

Abstract
The biochemical characterization of the bacterial transcription cycle has been greatly facilitated by the production and characterization of targeted RNA polymerase (RNAP) mutants. Traditionally, RNAP preparations containing mutant subunits have been produced by reconstitution of denatured RNAP subunits, a process that is undesirable for biophysical and structural studies. Although schemes that afford the production of in vivo-assembled, recombinant RNAP containing amino acid substitutions, insertions, or deletions in either the monomeric β or β′ subunits have been developed, there is no such system for the production of in vivo-assembled, recombinant RNAP with mutations in the homodimeric α-subunits. Here, we demonstrate a strategy to generate in vivo-assembled, recombinant RNAP preparations free of the α C-terminal domain. Furthermore, we describe a modification of this approach that would permit the purification of in vivo-assembled, recombinant RNAP containing any α-subunit variant, including those variants that are lethal. Finally, we propose that these related approaches can be extended to generate in vivo-assembled, recombinant variants of other protein complexes containing homomultimers for biochemical, biophysical, and structural analyses.
Keywords: RNA polymerase, α-subunit, macromolecular complex, purification
Introduction
The Escherichia coli (E. coli) RNA polymerase (RNAP) holoenzyme is a large (∼450 kDa), multisubunit enzyme consisting of an α-subunit homodimer, single β, β′, and ω subunits, and one of seven σ-subunits. The major form of holoenzyme, σ70-associated holoenzyme, locates promoters upstream of coding regions and at many promoters recognizes three elements: the −10 and the −35 elements, bound by the σ70 subunit, and the UP element, bound by the α subunit.1 The ∼37 kDa, 329 amino acid α-subunit (encoded by the rpoA gene), is organized into two domains, the ∼25 kDa N-terminal domain (αNTD; residues 8–231) and the ∼9 kDa C-terminal domain (αCTD; residues 249–329), 2 3 that are separated by a flexible linker of at least 13 amino acids in length.4 The αNTD is the scaffold for RNAP assembly, contains determinants for dimerization, and at some promoters facilitates transcription activation.5–7 By interacting with a plethora of transcription factors or by binding the A+T-rich UP element, the αCTD plays a major role in transcription initiation.8–11 The flexibility and length of the linker ensure that the αCTD can contact DNA and transcription factors at upstream positions distant from the transcription start site.4 8 9 11–14
Central to our understanding of the bacterial transcription cycle has been the preparation and in vitro characterization of E. coli RNAP variants, the most notable example being E. coli RNAP bereft of the αCTD (ΔαCTD RNAP).12 15 16 Traditionally, RNAP variants, including lethal RNAP mutants, have been prepared in functional form by reconstitution of denatured individual subunits.16–18 Although these methods are rapid and afford the production of multiple, biochemically active variants alongside one another, the preparations can suffer from assembly defects and loss of activity due to the use of denaturants.19 To ameliorate these complications, Landick and coworkers developed a recombinant E. coli RNAP coexpression system that permits the production of in vivo-assembled, recombinant E. coli RNAP preparations.20 Although more labor intensive, this method generates RNAP samples suitable for biochemical, biophysical, and structural studies because of the omission of a denaturation/renaturation step. Notwithstanding the usefulness of the coexpression system to generate recombinant wild-type (WT) RNAP and variants with mutations in the β and β′ subunits, in its current format it does suffer from one major limitation: the inability to generate E. coli RNAP preparations with homogenous α-subunit variants. As α is assembled as a homodimer in RNAP, the system will yield mutant α RNAP preparations containing substantial amounts of genomically encoded contaminating WT α. This contamination results in sample heterogeneity that is prohibitive to biochemical, biophysical, and structural studies, which are highly dependent on homogenous protein preparations.
Here, we demonstrate the use of a novel α variant, α-X234–241H, where residues 235–238, 240, and 241 of the interdomain linker are substituted to create a PreScission protease recognition site (encompassing residues 234–241) and a noncleavable decahistidine [(His)10] tag is appended to the C-terminus of the protein (Fig. 1), which permits efficient production of in vivo-assembled, recombinant ΔαCTD RNAP (an RNAP variant that contains a lethal α-mutant 21). We show by use of Western blot and in vitro transcription assays that the in vivo-assembled, recombinant ΔαCTD RNAP preparation is completely devoid of contaminating WT α or αCTD. Furthermore, we suggest a modification of this strategy that would afford the purification of in vivo-assembled, recombinant RNAP containing any α-subunit variant, including other α-variants that are lethal. Finally, these related approaches can be applied to generate variants of other macromolecular complexes that contain homomultimers for biochemical, biophysical, and structural analyses.
Figure 1.
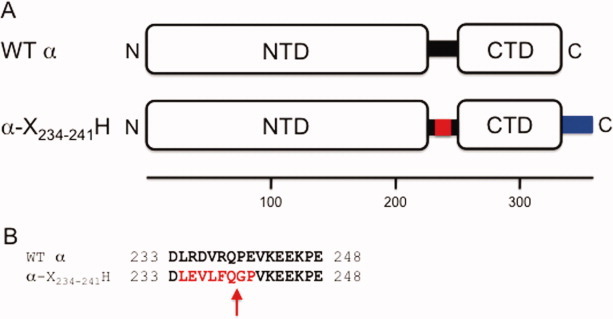
RNAP α-subunit variants. (A) Schematic diagram illustrating the WT α and α-X234–241H variants. WT α interdomain linker, black rectangle; the PreScission protease site substituted in the α-X234–241H interdomain linker, red rectangle; (His)10 tag, blue rectangle. The numbers beneath indicate the amino acid position. (B) Amino acid sequence alignment of the WT α and α-X234–241H interdomain linker. The numbers at the beginning and end of each line indicate the amino acid position. The PreScission protease recognition site in α-X234–241H is shown in emboldened red text, and the cleavage site is indicated with a red arrow.
Results
Overexpression of in vivo-assembled, recombinant ΔαCTD RNAP
To facilitate in vivo-assembly of recombinant ΔαCTD RNAP, we constructed a coexpression plasmid, pEcrpoA(-X234–241H)BCZ, that expresses α-X234–241H, β, β′, and ω from a single bacteriophage T7 RNAP promoter. In the α-X234–241H variant, a segment of the interdomain linker is substituted by a recognition site for the PreScission protease, 22 23 denoted here as X, and a noncleavable (His)10 tag is appended to the C-terminus, denoted here as H (Fig. 1). Critical to our strategy was the generation of a derivative of the E. coli BL21(DE3) overexpression strain, 24 BL21(DE3)T-X234–241H, where the WT chromosomal copy of rpoA was replaced by DNA encoding α-X234–241H using an rpoA allelic replacement technique.25 Thus, in BL21(DE3)T-X234–241H, the sole source of cellular-derived α is the α-X234–241H variant. Although some alterations of the linker are tolerated in vivo, gross alteration of the linker can result in significantly reduced growth rates.25 This would be an undesirable characteristic in a recombinant protein production strain. Therefore, we examined the effect of the α-X234–241H allele on bacterial growth in both minimal salts and rich media at 37°C (Table I).
