sulfone tazobactam has a 5-fold higher Ki for the Arg349Ala variant as compared to the WT β-lactamase. While this decreased apparent affinity suggests a role of the Arg349 residue in binding recognition, the kinact values for the two PDC-3 enzymes are very similar (0.082 ± 0.005 s−1 for WT and 0.071 ± 0.003 s−1 for PDC-3 Arg349Ala).
We next tested aztreonam, which behaves as a “slow substrate,” or “transient inhibitor” of class C β-lactamases.41 In competition assays with PDC-3 and nitrocefin, aztreonam demonstrated a Ki in the range of effective β-lactamase inhibition (1.31 ± 0.13 μM). In contrast, the Arg349Ala variant had a slightly increased Ki for this inhibitor (15.6 ± 1.1 μM), but the kinact value of 0.183 ± 0.011 s−1 was within error of the WT kinact, 0.180 ± 0.028 s−1 (kinact values not shown in Table III). This data is consistent with the small effect observed in the MIC substrate testing of aztreonam. Monobactams like aztreonam have an N-linked 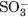 group that is oriented toward the pocket of the active site containing the 343, 346, and 349 residues in the Citrobacter freundii AmpC/aztreonam complex crystal structure.42 The elimination of the N-linked substituent following hydrolysis of the acyl-enzyme's sulfonamide bond was described for monobactams with other class C and A β-lactamases.37,43 Assuming that aztreonam adopts a similar conformation in PDC-3, elimination of the group following acylation could decrease the impact of the Arg349Ala substitution, explaining the similar kinact values between the variant and WT enzymes.
group that is oriented toward the pocket of the active site containing the 343, 346, and 349 residues in the Citrobacter freundii AmpC/aztreonam complex crystal structure.42 The elimination of the N-linked substituent following hydrolysis of the acyl-enzyme's sulfonamide bond was described for monobactams with other class C and A β-lactamases.37,43 Assuming that aztreonam adopts a similar conformation in PDC-3, elimination of the group following acylation could decrease the impact of the Arg349Ala substitution, explaining the similar kinact values between the variant and WT enzymes.
Boronates
To complete our analysis, we next screened boronic acid compounds that were synthesized to resemble the R1 substituents on either penicillin or cephalopsporin β-lactams. The relative Kis of the boronates provide insight into the “molecular preferences” or “substrate specificities” of the PDC-3 active site. Chiral boronate compounds incorporate an “R2” side chain, an additional functional group that resembles the thiazolidine and dihydrothiazine rings of penicillins and cephalopsorins, respectively, as well as the conserved C3/C4 β-lactam carboxylate. These reversible inhibitors form a covalent bond with Ser64 of class C β-lactamases, and the measured Kis are interpreted relative to a reference compound (compound R) which lacks these side chains.44,45 We note that the boronate's “R2” group is not equivalent to the R2 group found on the corresponding β-lactam, for example, the pyridinium ring found on ceftazidime. While we cannot probe the role of the β-lactam R2 group with the boronates, crystal structures of complexed AmpCs show minimal interactions between the cephalosporin's R2 group and the enzyme before acylation and elimination of the group following acylation.32,40
The boronate compounds in the series without an R2 group (compounds 3, 7, 8) display Ki values that are at least two orders of magnitude lower than the reference compound for both PDC-3 and PDC-3 Arg349Ala (Ki range = 0.004 ± 0.001 μM to 0.48 ± 0.05 μM). Boronates 3 and 7 demonstrate Kis that were similar for both the WT and variant β-lactamases. The ceftazidime R1 analog, compound 8, displayed the lowest Ki of the panel without R2 groups for the WT and variant enzymes. However, this Ki was 52-fold higher for the Arg349Ala variant than PDC-3. This result is somewhat unexpected as the compound lacks the R2 group, which is designed to introduce additional high-affinity substrate substituents. Therefore, retention of the positively charged Arg in this region must contribute to the Ki of this boronate. The mechanism of boronate recognition likely differs from the recognition of the same side chain as part of the substrate (where the Ki of ceftazidime is very similar for PDC-3 and the Arg349Ala variant).
In the series of boronates with R2 groups, the ampicillin analog (compound 1) has sub μM Ki for the PDC-3 β-lactamase, but this value increases more than five times for the Arg349Ala variant. Binding of the penicillin C3 carboxylate group may be particularly dependent on features of the carboxylate recognition region in this cephalosporinase. The nafcillin analog (compound 2) shows Kis of a similar magnitude to the cefotaxime and cephalothin boronates without an R2 group, and displays less than twofold difference between PDC-3 and PDC-3 Arg349Ala. When compared with the ampicillin boronate, the two rings comprising the nafcillin R1 may allow for additional enzyme–ligand interactions that help offset substitution of the Arg349 residue. In fact, cloxacillin and oxacillin are described as inhibitors of AmpC β-lactamases, and both possess two rings in their R1 group.46
Finally, cephalothin boronates with three different R2 side chains were tested (compounds 4, 5, and 6). Compound 4, which lacks the meta-carboxylate of compound 5, has a very similar (low nM Ki) for both β-lactamases. Compound 5 has a lower apparent affinity for PDC-3 Arg349Ala, but the Ki value is still very close to that for PDC-3. We can compare the binding energy of the boronates for the two enzymes by using Ki as an equilibrium constant in the Gibbs free energy equation:28
This calculation reveals a 0.78 kcal mol−1 difference between the energy associated with binding compound 5 in PDC-3 as compared to PDC-3 Arg349Ala, which is lower than expected for a hydrogen bond (∼ 2 kcal mol−1). Thus, the energy from the interaction observed between the carboxylate and Arg349 in the model of PDC-3 may be acquired by the hydrogen bond with Asn343, or otherwise compensated by a local rearrangement of the residues; Asn346 may move into closer approximation with the meta-carboxylate in the Arg349Ala variant.
The cephalothin analog boronate, compound 6, differs from compound 5 by the addition of a carbon on the linker between the boronate group and the R2 side chain. Both the PDC-3 and Arg349Ala AmpCs show a significant decrease in affinity for compound 6, suggesting that the extended linker introduces less favorable interactions in the P. aeruginosa β-lactamases.
Finally, compound 9 bearing both the ceftazidime R1 side chain and an R2 meta-carboxyphenyl shows a fourfold affinity loss for the Arg349Ala variant of PDC-3 β-lactamase. This loss contrasts the 52-fold drop observed for compound 8 possessing just the ceftazidime R1 side chain. Loss of the Arg349 residue is mitigated by the incorporation of the R2 group with additional enzyme binding groups.
Susceptibility testing with boronate compounds
Low nM Kis in kinetic assays do not necessarily translate to effective enzyme inhibition; multiple pharmacologic, biochemical, and microbiological characteristics affect the ability of a compound to restore susceptibility to a partner β-lactam in vivo.47 We determined the cephalosporin susceptibility of E. coli expressing blaPDC-3 and the blaPDC-3 variants with the addition of either boronate compound 5 or 8. The cephalothin analog 5 was selected because of its low Ki for both PDC-3 and PDC-3 Arg349Ala; in contrast, the ceftazidime boronate 8 was selected because of the 52-fold increase in Ki for PDC-3 Arg349Ala.
At 4 μg mL−1, both boronates lowered the MICs for cefotaxime in E. coli blaPDC-3 expressing cells (Table IV). For the cephalothin analog 5, the cefotaxime susceptibility was increased by two dilutions for PDC-3, the Asn343Ala, and the Asn346Ala variants, and one dilution for the Arg349Ala variant. Thus, the similar Ki value of compound 5 for PDC-3 and PDC-3 Arg349Ala was reflected in whole cell assays. For the ceftazidime analog 8, cefotaxime susceptibilities were improved by four dilutions for PDC-3, the Asn343Ala, and the Asn346Ala variants, but only two dilutions for the Arg349Ala variant. The decreased MIC effect for the Arg349Ala variant may result from compound 8's 52-fold higher Ki for this variant as compared to WT. However, the Ki of compound 8 for PDC-3 was equal to that for compound 5 (4 nM), yet a two-dilution difference exists in the inhibitors' abilities to lower cefotaxime MICs to E. coli expressing blaPDC-3.
Table IV.