Table I.
Effects of the α-Variants on the Growth Rate of E. coli
| Growth rate (doublings per hour) | ||
|---|---|---|
| Strain (α-variant) | Minimal medium (M9 minimal medium) | Rich medium (LB medium) |
| BL21(DE3)T | 1.10 | 1.96 |
| BL21(DE3)T-X234–241 | 0.90 | 1.66 |
| BL21(DE3)T-H | 1.08 | 1.80 |
| BL21(DE3)T-X234–241H | 0.92 | 1.56 |
The results show that the growth rate of the BL21(DE3)T-X234–241H strain is decreased by ∼16% and ∼20% in rich and minimal medium, respectively, when compared with the derivative harboring the WT rpoA (Table I). Comparison of the growth rate of the BL21(DE3)T-X234–241H strain with those of BL21(DE3)T encoding either α-X234–241 or α-H on the chromosome [generated during the same allelic replacement procedure used to construct the BL21[DE3]T-X234–241H strain] show that most, if not all, of the growth retardation is due to the six amino acid substitution in the α-interdomain linker. Regardless of the small decrease in the growth rate of BL21(DE3)T-X234–241H, we were able to use this strain containing the pEcrpoA(-X234–241H)BCZ plasmid to overexpress recombinant RNAP.
Purification of in vivo-assembled, recombinant ΔαCTD RNAP
In vivo-assembled, recombinant E. coli ΔαCTD RNAP was purified from BL21(DE3)T-X234–241H cells harboring pEcrpoA(-X234–241H)BCZ according to the scheme shown in Figure 2(A). As an initial purification step to remove nucleic acids and some protein contaminants, the clarified soluble fraction containing recombinant RNAP was treated with polyethyleneimine (PEI).26–28 Subsequent to elution from the pellet, the RNAP sample was applied to an immobilized metal affinity chromatography (IMAC) column 29 and developed with a step gradient of imidazole. Crucial to the success of our procedure was the overnight protease-dependent cleavage of the RNAP sample obtained from the first IMAC step followed by a second subtractive IMAC step. As the PreScission protease site is encoded within the interdomain linker, cleavage with the protease releases the ∼11 kDa αCTD-(His)10 fusion protein that, together with any uncleaved RNAP-(His)10, is removed from the ΔαCTD RNAP population by the subtractive IMAC step. Afterward, the partially purified ΔαCTD RNAP sample was applied to a Bio-Rex 70 anion exchange column to remove residual endogenous σ70. Finally, the peak fractions containing ΔαCTD RNAP eluted from the Bio-Rex 70 column were applied to a Sephadex 200 gel filtration column to remove any supramolecular mass aggregates prior to storage at −80°C. The resulting ΔαCTD RNAP preparation was judged to be ∼95% pure [Fig. 2(B)], and the yield was estimated to be ∼2.5 mg of protein per liter of bacterial culture.
Figure 2.
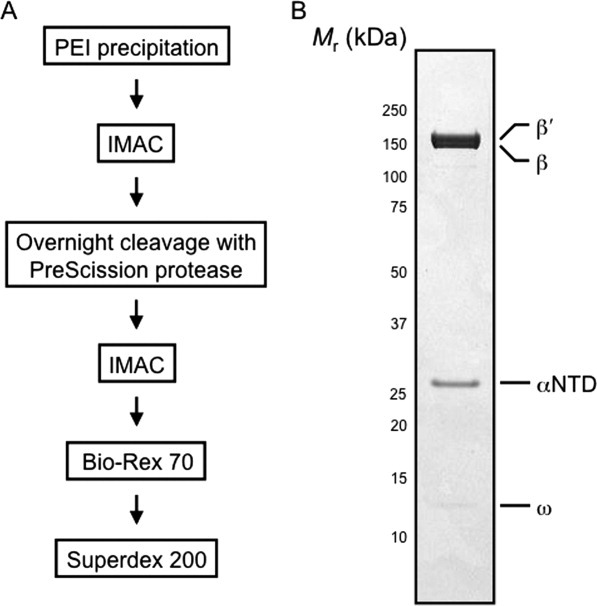
Purification and denaturing gel analysis of the in vivo-assembled, recombinant E. coli ΔαCTD RNAP preparation. (A) Flow diagram highlighting the steps used to purify the in vivo-assembled, recombinant ΔαCTD RNAP preparation. (B) A denaturing 4–12% (w/v) polyacrylamide Bis-Tris gel loaded with 2 μg of recombinant ΔαCTD RNAP. The gel was calibrated with molecular mass standards ranging from 250 to 10 kDa; numbers at the side of the gel indicate the molecular mass values of the standards in kilodaltons, and arrows indicate bands due to αNTD, β, β′, and ω.
Characterization of the ΔαCTD RNAP preparation
To ensure the absence of αCTD from the ΔαCTD RNAP preparation, samples were probed with antibodies specific for either αNTD or αCTD. A membrane blot of a denaturing gel loaded with in vivo-assembled, recombinant WT E. coli RNAP [purified from BL21[DE3] cells harboring pEcrpoABC(-XH)Z using the scheme shown in Fig. 2(A)] and ΔαCTD RNAP was stained with amido black to confirm equivalent protein transfer [Fig. 3(A), lanes 1 and 3]. The membrane was then probed with antibodies specific for αNTD [Fig. 3(A)]. A band corresponding in size to full-length α was observed in the WT E. coli RNAP sample [Fig. 3(A), lane 2], but not in the ΔαCTD RNAP sample [Fig. 3(A), lane 4]. Although these data are strongly suggestive of the absence of full-length α from the ΔαCTD RNAP preparation, they do not directly disclose the absence, or presence, of αCTD in the sample. Therefore, we repeated the experiment and probed the samples with antibodies specific for αCTD [Fig. 3(B)]. The results reveal the presence of a band due to full-length α in the lane loaded with WT E. coli RNAP [Fig. 3(B), lane 2], but not in the lane loaded with ΔαCTD RNAP [Fig. 3(B), lane 4]. Furthermore, no band due to αCTD alone was detected in any of the RNAP preparations [Fig. 3(B), lanes 2 and 4], although as a control, a band due to purified αCTD was observed [Fig. 3(B), lane 6].
Figure 3.