Contribution of Boronate Analogs 5 and 8 to MIC Values (μg mL−1) of P. aeruginosa 18SH and E. coli DH10B Expressing blaPDC-3 or blaPDC-3 Variants at Possible Carboxylate Binding Sites
| Isolates | Cefotaxime | Cefotaxime/5 | Cefotaxime/8 |
|---|---|---|---|
| P. aeruginosa 18SH | >1024 | >64 | >64 |
| E. coli DH10B | <0.125 | <0.125 | 0.125 |
| E. coli blaPDC-3 | 8 | 2 | 0.5 |
| E. coli blaPDC-3 Asn343Ala | 4 | 1 | 0.25 |
| E. coli blaPDC-3 Asn346Ala | 4 | 1 | 0.25 |
| E. coli blaPDC-3 Arg349Ala | 0.5 | 0.25 | 0.125 |
Boronates tested at a constant concentration of 4 μg mL−1.
Overall, these MIC data display some parallels with the Ki values, but also underscores the complexities of whole cell assays and microbiological properties such as periplasmic β-lactamase concentration of enzyme and synergy with the β-lactam partner.47
Conclusions
Our data represent one of the first in-depth examinations of the structure–function relationships of the P. aeruginosa AmpC β-lactamase, an enzyme of tremendous clinical importance. We chose to focus on exploring the mechanism in which the C3/C4 carboxylate binds in the active site. The ready recognition of this substituent by the four different β-lactamase classes provides a solid argument that these enzymes are adapted and evolved to identify this functionality. Previous work with class A β-lactamases often reveals active site locations with high-binding affinity for the C3/C4 carboxylate but attempts to identify a recognition region in class C β-lactamases have been more elusive.24,30,48,49 We have performed a careful analysis of a molecular model of the PDC-3 protein in complex with a boronate inhibitor bearing a side chain that mimics the thiazolidine/dihydrothiazine ring and the C3/C4 carboxylate characteristic of β-lactam substrates. Based on this analysis, we engineered PDC-3 variants at residues that appeared important in the recognition of the carboxylate moiety.
From our susceptibility testing and kinetic characterizations, we identify properties of the P. aeruginosa AmpC that suggest a significant degree of enzyme plasticity. First, our studies suggest that more than one C3/C4 carboxylate binding residue may exist in PDC-3. Herein, we examined the effects of three possible binding residues and observed decreases in the MICs of these variants for several specific substrates. While the magnitude of the MIC decrease was greatest for the Arg349Ala variant, Ala substitutions at Asn343 and Asn346 also have an impact on susceptibility. This data argue for more of a binding “region” rather than a binding “site,” supported by both the Asn and Arg residues. Loss of the positive charge at 349 may lead to the most dramatic change in enzyme architecture and electrostatics, but clearly 343 and 346 are also implicated in the topology of the pocket. Furthermore, the PDC-3 Arg349Ala β-lactamase maintained substantial hydrolytic activity for the tested substrates. Despite reductions in kcat for cephalothin and cephaloridine, the apparent affinity remains high, preserving a robust catalytic efficiency >1 μM−1 s−1. The decrease in ampicillin and cefotaxime MICs for the Arg349Ala variant is accompanied by a reduction in apparent Kis, but again the Kis remain in the low μM range.
The homology-based protein-modeling server used to generate the PDC-3 representation identified a P. fluorescens class C β-lactamase to serve as the highest similarity PDB template (at the time the modeling was performed, the P. aeruginosa AmpC structure was not yet reported by Blizzard et al.16 Michaux et al.50 defined and described the structure of this enzyme (which shares 76% sequence similarity to PDC-3) and identified biochemical properties that allow this cold-adapted enzyme to optimize catalytic activity at low temperatures. Specifically, psychrophilic enzymes enjoy increased structural flexibility at the price of decreased stability.51,52 P. aeruginosa is not a psychrophilic organism, but the similarity between the two class C β-lactamases helps generate hypotheses about the biochemical properties of PDC-3. In general, cold-adapted enzymes have decreased numbers of ion pairs and hydrogen bonds, low Arg content, and increased solvent accessibility to the active site.50,51 If PDC-3 shares attributes with its “cold-loving” counterpart, we may expect it to be a more “flexible” enzyme with binding regions comprised of residues capable of serving multiple recognition roles. These notions await testing of the thermal stability and conformational flexibility of PDC-3 through circular dichroism and thermal denaturation experiments.
Second, by probing the PDC-3 Arg349Ala active site with the boronate substrate analogs, we reveal the complexities of ligand binding in this β-lactamase. Compound 5 bearing the R1 of cephalothin and an R2 group designed to mimic the conserved cephalosporin dihydrothiazine ring and C4 carboxylate demonstrates very similar Ki values for PDC-3 and PDC-3 Arg349Ala. However, the incorporation of an R2 group on the ceftazidime boronate (compound 9) leads to a fourfold decrease in Ki for the Arg349Ala variant when compared with WT enzyme. Paradoxically, removing this R2 group from the ceftazidime boronate (compound 8) serves to increase the affinity for the PDC-3 WT β-lactamase. Considered alongside the PDC-3 Arg349Ala's perplexing increase in apparent affinity for the substrate ceftazidime, these data suggest important differences in how PDC-3 recognizes these β-lactams and their boronate analogs. β-Lactam C3/C4 carboxylate regions may be more substrate and inhibitor specific for this class of enzymes, that is, the same functional groups presented on different scaffolds can use unique areas of the enzyme for recognition.
In summary, our data lead us to assert that multiple “ancillary” interactions contribute to substrate and inhibitor binding in the P. aeruginosa AmpC. The individual residues in the PDC-3 active site exhibit a high degree of versatility, plasticity, and flexibility for recognition of both substrates and inhibitors. These notions of enzyme plasticity are consistent with theses developed by Todd et al.35 that key functional residues can exist in multiple active site locations, and this residue “hopping” may be a important property of class C and other β-lactamases. From an evolutionary and mechanistic standpoint, these dynamic features may lead to more opportunities for AmpC variants with increasing resistance profiles and novel catalytic properties, posing and enormous challenge to the medicinal chemist (e.g., extended-spectrum and carbapenemase AmpCs).17,53 This unique perspective introduces a new appreciation of how β-lactams are hydrolyzed in class C β-lactamases and helps explain, in part, the broad substrate profile of these enzymes. Such plasticity in substrate recognition has clear structural, mechanistic, and evolutionary implications for novel drug design.54
Materials and Methods
Antibiotics and commercially available inhibitors
The chemical structures of the substrates and commercially available inhibitors studied are shown in Figure 4. Nitrocefin was purchased from BD Biosciences (San Jose, CA), imipenem from U.S. Pharmacopeia (Rockville, MD) and meropenem from AstraZeneca Pharmaceuticals (Wilmington, DE). The remaining antibiotics were purchased from Sigma (St. Louis, MO). Lithium clavulanate was a kind gift from GlaxoSmithKline (Surrey, United Kingdom); tazobactam was obtained from Chem-Impex International (Wood Dale, IL).
Figure 4.
Substrates and commercially available inhibitors used in this study.
Synthesis of boronates
Figure 5 shows all tested boronates. All reactions were performed under argon using oven-dried glassware and dry solvents. Dry tetrahydrofuran (THF) and diethyl ether were obtained by standard methods and distilled freshly under argon from sodium benzophenone ketyl before use. The –100°C bath was prepared by addition of liquid nitrogen to a precooled (–78°C) mixture of ethanol/methanol (1:1). Reactions were monitored by thin layer chromatography (TLC), which were visualized by UV fluorescence and by Hanessian's cerium molybdate stain. Chromatographic purification of the compounds was performed on silica gel (particle size 0.05–0.20 mm). Melting points were measured on a Büchi 510 apparatus. Optical rotations were recorded at +20°C on a Perkin-Elmer 241 polarimeter and are expressed in 10−1 deg cm2 g−1. 1H NMR and 13C NMR spectra were recorded on a Bruker DPX-200 or Avance-400 spectrometer; chemical shifts (δ) are reported in ppm downfield from tetramethylsilane (TMS) as internal standard (s singlet, d doublet, t triplet, q quartet, m multiplet, and br broad signal); coupling constants (J) are given in Hz. Two-dimensional NMR techniques (correlation spectroscopy (COSY), heteronuclear multiple bond correlation (HMBC), heteronuclear single quantum coherence (HSQC)) were used to aid in the assignment of signals in 1H and 13C spectra. Mass spectra were determined on a gas chromatography HP 5890 associated with mass spectrometer detector HP 5972 (EI, 70 eV) or on an agilent technologies liquid chormatography-mass spectrometry (LC-MS)(n) Ion Trap 6310A (electrospray ionization (ESI), 70 eV). Elemental analyses were performed on a Carlo Erba Elemental Analyzer 1110. Compounds 3, 7, and 8 were synthesized as in55, compounds 4 and 5 as in28 and compound 9 as in (Miriagou et al., in revision, JBC). Compounds 1, 2, and 6 were synthesized starting from bromomethaneboronate of pinanaediol in a four-step synthesis described in detail below (Fig. 6).
Figure 5.
Boronate compounds used in this study.
Figure 6.
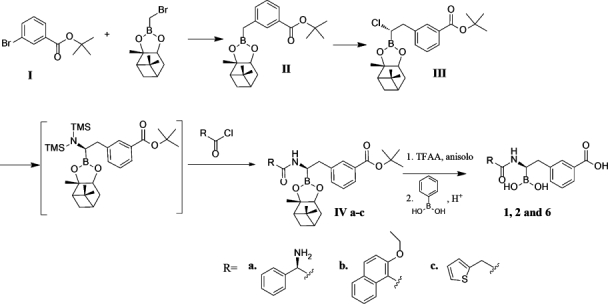
Four step synthesis for compounds 1, 2, and 6.
(+)-Pinanediol (3-tert.butoxycarbonylphenyl)methaneboronate (II)
n-BuLi (7.23 mmol of a 2.5M solution in hexane) was added dropwise with stirring to a solution of tert-butyl-3-bromobenzoate (1.77 mg, 6.88 mmol) in THF (2 mL) at −100°C under argon atmosphere. After 30 min, a solution of bromomethaneboronate of (+)-pinanaediol I (1.31 g, 4.82 mmol) in THF (10 mL) was added, and the stirred mixture was allowed to reach room temperature (RT) overnight.56 The reaction mixture was concentrated under vacuum to give a thick yellow oil, which was purified by chromatography (light petroleum/ethyl acetate 98:2), affording II (0.77 g, 43% yield) as a yellow oil, [α]D = +8.7 (c 1.42, CHCl3).
1H NMR (400 MHz, CDCl3): δH 0.87 (3H, s), 1.11 (1H, d, J = 11), 1.32 (3H, s), 1.43 (3H, s), 1.63 (9H, s), 1.84–2.39 (5H, m), 2.42 (2H, s, H), 4.32 (1H, dd, J = 8.8, 2.0), 7.33 (1H, t, J = 7.7), 7.40 (1H, brd, J = 7.7), 7.80 (1H, brd, J = 7.7), 7.86 (1H, brs).
13C NMR (200 MHz, CDCl3): δC 19.3 (br, C), 24.0, 26.5, 27.1, 28.2, 28.6, 35.4, 38.2, 39.5, 51.3, 78.0, 80.7, 86.0, 126.2, 128.1, 129.9, 132.0, 133.2, 139.0, 166.0. EI-MS: m/z 370 (M, 18%), 314 (71%), 297 (59%), 135 (55%), 57 (100%).
(+)-Pinanediol (1S)-1-chloro-2-(3-tert.butoxycarbonylphenyl)ethaneboronate (III)
n-BuLi (2.44 mmol of a 1.6M solution in hexane) was added dropwise to a solution of CH2Cl2 (0.21 mL, 3.25 mmol) in THF (4 mL) with stirring at −100°C under argon. At the end of the BuLi addition, a white microcrystalline precipitate (LiCHCl2) became evident. After 30 min, a solution of II (0.752 g, 2.03 mmol) in THF (5 mL) was slowly added at the same temperature. The white precipitate disappeared and the mixture was allowed to reach RT overnight. The reaction mixture was concentrated under vacuum, and the brownish residue was diluted with light petroleum (50 mL). The precipitate was filtered on an MgSO4 pad and washed with the same solvent (100 mL); the solution was then concentrated to give pale yellow oil (0.7 g, 82%). [α]D = +12.0 (c 2.44, CHCl3), de ≥ 98%, used as such in the following step.
1H NMR (200 MHz, CDCl3): δH 0.84 (3H, s), 1.03 (1H, d, J = 10.8), 1.29 (3H, s), 1.37 (3H, s), 1.61 (9H, s), 1.76–2.40 (5H, m), 3.14 (1H, dd, J = 13.9, 8.4), 3.27 (1H, dd, J = 13.9, 7.5), 3.67 (1H, dd, J = 8.4, 7.5), 4.37 (1H, dd, J = 8.7, 2.0), 7.30–7.47 (2H, m), 7.85–7.90 (2H, m).
13C NMR (200 MHz, CDCl3): δC 25.3, 27.0, 28.4, 29.6, 29.7, 36.5, 39.6, 40.7, 41.5 (br, C-B), 44.1, 52.5, 80.0, 82.3, 88.3, 129.3, 129.6, 131.6, 133.5, 134.8, 139.9, 167.0.
(+)-Pinanediol (1R)-1-[(2-tert-butoxycarbonylamino-2-phenyl)acetylamino]-2-[3(tert.Butoxycarbonyl)phenyl] ethane boronate (IVa)
Lithiumhexamethyldisilazane (1.6 mmol of a 1M solution in THF) was added dropwise to a stirred solution of III (0.618 g, 1.47 mmol) in THF (6 mL) at −78°C under argon atmosphere and the mixture was allowed to warm gradually at RT overnight. The resulting solution was concentrated under vacuum, and light petroleum (100 mL) was added. The precipitate was filtered on a MgSO4 pad and washed with the same solvent, and the solution concentrated to give pale yellow oil (0.255 g, 32% yield). The silylamine (0.255 g, 0.47 mmol) was diluted with THF (6 mL) and MeOH (0.224 mL of a 2.5M solution in THF) was added dropwise at 0°C. In the meantime, the acylating mixture was prepared as follows: isobutylchloroformiate (0.061 mL, 0.47 mmol) was added via syringe to a precooled solution of Boc-d-PheGly (0.118 g, 0.47 mmol) in THF (6 mL) with stirring at –20°C, followed by addition of N-Methylmorfoline (NMM) (0.048 mL, 0.47 mmol). After 30 min at 0°C, the solution of silylamine was added dropwise via syringe to the acylating mixture; the temperature was then allowed to reach r.t. gradually, and the resulting mixture stirred for additional 16 h. The white suspension was then partitioned between ethyl acetate (60 mL) and water (20 mL), and the organic phase was washed with saturated NaHCO3 solution (30 mL). The organic phase was dried (MgSO4), filtered and concentrated to give a grey solid (0.305g), which was crystallized by diethylether and n-hexane affording IVa (0.184 g, 62% yield) as a white solid (mp 81–85°C dec). [α]D = –10.0 (c 1.05%; CDCl3), de ≥ 98%.
1H NMR (400 MHz, CDCl3): δH 0.86 (3H, s), 1.09 (1H, d, J = 10.9), 1.31 (3H, s), 1.34 (3H, s), 1.43 (9H, s), 1.62 (9H, s), 1.78–2.43 (5H, m), 2.87 (1H, dd, J = 13.9, 7.8), 2.98 (1H, dd, J = 13.9, 5.3), 3.57 (1H, m), 4.37 (1H, dd, J = 8.9, 1.7), 5.11 (1H,s), 5.74 (1H, b), 5.88 (1H, b), 6.90 (1H, d, J 6.6), 7.15 (1H, t, J = 7.6), 7.30–7.34 (5H, m), 7.77 (1H, s), 7.81 (1H, d, J = 7.6).
13C NMR (400 MHz, CDCl3): δC 24.0, 26.3, 27.1, 28.2, 28.3, 28.4, 35.3, 36.5, 38.1, 38.8(br, C-B), 39.5, 51.3, 58.1, 78.2 79.9, 80.1, 86.4, 127.3, 127.5, 128.2, 128.3, 128.9, 129.9, 132.1, 133.3, 138.6, 139.1, 155.0, 165.7, 170.4.
LC-MS (ESI, Ion trap): m/z 655 (60%, [M + Na]+), 633 (100%, [M + H]+, 533 (24%). MS/MS of 633 m/z: 533 (100%), 477 (5%), 381 (9%), 325 (2%).