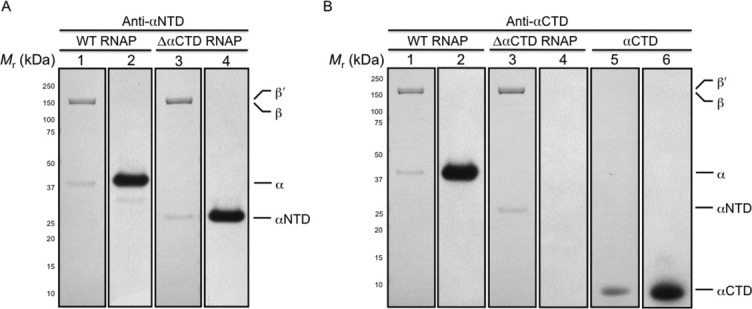
Western blot analysis of the in vivo-assembled, recombinant E. coli WT and ΔαCTD RNAP preparations. Purified recombinant RNAPs were fractionated by denaturing gel electrophoresis and electroblotted onto membrane. (A) The figure shows a membrane stained with amido black post-transfer of WT and ΔαCTD RNAP preparations and a photographic film developed after exposure to the membrane probed with anti-αNTD antibody and horse radish peroxidase conjugated anti-mouse IgG antibodies. Lanes were loaded as follows: (1) 1 μg of recombinant WT RNAP (membrane); (2) 1 μg of recombinant WT RNAP (photographic film); (3) 1 μg of recombinant ΔαCTD RNAP (membrane); (4) 1 μg of recombinant ΔαCTD RNAP (photographic film). (B) The figure shows a membrane stained with amido black post-transfer of WT RNAP, ΔαCTD RNAP and αCTD preparations and a photographic film developed after exposure to the membrane probed with anti-αCTD antibody and horse radish peroxidase conjugated anti-mouse IgG antibodies. Lanes were loaded as follows: (1) 1 μg of recombinant WT RNAP (membrane); (2) 1 μg of recombinant WT RNAP (photographic film); (3) 1 μg of recombinant ΔαCTD RNAP (membrane); (4) 1 μg of recombinant ΔαCTD RNAP (photographic film); (5) 1 μg of recombinant αCTD (membrane); (6) 1 μg of recombinant αCTD (photographic film). The membranes were calibrated with molecular mass standards ranging from 250 to 10 kDa; numbers at the side indicate the molecular mass values of the standards in kilodaltons, and arrows indicate bands due to α, αNTD, αCTD, β, and β′.
The αCTD is the target for both the cis-acting UP element and myriad transcription factors, including CRP.8–11 Therefore, we used multiple-round in vitro transcription experiments at promoters dependent on the UP element or CRP to further corroborate the absence of αCTD from our in vivo-assembled, recombinant ΔαCTD RNAP preparation. Initially, the activities of recombinant WT and ΔαCTD RNAP preparations [purified using the same scheme; Fig. 2(A)] were compared with the activity of endogenous WT RNAP with a defined specific activity at the constitutive (i.e., αCTD-independent) lacUV5 promoter to ensure comparable activities of RNAP enzyme were used in subsequent experiments [Fig. 4(A)]. To investigate the activity of the recombinant WT and ΔαCTD RNAP samples, UP element-dependent stimulation of transcription from an rrnB P1 promoter derivative that contains the consensus UP element was examined.8 The results are shown in Figure 4(B). The samples of RNAP containing WT α exhibited high levels of UP-dependent transcription at the WT rrnB P1 promoter [Fig. 4(B), lane 1], whereas the ΔαCTD RNAP preparation exhibited ∼10% of the activity of WT RNAP [Fig. 4(B), lane 2], indicating that the αCTD is absent from our ΔαCTD RNAP preparation, but not from the WT RNAP sample. As a control to check the contribution of the UP element to stimulation of rrnB P1 transcription, the transcriptional activity of RNAP containing either WT α or lacking αCTD was determined at the rrnB P1 core promoter that has the UP element replaced by a sequence devoid of UP element-like activity.8 At this promoter, both WT and ΔαCTD RNAP samples exhibited similar activities, which in the case of WT RNAP was significantly reduced in comparison with its activity at rrnB P1, that is, ∼10% of the activity at UP element-dependent rrnB P1, whereas the activity of ΔαCTD RNAP was similar at both promoters [Fig. 4(B), lanes 3 and 4]. Next, we determined the activity of the RNAP samples at the synthetic CC(−61.5) promoter that is dependent on both αCTD and the cAMP/CRP complex.11 30 The CRP dependence of CC(−61.5) was substantiated as neither RNAP preparation elicited transcriptional activity in the absence of the cAMP/CRP complex [Fig. 4(C), lanes 1 and 3]. However, on addition of cAMP/CRP to the reaction mixtures, CRP-dependent transcription was observed in the presence of WT RNAP [Fig. 4(C), lane 2], whereas no enhancement of transcriptional activity was observed in the presence of ΔαCTD RNAP [Fig. 4(C), lane 4].
Figure 4.
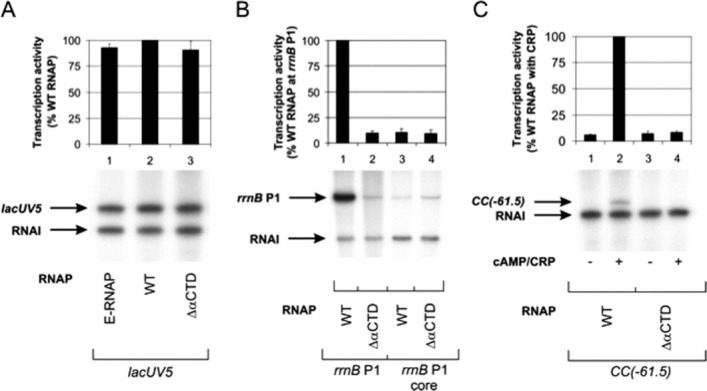
In vitro activity of in vivo-assembled, recombinant E. coli WT and ΔαCTD RNAP preparations. Multiple-round transcription reactions were performed to assess the response of recombinant WT and ΔαCTD σ70-associated holoenzymes to the promoter UP element and the transcription factor CRP. (A) Multiple-round transcription assays were performed at the lacUV5 promoter to determine the concentration of recombinant WT and ΔαCTD RNAPs that gave rise to levels of transcript equivalent to WT endogenous RNAP [E-RNAP (Epicenter); specific activity, 1.4 × 103 U/mg]. (B) Multiple-round transcription assays were performed at the WT rrnB P1 promoter and a derivative of the rrnB P1 promoter lacking an UP element. (C) Multiple-round transcription assays were performed at the synthetic CRP-dependent CC(−61.5) promoter. The different RNAPs are indicated below the gel in each panel. Concentrations of RNAP used are as follows: endogenous WT RNAP, 9.2 nM; in vivo-assembled, recombinant WT RNAP, 9.2 nM; and in vivo-assembled, recombinant ΔαCTD RNAP, 20 nM. Concentrations of supercoiled DNA template used are as follows: lacUV5, 0.2 nM; rrnB P1, 0.6 nM; rrnB P1 core, 0.6 nM; and CC(−61.5), 0.2 nM. The identities of specific transcripts are indicated by arrows [the vector-encoded replication repressor, RNA-I, 108 nucleotides; lacUV5, 131 nucleotides; rrnB P1 promoters, 202 nucleotides; and CC[−61.5], 123 nucleotides]. The abundance of transcripts originating from the lacUV5, rrnB P1, rrnB P1 core, and CC(−61.5) promoters was quantified from three experiments and plotted. In (A), the values were calculated as a percentage of transcript obtained with E-RNAP, whereas in (B) and (C), the values were calculated as a percentage of transcript obtained with WT RNAP and in all cases are presented (with standard deviations) above the appropriate gel, aligned with the corresponding gel lane. Transcription was measured from promoters harbored in the following plasmids: pSR/lacUV5, lacUV5 promoter; pRLG3278, rrnB P1 promoter; pRLG4210, rrnB P1 core promoter; and pSR/CC(−61.5), CC(−61.5) promoter.