Anal. Calcd. for C36H49BN2O7; C, 68.35; H, 7.81; N, 4.43. Found: C, 68.06; H, 8.08; N, 4.69.
(+)-Pinanediol (1R)-1-[(2-ethoxynaphtalene-1-carbonyl)amino]-2-[3-(tert.butoxycarbonyl)phenyl] ethaneboronate (IVb)
Lithiumhexamethyldisilazane (1.71 mmol of a 1M solution in THF) was added dropwise to a stirred solution of III (0.622 g, 1.49 mmol) in THF (7 mL) at −78°C under argon atmosphere, and the mixture was allowed to warm gradually at r.t. overnight. The resulting solution containing the silylamine was used as such for the following step. A mixture of 2-ethoxynaphtoic acid (0.387 g, 1.79 mmol) and 2-ethoxynaphtoylchloride (0.490 mL, 2.09 mmol) in THF (6 mL) was added dropwise to the sylilamine solution at −78°C and allowed to reach RT overnight. After 16 h, the brownish suspension was partitioned between ethyl acetate (70 mL) and water (20mL); the organic phase was washed with saturated NaHCO3 solution (25 mL), and the aqueous phase extracted with EtOAc (60, 40 mL). The organic phases were dried (MgSO4), and concentrated to give a viscous yellowish oil, which was purified by gradient chromatography (light petroleum/ethyl acetate from 70/30) affording IVb (0.123 g, 14% yield) as yellow viscous oil. [α]D = –17.1 (c 1.15%; CHCl3), de ≥ 98%.
1H NMR (400 MHz, CDCl3): δH 0.95 (3H, s), 1.27 (3H, t, J = 7.0), 1.35 (3H, si), 1.57 (3H, s), 1.62 (9H, s), 1.94–2.48 (5H, m), 3.05 (1H, dd, J = 14.7, 11.3), 3.25 (1H, dd, J = 14.7, 4.4), 3.50 (1H, m), 4.21 (2H, m), 4.46 (1H, dd, J = 8.7, 2.2), 7.23 (1H, d, J = 9.2.), 7.39–7.46 (2H, m.), 7.55–7.61 (2H, m.), 7.72 (1H, s), 7.79 (1H, d, J = 8.1.), 7.90 (1H, d, J = 7.8), 7.95–7.99 (2H, m.), 8.64 (1H, d, J = 8.9.).
13C NMR (400 MHz, CDCl3): δC 14.7, 24.2, 26.6, 27.4, 28.2 (3C), 29.1, 36.5, 37.3, 38.3, 40.1, 43.8 (br, C-B), 52.3, 65.6, 77.2, 81.0, 84.0, 113.3, 124.7, 125.6, 127.5, 127.1, 128.3, 128.5, 128.9, 129.7, 132.4 (2C), 133.1, 134.0, 134.5, 140.9, 156.2, 165.8, 170.9.
Anal. Calcd. for C36H44BNO6: C, 72.36; H, 7.42; N, 2.34. Found: C, 72.52; H, 7.36; N, 2.18.
(+)-Pinanediol (1R)-1-[(2-thiophene-2-yl)acetylamino]-2-[3-(tert.butoxycarbonyl)phenyl]ethaneboronate (IVc)
Lithiumhexamethyldisilazane (1.63 mmol of a 1M solution in THF) was added dropwise to a stirred solution of III (0.568 g, 1.36 mmol) in THF (6 mL) at −78°C under argon atmosphere and the mixture was allowed to warm gradually at RT overnight. The resulting solution containing the silylamine was used as such for the following step. A mixture of thiopheneacetic acid (0.232 g, 1.63 mmol) and thiopheneacetyl chloride (0.201 mL, 1.63 mmol) in THF (3 mL) was added dropwise to the sylilamine solution at −78°C and allowed to reach RT overnight. After 16 h, the brownish suspension was partitioned between ethyl acetate (70 mL) and water (25 mL); the organic phase was washed with saturated NaHCO3 solution (25 mL), and the aqueous phase extracted with EtOAc (60, 40 mL). The organic phases were dried (MgSO4), and concentrated to give viscous yellowish oil, which was purified by gradient chromatography (light petroleum/ethyl acetate from 1:1) affording IVc (0.097 g, 14% yield) as white solid (mp 154°C). [α]D = –25.5 (c 0.84%; CHCl3), de ≥ 98%.
1H NMR (400 MHz, CDCl3): δ 0.90 (3H, s), 1.30 (1H, d, J = 10.7), 1.32 (3H, s), 1.44 (3H, s), 1.64 (9H, s), 1.87–2.40 (5H, m), 2.82 (1H, dd, J = 13.7, 10.0), 3.03 (2H, m), 3.87 (3H, m), 4.34 (1H, dd, J = 8.7, 2.0), 6.04 (1H, br), 6.90 (1H, dd, J = 3.5, 1.1), 6.95 (1H, dd, J = 5.2, 3.5), 7.23 (1H, dd, J = 5.2, 1.2), 7.24 (1H, t, J = 1.5), 7.30 (1H, t, J = 7.5), 7.78 (1H, t, J = 1.5), 7.84 (1H, dt, J = 7.6, 1.5).
13C NMR (400 MHz, CDCl3): δ 2.24, 26.4, 27.3, 28.3, 28.8, 34.7, 35.9, 37.0, 38.2, 39.8, 41.8 (C-B), 51.9, 77.3, 81.0, 84.9, 125.9, 127.5, 127.9, 128.4, 129.9, 132.3, 133.0, 134.0, 139.9, 165.7, 172.9.
EI-MS: m/z 523 (M, 4%), 371 (27%), 315 (19%), 208 (30%), 135 (61%), 97 (100%), 93 (38%), 57 (45%).
Anal. Calcd. for C29H38BNO5S: C, 66.54; H, 7.32; N, 2.68; S, 6.13. Found: C, 66.38; H, 7.42; N, 2.75; S, 6.09.
Deprotection of the carboxy and boronic functionalities: general procedure
Trifluoroacetic acid (0.4 mL, 5.3 mmol) and catalytic anisol were added to a solution of IVa–c (0.18 mmol) in anydrous CH2Cl2 (4mL) and after 2 h, the mixture was concentrated under vacuum to give a yellowish solid which was directly dissolved in CH3CN (4 mL). To this solution HCl (0.18 mmol of a 1M solution in H2O), phenylboronic acid (0.022 g, 0.18 mmol) and n-exane (4 mL) were sequentially added, and the resulting biphasic solution was vigorously stirred. After 30 min, the n-hexane solution (containing the pinanediol phenylboronate) was removed, and n-hexane (4 mL) was added. This last procedure was repeated several times until a TLC analysis revealed no more presence of IVa–c. The acetonitrile phase was concentrated affording the desired compounds 1, 2, and 6 with overall yields of 5.2, 4.2, and 3.5%, respectively.
(1R)-1-(2-Amino-2-phenylacetylamino)-2-(3-carboxyphenyl)ethaneboronic acid (1)
Compound 1 was obtained (0.048 g, yield 74%) as white solid (mp 226°C dec.), [α]D = –59.3 (c 0.96%; MeOD), de ≥ 98%.
1H NMR (400 MHz, DMSO): δH 2.65 (1H, dd, J = 13.7, 8.9), 2.81 (1H, dd, J = 13.7, 4.3), 3.58 (1H, m), 4.95 (1H,s), 6.83 (1H, d, J 7.4), 7.15 (1H, t, J = 7.6), 7.14–7.55 (5H, m), 7.63 (1H, d, J = 7.6). 7.67 (1H, s), 8.28 (1H, b), 8.65 (2H, br), 12.82 (1H, br).
13C NMR (400 MHz, DMSO): δC 36.6, 41.5 (br, C-B), 55.8, 127.2, 128.0, 128.3, 128.9, 129.2, 130.2, 130.9, 133.7, 134.7, 140.8, 167.0, 167.8.
(1R)-1-[(2-Ethoxynaphtalene-1-carbonyl)amino]-2-(3-carboxyphenyl)ethaneboronic acid (2)
Compound 2 was obtained (0.064 g, yield 87%) as white solid (mp 79–81°C), [α]D = –55.8 (c 0.998%; MeOD), de ≥ 98%.
1H NMR (400 MHz, MeOD): δH 1.45 (3H, t, J = 7.0), 2.98 (1H, dd, J = 14.5, 10.3), 3.27 (1H, dd, J = 14.5, 5.2), 3.75 (1H, dd, J = 10.2, 5.3), 4.29–4.39 (2H, m), 7.42–7.50 (3H, m), 7.55–7.60 (2H, m.), 7.89 (1H, d, J = 8.0.), 7.93 (1H, d, J = 7.8), 8.02 (1H, br), 8.07 (1H, d, J = 8.7.), 8.14 (1H, d, J = 9.2.).
13C NMR (400 MHz, MeOD): δ 13.7, 35.3, 51.4 (br, C-B), 65.5, 113.4, 123.3, 124.4, 126.2, 127.7, 128.2, 128.3, 128.5, 128.9, 129.7, 130.9, 131.4, 133.1, 135.4, 157.1, 167.4.
(1R)-1-(2-Thiophene-2-yl)-2-(3-carboxyphenyl)ethaneboronic acid (6)
Compound 6 was obtained (0.042 g, yield 70%) as white solid (mp 233°C dec.), [α]D = –124.5 (c 0.22%; MeOH), de ≥ 98%.
1H NMR (400 MHz, MeOD): δ 2.62–2.73 (1H, m), 2.89–2.99 (2H, m), 3.96 (2H, s), 6.04 (1H, s), 6.98–7.02 (2H, m), 7.40 (1H, d, J = 4.0), 7.41 (1H, t, J = 7.6), 7.49 (1H, d, J = 7.6), 7.88 (1H, d, J = 7.6), 7.92 (1H, s, H2).
13C NMR (400 MHz, MeOD): δ 30.7, 36.4 (Cβ), 47.7 (br, C-B), 125.3, 126.7, 127.2, 127.3, 128.2, 129.9, 130.6, 133.4, 133.6, 141.1, 168.7, 176.4.
Molecular representations
The PDC-3 model was generated by the SWISS-MODEL automated protein structure homology-modeling server, available at http://swissmodel.expasy.org.57 The determined PDC-3 protein sequence was entered, and a model generated by the software using the Pseudomonas fluorescens class C β-lactamase template (Protein Data Bank entry 2QZ6).50 PDC-3 and the P. fluorescens protein share 76% sequence similarity; accurate high resolution protein models can be generated from templates with greater than 50% sequence similarity.58
The generated PDC-3 model was optimized by energy minimization using Discovery Studio 2.1 software (Accelrys, San Diego, CA). The minimization was performed in several steps, using Steepest Descent and Conjugate Gradient algorithms to reach the minimum convergence (0.02 kcal mol−1*Å). The protein was immersed in a water box, 7 Å from any face of the box, and the solvation model used was with periodic boundary conditions. The force-field parameters of CHARMm were used for minimization and the Particle Mesh Ewald method addressed long-range electrostatics. The bonds that involved hydrogen atoms were constrained with the SHAKE algorithm. Following equilibration, two separate 2 ps molecular dynamics simulations (heating/cooling and production) at constant pressure and temperature (300 K) were carried out for the PDC-3 model. The trajectories were analyzed, and the minimum energy conformation was chosen.
The minimized and equilibrated PDC-3 model was used for constructing the acylation complexes of the PDC-3 β-lactamase and the chiral cephalothin analog 5. The ligand structure was built using Discovery Studio Fragment Builder tools. The CHARMm force field was applied; the molecule was solvated with periodic boundary conditions and minimized using a Standard Dynamics Cascade protocol (one minimization using Steepest Descent algorithm, followed by Adopted Basis Newton-Raphson algorithm and three subsequent dynamics stages at NVT and 300 K).
The minimized ligand was docked in the active site of the enzyme using LibDock.59 The generated conformations (30–40 poses) were visually inspected, and the most favorable ones chosen based on minimum energy. The complex between the ligand and the enzyme was created, solvated, and energy minimized. The acyl-enzyme complex was created by making a bond with Ser64 of PDC-3, and the assembly was further minimized using conjugate gradient algorithm with periodic boundary conditions to 0.001 minimum derivatives. To reach the minimum equilibrium, the complexes were equilibrated using molecular dynamic simulations.
Antimicrobial susceptibility (MICs)
Susceptibility profiles were determined by cation-adjusted Mueller-Hinton agar dilution MICs according to the Clinical and Laboratory Standard Institute (CLSI) standards.60 For the piperacillin/tazobactam and cefotaxime/boronate combinations, the substrate concentrations were varied, whereas the inhibitors were tested at a constant concentration of 4 μg mL−1.
Cloning of blaPDC-3 into pBC SK(-) vector
The P. aeruginosa blaampC gene used in these studies was cloned from the laboratory strain P. aeruginosa 18SH, a kind gift from Dr. M. G. P. Page (Basilea). The AmpC β-lactamase from P. aeruginosa 18SH is produced at constitutively high levels as its regulation is “derepressed,” that is, the expression does not increase with induction by β-lactams.61,62 The AmpC from the PAO1 comparator strain has only one amino acid that is different from the P. aeruginosa 18SH AmpC, and has been designated PDC-1 in the classification system proposed by Rodríguez-Martínez and colleagues.17 By this nomenclature, the P. aeruginosa 18SH AmpC is designated PDC-3.
To subclone the P. aeruginosa 18SH blaPDC-3 gene into the pBC SK(−) vector, we designed Polymerase Chain Reaction (PCR) primers to the conserved regions of the blaPDC gene, including approximately 50 base pairs on either side of the start and stop codons (PDC upstream and PDC downstream, 5′ CGT CGT TTG CGG CAA ATC CTG CGC 3′ and 5′ GCG GAG GGG CGG GGA AGC GCT CAT 3′, respectively).63
The PCR template was prepared from genomic DNA isolated from overnight cultures of the P. aeruginosa 18SH strain.39 The PDC upstream and PDC downstream primers successfully amplified a 1400-base pair amplicon from the P. aeruginosa 18SH DNA. The amplicon was ligated into the pCR 2.1-TOPO vector (Invitrogen, Carlsbad, CA) and transformed into E. coli DH10B cells (Invitrogen). Following sequence verification of the P. aeruginosa 18SH blaPDC-3 gene, we performed a restriction enzyme digest of the recombinant plasmid using the BamHI and XbaI restriction enzymes (Promega, Madison, WI). The digestion product was ligated to the BamHI- and XbaI-cut pBC SK(−) vector and E. coli DH10B cells were transformed with the recombinant plasmid. The M13 Universal, M13 Reverse primers, PDC upstream, PDC downstream, and primers designed to the PAO1 active site were used to verify the sequence of P. aeruginosa 18SH blaPDC-3 in pBC SK(−).64
This blaPDC-3 gene was also directionally subcloned into the pET 24a(+) vector (Novagen, Madison, WI) for “large scale” protein expression from E. coli BL21(DE3) RP CodonPlus cells (Stratragene, La Jolla, CA). CodonPlus cells contain a pACYC plasmid with extra copies of the argU and proL tRNA genes to help overcome codon bias encountered during expression of P. aeruginosa protein in E. coli. We designed PCR amplification primers to introduce NdeI and BamH1 restriction sites into the beginning of the gene sequence that coded for the mature protein (i.e., the β-lactamase leader sequence was not included) and the end of the coding sequence, respectively. PCR mutagenesis was performed on the blaPDC-3 in the pCR 2.1-TOPO vector, and the successful introduction of the restriction sites confirmed by digesting the resultant plasmids with NdeI and BamH1. The insert was purified from agarose gel, ligated to a NdeI- and BamH1-digested plasmid prep of the pET 24a(+) vector, and E. coli DH10B cells were transformed with the purified insert. Plasmids were isolated and sequenced, and E. coli BL21(DE3) RP CodonPlus cells were transformed with plasmids of confirmed blaPDC-3 sequence according to the manufacturer's protocol.
Mutagenesis and sequencing
Site-directed mutagenesis primers were designed to replace the blaPDC-3 wild-type (WT) amino acid codon with the alanine codon at amino acid positions 343, 346, and 349 (based on sequence alignment with P99, E. coli AmpC, and Acinteobacter-derived cephalosporinase).65 Using the template blaPDC-3 gene in the pBC SK(−) vector, we used Stratagene's QuikChange Mutagenesis Kit® to introduce the alternate codon. For the Arg349Ala variant, mutagenesis was also performed in the pET 24a(+) vector using the same primers. DNA sequencing of the blaPDC genes was performed to confirm introduction of the desired mutation.