The results of the multiple-round in vitro transcription assays clearly indicate that our ΔαCTD RNAP preparation is devoid of αCTD and together with the results of the Western blot analysis unequivocally demonstrate that the purification procedure described herein yields active in vivo-assembled, recombinant RNAP bereft of αCTD.
Discussion
Biochemical characterization of the bacterial transcription cycle has been greatly facilitated by the production and characterization of targeted RNAP mutants. Traditionally, RNAP preparations containing mutant α-subunits have been produced by reconstitution and renaturation of denatured RNAP subunits, a process that is highly undesirable for biophysical and structural studies. Although systems that afford the production of in vivo-assembled, recombinant RNAP encoding mutations in either the β or β′ subunits have been developed, 20 31 these systems are not amenable to the generation of in vivo-assembled, recombinant RNAP samples containing homodimeric mutant α-subunits. Here, we have described a scheme for the production of in vivo-assembled, recombinant ΔαCTD RNAP that overcomes these restrictions and demonstrated that the sample is free of detectable αCTD.
Considering E. coli RNAP further, in vivo-assembled, recombinant enzyme containing any type of homodimeric α-variant could be obtained by introducing the desired substitution into the rpoA gene contained on the expression plasmid pEcrpo(HX-)ABCZ (unpublished data). In this plasmid, α is produced with a PreScission protease-cleavable N-terminal (His)10 tag. Following recombinant protein overexpression in the BL21(DE3)T-H strain [that encodes WT α with a noncleavable (His)10 tag appended to the C-terminus as the sole source of cellular alpha], RNAP containing either or both chromosome- and plasmid-encoded α is purified by IMAC. Subsequently, PreScission protease cleavage of the RNAP preparation and a subtractive IMAC step is performed to remove RNAP molecules containing WT α homodimers and WT/mutant α heterodimers, thus, yielding homogenous preparations of in vivo-assembled, recombinant RNAP containing any desired α-subunit variant, including variants that would be lethal if expressed in the absence of WT α (Fig. 5).
Figure 5.
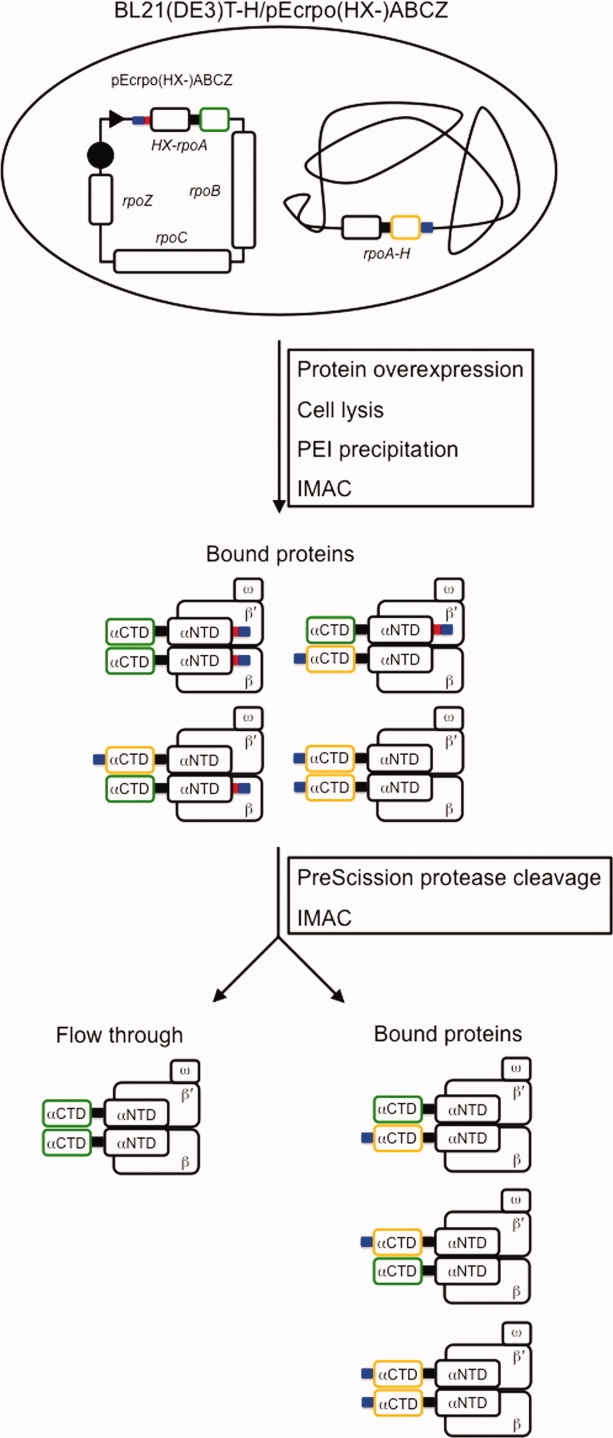
Schematic diagram illustrating the procedure to purify in vivo-assembled, recombinant E. coli RNAP containing any homodimeric α-variant, including those variants that are lethal in vivo. In the example shown above, a variant of E. coli RNAP harboring a mutant αCTD is overexpressed and isolated from other E. coli RNAP variants that arise due to in vivo assembly between plasmid- and chromosome-encoded α. The desired rpoA mutation is introduced into the bacteriophage T7 RNAP-dependent overexpression plasmid, pEcrpo(HX-)ABCZ, that encodes N-terminal (His)10-tagged α with a PreScission protease tag between the (His)10 tag and the α ATG start codon; β, β′, and ω. The resultant plasmid is used to transform the E. coli overexpression strain, BL21(DE3)T-H, that encodes WT α with an uncleavable C-terminal (His)10 tag as the sole source of cellular α. Cells containing overexpressed recombinant RNAP are lysed, and the resultant cell lysate is treated with PEI to precipitate nucleic acids and proteins. After an initial IMAC step to enrich the (His)10-tagged RNAP complexes, the sample is incubated with PreScission protease to remove the (His)10 tag appended to the N-terminus of the plasmid-encoded α-mutant. Subsequently, a subtractive IMAC step is used to fractionate the different RNAP complexes; RNAP harboring the desired mutant α homodimer will flow through the column, whereas the RNAP enzymes containing either WT/mutant α heterodimers or WT α homodimers will be retained on the column. WT αCTD, orange outlined rectangle; mutant αCTD, green outlined rectangle; (His)10 tag, blue-colored rectangle; PreScission protease site, red-colored rectangle; α interdomain linker, black-colored rectangle; bacteriophage T7 RNAP promoter, black dart; bacteriophage T7 RNAP terminator, black circle.
Finally, these approaches can be easily manipulated to generate homogenous recombinant, in vivo-assembled variants of other homomultimers for biochemical, biophysical, and structural analyses.