β-Lactamase purification
The PDC-3 and PDC-3 Arg349Ala β-lactamases were purified from the E. coli BL21(DE3) RP CodonPlus cells by induction with isopropyl β-d-thiogalactopyranoside. Briefly, 12 mL of an overnight culture grown in lysogeny broth were used to inoculate 500 mL of super optimal broth and grown at 37°C for 2 h. We added isopropyl β-d-thiogalactopyranoside at a final concentration of 1 mM, and grew the cultures for an additional 2.5 h. For all the P. aeruginosa and E. coli cultures, the cells were pelleted, resuspended in 50 mM Tris (pH 7.4), and β-lactamase liberated with lysozyme and ethylenediaminetetraacetic acid per established methods.66 Accordingly, PDC-3 enzyme was purified by preparative isoelectric focusing and fast protein liquid chromotography with a Sephadex Hi Load 16/60 column and a HiTrap High Performance sulfopropyl strong cation exchanger (Pharmacia, Uppsala, Sweden).67 The protein was quantified by BSA assay and purity was assessed by 5% stacking, 12% resolving SDS-PAGE.39
Kinetics
Steady-state kinetics were performed on an Agilent 8453 diode array spectrophotometer (Palo Alto, CA). Each continuous assay was performed in 10 mM phosphate-buffered saline at pH 7.4 at RT in a quartz cuvette with a 1-cm pathlength. Measurements were obtained using nitrocefin (Δɛ482 = 17,400 M−1 cm−1), cephalothin (Δɛ262 = −7660 M−1 cm−1), and cephaloridine (Δɛ260 = −10,200 M−1 cm−1).
The kinetic parameters, Vmax and Km, were obtained with non-linear least squares fit of the data (Henri Michaelis-Menten equation) using Origin 7.5® (OriginLab, Northampton, MA):
As previously reported, direct hydrolysis of the substrates ampicillin, ceftazidime, and cefotaxime could not be measured at this time (in assays with up to 5 μg of protein), and thus the apparent affinity values were obtained in competition assays with nitrocefin.37 Similarly, competition assays were employed to measure the relative affinity of PDC-3 Arg349Ala variant for cephalothin, whereas kcat was estimated from direct hydrolysis assays.
For the boronic acid inhibitors, Ki values were calculated by measuring the initial velocity (0–10 s) in the presence of a constant concentration of enzyme (3 nM) and increasing concentrations of the inhibitors competed against the indicator substrate, nitrocefin. Because of the time-dependent inhibition observed previously with some of the chiral boronates, all boronates were preincubated with enzyme for 5 min in phosphate-buffered saline before initiating the reaction with the addition of substrate.28,49,68–70
In these competition assays, initial velocities (v0) can be represented by the following equation71:
To determine Ki, the initial velocities immediately after mixing (1/v0) were plotted as a function of inhibitor concentration:
Rearrangement of the equation shows:
where I is Ki obs, the equation is solved by setting 1/v0 to 0.
Thus, the Ki obs is the concentration of I that reduces the velocity by 50%, and can be calculated from the x-intercept times −1.72
For tazobactam and aztreonam, the first-order rate constant for enzyme and inhibitor complex inactivation, kinact, was obtained by monitoring the reaction time courses in the presence of inhibitor. Fixed concentrations of enzyme (10 nM) and nitrocefin (100 μM) and increasing concentrations of inhibitor were used in each assay. The kobs was determined using a nonlinear least squares fit of the data using Origin 7.5®:
Here, A is absorbance, v0 (expressed in variation of absorbance per unit time) is initial velocity, vf is final velocity, and t is time. Each kobs was plotted versus I and fit to determine kinact.
Electrospray Ionization Mass Spectrometry
Mass spectrometry (MS) was performed to ascertain the molecular weight of the mature PDC-3 and PDC-3 Arg349Ala proteins expressed in E. coli BL21(DE3) RP CodonPlus cells. We prepared 14 μM of each protein in phosphate-buffered saline. The sample was acidified by the addition of 0.1% trifluoroacetic acid and immediately desalted and concentrated using a C18 ZipTip (Millipore, Bedford, MA) according to the manufacturer's protocol. Samples were then placed on ice and analyzed within 1 h.
Spectra of the intact proteins were generated on a Q-STAR XL Quadrupole-Time-of-Flight mass spectrometer (Applied Biosystems, Framingham, MA) equipped with a nanospray source. Experiments were performed by diluting the protein sample with 50% acetonitrile/0.1% trifluoroacetic acid to a concentration of 10 μM. This protein solution was then infused at a rate of 0.5 μl min−1 and the data were collected for 2 min. Spectra were deconvoluted using the Analyst program (Applied Biosystems). We assign an error of ±3 amu to each measurement.
Acknowledgments
The authors thank Arne Rietsch and Krisztina Papp-Wallace for helpful suggestions concerning the purification of the P. aeruginosa proteins and manuscript preparation.
REFERENCES
- 1.Page MG, Heim J. Prospects for the next anti-Pseudomonas drug. Curr Opin Pharmacol. 2009;9:558–565. doi: 10.1016/j.coph.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect Control Hosp Epidemiol. 2008;29:996–1011. doi: 10.1086/591861. [DOI] [PubMed] [Google Scholar]
- 3.Gaynes R, Edwards JR. Overview of nosocomial infections caused by Gram-negative bacilli. Clin Infect Dis. 2005;41:848–854. doi: 10.1086/432803. [DOI] [PubMed] [Google Scholar]
- 4.Vidal F, Mensa J, Almela M, Martinez JA, Marco F, Casals C, Gatell JM, Soriano E, Jimenez de Anta MT. Epidemiology and outcome of Pseudomonas aeruginosa bacteremia, with special emphasis on the influence of antibiotic treatment. Analysis of 189 episodes. Arch Intern Med. 1996;156:2121–2126. [PubMed] [Google Scholar]
- 5.Rahal JJ. Novel antibiotic combinations against infections with almost completely resistant Pseudomonas aeruginosa and Acinetobacter species. Clin Infect Dis. 2006;43(Suppl 2):S95–S99. doi: 10.1086/504486. [DOI] [PubMed] [Google Scholar]
- 6.Goossens H. Susceptibility of multi-drug-resistant Pseudomonas aeruginosa in intensive care units: results from the European MYSTIC study group. Clin Microbiol Infect. 2003;9:980–983. doi: 10.1046/j.1469-0691.2003.00690.x. [DOI] [PubMed] [Google Scholar]
- 7.Tam VH, Chang KT, Abdelraouf K, Brioso CG, Ameka M, McCaskey LA, Weston JS, Caeiro JP, Garey KW. Prevalence, mechanism and susceptibility of multidrug resistant bloodstream isolates of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2010;54:1160–1164. doi: 10.1128/AAC.01446-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rice LB. The clinical consequences of antimicrobial resistance. Curr Opin Microbiol. 2009;12:476–481. doi: 10.1016/j.mib.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 9.Livermore DM. Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: our worst nightmare? Clin Infect Dis. 2002;34:634–640. doi: 10.1086/338782. [DOI] [PubMed] [Google Scholar]
- 10.Walsh TR. Emerging carbapenemases: a global perspective. Int J Antimicrob Agents. 2010;36(Suppl 3):S8–S14. doi: 10.1016/S0924-8579(10)70004-2. [DOI] [PubMed] [Google Scholar]
- 11.Hanson ND, Sanders CC. Regulation of inducible AmpC β-lactamase expression among Enterobacteriaceae. Curr Pharm Des. 1999;5:881–894. [PubMed] [Google Scholar]
- 12.Juan C, Gutierrez O, Oliver A, Ayestaran JI, Borrell N, Perez JL. Contribution of clonal dissemination and selection of mutants during therapy to Pseudomonas aeruginosa antimicrobial resistance in an intensive care unit setting. Clin Microbiol Infect. 2005;11:887–892. doi: 10.1111/j.1469-0691.2005.01251.x. [DOI] [PubMed] [Google Scholar]