Materials and Methods
Bacterial strains and plasmids
The bacterial strains and plasmids used or described are listed in Table II. Standard molecular biology techniques for plasmid isolation and DNA manipulation were used throughout. The rpoA allelic replacement plasmid, pRecEcrpoA-X234–241H, encodes an E. coli RNAP α-subunit variant (α-X234–241H), in which residues 235–238, 240, and 241 of the interdomain linker are substituted to create a PreScission protease (GE Healthcare) recognition site encompassing residues 234–241 (Fig. 1), and 12 amino acids, that include a (His)10 tag, are appended to the C-terminus. Located downstream of rpoA on this plasmid is the rplQ gene, which is also located downstream of rpoA on the E. coli chromosome and is required to provide homologous sequences for the allelic replacement technique (see below). The pRecEcrpoA-X234–241H plasmid was constructed in five steps. First, megaprimer polymerase chain reaction (PCR) 32 was used to substitute the DNA encoding amino acid residues 235–238, 240, and 241 of the RNAP α-subunit with the DNA encoding a PreScission protease recognition site and append NdeI and BamHI sites at the 5′ and 3′ ends of the PCR product, respectively. The resultant PCR product was cleaved with NdeI and BamHI and cloned between the NdeI and BamHI sites of a pET21a-based plasmid that encodes both ampicillin and kanamycin resistance cassettes, creating pET21aEcrpoA-X234–241. Second, the rpoA allele encoded by pET21aEcrpoA-X234–241 was PCR amplified using primers that retained the NdeI site at the 5′ end and appended DNA encoding an Ala-Ser dipeptide linker and a (His)10 tag, a TAA translation termination codon, and NcoI and BamHI sites to codon 329 of rpoA-X234–241. The resultant PCR product (rpoA-X234–241H) was cleaved with NdeI and BamHI and cloned between the NdeI and BamHI sites of pET21aEcrpoA-X234–241, creating pET21aEcrpoA-X234–241H. Third, PCR was used to amplify 500 bp of the E. coli chromosome immediately downstream of rpoA using primers that appended a KpnI site to the 5′ end and I-SceI and BlpI sites to the 3′ end. The PCR product, containing the rplQ gene encoding ribosomal protein L17, was cleaved with KpnI and BlpI and cloned between the KpnI and BlpI sites of pET21aEcrpoA-X234–241H, creating pET21aEcrpoA-X234–241H-rplQ. Fourth, the kanamycin resistance cassette, which separates the rpoA and rplQ sequences and was not required for the rpoA allelic replacement step, was removed from pET21aEcrpoA-X234–241H-rplQ by cleavage with BamHI and KpnI, followed by treatment with DNA polymerase I Klenow fragment, resulting in pET21aEcrpoA-X234–241HΔBK-rplQ. Finally, the first 157 codons of rpoA in pET21aEcrpoA-X234–241HΔBK-rplQ was removed by cleavage with XbaI and EcoRI, treatment with DNA polymerase I Klenow fragment, and self-ligation, creating pRecEcrpoA-X234–241H. The plasmid encoding WT E. coli (His)10-tagged core RNAP, pEcrpoABC(-XH)Z [in which a PreScission protease cleavage site, -Ser-Ser-Gly- linker and (His)10 tag are appended to the C-terminus of the β′ subunit], was constructed from plasmid pEcRNAP1 (Ref. 31) by introduction of an annealed pair of complementary oligonucleotides encoding the protease recognition site, Ser-Ser-Gly linker and (His)10 tag between the XhoI and HindIII sites of pEcRNAP1. A protein overexpression plasmid encoding α-X234–241H, β, β′, and ω was constructed by cleaving pET21aEcrpoA-X234–241H with NdeI and NcoI and cloning the NdeI-NcoI fragment (encoding α-X234–241H) between the NdeI and NcoI sites of pEcRNAP1, creating pEcrpoA(-X234–241H)BCZ. All DNA manipulations were confirmed to be correct by DNA sequencing.
Table 2.
Bacterial Strains and Plasmids
| Strain/plasmid | Relavent characteristics | Source or reference |
|---|---|---|
| Strains | ||
| VH1000 | MG1655 lacZ lacI pyrE+ | 25 |
| VH1000T | VH1000 rpoA+ zhc::Tn10 | This study |
| VH1000T-K271E | VH1000 rpoA341 zhc::Tn10 | This study |
| VH1000T-X234–241 | VH1000 rpoA-X234–241 zhc::Tn10 | This study |
| VH1000T-H | VH1000 rpoA-H zhc::Tn10 | This study |
| VH1000T-X234–241H | VH1000 rpoA-X234–241H zhc::Tn10 | This study |
| BL21(DE3) | F−ompT hsdS (rB−mB−) gal λDE3 lysogen | 24 |
| BL21(DE3)T | BL21(λDE3) rpoA+zhc::Tn10 | This study |
| BL21(DE3)T-X234–241 | BL21(λDE3) rpoA-X234–241 zhc::Tn10 | This study |
| BL21(DE3)T-H | BL21(λDE3) rpoA-H zhc::Tn10 | This study |
| BL21(DE3)T-X234–241H | BL21(λDE3) rpoA-X234–241H zhc::Tn10 | This study |
| Plasmids | ||
| pET21aEcrpoA-X234–241 | pET21a derivative encoding E. coli α with residues 234–241 of the interdomain linker specifying a PreScission protease site | This study |
| pET21aEcrpoA-X234–241H | pET21aEcrpoA-X234–241 derivative with the DNA encoding a (His)10 tag appended to the C-terminus of α-X234–241 | This study |
| pET21aEcrpoA-X234–241H-rplQ | pET21aEcrpoA-X234–241H derivative with the DNA encoding RplQ (L17) immediately downstream of rpoA-X234–241H | This study |
| pET21aEcrpoA-X234–241HΔBK-rplQ | pET21aEcrpoA-X234–241H-rplQ derivative with the kanamycin resistance cassette removed | This study |
| pRecEcrpoA-X234–241H | pET21aEcrpoA-X234–241HΔBK-rplQ derivative with the first 157 codons of rpoA removed | This study |
| pEcRNAP1 | pET21c derivative encoding E. coli α, β, β′, and ω. | 31 |
| pEcrpo(HX-)ABCZ | pEcRNAP1 derivative with the DNA encoding a (His)10 tag and PreScission protease site appended to the N-terminus of α | This study |
| pEcrpoA(-X234–241H)BCZ | pEcRNAP1 derivative encoding E. coli α-X234–241H, β, β′, and ω | This study |
| pEcrpoABC(-XH)Z | pEcRNAP1 derivative with the DNA encoding a PreScission protease site and (His)10 tag appended to the C-terminus of β′ | This study |
| pSR/lacUV5 | pBR322 derivative carrying lacUV5 (−60 to +40) promoter | 11 |
| pSR/CC(−61.5) | pBR322 derivative carrying CC(−61.5) promoter | 11 |
| pRLG4210 | pRLG770 containing core rrnB P1 (−66 to +52) promoter | 8 |
| pRLG4238 | pRLG770 containing the rrnB P1 promoter, including the UP element | 8 |
rpoA allelic replacement
The procedure was performed essentially as described previously 25 except that strain VH1000T-K271E was used to select recombinants. VH1000T-K271E was constructed by bacteriophage P1 transduction of the rpoA341 allele from WAM123 (Ref. 33) using selection for the linked Tn10 (20 μg/mL tetracycline). Transductants containing the rpoA341 allele were identified by virtue of their inability to use melibiose as a carbon source. Subsequently, VH1000T-K271E was transformed with pRecEcrpoA-X234–241H, and the transformants were streaked on M9 minimal salts agar supplemented with 0.2% (w/v) melibiose and 5 μg/mL thiamine and grown at 30°C for 2 to 3 days without antibiotic selection. Melibiose-positive recombinants were screened for the transfer of the rpoA-X234–241H allele to the chromosome (resulting in VH1000T-X234–241H) by PCR amplification of full-length rpoA. The presence of the PreScission cleavage site and (His)10 tag was confirmed by DNA sequencing. During the screening procedure, recombinants were also identified in which rpoA sequences encoding either the PreScission cleavage site or (His)10 tag were transferred to the E. coli chromosome (VH1000T-X234–241 and VH1000T-H, respectively). Again, by selecting for the linked Tn10, bacteriophage P1 transduction was used to transfer the mutant alleles from the VH1000T background into BL21(DE3), resulting in BL21(DE3)T-X234–241, BL21(DE3)T-H, and BL21(DE3)T-X234–241H [although it was necessary to reduce the selection concentration to 2 μg/mL tetracycline for the BL21(DE3) strains]. To confirm the presence of the desired allele, in each case, the chromosomal rpoA gene was amplified and the resultant PCR product was sequenced.