- 13.Bush K, Macalintal C, Rasmussen BA, Lee VJ, Yang Y. Kinetic interactions of tazobactam with β-lactamases from all major structural classes. Antimicrob Agents Chemother. 1993;37:851–858. doi: 10.1128/aac.37.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monnaie D, Frere JM. Interaction of clavulanate with class C β-lactamases. FEBS Lett. 1993;334:269–271. doi: 10.1016/0014-5793(93)80692-n. [DOI] [PubMed] [Google Scholar]
- 15.Kazmierczak A, Cordin X, Duez JM, Siebor E, Pechinot A, Sirot J. Differences between clavulanic acid and sulbactam in induction and inhibition of cephalosporinases in enterobacteria. J Int Med Res. 1990;18(Suppl 4):67D–77D. [PubMed] [Google Scholar]
- 16.Blizzard TA, Chen H, Kim S, Wu J, Young K, Park YW, Ogawa A, Raghoobar S, Painter RE, Hairston N, Lee SH, Misura A, Felcetto T, Fitzgerald P, Sharma N, Lu J, Ha S, Hickey E, Hermes J, Hammond ML. Side chain SAR of bicyclic β-lactamase inhibitors (BLIs). 1. Discovery of a class C BLI for combination with imipinem. Bioorg Med Chem Lett. 2010;20:918–921. doi: 10.1016/j.bmcl.2009.12.069. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez-Martinez JM, Poirel L, Nordmann P. Extended-spectrum cephalosporinases in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2009;53:1766–1771. doi: 10.1128/AAC.01410-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacob-Dubuisson F, Lamotte-Brasseur J, Dideberg O, Joris B, Frere JM. Arginine 220 is a critical residue for the catalytic mechanism of the Streptomyces albus G β-lactamase. Protein Eng. 1991;4:811–819. doi: 10.1093/protein/4.7.811. [DOI] [PubMed] [Google Scholar]
- 19.Knox JR, Moews PC. β-lactamase of Bacillus licheniformis 749/C. Refinement at 2 A resolution and analysis of hydration. J Mol Biol. 1991;220:435–455. doi: 10.1016/0022-2836(91)90023-y. [DOI] [PubMed] [Google Scholar]
- 20.Imtiaz U, Billings EM, Knox JR, Manavathu EK, Lerner SA, Mobashery S. Inactivation of class A β-lactamases by clavulanic acid: the role of Arginine 244 in a proposed noncerted sequence of events. J Am Chem Soc. 1993;115:4435–4442. [Google Scholar]
- 21.Zafaralla G, Manavathu EK, Lerner SA, Mobashery S. Elucidation of the role of Arginine-244 in the turnover processes of class A β-lactamases. Biochemistry. 1992;31:3847–3852. doi: 10.1021/bi00130a016. [DOI] [PubMed] [Google Scholar]
- 22.Thomson JM, Distler AM, Bonomo RA. Overcoming resistance to β-lactamase inhibitors: comparing sulbactam to novel inhibitors against clavulanate resistant SHV enzymes with substitutions at Ambler position 244. Biochemistry. 2007;46:11361–11368. doi: 10.1021/bi700792a. [DOI] [PubMed] [Google Scholar]
- 23.Moews PC, Knox JR, Dideberg O, Charlier P, Frere JM. β-lactamase of Bacillus licheniformis 749/C at 2 A resolution. Proteins. 1990;7:156–171. doi: 10.1002/prot.340070205. [DOI] [PubMed] [Google Scholar]
- 24.Marciano DC, Brown NG, Palzkill T. Analysis of the plasticity of location of the Arg244 positive charge within the active site of the TEM-1 β-lactamase. Protein Sci. 2009;18:2080–2089. doi: 10.1002/pro.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perez-Llarena FJ, Cartelle M, Mallo S, Beceiro A, Perez A, Villanueva R, Romero A, Bonnet R, Bou G. Structure-function studies of arginine at position 276 in CTX-M b-lactamases. J Antimicrob Chemother. 2008;61:792–797. doi: 10.1093/jac/dkn031. [DOI] [PubMed] [Google Scholar]
- 26.Beadle BM, Shoichet BK. Structural basis for imipenem inhibition of class C β-lactamases. Antimicrob Agents Chemother. 2002;46:3978–3980. doi: 10.1128/AAC.46.12.3978-3980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lobkovsky E, Billings EM, Moews PC, Rahil J, Pratt RF, Knox JR. Crystallographic structure of a phosphonate derivative of the Enterobacter cloacae P99 cephalosporinase: mechanistic interpretation of a β-lactamase transition-state analog. Biochemistry. 1994;33:6762–6772. doi: 10.1021/bi00188a004. [DOI] [PubMed] [Google Scholar]
- 28.Morandi F, Caselli E, Morandi S, Focia PJ, Blazquez J, Shoichet BK, Prati F. Nanomolar inhibitors of AmpC β-lactamase. J Am Chem Soc. 2003;125:685–695. doi: 10.1021/ja0288338. [DOI] [PubMed] [Google Scholar]
- 29.Dubus A, Wilkin JM, Raquet X, Normark S, Frere JM. Catalytic mechanism of active-site serine β-lactamases: role of the conserved hydroxy group of the Lys-Thr(Ser)-Gly triad. Biochem J. 1994;301(Pt 2):485–494. doi: 10.1042/bj3010485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Z, Yu Y, Musser JM, Palzkill T. Amino acid sequence determinants of extended spectrum cephalosporin hydrolysis by the class C P99 β-lactamase. J Biol Chem. 2001;276:46568–46574. doi: 10.1074/jbc.M102757200. [DOI] [PubMed] [Google Scholar]
- 31.Patera A, Blaszczak LC, Shoichet B. Crystal structures of substrate and inhibitor complexes with AmpC β-lactamase: possible implications for substrate-assisted catalysis. J Am Chem Soc. 2000;122:10504–10512. [Google Scholar]
- 32.Beadle BM, Trehan I, Focia PJ, Shoichet BK. Structural milestones in the reaction pathway of an amide hydrolase: substrate, acyl, and product complexes of cephalothin with AmpC β-lactamase. Structure. 2002;10:413–424. doi: 10.1016/s0969-2126(02)00725-6. [DOI] [PubMed] [Google Scholar]
- 33.Powers RA, Shoichet BK. Structure-based approach for binding site identification on AmpC β-lactamase. J Med Chem. 2002;45:3222–3234. doi: 10.1021/jm020002p. [DOI] [PubMed] [Google Scholar]
- 34.Tomatis PE, Fabiane SM, Simona F, Carloni P, Sutton BJ, Vila AJ. Adaptive protein evolution grants organismal fitness by improving catalysis and flexibility. Proc Natl Acad Sci USA. 2008;105:20605–20610. doi: 10.1073/pnas.0807989106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Todd AE, Orengo CA, Thornton JM. Plasticity of enzyme active sites. Trends Biochem Sci. 2002;27:419–426. doi: 10.1016/s0968-0004(02)02158-8. [DOI] [PubMed] [Google Scholar]
- 36.Brown NG, Pennington JM, Huang W, Ayvaz T, Palzkill T. Multiple global suppressors of protein stability defects facilitate the evolution of extended-spectrum TEM β-lactamases. J Mol Biol. 2010;404:832–846. doi: 10.1016/j.jmb.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Endimiani A, Doi Y, Bethel CR, Taracila M, Adams-Haduch JM, O'Keefe A, Hujer AM, Paterson DL, Skalweit MJ, Page MG, Drawz SM, Bonomo RA. Enhancing resistance to cephalosporins in class C β-lactamases: impact of Gly214Glu in CMY-2. Biochemistry. 2010;9:1014–1023. doi: 10.1021/bi9015549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galleni M, Amicosante G, Frere JM. A survey of the kinetic parameters of class C β-lactamases. Cephalosporins and other β-lactam compounds. Biochem J. 1988;255:123–129. doi: 10.1042/bj2550123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hujer KM, Hamza NS, Hujer AM, Perez F, Helfand MS, Bethel CR, Thomson JM, Anderson VE, Barlow M, Rice LB, Tenover FC, Bonomo RA. Identification of a new allelic variant of the Acinetobacter baumannii cephalosporinase, ADC-7 β-lactamase: defining a unique family of class C enzymes. Antimicrob Agents Chemother. 2005;49:2941–2948. doi: 10.1128/AAC.49.7.2941-2948.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Powers RA, Caselli E, Focia PJ, Prati F, Shoichet BK. Structures of ceftazidime and its transition-state analogue in complex with AmpC β-lactamase: implications for resistance mutations and inhibitor design. Biochemistry. 2001;40:9207–9214. doi: 10.1021/bi0109358. [DOI] [PubMed] [Google Scholar]
- 41.Bush K, Freudenberger JS, Sykes RB. Interaction of azthreonam and related monobactams with β-lactamases from gram-negative bacteria. Antimicrob Agents Chemother. 1982;22:414–420. doi: 10.1128/aac.22.3.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oefner C, D'Arcy A, Daly JJ, Gubernator K, Charnas RL, Heinze I, Hubschwerlen C, Winkler FK. Refined crystal structure of β-lactamase from Citrobacter freundii indicates a mechanism for β-lactam hydrolysis. Nature. 1990;343:284–288. doi: 10.1038/343284a0. [DOI] [PubMed] [Google Scholar]
- 43.Mourey L, Miyashita K, Swaren P, Bulychev A, Samama JP, Mobashery S. Inhibition of the NMC-A β-lactamase by a penicillanic acid derivative and the structural bases for the increase in substrate profile of this antibiotic resistance enzyme. J Am Chem Soc. 1998;120:9382–9383. [Google Scholar]
- 44.Chen CC, Rahil J, Pratt RF, Herzberg O. Structure of a phosphonate-inhibited β-lactamase. An analog of the tetrahedral transition state/intermediate of β-lactam hydrolysis. J Mol Biol. 1993;234:165–178. doi: 10.1006/jmbi.1993.1571. [DOI] [PubMed] [Google Scholar]
- 45.Crompton IE, Cuthbert BK, Lowe G, Waley SG. β-lactamase inhibitors. The inhibition of serine β-lactamases by specific boronic acids. Biochem J. 1988;251:453–459. doi: 10.1042/bj2510453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bush K, Jacoby GA, Medeiros AA. A functional classification scheme for β-lactamases and its correlation with molecular structure. Antimicrob Agents Chemother. 1995;39:1211–1233. doi: 10.1128/aac.39.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drawz SM, Bonomo RA. Three decades of β-lactamase inhibitors. Clin Microbiol Rev. 2010;23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lobkovsky E, Moews PC, Liu H, Zhao H, Frere JM, Knox JR. Evolution of an enzyme activity: crystallographic structure at 2-A resolution of cephalosporinase from the ampC gene of Enterobacter cloacae P99 and comparison with a class A penicillinase. Proc Natl Acad Sci USA. 1993;90:11257–11261. doi: 10.1073/pnas.90.23.11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morandi S, Morandi F, Caselli E, Shoichet BK, Prati F. Structure-based optimization of cephalothin-analogue boronic acids as β-lactamase inhibitors. Bioorg Med Chem. 2008;16:1195–1205. doi: 10.1016/j.bmc.2007.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Michaux C, Massant J, Kerff F, Frere JM, Docquier JD, Vandenberghe I, Samyn B, Pierrard A, Feller G, Charlier P, Van Beeumen J, Wouters J. Crystal structure of a cold-adapted class C β-lactamase. FEBS J. 2008;275:1687–1697. doi: 10.1111/j.1742-4658.2008.06324.x. [DOI] [PubMed] [Google Scholar]
- 51.Feller G, Gerday C. Psychrophilic enzymes: hot topics in cold adaptation. Nat Rev Microbiol. 2003;1:200–208. doi: 10.1038/nrmicro773. [DOI] [PubMed] [Google Scholar]
- 52.Wang X, Minasov G, Shoichet BK. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J Mol Biol. 2002;320:85–95. doi: 10.1016/S0022-2836(02)00400-X. [DOI] [PubMed] [Google Scholar]
- 53.Kim JY, Jung HI, An YJ, Lee JH, Kim SJ, Jeong SH, Lee KJ, Suh PG, Lee HS, Lee SH, Cha SS. Structural basis for the extended substrate spectrum of CMY-10, a plasmid-encoded class C β-lactamase. Mol Microbiol. 2006;60:907–916. doi: 10.1111/j.1365-2958.2006.05146.x. [DOI] [PubMed] [Google Scholar]
- 54.Khersonsky O, Tawfik DS. Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu Rev Biochem. 2010;79:471–505. doi: 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- 55.Caselli E, Powers RA, Blasczcak LC, Wu CY, Prati F, Shoichet BK. Energetic, structural, and antimicrobial analyses of β-lactam side chain recognition by β-lactamases. Chem Biol. 2001;8:17–31. doi: 10.1016/s1074-5521(00)00052-1. [DOI] [PubMed] [Google Scholar]
- 56.Davoli P, Fava R, Spaggiari A, Morandi S, Prati F. Enantioselective total synthesis of (-)-microcarpalide. Tetrahedron. 2005;61:4427–4436. [Google Scholar]
- 57.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 58.Kryshtafovych A, Fidelis K. Protein structure prediction and model quality assessment. Drug Discov Today. 2009;14:386–393. doi: 10.1016/j.drudis.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Diller DJ, Merz KM., Jr High throughput docking for library design and library prioritization. Proteins. 2001;43:113–124. doi: 10.1002/1097-0134(20010501)43:2<113::aid-prot1023>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 60.Clinical and Laboratory Standard Institute (2006) 7th ed. Wayne, PA: Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Approved Standard, CLSI document M7-A7; [Google Scholar]
- 61.Berks M, Redhead K, Abraham EP. Isolation and properties of an inducible and a constitutive β-lactamase from Pseudomonas aeruginosa. J Gen Microbiol. 1982;128:155–159. doi: 10.1099/00221287-128-1-155. [DOI] [PubMed] [Google Scholar]
- 62.Flett F, Curtis NA, Richmond MH. Mutant of Pseudomonas aeruginosa 18S that synthesizes type Id β-lactamase constitutively. J Bacteriol. 1976;127:1585–1586. doi: 10.1128/jb.127.3.1585-1586.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lodge JM, Minchin SD, Piddock LJ, Busby JW. Cloning, sequencing and analysis of the structural gene and regulatory region of the Pseudomonas aeruginosa chromosomal ampC β-lactamase. Biochem J. 1990;272:627–631. doi: 10.1042/bj2720627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Knott-Hunziker V, Petursson S, Jayatilake GS, Waley SG, Jaurin B, Grundstrom T. Active sites of β-lactamases. The chromosomal β-lactamases of Pseudomonas aeruginosa and Escherichia coli. Biochem J. 1982;201:621–627. doi: 10.1042/bj2010621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Drawz SM, Babic M, Bethel CR, Taracila M, Distler AM, Ori C, Caselli E, Prati F, Bonomo RA. Inhibition of the class C β-lactamase from Acinetobacter spp.: insights into effective inhibitor design. Biochemistry. 2010;49:329–340. doi: 10.1021/bi9015988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hujer AM, Hujer KM, Helfand MS, Anderson VE, Bonomo RA. Amino acid substitutions at Ambler position Gly238 in the SHV-1 β-lactamase: exploring sequence requirements for resistance to penicillins and cephalosporins. Antimicrob Agents Chemother. 2002;46:3971–3977. doi: 10.1128/AAC.46.12.3971-3977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pattanaik P, Bethel CR, Hujer AM, Hujer KM, Distler AM, Taracila M, Anderson VE, Fritsche TR, Jones RN, Pagadala SR, van den Akker F, Buynak JD, Bonomo RA. Strategic design of an effective β-lactamase inhibitor: LN-1–255, A 6-alkylidene-2′-substituted penicillin sulfone. J Biol Chem. 2009;284:945–953. doi: 10.1074/jbc.M806833200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thomson JM, Distler AM, Prati F, Bonomo RA. Probing active site chemistry in SHV β-lactamase variants at Ambler position 244. Understanding unique properties of inhibitor resistance. J Biol Chem. 2006;281:26734–26744. doi: 10.1074/jbc.M603222200. [DOI] [PubMed] [Google Scholar]
- 69.Chen Y, Shoichet B, Bonnet R. Structure, function, and inhibition along the reaction coordinate of CTX-M β-lactamases. J Am Chem Soc. 2005;127:5423–5434. doi: 10.1021/ja042850a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang X, Minasov G, Blazquez J, Caselli E, Prati F, Shoichet BK. Recognition and resistance in TEM β-lactamase. Biochemistry. 2003;42:8434–8444. doi: 10.1021/bi034242y. [DOI] [PubMed] [Google Scholar]
- 71.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery. John Wiley & Sons, Inc; 2005. [PubMed] [Google Scholar]
- 72.Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]