Measurement of bacterial growth rate
BL21(DE3) derivatives, each encoding a chromosomal rpoA allele linked to zhc::Tn10, were inoculated from freshly struck colonies into either Luria-Bertani (LB) medium or M9 minimal medium containing 0.2% (w/v) glucose and thiamine (5 μg/μL). Cultures were inoculated at an A600 nm of 0.015 and grown with vigorous agitation at 37°C. Growth was monitored by periodically measuring the A600 nm, and the rate of growth was calculated during the exponential phase of growth.
Overexpression and purification of proteins
To purify WT or ΔαCTD RNAP preparations, pEcrpoABC(-XH)Z and pEcrpoA(-X234–241H)BCZ were introduced into BL21(DE3) or BL21(DE3)T-X234–241H cells, respectively. BL21(DE3)/pEcrpoABC(-XH)Z transformants were selected in the presence of ampicillin (100 μg/mL), whereas BL21(DE3)T-X234–241H/pEcrpoA(-X234–241H)BCZ transformants were selected in the presence of ampicillin (100 μg/mL) and tetracycline (10 μg/mL). Cultures supplemented with ampicillin (200 μg/mL) were grown at 37°C to an A600 nm ∼0.6, and recombinant protein expression was induced with 1 mM isopropyl β-d-thiogalactopyranoside for 4 h at 30°C. Cells containing overexpressed recombinant proteins were harvested by centrifugation and stored as pellets at −80°C.
Cell pellets were resuspended in lysis buffer [50 mM Tris-HCl (pH 8), 5% (v/v) glycerol, 1 mM ethylenediaminetetraacetic acid (EDTA), 5 mM dithiothreitol (DTT), 1 mM phenylmethanesulfonyl fluoride] supplemented with complete, EDTA-free protease inhibitor cocktail tablets (Roche Applied Science) and lysed using a continuous-flow homogenizer (Avestin). The cell lysate was clarified by centrifugation to remove insoluble debris, and 10% (v/v) PEI (pH 7.9) was added to a final concentration of 0.6% (v/v) to induce the precipitation of nucleic acids and proteins. The mixture was centrifuged, and the pellet was washed twice with TGED buffer [10 mM Tris-HCl (pH 8), 5% (v/v) glycerol, 0.1 mM EDTA, 1 mM DTT] + 0.5M NaCl. RNAP was eluted from the pellet by washing the pellet with TGED buffer + 1M NaCl. Solid ammonium sulfate was added to the eluted sample to a final concentration of 350 g/L, and then the precipitate was pelleted by centrifugation. The pellet was resuspended in IMAC buffer A [20 mM Tris-HCl (pH 8), 5% (v/v) glycerol, 0.5 mM β-mercaptoethanol, 1M NaCl] and applied to a nickel-charged HiTrap column (GE Healthcare) equilibrated in IMAC buffer A. The column was washed with 20 column volumes (cv) of IMAC buffer A + 5 mM imidazole, 5 cv of IMAC buffer A + 20 mM imidazole, 5 cv IMAC buffer A + 40 mM imidazole, 5 cv IMAC buffer A + 60 mM imidazole, 5 cv IMAC buffer A + 80 mM imidazole, and 5 cv IMAC buffer A + 100 mM imidazole. Proteins bound to the column were eluted with IMAC buffer A + 250 mM imidazole. Subsequently, during dialysis against IMAC buffer A, fractions containing in vivo-assembled, recombinant RNAP eluted from the nickel-charged HiTrap column were incubated overnight with PreScission protease (GE Healthcare), a fusion of glutathione S-transferase and the rhinovirus 3C protease, at a ratio of 1:5 (w/w) PreScission protease to RNAP, to enzymatically remove either the (His)10 tag or the αCTD-(His)10. Afterward, a subtractive IMAC chromatographic step removed uncleaved RNAP and, depending on the RNAP preparation, either the cleaved (His)10 tag or the cleaved αCTD-(His)10 fusion protein. The flow through the column, containing RNAP, was dialysed against TGED buffer + 0.1M NaCl and applied to a Bio-Rex 70 column (Bio-Rad) equilibrated in TGED buffer + 0.1M NaCl, and the column was developed with a linear gradient from 0.2 to 1M NaCl. RNAP in the peak fractions was precipitated with ammonium sulfate (added to a final concentration of 350 g/L), and then the precipitate was pelleted by centrifugation. The pellet was resuspended in TGED buffer + 0.5M NaCl to give a final protein concentration of ∼10 mg/mL. The protein sample was applied to a Superdex 200 gel filtration column (GE Healthcare) equilibrated in TGED buffer + 0.5M NaCl, and the peak fractions containing RNAP eluted from the column were dialysed against RNAP storage buffer (TGED + 0.1M NaCl) prior to storage at −80°C.
Recombinant full-length σ70 was purified using a combination of IMAC, anion-exchange, and gel filtration resins, whereas recombinant αCTD was purified as described previously.21
Western blot analysis
Purified preparations of WT RNAP (1 μg), ΔαCTD RNAP (1 μg), and αCTD (1 μg) were resolved on gradient 4–12% (w/v) polyacrylamide Bis-Tris gels (Invitrogen) and transferred to membrane overnight at 4°C. The membrane was stained with Amido black to ensure that protein transfer had been successful and subsequently incubated for 1 h at room temperature in blocking buffer [20 mM Tris-HCl (pH 7.6), 137 mM NaCl, 0.1% (v/v) Tween 20, 5% (w/v) dry milk]. The blocking buffer was decanted, and the membrane was incubated for 1 h at room temperature with the primary antibody, either anti-αNTD or anti-αCTD (Neoclone), diluted 1:1,000 in blocking buffer. The primary antibody was decanted, and the membrane was washed with wash buffer [20 mM Tris-HCl (pH 7.6), 137 mM NaCl, 0.1% (v/v) Tween 20] three times for 5 min each wash. The secondary antibody, horse radish peroxidase conjugated anti-mouse IgG (GE Healthcare), was diluted 1:10,000 in blocking buffer and incubated with the membrane for 45 min at room temperature. After secondary antibody incubation, the membrane was washed with wash buffer (three times for 5 min each wash) and then incubated in SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) for 5 min prior to exposure to photographic film.
In vitro transcription assays
All RNAP holoenzymes were reconstituted from the corresponding core enzyme and purified σ70 in the ratio 1:5 (RNAP:σ70) in holoenzyme storage buffer [10 mM Tris-HCl (pH 8.0), 50% (v/v) glycerol, 0.1 mM EDTA, 1 mM DTT, 0.1M NaCl], as described previously.34 Multiple-round transcription reactions were performed in a final volume of 25 μL and contained either KCl or NaCl (see below). Reconstituted RNAPs were titrated at the lacUV5 promoter: RNAP was incubated with 0.2 nM supercoiled pSRlacUV5 plasmid DNA in reaction buffer [Tris-acetate (pH 7.9), 100 mM KCl, 10 mM MgCl2, 1 mM DTT, 100 μg/mL acetylated BSA] for 15 min at 30°C. The in vitro transcription reactions were initiated by the addition of 200 μM each of CTP, GTP, and ATP, 10 μM of UTP, and 5 μCi of α-32P UTP [800 Ci/mmol (10 mCi/mL)], and incubated for 20 min at 30°C. The activities of recombinant WT and ΔαCTD RNAP preparations were compared with the activity of endogenous WT RNAP (Epicenter; specific activity, 1.4 × 103 U/mg); 9.2 nM of endogenous WT RNAP, 9.2 nM of recombinant WT RNAP, and 20 nM of recombinant ΔαCTD RNAP resulted in equivalent activities at the constitutive lacUV5 promoter, and these concentrations were used in subsequent experiments at the rrnB P1 and CC(−61.5) promoters. Multiple-round transcription from rrnB P1 promoters was performed exactly as described previously 35 in reaction buffer supplemented with 150 mM NaCl. Transcription from the CC(−61.5) promoter was performed in reaction buffer supplanted with 100 mM KCl as described previously 36; except template DNA was preincubated for 5 min with cAMP (0.2 mM), CRP (20 nM), and NTPs. All reactions were terminated by the addition of 25 μL of stop solution [95% (v/v) deionized formamide, 20 mM EDTA, 0.05% (w/v) bromophenol blue, 0.05% (w/v) xylene cyanol]. Samples were electrophoresed in a denaturing 5.5% (w/v) polyacrylamide gel containing 7M urea, and transcript abundance was quantified using a FujiFilm FLA-3000 Phosphorimager.
Acknowledgments
The authors are grateful to Wilma Ross, Rick Gourse, Brian Chait, and Michael Rout for advice and reagents.
References
- 1.Browning DF, Busby SJ. The regulation of bacterial transcription initiation. Nat Rev Microbiol. 2004;2:57–65. doi: 10.1038/nrmicro787. [DOI] [PubMed] [Google Scholar]
- 2.Blatter EE, Ross W, Tang H, Gourse RL, Ebright RH. Domain organization of RNA polymerase alpha subunit: C-terminal 85 amino acids constitute a domain capable of dimerization and DNA binding. Cell. 1994;78:889–896. doi: 10.1016/s0092-8674(94)90682-3. [DOI] [PubMed] [Google Scholar]
- 3.Negishi T, Fujita N, Ishihama A. Structural map of the alpha subunit of Escherichia coli RNA polymerase: structural domains identified by proteolytic cleavage. J Mol Biol. 1995;248:723–728. doi: 10.1006/jmbi.1995.0254. [DOI] [PubMed] [Google Scholar]
- 4.Jeon YH, Yamazaki T, Otomo T, Ishihama A, Kyogoku Y. Flexible linker in the RNA polymerase alpha subunit facilitates the independent motion of the C-terminal activator contact domain. J Mol Biol. 1997;267:953–962. doi: 10.1006/jmbi.1997.0902. [DOI] [PubMed] [Google Scholar]
- 5.Igarashi K, Fujita N, Ishihama A. Identification of a subunit assembly domain in the alpha subunit of Escherichia coli RNA polymerase. J Mol Biol. 1991;218:1–6. [PubMed] [Google Scholar]
- 6.Niu W, Kim Y, Tau G, Heyduk T, Ebright RH. Transcription activation at class II CAP-dependent promoters: two interactions between CAP and RNA polymerase. Cell. 1996;87:1123–1134. doi: 10.1016/s0092-8674(00)81806-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang G, Darst SA. Structure of the Escherichia coli RNA polymerase alpha subunit amino-terminal domain. Science. 1998;281:262–266. doi: 10.1126/science.281.5374.262. [DOI] [PubMed] [Google Scholar]
- 8.Estrem ST, Gaal T, Ross W, Gourse RL. Identification of an UP element consensus sequence for bacterial promoters. Proc Natl Acad Sci USA. 1998;95:9761–9766. doi: 10.1073/pnas.95.17.9761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Estrem ST, Ross W, Gaal T, Chen ZW, Niu W, Ebright RH, Gourse RL. Bacterial promoter architecture: subsite structure of UP elements and interactions with the carboxy-terminal domain of the RNA polymerase alpha subunit. Genes Dev. 1999;13:2134–2147. doi: 10.1101/gad.13.16.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Busby S, Ebright RH. Transcription activation by catabolite activator protein (CAP) J Mol Biol. 1999;293:199–213. doi: 10.1006/jmbi.1999.3161. [DOI] [PubMed] [Google Scholar]
- 11.Savery NJ, Lloyd GS, Busby SJ, Thomas MS, Ebright RH, Gourse RL. Determinants of the C-terminal domain of the Escherichia coli RNA polymerase alpha subunit important for transcription at class I cyclic AMP receptor protein-dependent promoters. J Bacteriol. 2002;184:2273–2280. doi: 10.1128/JB.184.8.2273-2280.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ross W, Gosink KK, Salomon J, Igarashi K, Zou C, Ishihama A, Severinov K, Gourse RL. A third recognition element in bacterial promoters: DNA-binding by the alpha subunit of RNA polymerase. Science. 1993;262:1407–1413. doi: 10.1126/science.8248780. [DOI] [PubMed] [Google Scholar]
- 13.Ross W, Aiyar SE, Salomon J, Gourse RL. Escherichia coli promoters with UP elements of different strengths: modular structure of bacterial promoters. J Bacteriol. 1998;180:5375–5383. doi: 10.1128/jb.180.20.5375-5383.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naryshkin N, Revyakin A, Kim Y, Mekler V, Ebright RH. Structural organization of the RNA polymerase-promoter open complex. Cell. 2000;101:601–611. doi: 10.1016/s0092-8674(00)80872-7. [DOI] [PubMed] [Google Scholar]
- 15.Igarashi K, Ishihama A. Bipartite functional map of the E. coli RNA polymerase alpha subunit: involvement of the C-terminal region in transcription activation by cAMP-CRP. Cell. 1991;65:1015–1022. doi: 10.1016/0092-8674(91)90553-b. [DOI] [PubMed] [Google Scholar]
- 16.Tang H, Severinov K, Goldfarb A, Ebright RH. Rapid RNA polymerase genetics: one-day, no-column preparation of reconstituted recombinant Escherichia coli RNA polymerase. Proc Natl Acad Sci USA. 1995;92:4902–4906. doi: 10.1073/pnas.92.11.4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishihama A, Fukuda R, Ito K. Subunits of RNA polymerase in function and structure. IV. Enhancing role of sigma in the subunit assembly of Escherichia coli RNA polymerase. J Mol Biol. 1973;79:127–136. doi: 10.1016/0022-2836(73)90274-x. [DOI] [PubMed] [Google Scholar]
- 18.Fukuda R, Ishihama A. Subunits of RNA polymerase in function and structure; maturation in vitro of core enzyme from Escherichia coli. J Mol Biol. 1974;87:523–540. doi: 10.1016/0022-2836(74)90102-8. [DOI] [PubMed] [Google Scholar]
- 19.Zillig W. On the role of RNA polymerase subunits in RNA synthesis and the gross regulation of transcription by specific modifications of RNA polymerase subunits. In: Hidvegi EJ, Sumegi J, Venetianer P, editors. Biochemistry of the cell nucleus. Mechanism and regulation of gene expression. Amsterdam: North-Holland; 1974. pp. 239–251. [Google Scholar]
- 20.Artsimovitch I, Svetlov V, Murakami KS, Landick R. Co-expression of Escherichia coli RNA polymerase subunits allows isolation and analysis of mutant enzymes lacking lineage-specific sequence insertions. J Biol Chem. 2003;278:12344–12355. doi: 10.1074/jbc.M211214200. [DOI] [PubMed] [Google Scholar]
- 21.Gaal T, Ross W, Blatter EE, Tang H, Jia X, Krishnan VV, Assa-Munt N, Ebright RH, Gourse RL. DNA-binding determinants of the alpha subunit of RNA polymerase: a novel DNA-binding domain architecture. Genes Dev. 1996;10:16–26. doi: 10.1101/gad.10.1.16. [DOI] [PubMed] [Google Scholar]
- 22.Cordingley MG, Callahan PL, Sardana VV, Garsky VM, Colonno RJ. Substrate requirements of human 3C protease for peptide cleavage in vitro. J Biol Chem. 1990;265:9062–9065. [PubMed] [Google Scholar]
- 23.Walker PA, Leong LE, Ng PW, Tan SH, Waller S, Murphy D, Porter AG. Efficient and rapid affinity purification of proteins using recombinant fusion proteases. Biotechnology (NY) 1994;12:601–605. doi: 10.1038/nbt0694-601. [DOI] [PubMed] [Google Scholar]
- 24.Studier FW, Moffatt BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 25.Husnain SI, Meng W, Busby SJ, Thomas MS. Escherichia coli can tolerate insertions of up to 16 amino acids in the RNA polymerase subunit inter-domain linker. Biochim Biophys Acta. 2004;1678:47–56. doi: 10.1016/j.bbaexp.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 26.Burgess RR, Jendrisak JJ. A procedure for the rapid, large-scale purification of Escherichia coli DNA-dependent RNA polymerase involving Polymin P precipitation and DNA-cellulose chromatography. Biochemistry. 1975;14:4634–4638. doi: 10.1021/bi00692a011. [DOI] [PubMed] [Google Scholar]
- 27.Murakami KS, Masuda S, Darst SA. Crystallographic analysis of Thermus aquaticus RNA polymerase holoenzyme and a holoenzyme/promoter DNA complex. Methods Enzymol. 2003;370:42–53. doi: 10.1016/S0076-6879(03)70004-4. [DOI] [PubMed] [Google Scholar]
- 28.Fong BA, Gillies AR, Ghazi I, LeRoy G, Lee KC, Westblade LF, Wood DW. Purification of Escherichia coli RNA polymerase using a self-cleaving elastin-like polypeptide tag. Protein Sci. 2010;19:1243–1252. doi: 10.1002/pro.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porath J, Carlsson J, Olsson I, Belfrage G. Metal chelate affinity chromatography, a new approach to protein fractionation. Nature. 1975;258:598–599. doi: 10.1038/258598a0. [DOI] [PubMed] [Google Scholar]
- 30.Gaston K, Bell A, Kolb A, Buc H, Busby S. Stringent spacing requirements for transcription activation by CRP. Cell. 1990;62:733–743. doi: 10.1016/0092-8674(90)90118-x. [DOI] [PubMed] [Google Scholar]
- 31.Feklistov A, Mekler V, Jiang Q, Westblade LF, Irschik H, Jansen R, Mustaev A, Darst SA, Ebright RH. Rifamycins do not function by allosteric modulation of the binding of Mg2+ to the RNA polymerase active center. Proc Natl Acad Sci USA. 2008;105:14820–14825. doi: 10.1073/pnas.0802822105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burke E, Barik S. Megaprimer PCR: application in mutagenesis and gene fusion. Methods Mol Biol. 2003;226:525–532. doi: 10.1385/1-59259-384-4:525. [DOI] [PubMed] [Google Scholar]
- 33.Wegrzyn G, Glass RE, Thomas MS. Involvement of the Escherichia coli RNA polymerase alpha subunit in transcriptional activation by the bacteriophage lambda CI and CII proteins. Gene. 1992;122:1–7. doi: 10.1016/0378-1119(92)90025-k. [DOI] [PubMed] [Google Scholar]
- 34.Colland F, Barth M, Hengge-Aronis R, Kolb A. Sigma factor selectivity of Escherichia coli RNA polymerase: role for CRP, IHF and lrp transcription factors. EMBO J. 2000;19:3028–3037. doi: 10.1093/emboj/19.12.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meng W, Belyaeva T, Savery NJ, Busby SJ, Ross WE, Gaal T, Gourse RL, Thomas MS. UP element-dependent transcription at the Escherichia coli rrnB P1 promoter: positional requirements and role of the RNA polymerase alpha subunit linker. Nucleic Acids Res. 2001;29:4166–4178. doi: 10.1093/nar/29.20.4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Husnain SI, Busby SJ, Thomas MS. Downregulation of the Escherichia coli guaB promoter by upstream-bound cyclic AMP receptor protein. J Bacteriol. 2009;191:6094–6104. doi: 10.1128/JB.00672-09. [DOI] [PMC free article] [PubMed] [Google Scholar]